

Abstract
We report a direct C–H aminoalkylation reaction using two light-activated H-atom transfer catalyst systems that enable the introduction of protected amines to native adamantane scaffolds with C–C bond formation. The scope of adamantane and imine reaction partners is broad and deprotection provides versatile amine and amino acid building blocks. Using readily available chiral imines, the enantioselective synthesis of the saxagliptin core and rimantadine derivatives is also described.
Chiral amines are an important class of molecules found in bioactive natural products and pharmaceuticals, making them desirable synthetic targets. Many methods have been developed for their synthesis from classical approaches (e.g. Strecker, Ugi reactions1 to modern stereoselective catalytic strategies.2 Aminoalkylation reactions that simultaneously introduce a C–C bond and amine are particularly powerful (Figure 1A). Three main strategies for radical-mediated aminoalkylation are commonly used:3 addition of an alkyl radical to an imine derivative (disconnection i),4,5,6 radical conjugate addition into an ɑ,β-dehydroamino acid (discon-nection ii),7 or the addition of a captodative ɑ-amino radical to a radical acceptor.8 The use of imine derivatives as radical acceptors has grown in recent years and typically involves the use an alkyl halide in conjunction with a radical initiator. Improvements on traditional initiators (e.g. Et3B/O2, Bu3SnH/AIBN, etc.) with their associated drawbacks have been reported, such as photoredox activation of organosilicon and organoboron compounds by Molander, Gong and Friestad.9 The importance of chiral amines highlights an ongoing need for efficient, stereoselective methods for their synthesis.
Figure 1.

Bioactive aminoadamantanes and synthetic approaches for the preparation of aminoalkylated molecules.
The direct aminoalkylation of hydrocarbons is an attractive but challenging method for direct C–H functionalization. The high reactivity required to perform H-atom transfer (HAT) of unactivated C–H bonds often leads to incompatibility with redox-sensitive functional groups and site selectivity issues on complex substrates.10 As a result, most examples have focused on more reactive substrates such as ethers with activated α-heteroatom-C–H bonds that can be selectively targeted.11. The C–H functionalization of diamondoids is challenging due to unusually high bond dissociation energies (BDEs) of these hydrocarbons (96 and 99 kcal/mol for 2º and 3º C–H bonds, respectively) and potential regioselectivity issues.12 The amino-alkyl derivatives typified by rimantadine (2, Figure 1B) are especially prevalent in a variety of anti-virals,13 HDAC inhibitors (martinostat)14 and anti-diabetic agents (saxagliptin 3),15a high-lighting the utility of this pharmacophore.15b A direct amino-alkylation of diamondoids would provide an ideal method for their synthesis, which often relies on the reductive amination of acyl-derivatives such as 4, as shown in Figure 1C.13a The adamantyl-glycine core of saxagliptin (3) has been synthesized using asymmetric reductive amination or Strecker approaches.15 This strategy necessitates pre-functionalization of adamantane and complicates the introduction of additional substituents on the adamantane core. Herein, we describe a photocatalytic method enabling the direct aminoalkylation of adamantanes without these drawbacks (Figure 1D).
Key to the efficient substitution of adamantanes is an HAT step that can overcome the strong C–H BDEs while maintaining high chemo- and regioselectivity. We recently reported a method for the alkylation of diamondoids with alkenes using a photoredox/HAT dual catalytic system.16 This alkylation reaction displays unusual selectivity for the 3º C–H bonds of adamantanes over weaker C–H bonds. Using the combination of an oxidizing photocatalyst (Ir(dF(CF3)ppy)2(d(CF3)bpy)PF6, Ir-1) with an electron-deficient quinuclidine catalyst (quinuclidin-3-yl benzenesulfonate, Q-1),†16 we explored the reaction of adamantane with various imine derivatives under blue-light irradiation (Table 1).17,18 We found that Ts-imines and Boc-imines were efficient coupling partners, giving the aminoalkylated products 9 and 10 in 75% and 72% yield, respectively. Hydrazones were also competent radical acceptors in this reaction, however with lower efficiency (12 and 14, 22–37% yield). Substitution of the phenyl ring with an electron-withdrawing substituent such as p-F and p-CN led to higher yields, as shown for N-Boc 11 (80% yield) and N-Bz-hydrazide 13 (78% yield), consistent with a LUMO-lowering effect and the nucleophilic character of the adamantyl radical.19 A Ns-protected imine was less efficient due to competing decomposition pathways (see ESI†). Other imines such as a glyoxalate derivative and a cyclic, trisubstituted sulfonylimine were excellent partners, giving esters 15 and 16 in 67% and 83% yield, respectively. Sulfonamide product 16 has adjacent fully substituted carbons, highlighting the power of radical addition reactions to generate congested C–C bonds.
Table 1.
Survey of imine and hydrazone derivatives in aminoalkylation of adamantane.
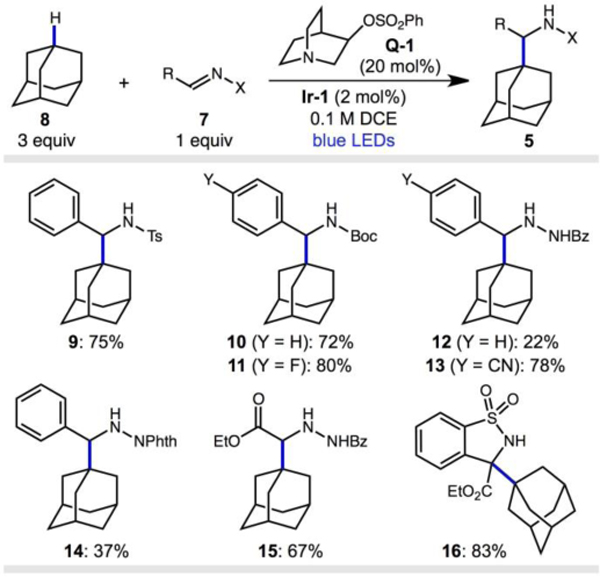 |
We next explored the scope of imine and adamantane partners. As shown in Table 2, both electron-deficient and electron-rich Ts-imines are coupled with moderate to high yield. A pyridine substituent was tolerated with some substrate degradation observed (22, 48% yield). Electron-rich adamantanes were high yielding while electron-withdrawn adamantanes with a chloro and NHBoc substituent were incor-porated with lower efficiency. The scope of hydrazide products was broad, provided an electron-withdrawing substituent was present on the aryl group. Both ortho- and para-substitution was tolerated. The highest yields were achieved with electron-rich adamantanes and yields were diminished for acetyl- and hydroxyadamantane substrates (compare 34 and 35, 28 and 36). In general, the reaction rate was slower for hydrazone substrates. The scope of cyclic, alpha-3º amine products was also broad. Electron-rich and moderately electron-deficient adamantanes led to the highest yields for 37–39 (74–80% yield) while acetyl- and cyano-substituents showed more significant decreases (66% for 40, 40% for 41).
Table 2.
Scope of imine and hydrazone derivatives in aminoalkylation of adamantanes.
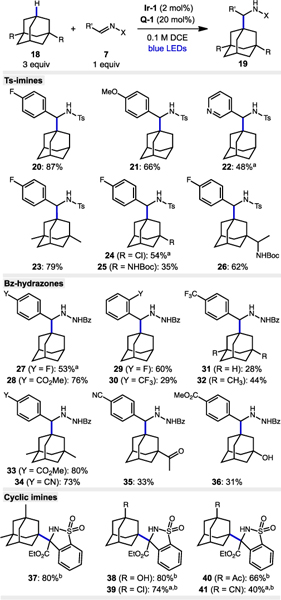 |
Notes. Reactions performed on a 0.5 mmol scale using 2 × 40W 456nm lamps over 24–72 h. All yields are isolated yields.
Solvent is CH3CN.
2 equiv H2O added.
We then turned to the synthesis of enantioenriched ɑ-adamantyl amines using our dual catalytic system in conjunction with chiral N-sulfinyl imines. The use of N-sulfinyl imines as chiral auxiliaries is a well-established strategy for the asymmetric synthesis of ɑ-branched amines and ɑ-amino acids.20 There are fewer but nonetheless important examples of stereocontrolled radical additions.4a,b,21 Unfortunately, we found that using our Ir-1/Q-1 system with a N-tolylsulfinimine 43 (R” = p-Tol) resulted in poor conversion and some decom-position, therefore we looked for possible alternative catalysts. Kamijo and coworkers have shown that using 5,7,12,14-pentacenetetraone (PT) as a photocatalyst provides a viable method for generating the desired 1-adamantyl radical 6.22,6e
Gratifyingly, we found that the use of PT with 390 nm LEDs gave benzylic N-tolylsulfinamide 45 in good yield with mode-rate stereoselectivity (9:1 d.r., Table 3). A glyoxalate-derived imine also proceeded with 9:1 d.r., albeit in lower yield (47, 41% yield). Only marginal asymmetric induction was observed using the Ellman N-tert-butylsulfinyl auxiliary (48, 1.3:1 d.r.). The stereocontrol was significantly improved by switching to N-mesitylsulfinimines, providing 46 and 49 in good yields and >20:1 d.r.. Notably, use of N-mesitylsulfinimines provides a direct route to enantiopure adamantyl glycine precursor 49 and the 3-hydroxyadmantyl glycine core of saxagliptin 50 in a direct and highly selective manner, enabling further exploration of these unnatural building blocks.15
Table 3.
Scope of chiral imines acceptors for preparation of enantioenriched amines.
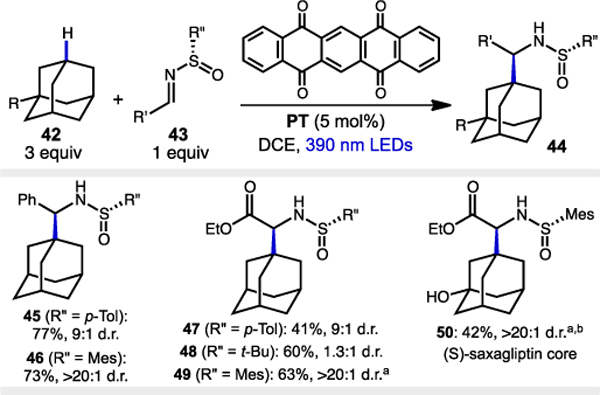 |
Notes. Reactions performed on a 0.3 mmol scale using 2 × 40W 390nm lamps over 24–48 h. Reaction temperature is approx. 34 °C. All yields are isolated yields. d.r. values obtained via 1H NMR analysis of crude product mixture (see ESI† for details).
Reaction performed on a 0.5 mmol scale.
Solvent is 1:1 DCE:PhCl.
Next, we explored the performance of the aminoalkylation method with cyclohexane and tetrahydrofuran (Scheme S2†). Using the PT system, cyclohexane reacted with an N-tosyl imine in 31% yield. Tetrahydrofuran was selectively amino-alkylated at the α-oxy-C–H bond using Ir-1/Q-1 in 50% yield. While the yields of these reactions are lower than related transformations by Gong6e and Dilman’s recent report6d with a decatungstate HAT catalyst, we have found that TBADT is ineffective for activating adamantanes in this reaction (see ESI† for details). As a result, the catalyst systems described here provide a complementary substrate scope. Efforts to ge-neralize this reaction manifold are ongoing in our laboratory.
Finally, we moved to the deprotection of representative reaction products to provide the free amines (Scheme 1). The use of samarium diiodide allowed efficient cleavage of the N–NHBz bond of 27 to afford free amine 51 (79% yield). Similarly, deprotection of the tosyl group was very efficient (92% yield). Cleavage of the sulfinyl group in sulfinamide 49 proceeded smoothly under acidic conditions to afford the corresponding adamantylglycine ethyl ester 52 in 53% yield.
Scheme 1.

Deprotection reactions with SmI2 and TFA to afford free amines.
Based on related mechanistic work, we propose that this transformation parallels the previously reported alkylation reaction via either a direct HAT process for PT (Scheme 2) or an indirect HAT process for Ir-1/Q-1 (Scheme S1†).16,11b For the direct HAT process, excitation of PT with light generates an excited state capable of H-atom abstraction to give radical 6.22 Addition of the radical to the imine or hydrazone gives an N-centered radical 53. Turnover would either proceed via HAT from semiquinone PT-H to aminyl radical 53 or single electron reduction by PT-H followed by proton transfer. For the indirect process, excitation of the photocatalyst Ir-1 followed by oxidation of quinuclidine Q-1 yields the corresponding radical cation which can undergo HAT to give the adamantyl radical 6.16,17 Addition of the radical gives aminyl radical 53, which can be reduced by Ir(II) to the corresponding anion. Proton transfer from the quinuclidinium ion gives the final product 5 and closes the catalytic cycle. While we cannot rule out an alternative mechanism proceeding via reduction of the imine or hydrazone to a radical anion at this time, the similar efficiency observed for a variety of N-substituents (Tables 1 and 3) with a range of reduction potentials is less consistent with literature examples that proceed via this mechanism.6e,24
Scheme 2.

Mechanism of PT-catalyzed aminoalkylation reaction.
In conclusion, we have described a direct aminoalkylation reaction promoted by selective hydrogen atom abstraction. A dual catalytic system consisting of an Ir-photocatalyst and quinuclidine co-catalyst enables the efficient coupling of diverse imines, hydrazones and adamantane coupling partners with high chemoselectivity. In addition, a quinone catalyst PT provides high yields for the enantioselective synthesis of aminoalkylated derivatives of known bioactive molecules and unnatural amino acids. The catalyst systems described tolerate other substrate classes beyond adamantanes, albeit in lower yield, providing a complementary method to other HAT catalysts. Applications to the synthesis of aminoalkylated analogs of the antiviral rimantadine pharmacophore and amino acid building blocks are under active investigation in our laboratory and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by generous start-up funds from UC Riverside and the University of Iowa. NMR instrumentation was supported by funding from the NSF (CHE-1626673, UCR) and the U.S. Army (W911NF-16-1-0523, UCR) and from the NIH (S10-RR025500, UI). HRMS was supported by funding from the NSF (CHE-0541848, UCR; CHE-0946779, UI).
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, optimization studies and analytical data of the products. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.(a) Duthaler RO, Tetrahedron, 1994, 50, 1539–1650; [Google Scholar]; (b) Kobayashi S. and Ishitani H, Chem. Rev, 1999, 99, 1069–1094; [DOI] [PubMed] [Google Scholar]; (c) Friestad GK and Mathies AK, Tetrahedron, 2007, 63, 2541–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Nájera C. and Sansano JM, Chem. Rev, 2007, 107, 4584–4671; [DOI] [PubMed] [Google Scholar]; (b) Nugent TC and El-Shazly M, Adv. Synth. Catal, 2010, 352, 753–819; [Google Scholar]; (c) Li W, Zhang X, Topics In Current Chemistry: Stereoselective Formation of Amines, Springer-Verlag, Berlin, 2014. [Google Scholar]
- 3.For a detailed overview of these strategies see:Deska J, in Amino Acids, Peptides and Proteins in Organic Chemistry, Ed. Hughes AB, Wiley-VCH: Weinheim, 2011, Vol. 3, pp. 115–141.
- 4.For reviews, see:Friestad GK, Tetrahedron, 2001, 57, 5461–5496;Friestad GK, Top. Curr. Chem, 2014, 343, 1–32;
- 5.(a) Miyabe H, Ueda M. and Naito T, Chem. Commun, 2000, 2059–2060; [Google Scholar]; (b) Deng G. and Li C-J, Tetrahedron Lett, 2008, 49, 5601–5604; [Google Scholar]; (c) Yamada K. and Tomioka K, Chem. Rec, 2015, 15, 854–871; [DOI] [PubMed] [Google Scholar]; (d) Ueda M, Miyabe H, Miyata O. and Naito T, Tetrahedron, 2009, 65, 1321–1326. [Google Scholar]
- 6.For light-promoted examples, see:Zhang L, Deng Y. and Shi F, Tetrahedron Lett., 2013, 54, 5217–5219;Lahm G. and Opatz T, J. Org. Chem, 2015, 80, 12711–12717;Yang S, Zhu S, Lu D. and Gong Y, Org. Lett, 2019, 21, 2019–2024;Supranovich VI, Levin VV, and Dilman AD, Org. Lett, 2019, 21, 4271–4274;Li Y, Lei M. and Gong L, Nat. Catal, 2019, 2, 1016–1026.
- 7.(a) Axon JR and Beckwith J ALJ. Chem. Soc., Chem Commun, 1995, 549–550; [Google Scholar]; (b) Aycock RA, Vogt DB, and Jui NT, Chem. Sci, 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Nakajima K, Miyake Y. and Nishibayashi Y, Acc. Chem. Res, 2016, 49, 1946–1956; [DOI] [PubMed] [Google Scholar]; (b) Rossolini T, Leitch JA, Grainger R, and Dixon DJ, Org. Lett, 2018, 20, 6794–6798. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zheng S, Primer DN and Molander GA, ACS Catal., 2017, 7, 7957–7961; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li Y, Zhou K, Wen Z, Cao S, Shen X, Lei M. and Gong L., J. Am. Chem. Soc, 2018, 140, 15850–15858; [DOI] [PubMed] [Google Scholar]; (c) Han B, Li Y, Yu Y. and Gong L, Nat. Commun, 2019, 10, 3804; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cullen STJ and Friestad GK, Org. Lett, 2019, 21, 8290–8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Newhouse T. and Baran PS, Angew. Chem. Int. Ed, 2011, 50, 3362–3374; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutekunst WR and Baran PS, Chem. Soc. Rev, 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hydrogen-Transfer Reactions, Eds. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, Wiley-VCH, Weinheim, 2007. [Google Scholar]; (b) Capaldo L, and Ravelli D, Eur. J. Org. Chem, 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng H, Yu J-T, Jiang Y, Yang H, Cheng J, J. Org. Chem 2014, 79, 9847–9853. [DOI] [PubMed] [Google Scholar]; (d) Zhao H, Li Z, Jin J, New J. Chem 2019, 43, 12533–12537. [Google Scholar]; (e) Sun M-X, Wang Y-F, Xu B-H, Xu X-Q, Zhang S-J, Org. Biomol. Chem 2018, 16, 1971–1975. [DOI] [PubMed] [Google Scholar]
- 12.(a) Fokin AA and Schreiner PR, Chem. Rev, 2002, 102, 1551–1594; [DOI] [PubMed] [Google Scholar]; (b) Bagrii EI, Nekhaev AI and Maksimov AL, Pet. Chem, 2017, 57, 183–197; [Google Scholar]; (c) Kruppa GH and Beauchamp JL, J. Am. Chem. Soc, 1986, 108, 2162–2169. [DOI] [PubMed] [Google Scholar]
- 13.(a) Grava IY, Polis YY, Lidak MY, Indulen MK and Zamyatina NA, Pharm. Chem. J, 1980, 33, 372–376; [Google Scholar]; (b) Balzarini J, Orzeszko-Krzesińska B, Maurin JK and Orzeszko A, Eur. J. Med. Chem, 2009, 44, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cincinelli R, Musso L, Giannini G, Zuco V, De Cesare M, Zunino F. and Dallavalle S, Eur. J. Med. Chem, 2014, 79, 251–259. [DOI] [PubMed] [Google Scholar]
- 15.(a) Villhauer EB, Brinkman JA, Naderi GB, Burkey BF, Dunning BE, Prasad K, Mangold BL, Russell ME and Hughes TE, J. Med. Chem, 2003, 46, 2774–2789. [DOI] [PubMed] [Google Scholar]; (b) Hanson RL, Goldberg SL, Brzozowski DR, Tully TP, Cazzulino D, Parker WL, Lungberg OK, Vu TC, Wong MK and Patel RN, Adv. Synth. Catal, 2007, 349, 1369–1378; [Google Scholar]; (c) US Pat., 20130023671A1, 2013.
- 16.Wanka L, Iqbal K. and Schreiner PR, Chem. Rev, 2013, 113, 3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H-B, Feceu A. and Martin DBC, ACS Catal., 2019, 9, 5708–5715. [Google Scholar]
- 18.For selected examples, see:Jeffrey JL, Terrett JA and MacMillan DWC, Science, 2015, 349, 1532–1536;Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD and MacMillan DWC, Science, 2016, 352, 1304–1308;Kawamata Y, Yan M, Liu Z, Bao D-H, Chen J, Starr JT and Baran PS, J. Am. Chem. Soc, 2017, 139, 7448–7451;Ye J, Kalvet I, Schoenebeck F, Rovis T, Nat. Chem, 2018, 10, 1037–1041;Dimakos V, Su HY, Garrett GE and Taylor MS, J. Am. Chem. Soc, 2019, 141, 5149–5153.
- 19. For dual catalysis reviews, see:Skubi KL, Blum TR and Yoon TP, Chem. Rev, 2016, 116, 10035–10074;Shaw MH, Twilton J. and MacMillan DWC, J. Org. Chem, 2016, 81, 6898–6926.
- 20.Recupero F, Bravo A, Byorsvik H-R, Fontana F, Minisci F, and Piredda M, J. Chem. Soc., Perkin Trans 2, 1997, 2399–2406. [Google Scholar]
- 21.(a) Davis FA and Chen B-C, Chem. Soc. Rev, 1998, 27, 13–18; [Google Scholar]; (b) Ellman JA, Owens TD and Tang TP, Acc. Chem. Res, 2002, 35, 984–995; [DOI] [PubMed] [Google Scholar]; (c) Zhou P, Chen B-C and Davis FA, Tetrahedron, 2004, 60, 8003–8030; [Google Scholar]; (d) Robak MT, Herbage MA and Ellman JA, Chem. Rev, 2010, 110, 3600–3740. [DOI] [PubMed] [Google Scholar]
- 22.(a) Akindele T, Yamada K, Sejima T, Maekawa M, Yamamoto Y, Nakano M. and Tomioka K, Chem. Pharm. Bull, 2010, 58, 265–269; [DOI] [PubMed] [Google Scholar]; (b) Huang W, Ye J-L, Zheng W, Dong H-Q and Wei B-G, J. Org. Chem, 2013, 78, 11229–11237; [DOI] [PubMed] [Google Scholar]; (c) Rochette EM, Lewis W, Dossetter AG and Stockman RA, Chem. Commun, 2013, 49, 9395–9397; [DOI] [PubMed] [Google Scholar]; (d) Garrido-Castro AF, Choubane H, Daaou M, Carmen Maestro M.and Alemán J, Chem. Commun, 2017, 53, 7764–7767; [DOI] [PubMed] [Google Scholar]; (e) Ni S, Garrido‐Castro AF, Merchant RR, de Gruyter JN, D. C. Schmitt, Mousseau JJ, Gallego GM, Yang S, Collins MR, Qiao JX, Yeung K-S, Langley DR, Poss MA, Scola PM, Qin T. and Baran PS, Angew. Chem. Int. Ed, 2018, 57, 14560–14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamijo S, Kamijo K, Maruoka K. and Murafuji T, Org. Lett, 2016, 18, 6516–6519. [DOI] [PubMed] [Google Scholar]
- 24.(a) Hager D, MacMillan DWC, J. Am. Chem. Soc, 2014, 136, 16986–16989; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jeffrey J, Petronijevic FR, MacMillan DWC, J. Am. Chem. Soc, 2015, 137, 8404–8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.