

Abstract
Estrogen receptor-positive (ER+) breast cancer can recur up to 20 years after initial diagnosis. Delayed recurrences arise from disseminated tumors cells (DTCs) in sites such as bone marrow that remain quiescent during endocrine therapy and subsequently proliferate to produce clinically-detectable metastases. Identifying therapies that eliminate DTCs and/or effectively target cells transitioning to proliferation promises to reduce risk of recurrence. To tackle this problem, we utilized a 3D co-culture model incorporating ER+ breast cancer cells and bone marrow mesenchymal stem cells to represent DTCs in a bone marrow niche. 3D co-cultures maintained cancer cells in a quiescent, viable state as measured by both single-cell and population-scale imaging. Single-cell imaging methods for metabolism by fluorescence lifetime (FLIM) of NADH and signaling by kinases Akt and ERK revealed that breast cancer cells utilized oxidative phosphorylation and signaling by Akt to a greater extent both in 3D co-cultures and a mouse model of ER+ breast cancer cells in bone marrow. Using our 3D co-culture model, we discovered that combination therapies targeting oxidative phosphorylation via the thioredoxin reductase (TrxR) inhibitor, D9, and the Akt inhibitor, MK-2206, preferentially eliminated breast cancer cells without altering viability of bone marrow stromal cells. Treatment of mice with disseminated ER+ human breast cancer showed that D9 plus MK-2206 blocked formation of new metastases more effectively than tamoxifen. These data establish an integrated experimental system to investigate DTCs in bone marrow and identify combination therapy against metabolic and kinase targets as a promising approach to effectively target these cells and reduce risk of recurrence in breast cancer.
Introduction
Delayed recurrence of estrogen receptor-positive (ER+) breast cancer, the predominant clinical subtype, poses an ongoing threat to patients as risks of recurrence and death steadily increase between 5 and 20 years post-diagnosis (1). Recurrent disease arises from disseminated tumor cells (DTCs) that are suppressed but not eliminated by standard endocrine therapy (1–3). DTCs typically persist with a non- or minimally proliferative, cancer stem cell-like phenotype that promotes drug resistance and long-term survival while retaining the capacity to resume proliferation to form clinically detectable metastases even years later (4, 5). Bone marrow is the leading site for metastases in ER+ disease and only clinically-accessible site to detect DTCs (6). Patients presenting with bone marrow DTCs have significantly lower overall, breast-cancer-specific, disease-free, and distant metastasis-free survival probabilities than patients without DTCs (1, 3).
Although rich in growth factors, bone marrow presents a hostile microenvironment for DTCs due to relative hypoxia and shifting nutrient supplies (7). DTCs must exhibit metabolic plasticity between glycolysis and oxidative phosphorylation (OXPHOS) to survive in the bone marrow environment. Targeting metabolic dependencies of DTCs presents opportunities for novel therapies to eliminate these cells and prevent recurrent cancer (8). In addition to metabolism, cancer cells also reprogram tumorigenic pathways by activating kinase signaling pathways (9). Two of the most commonly activated signaling pathways in cancer are Raf/MEK/ERK and PI3K/Akt/mTOR, which drive many cellular processes including proliferation, survival, and drug resistance (10, 11). Although inhibiting these key oncogenic signaling pathways shows promise across multiple types of cancer, resistance commonly emerges to molecularly-targeted drugs. Exploiting metabolic dependencies of DTCs in bone marrow in combination with kinase inhibitors presents a potential treatment strategy to limit drug resistance and eliminate these malignant cells.
Discovery of possible treatments against bone marrow DTCs requires a model system that recapitulates quiescence of DTCs, the bone marrow microenvironment, and heterogeneous drug and nutrient profiles (12, 13). Incorporating bone marrow stromal cells into a multicellular spheroid with cancer cells presents an opportunity to study DTCs in a physiologically relevant environment known to regulate tumor dormancy (14). In this study, we applied our previously described system for modeling bone marrow DTCs (15) to focus on interactions between ER+ breast cancer and bone marrow stromal cells. This model represents quiescent ER+ breast cancer cells in a bone marrow microenvironment using bone marrow mesenchymal stromal cells (MSCs) as a surrogate for normal bone marrow stroma. We used single-cell imaging techniques to quantify differences in metabolic reliance on OXPHOS versus glycolysis in parallel with activation of ERK and Akt between cancer and stromal cells. Next, we simultaneously quantified viability and drug targeting of cancer and stromal cell populations in response to compounds that capitalized on reliance of breast cancer cells on OXPHOS and Akt signaling to selectively target cancer cells. Lastly, we verified our novel combinatory treatment of OXPHOS and Akt inhibitors in a mouse model of ER+ breast cancer bone metastasis. These results suggest a new treatment paradigm against DTCs in ER+ breast cancer with potential to reduce delayed recurrences.
Results
Co-culture Spheroids Promote Quiescence of ER+ Breast Cancer Cells
To model bone marrow DTCs, we used co-cultures of ER+ breast cancer cells and MSCs to model how intercellular interactions between these cells regulate quiescence and drug resistance in bone marrow. We co-cultured ER+ breast cancer cell lines MCF7 and T47D with two bone marrow MSC lines, HS5 and HS27a (16), first in 2D and then in spheroids. These bone marrow stromal cell lines act as surrogates for normal cells in a bone marrow microenvironment. We cultured stromal cells with 5% cancer cells to model low numbers of DTCs in bone marrow. We further refined these systems with physiologically relevant low glucose (4 mM) and low FBS (1%) media conditions. For both model systems, we measured relative numbers of viable cells with bioluminescence imaging for click beetle green and red luciferases stably expressed in cancer and stromal cells, respectively. Separating emitted green and red light with optical filters allowed us to independently measure cancer and stromal cell viabilities in co-cultures. We also determined cell cycle status of cancer cells with a fluorescent ubiquitination-based cell cycle indicator (FUCCI) system (17) or propidium iodide (PI) staining (18).
In 2D co-culture conditions, cancer cells exhibited a 1.1 – 2.2-fold growth increase between days one and three based on cellular bioluminescence (Fig 1A). We confirmed this result with FUCCI imaging for cell cycle analysis of breast cancer cell lines (Fig 1B). We detected between 20–48% of breast cancer cells undergoing cell division in the 2D co-culture model (Fig 1C), verifying that 2D cultures with MSCs did not arrest proliferation of breast cancer cells. Furthermore, we verified that both stromal cell lines remained in viable, non-growth-arrested states in 2D culture by bioluminescence, FUCCI, and PI stain analyses (Fig 1 A, B, C; Fig S1).
Figure 1: ER+ breast cancer cells in co-culture spheroids with bone marrow stromal cells exhibit cellular quiescence.
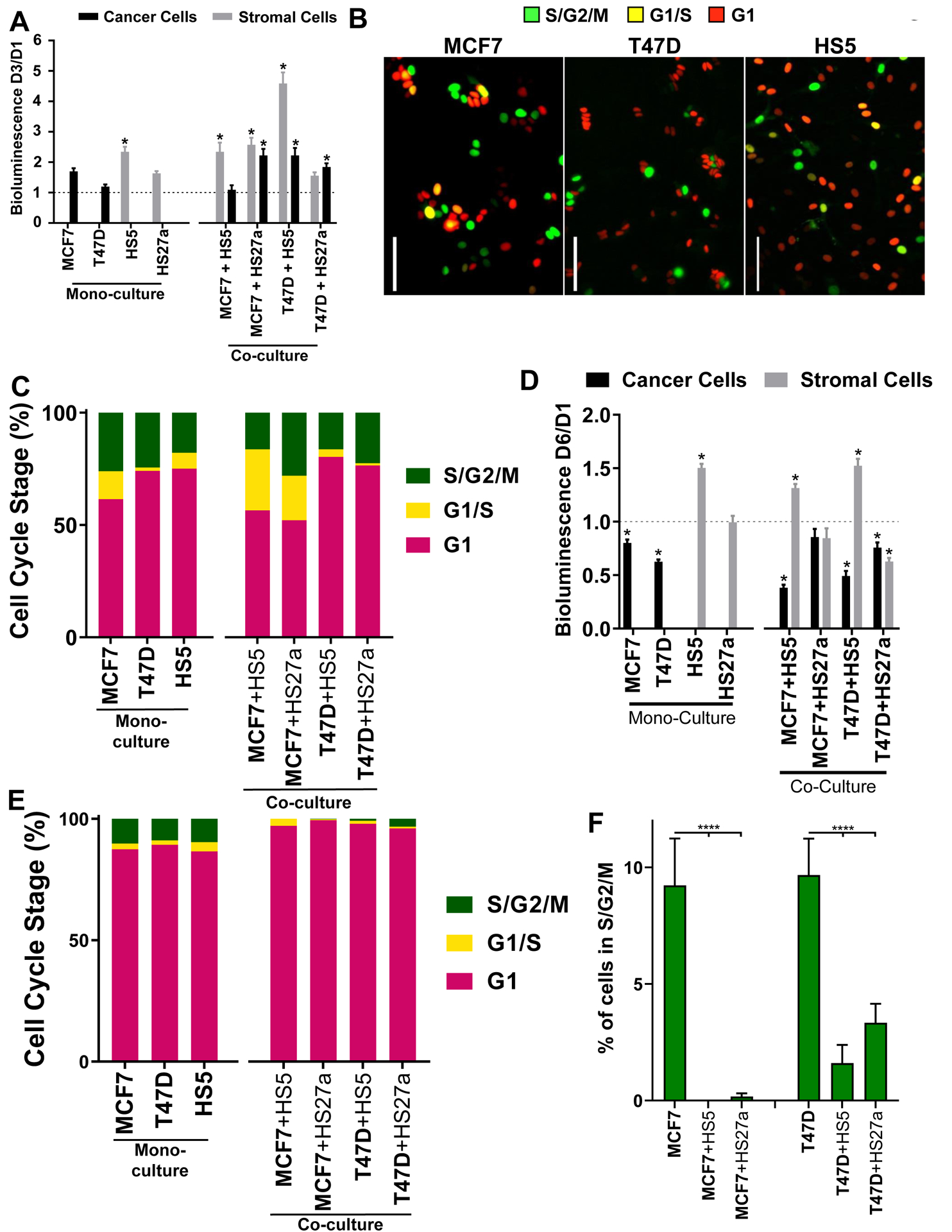
A. color cellular bioluminescence of both breast cancer (CBG, black) and stromal cells (CBRed, grey) at day three normalized to day one. Dashed line at y = 1 represents no change between days one and three. Graphs show mean values + SEM. * = p < 0.05 versus y = 1. N = 11 per condition. B. Representative images of FUCCI labeled cells in 2D culture. Pseudocolor represents S/G2/M phases (green), G1/S phases (yellow, double-positive red/green), and G1 phase (red) of the cell cycle. Scale bar = 100 μm, C. Fraction of FUCCI labeled cells in each phase of the cell cycle in 2D culture. Cell type of interest is bolded in x-axis label. N = 238–779 cells over 3–27 images. D. Dual-color cellular bioluminescence of both breast cancer (CBG, black) and stromal cells (CBRed, grey) at day six normalized to day one. Dashed line at y = 1 represents no change between days one and six. Graphs show mean values + SEM. * = p < 0.05 versus y = 1. N = 48 spheroids per condition. E. of FUCCI labeled cells in each phase of the cell cycle in spheroid co-cultures. Cell type of interest is bolded in x-axis label. N = 167–1328 cells over 5–18 spheroids. F. Fraction of FUCCI labeled cancer cells in S/G2/M phases in spheroid cultures. Cell type of interest is bolded in x-axis label. Graphs show mean values + SEM. Lines show pair-wise comparisons with **** = p < 0.0001. N = 167–1328 cells over 5–18 spheroids.
We hypothesized that a 3D co-culture environment would promote quiescence in ER+ breast cancer cells, reproducing quiescence of dormant DTCs in bone marrow. In our 3D co-culture system, bioluminescence imaging revealed no increase in numbers of cancer cells between days one and six, while remaining viable (Fig 1D). We verified bioluminescence results for quiescence of cancer cells by FUCCI imaging. In marked contrast to 2D culture, nearly all cancer cells in co-culture spheroids remained in G1 (Fig 1E; Fig S2C). Furthermore, cancer cells in 3D co-culture exhibited a lower number of cells undergoing cell division than monoculture, further substantiating the utility of the co-culture system to represent quiescent bone marrow DTCs (Fig 1F; Fig S2B, C). Stromal cells in 3D spheroids remained cycling and viable as confirmed by bioluminescence, FUCCI, and PI stain analyses (Fig 1D, E; Fig S2A). These data support that 3D spheroid co-cultures with bone marrow stromal cells shift ER+ breast cancer cells to quiescence.
Breast Cancer Cells Exhibit Increased Reliance on OXPHOS Relative to Surrounding Stromal Cells
Since metabolism presents a potential therapeutic target for DTCs, we investigated differences in utilization of OXPHOS versus glycolysis between DTCs and surrounding stromal cells. We used fluorescence lifetime imaging (FLIM) of endogenous NADH (15, 19, 20) to analyze metabolic states of cells alone in spheroids (monoculture) or combined bone marrow MSCs with breast cancer cells (co-culture). In monoculture spheroids, both HS5 and HS27a stromal cells exhibited glycolytic metabolism while MCF7 and T47D breast cancer cells exhibited OXPHOS (Fig 2B; Fig S3). In co-culture spheroids, both MCF7 and T47D cells showed 1.2 – 1.3-fold greater dependence on OXPHOS than stromal cells in the same spheroid (Fig 2A, B). Both cancer cell types exhibited higher OXPHOS when cultured with HS5 versus HS27a stromal cells. We compared metabolic states of stromal cells with or without immediately adjacent breast cancer cells and detected no significant differences based on proximity to cancer cells (Fig 2C). In all breast cancer-stromal cell combinations, cancer cells relied on OXPHOS, while stromal cells utilized glycolytic metabolism to a greater extent.
Figure 2: ER+ breast cancer cells show increased reliance on OXPHOS versus surrounding bone marrow stromal cells.

A. Representative images of NADH lifetimes in co-culture spheroids. Red and purple on the pseudocolor scale indicate longer and shorter NADH lifetimes, respectively. A longer NADH lifetime represents increased usage of cellular NADH for OXPHOS over glycolysis and a shorter NADH lifetime represents a preferential use of NADH for glycolysis over OXPHOS. White boxes show cancer cells marked with mCherry NLS (pseudocolored grey in presented images). Scale bar = 30 μm, B. Box and whisker plots show distributions of NADH lifetimes for cancer (black) and stromal (grey) cells in mono and co-culture spheroids. Lines represent a significant (**** = p < 0.0001) difference in NADH lifetime. N = 26–67 images over 5–10 spheroids, C. Box and whisker plots show distributions of NADH lifetimes for stromal cells non-adjacent (magenta) and adjacent (green) to cancer cells. Cell type of interest is bolded in x-axis label. There were no significant differences between non-adjacent and adjacent cells at p = 0.05. N = 26–67 images over 5–10 spheroids
Breast Cancer Cells Display Minimal, Heterogeneous Activation of ERK and High Activation of Akt
Building on single-cell differences in metabolism between DTCs and bone marrow MSCs, we probed for differences in signaling pathway activation between cancer and stromal cells. We used fluorescent kinase translocation reporters (KTRs) to quantify activation of ERK and Akt in single cells (21–23). KTRs reversibly move between the nucleus and cytoplasm based upon phosphorylation by a kinase of interest. Phosphorylation of the KTR shifts the reporter to the cytoplasm (darker nucleus, kinase “on”). The ratio of fluorescence intensities in the cytoplasm to nucleus quantifies the magnitude of kinase activity in an individual cell. Representative images of breast cancer cells stably expressing these imaging reporters in co-culture spheroids with bone marrow stromal cells exhibited heterogeneous signaling through ERK and high Akt activation (Fig 3A). Quantified data show 3.2- to 13.7-fold greater activity of Akt versus ERK in breast cancer cells, consistent with activating mutations in phosphatidylinositol-3-kinase (PI3K) driving Akt signaling in MCF7 and T47D cells (24) (Fig 3B). MCF7 and T47D cells both demonstrated a 2.5- to 2.9-fold-change higher activity of Akt than HS5 stromal cells. Bone marrow stromal cells stably expressing KTRs for ERK and Akt and nuclear marker H2B-mCherry minimally activate ERK and Akt (Fig S4A; Fig 3B) while MCF7 and T47D cells in monoculture activate Akt to a higher extent than ERK (Fig S4B; Fig 3B). We could not stably express KTRs in HS27a cells, precluding single-cell imaging of Akt and ERK in these cells. Higher activity of Akt in cancer versus stromal cells suggests this kinase as a therapeutic target to preferentially eliminate DTCs.
Figure 3: ER+ breast cancer cells activate Akt to a greater extent than ERK in co-culture spheroids with bone marrow stromal cells.

A. Representative images show breast cancer cells (defined by mCherry-H2B fusion) expressing both ERK and Akt KTRs grown in spheroids with stromal cells. Regions of interest define cancer cell nuclei in all images. Cell type of interest is bolded in image label. Scale bar = 15 μm, B. Box and whisker plots show quantified activities of ERK and Akt KTRs presented as the log base 2 of the ratios of cytoplasmic to nuclear fluorescence intensities (CNR) for each KTR in single cells. A higher ratio defines greater kinase activation. Cell type of interest is bolded in x-axis label. Lines show pair-wise comparisons with * = p < 0.05 or **** = p < 0.0001. N = 125–420 cells from 8 spheroids.
Ex Vivo Imaging of Bone Marrow Metastases Recapitulates Metabolic and Signaling Profiles in 3D co-culture
To validate metabolic and signaling profiles of DTCs measured in our 3D co-culture model, we used a mouse model for bone metastases to measure both metabolism and signaling of DTCs in vivo. We injected MCF7 cells into the left femoral artery of mice to preferentially disseminate breast cancer cells to bone marrow (25). Bioluminescence imaging showed MCF7 cells in both right and left ovaries, distal femur and proximal tibia (knees), and hind paws (locations shown in Fig 4A) four-weeks post-injection (Fig 4B). Metastases occurred most commonly in bilateral knees, consistent with our previous findings that cancer cells hone to developing growth plates in skeletally immature mice (26) (Fig 4C). Mice presented with metastases on average in 4 out of the six analyzed sites (Fig 4A, C) per animal (SD = 1 site). Presence of metastases in sites other than the left hindlimb indicates that some cancer cells did not arrest in this hindlimb after injection and/or secondarily disseminated to other bones and organs.
Figure 4: Ex vivo imaging of cancer cells in bone marrow recapitulates metabolic and signaling profiles from 3D co-culture spheroids.

A. Red boxes denote metastatic locations of interest on both the left (L) and right (R) sides of the mouse being the foot (F), knee (K), and ovary (O). B. Representative bioluminescence images of mice four weeks post femoral artery injection. Pseudocolor scale depicts range of values with red being highest and blue lowest values, respectively. C. Metastatic incidence of MCF7 cells in mice 4 weeks post-injection. N = 19. D. Representative images of MCF7 cells marked by red nuclei (top) and corresponding pseudocolor map of NADH lifetimes (bottom) in bone marrow of a mouse. Longer NADH lifetimes (red) represent increased usage of NADH for OXPHOS over glycolysis. Scale bar = 30 μm. E. Histogram displays NADH lifetimes for MCF7 breast cancer cells in bone marrow of mice. N = 35 images from 3 mice, F. Representative images of MCF7 cells (red nuclei) expressing ERK and Akt KTRs in bone marrow of mice. Regions of interest define cancer cell nuclei. Scale bar = 15 μm, G Quantification of ERK and Akt KTR activation displayed as the log base 2 of the cytoplasmic to nuclear ratios of fluorescence intensities (CNR) for each channel in single cells. **** = p < 0.0001. N = 393 cells over 3 mice
To investigate metabolic and signaling profiles of breast cancer cells within bone marrow, we performed ex vivo imaging of mice with bone marrow metastases immediately following euthanasia. Prior research established that the metabolic status of cells remains unchanged in ex vivo samples for 8 hours of culture (27). We measured NADH lifetime to quantify the metabolic state of MCF7 cells inside the bone marrow environment (Fig 4D). Although cancer cells displayed heterogeneous lifetimes of NADH, the distribution shifted toward OXPHOS metabolism to an even greater extent than co-culture spheroids by 1.1- to 1.2-fold (Fig 4E). Next, we imaged both ERK and Akt signaling in cancer cells inside bone marrow (Fig 4F). Cancer cells in bone marrow demonstrated greater activation of Akt than ERK where malignant cells typically showed pairwise active Akt and inactive ERK (Fig 4F, G). These results demonstrate that breast cancer cells in bone marrow exhibit an OXPHOS metabolic profile, low ERK activation, and high Akt activation similar to our 3D bone marrow spheroid model.
Targeted Treatment and response of OXPHOS and Akt Signaling in Spheroids
After observing that selected ER+ breast cancer cells rely on OXPHOS and activate Akt to a greater extent than stromal cells, we compared treatments targeting OXPHOS and Akt with tamoxifen, a standard hormonal therapy for ER+ breast cancer. We measured responses of cancer and stromal cells to treatment with dual-color bioluminescence imaging of all four cell combinations (MCF7 + HS5, MCF7 + HS27a, T47D + HS5, T47D + HS27a). We quantified viability of both cancer and stromal cells, using the stromal population as surrogates for in vivo toxicity to normal cells. Figure 5A shows possible responses to treatment. When tested alone, specific inhibition of Akt by MK-2206 or treatment with tamoxifen reduced viability of cancer cells by 40% and had limited effects on stromal cells (Fig 5B). However, blocking activation of Akt with GSK458, an inhibitor of PI3-kinase, showed reduced efficacy compared to MK-2206. We indirectly targeted mitochondrial OXPHOS with compound D9, an inhibitor of the mitochondrial enzyme thioredoxin reductase (TrxR). TrxR catalyzes reduction of thioredoxin, a key scavenger of reactive oxygen species (ROS) produced as a by-product of OXPHOS (28). Treatment with D9 alone produced minimally more toxicity to cancer versus stromal cells. Next, we used treatments combining targeted signaling inhibitors, tamoxifen, and D9 (Fig 5C). Combination treatment with D9 plus MK-2206 showed the greatest efficacy for our desired endpoint of killing cancer cells, reducing numbers of breast cancer cells by 110% relative to control, with no decrease in viability of stromal cells. Furthermore, the combination of D9 plus MK-2206 markedly boosted toxicity toward cancer cells as compared with each compound alone. Other tested combinations minimally affected cancer and stromal cells relative to control or decreased numbers of both cancer and stromal cells in co-cultures (MK-2206 plus tamoxifen).
Figure 5: Simultaneous treatment of co-culture spheroids with inhibitors of OXPHOS and Akt decreases growth of cancer cells while maintaining viability of stromal cells.

A. Drug responses are shown as the log base 2 of treatment divided by control responses. A decrease in ratio along either axis represents bioluminescence less than control and an increase in L2R along either axis represents bioluminescence greater than control The four quadrants of the graph represent cancer cell growth and stromal cell death (red), cancer cell growth and stromal cell growth (orange), cancer cell death and stromal cell death (yellow), and cancer cell death and stromal cell growth, the desired outcome, (green). B. Graph shows responses of cancer (y-axis) and stromal (x-axis) cells in co-culture spheroids to treatment with single compounds. We calculated growth of cells as the ratio of bioluminescence on day six relative to day 1 and present mean values ± SEM for log2 of this ratio. Error bars for SEM are smaller than the symbol for some conditions if not evident. Dotted lines at X and Y = 0 represent no change from control spheroids. We pooled data from all four combinations of co-culture spheroids into a single data point per treatment regime. N = 48 spheroids per treatment. C. Mean values ± SEM for cancer and stromal cell responses to combination treatments as described in panels A and B. Drug concentrations are as follows: 0.1 μM D9 + 0.1 nM GSK458, 0.1 μM D9 + 1 μM Tamoxifen, 0.1 nM GSK458 + 1 μM Tamoxifen, 1 μM D9 + 100 nM MK-2206, and 10 nM MK-2206 + 1 μM Tamoxifen. N = 48 spheroids per treatment. D. Metabolic profiles of both cancer (black) and stromal (grey) cells in co-culture spheroids of T47D plus HS27a cells when treated with control, 1 μM Tamoxifen, or 1 μM D9 plus 100 nM MK-2206. Lines indicate pairwise comparisons at **** = p <0.0001. N = 38 images per 10 spheroids per treatment. E. Signaling activation of ERK (yellow) and Akt (cyan) in co-culture spheroids of T47D plus HS27a cells when treated with control, 1 μM Tamoxifen, or 1 μM D9 plus 100 nM MK-2206. **** = p < 0.0001. N = 463–523 cells per 10 spheroids per treatment. Abbreviations: Tam as Tamoxifen.
Next, we compared metabolic and signaling responses of both cancer and stromal cells treated with D9 plus MK-2206 versus tamoxifen in spheroids with T47D and HS27a cells. D9 plus MK-2206 significantly lowered the lifetime of NADH after 96 hours of treatment relative to control and tamoxifen treatments, reflecting that D9 plus MK-2206 shifted metabolism from OXPHOS to glycolysis (Fig 5D). Combined D9 plus MK-2206 treatment lowered the lifetime of NADH by 14.7% in cancer cells versus only 8.7% in stromal cells, suggesting greater effects of this treatment against cancer rather than stromal cells. Accordingly, D9 plus MK-2206 also reduced the lifetime of NADH to a greater extent in cancer than stromal cells in the other three cell combinations (Fig S5). Treatment with D9 plus MK-2206 also decreased signaling through Akt by 24% as compared with tamoxifen or control (Fig 5E). These studies identified D9 plus MK-2206 as a treatment with preferential toxicity to cancer versus stromal cells and verified that this combination produced expected effects on metabolism and signaling.
Treatment of OXPHOS and Akt Signaling in a Model of Mouse Bone Marrow Metastases
We compared the most effective treatment combination from our 3D co-culture model, D9 plus MK-2206, with tamoxifen in a mouse model of bone metastasis. Using femoral artery injection of MCF7 cells to establish bone metastases, we treated mice with D9 plus MK-2206, tamoxifen, or vehicle for 16 days. Mice did not exhibit significant differences in weight throughout treatment (Fig S6A). Treatment with D9 plus MK-2206 inhibited growth of breast cancer cells to a greater extent than vehicle control on both a whole mouse (Fig 6A, B; Fig S6B) and individual metastases scale (Fig 6C). Mice receiving D9 plus MK-2206 demonstrated significantly fewer new metastases than tamoxifen at both days 7 and 17 of treatment (Fig 6D). Overall, these data provide evidence for improved treatment response using combination therapy targeting OXPHOS and Akt signaling concurrently with compounds D9 plus MK-2206.
Figure 6: In vivo dual targeting of OXPHOS and Akt signaling of MCF7 cancer cells injected into the femoral artery of mice.

A. Representative bioluminescence images of mice before and after 17 days of treatment with vehicle, 5mg/kg tamoxifen, or combination 5 mg/kg D9 plus 120 mg/kg MK-2206. Pseudocolor scale depicts range of values with red being highest and blue lowest values, respectively. B. Total cumulative body bioluminescence AUC of each group over treatment normalized to the initial day. Data are shown as mean ± SEM. * = p < 0.05 versus control. N = 4–5 mice/group. C. Log2 fold change in bioluminescence of individual metastatic lesions between pre (Day −1) and post (Day 17) treatment. ** = p < 0.01. N = 4–5 mice/group. D. Total number of new metastasis formed per mouse over the course of treatment. * = p < 0.05. N = 4–5 mice/group.
Discussion
Effective treatment to target bone marrow DTCs and prevent metastatic recurrence in ER+ breast cancer patients remains a clinically unmet need. Standard hormonal therapies fail to eliminate DTCs, evidenced by the fact that recurrence rates escalate 5 to 20 years-post-diagnosis. Since patients with DTCs typically exhibit no symptoms and not all DTCs ultimately resume proliferation, these patients require effective treatments with milder toxicity toward normal cells than standard cancer therapeutics. Identifying novel therapies to prevent metastatic disease requires a versatile model system to study interactions between cancer cells and the bone marrow microenvironment and capitalize on vulnerabilities of bone marrow DTCs. Our system has the potential to benefit patients by recapitulating fundamental conditions of bone marrow DTCs in physiological, 3D co-culture spheroids of cancer and stromal cells, enabling screening for therapies that selectively eliminate cancer cells.
This work has several major observations that advance knowledge of metabolism and signaling by DTCs in bone marrow. First, our model system reflects metabolic and signaling profiles of DTCs observed in a mouse xenograft model of cancer cells in bone marrow. Cancer cells rely heavily on OXPHOS and Akt signaling compared to bone marrow MSCs. Next, heterogeneity among stromal cells has the potential to drive niche-specific variations in the vulnerability of cancer cells to therapy. Despite originating from the same donor, HS5 and HS27a bone marrow stromal cells have distinct effects on both metabolism and signaling of cancer cells. Lastly, targeting metabolic and signaling dependencies of DTCs provides a novel, exciting exploratory treatment option as established in both our 3D co-culture and mouse xenograft models.
In our 3D model of quiescent DTCs, cancer cells exhibit increased reliance on OXPHOS, substantiating previous reports that migratory/invasive cells, such as DTCs, favor OXPHOS metabolism (29, 30). We chose to inhibit TrxR, an NADH-binding protein involved in OXPHOS metabolism, as it is a potential contributor to the increase in OXPHOS. Inhibiting TrxR already has been identified as a potential therapeutic target in cancer because this molecule functions to protect cells against oxidative stress by ROS (31, 32). A prior study demonstrated that D9, an inhibitor of TrxR, slowed tumor growth and preferentially killed tumor cells (33). We have previously targeted ROS in triple-negative breast cancer stem cell states where inhibition of Nuclear factor erythroid 2-related factor 2 (Nrf2) or downstream antioxidant pathways suppressed tumor growth and metastases by directly targeting BCSC reliance on Nrf2-mediated antioxidant pathways (34). These data broadly implicate targeting metabolism and ROS as a breast cancer therapy. Since ROS and reprogramming of intracellular metabolism regulate signaling and activation of T lymphocytes and other immune cells (35–37), therapies targeting these pathways must balance effects on immunity versus elimination of DTCs.
Complementarily, cancer cells exhibited significantly higher basal activation of Akt than ERK and higher activation of Akt than surrounding stromal cells. Consistent with our findings, quiescent tumor cells typically show a low activation of ERK signaling (38), while Akt stimulates epithelial-to-mesenchymal transition and survival of DTCs (39–41). Akt regulates anti-oxidant signaling and targeting Akt signaling has emerged an encouraging treatment strategy against breast cancer by overcoming chemoresistance and immunosuppression (42). Interestingly, PI3K-Akt pathway inhibition upregulates antitumor immune responses in tumors, further supporting efforts to target this pathway as a therapy for DTCs (43–45). In a phase II clinical trial using an allosteric Akt inhibitor, MK-2206, concurrently with neoadjuvant chemotherapy, MK-2206 improved pathological complete response rates versus chemotherapy alone (46). Treatment with OXPHOS or Akt inhibitors selectively decreased viability of cancer versus stromal cells in our spheroid model, and combination treatment with both inhibitors amplified effects of these therapies. These data indicate that other existing or new compounds targeting metabolic and signaling vulnerabilities of DTCs with acceptable toxicity profiles could be developed or repurposed to incorporate in combination therapies against DTCs (47, 48).
Our study highlights bi-directional metabolic and signaling interactions between cancer and stromal cells in the bone marrow microenvironment. OXPHOS and Akt are upregulated in cancer cells, while stromal cells shift towards glycolytic metabolism in our model of DTCs. While we did not focus on specific mechanisms of crosstalk, prior studies suggest key processes regulating these interactions. Once DTCs arrive in bone marrow, stroma becomes a part of the tumor microenvironment, and both cancer and stromal cells begin to regulate one another (49). Cancer cells may begin to condition bone marrow stroma to promote drug resistance and survival (50). In parallel, MSCs secrete extracellular vesicles containing various metabolites, cytokines, and chemokines that interact with neighboring cancer cells to modify both metabolic and signaling pathways (51). Secretion of metabolites and signaling ligands by MSCs affects energy pathways leading to a functional change of cancer cell reliance on OXPHOS and activation of Akt, respectively (49, 52). These data highlight the need to incorporate multiple cell types into studies of DTCs to replicate functions of MSCs and the tumor microenvironment in disease progression and resistance to cancer therapeutics.
While our femoral artery injection model preferentially disseminates breast cancer cells to bone marrow, at least some ER+ cancer cells proliferated instead of remaining dormant in this environment. This limitation of our study reflects the challenge of generating mouse models of tumor dormancy and reactivation, particularly for ER+ breast cancer (53). Using a combination of intra-cardiac injection to disseminate breast cancer cells to bone marrow and ovariectomy to suppress hormonal stimulation, investigators have generated models with quiescent MCF-7 or T47D cells that may resume proliferation after weeks of latency (54–58). Early outgrowth of tumors in NSG mice as compared with more delayed kinetics in nude mice highlight functions of the immune system to maintain tumor mass dormancy. The fact that D9 plus MK-2206 targeted quiescent breast cancer cells in bone marrow spheroids and mouse metastases supports the use of this combination for both DTCs and actively proliferating cancer cells.
This work provides the first evidence highlighting dual targeting of metabolism and signaling as an effective approach to targeting DTCs. Our spheroid model provides an adaptable framework to further investigate metabolic and signaling differences between cancer and stromal cells. Treatments that preferentially target cancer versus stromal cells in this model show success in mouse models of bone marrow metastases as evidenced by the current study and past work. Future studies with this model system will focus on identifying cellular and molecular mechanisms of crosstalk between breast cancer and stromal cells that alter signaling and metabolism. In particular, identifying specific components of host immunity that control kinetics of outgrowth of bone marrow metastases for ER+ breast cancer cells will highlight therapeutic effects on the immune compartment. These mechanistic studies will reveal new targets and approaches for therapy to successfully eliminate DTCs and reduce the risk of recurrence in ER+ breast cancer. We plan to introduce additional cell types critical to bone marrow niches, such as endothelial cells and immune components, to better model a metastatic niche. In addition, we envision expanding our model system to incorporate patient-derived cancer and/or stromal cells, providing an opportunity to define precision therapies for individual patients with DTCs. These planned future directions will advance understanding of cancer cell quiescence in bone marrow and expand the landscape of options to prevent tumor dormancy and delayed recurrence in ER+ breast cancer and other malignancies.
Materials and methods
Cell culture
We obtained immortalized human bone marrow mesenchymal stromal cell lines HS5 (CRL-11882) and HS27a (CRL-2496) and breast cancer cell lines MCF7 (HTB-22) and T47D (HTB-133) from the American Type Culture Collection (ATCC, Manassus, VA, USA). We maintained HS5, MCF7, and T47D cells in DMEM (#10313, Gibco, Thermo Fisher, Grand Island, NY, USA), supplemented with 10% FBS (HyClone, ThermoScientific, Waltham, MA, USA), 1% GlutaMAX (#35050, Gibco), and 1% Penicillin-Streptomycin (P/S, #15140, Gibco). We maintained HS27a cells in RPMI (#11875, Gibco) supplemented with 10% FBS and 1% P/S. We maintained cells in a 5% CO2, 37°C, humidified incubator and passaged cells by trypsinizing and re-suspending every 3 days. We performed short tandem repeats analysis to authenticate cell lines and mycoplasma testing at the time of initial passaging.
Lentiviral and transposon vectors
We transduced HS5 and HS27a lines to stably express click beetle red-Flag ires pLVX (CBRed) and ER+ Breast cancer cell lines MCF7 and T47D to stably express click beetle green-Flag ires Blast pLVX (CBG) and selected with blasticidin as we previously described (59). We engineered MCF7 and T47D lines to stably express either mCherry fused to a nuclear localization sequence (mCherry NLS) or FUCCI (15) using recombinant lentiviral vectors. We used a transposon vector system (PiggyBac Transposon, System Biosciences, Palo Alto, CA, USA) with H2BmCherry-AKT KTR-Aqua-ERK KTR-mCitrine-puro piggyback (pHAEP) to express KTRs for Akt and ERK (22). We selected for positive mCherry NLS cells using FACS, FUCCI cells by FACS for red, and pHAEP cells by selection with puromycin.
2D culture model system assays
For 2D bioluminescence growth studies, we seeded 4 000 cells total per well in a black-walled 96 well plate (#3603, Corning, Kennebunk, ME, USA). For FUCCI analysis, we seeded cells at a density of 2×105 cells per 35mm glass-bottom dish (#D35–20–1.5P, Cellvis, Mountain View, CA, USA) coated with Collagen Type I (#50203, Ibidi, Martinsried, Germany). For co-culture conditions, we seeded 5% cancer cells with 95% stromal cells. We seeded cells in low glucose, phenol-red free DMEM (#11054, Gibco) with 10% FBS, 1% GlutaMAX, 1% P/S, and 10 nM β-Estradiol (#E2758, Sigma-Aldrich, Millipore Sigma, Saint Louis, MO, USA). We changed media to 1% FBS one day after seeding.
Bioluminescence imaging and analysis
For cell-based assays, we performed bioluminescence imaging using an IVIS Lumina Series III (Perkin Elmer, Waltham, MA, USA) and exported photon flux data from regions of interest defined in Living Image 4.3.1 (Perkin Elmer). We performed dual-color bioluminescence imaging and analysis of both CBRed and CBG as previously described (60). We performed bioluminescence imaging both one and three days after seeding for 2D studies and one and six days after seeding for 3D studies.
FLIM and two-photon microscopy and analysis
We performed all 3D, in vitro imaging studies after 6 days of culture in transfer, imaging, and microscopy plates (61). We used an Olympus FVMPE-RS upright microscope with Olympus Fluoview software, Spectra-Physics Insight DeepSee laser, and ISS Fast FLIM, when applicable, for all two-photon and FLIM studies. We acquired and analyzed FLIM data as we have previously described (15, 19). We acquired and analyzed pHAEP images by two-photon microscopy as described by our group (22). Briefly, we exported images as tiffs from ISS VistaVision or Fluoview software and used custom MATLAB scripts to segment and quantify images.
Cell cycle imaging and analysis
We imaged or performed FACS on cells for cell cycle analysis three days (2D) or six days (3D) after seeding. We acquired 2D FUCCI Images using an Olympus IX73 microscope and DP80 CCD camera (Olympus) in both red and green channels using CellSens Software (Olympus). We acquired and analyzed FUCCI images as we have previously described (15). We analyzed the number of red (G1), green (S/G2/M), and double-positive (G1/S) cells using FIJI. We binarized images using the auto local threshold Phalskanar function and used the particle analyzer to capture nuclei (62). We measured color intensities in both red and green channels for each nucleus and used standardized cutoffs for positive expression to determine phase of the cell cycle for single cells. For propidium iodide stain (#ab14083, Abcam, Cambridge, MA, USA) we followed manufacturers protocols for staining and analysis and dissociated cells or spheroids using Accumax (STEMCELL, Vancouver, BC, Canada). We analyzed cell cycle stages using FCS Express (De Novo Software, Pasadena, CA, USA).
Spheroid formation and culture
We formed spheroids using a low-binding 384 round bottom plate (#3676, Corning) as we previously described (15). Briefly, we seeded a total of 3 000 cells per well in 20μL of low glucose, phenol-red free DMEM with 1% FBS, 1% GlutaMAX, 1% P/S, and 10 nM β-Estradiol. For co-culture conditions, we seeded 5% cancer cells with 95% stromal cells. We changed media every two days.
Testing treatments in bone marrow spheroid model
For 3D co-culture treatment studies, we dissolved D9 (#5921, Tocris, Bristol, UK), MK-2206 (#S1078, Selleckchem, Houston, TX, USA), GSK458 (#S2658, Selleckchem), and Tamoxifen (#T5648, Sigma-Aldrich) in DMSO at stock concentrations of 10mM. We randomly assigned spheroids to each treatment group. We treated control wells with 0.01% v/v DMSO. 48 hours after seeding spheroids, we changed spheroid media to compound-containing media and changed the compound-containing media to fresh compound-containing media again 48 hours later. To analyze treatment data, we first subtracted background bioluminescence from each ROI. Next, we normalized bioluminescence of each spheroid to that of untreated control spheroids for both green and red channels. We then took the log2 of those data and plotted the mean and SEM for each treatment.
Femoral artery injection (FAI) and mouse imaging studies
The University of Michigan Institutional Animal Care and Use Committee approved all experimental procedures involving animals, and we housed mice as previously described (63). We used FAI to introduce breast cancer cells into hindlimb bone marrow as detailed previously (26). Briefly, we injected 1 × 106 cancer cells into the left femoral artery of 10–12-week old female NSG mice (The Jackson Laboratory, Bar Harbor, ME, USA). We performed bioluminescence imaging weekly using an IVIS Spectrum (Perkin Elmer). 28 days after FAI, we euthanized mice and removed the left tibia and femur. After removing all skin and muscle tissue, we used a Dremel to expose bone marrow and covered the site with clear ultrasound gel before performing ex vivo FLIM or pHAEP microscopy.
Treatment and analysis in a mouse model of metastases
We began treatment 28 days after FAI and randomly assigned mice to each treatment group. We selected group sizes to have a power of 0.85 to detect 25% differences in imaging signals at p < 0.05. We dissolved D9 in corn oil (#C8267, Sigma-Aldrich) to a concentration of 0.125mg/150μL and administered 5mg/kg via intraperitoneal (IP) injection (33). We dissolved MK-2206 in 30% Captisol (Captisol, Lawrence, KS, USA) to a concentration of 3mg/100μL and administered 120mg/kg via oral gavage (64). We dosed mice with D9 plus MK-2206 simultaneously. We dissolved Tamoxifen in corn oil at a concentration of 0.125mg/100μL and administered 5mg/kg every other day via IP injection (65). For the vehicle control, we injected 150μL corn oil IP and administered 100uL of 30% Captisol via oral gavage. We monitored weights of mice every other day and performed bioluminescence imaging 1 day before and 3, 7, and 17 days after beginning treatment. To determine the number of metastasis each imaging day, a person blinded to experimental groups counted foci with signal above background bioluminescence.
Statistical Analysis
Data are representative of experiments repeated independently three times. We analyzed data using GraphPad Prism (San Diego, CA) and MATLAB (Mathworks). We used a two-sided, unpaired student’s t-test to compare bioluminescence data for cell proliferation to the starting condition. We used a two-sided one-way ordinary ANOVA with Tukey correction for multiple comparisons to analyze differences between groups. We used a two-sided, two-way ANOVA with Geisser-Greenhouse correction as we did not assume equal variability of differences and Bonferroni correction to compare treatment responses over time. We used a value of p < 0.05 and bars on plots to infer significance between pairs. We prepared bar graphs (mean values +/± SD or SEM as denoted in figure legends) and box plots and whiskers using GraphPad Prism 8.3.0. For box plots and whiskers, the bottom and top of a box define the first and third quartiles, and the band inside the box marks the second quartile (the median). The ends of the whiskers represent the 5th percentile and the 95th percentiles, respectively.
Code availability
All custom MATLAB code including the image processing files require a material transfer agreement from the University of Michigan.
Supplementary Material
Acknowledgments
We thank Ayşe J. Muñiz and Max Wicha for manuscript feedback. We thank Michael Pihalja for assistance with flow cytometry. The authors acknowledge funding from United States National Institutes of Health grants R01CA238042, R01CA196018, U01CA210152, R01CA238023, R33CA225549, R50CA221807, and R37CA222563. Brock Humphries, Ph.D., was supported by an American Cancer Society - Michigan Cancer Research Fund Postdoctoral Fellowship, PF-18-236-01-CCG. We acknowledge support to the University of Michigan Rogel Cancer Center through National Institutes of Health grant P30CA046592 for flow cytometry and animal imaging studies. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1256260 to Johanna Buschhaus.
Footnotes
Conflict of Interest
The authors declare that they have no competing interests.
References
- 1.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. New England Journal of Medicine. 2017;377(19):1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Schnabel CA, Schroeder BE, Jerevall P-L, Jankowitz RC, Fornander T, et al. Breast Cancer Index Identifies Early-Stage Estrogen Receptor–Positive Breast Cancer Patients at Risk for Early- and Late-Distant Recurrence. Clinical Cancer Research. 2013;19(15):4196. [DOI] [PubMed] [Google Scholar]
- 3.Zhang XHF, Giuliano M, Trivedi MV, Schiff R, Kent Osborne C. Metastasis Dormancy in Estrogen Receptor-Positive Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(23): 10.1158/078-0432.CCR-13-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pantel K, Alix-Panabieres C. Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep. 2014;3:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chambers A, Groom A, MacDonald I. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–72. [DOI] [PubMed] [Google Scholar]
- 6.Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, et al. Metastatic Behavior of Breast Cancer Subtypes. Journal of Clinical Oncology. 2010;28(20):3271–7. [DOI] [PubMed] [Google Scholar]
- 7.Bartkowiak K, Riethdorf S, Pantel K. The interrelating dynamics of hypoxic tumor microenvironments and cancer cell phenotypes in cancer metastasis. Cancer microenvironment : official journal of the International Cancer Microenvironment Society. 2012;5(1):59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell chemical biology. 2017;24(9):1161–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. The Journal of clinical investigation. 2015;125(5):1780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Annals of Medicine. 2006;38(3):200–11. [DOI] [PubMed] [Google Scholar]
- 11.Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Annals of Medicine. 2014;46(6):372–83. [DOI] [PubMed] [Google Scholar]
- 12.Pradhan S, Sperduto JL, Farino CJ, Slater JH. Engineered In Vitro Models of Tumor Dormancy and Reactivation. Journal of Biological Engineering. 2018;12(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta G, Hsiao A, Ingram M, Luker G, Takayama S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J Control Release. 2012;May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Widner DB, Park SH, Eber MR, Shiozawa Y. Interactions Between Disseminated Tumor Cells and Bone Marrow Stromal Cells Regulate Tumor Dormancy. Curr Osteoporos Rep. 2018;16(5):596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavnar S, Rickelmann A, Meguiar K, Xiao A, Dosch J, Leung B, et al. Modeling selective elimination of quiescent cancer cells from bone marrow. Neoplasia. 2015;17:625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwata M, Sandstrom R, Delrow J, Stamatoyannopoulos J, Torok-Storb B. Functionally and phenotypically distinct subpopulations of marrow stromal cells are fibroblast in origin and induce different fates in peripheral blood monocytes. Stem Cells Dev. 2014;23(7):729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakaue-Sawano A, Kuorkawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–98. [DOI] [PubMed] [Google Scholar]
- 18.Pozarowski P, Darzynkiewicz Z. Analysis of cell cycle by flow cytometry. Methods Mol Biol. 2004;281:301–11. [DOI] [PubMed] [Google Scholar]
- 19.Humphries BA, Buschhaus JM, Chen YC, Haley HR, Qyli T, Chiang B, et al. Plasminogen Activator Inhibitor 1 (PAI1) Promotes Actin Cytoskeleton Reorganization and Glycolytic Metabolism in Triple-Negative Breast Cancer. Mol Cancer Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stringari C, Nourse JL, Flanagan LA, Gratton E. Phasor Fluorescence Lifetime Microscopy of Free and Protein-Bound NADH Reveals Neural Stem Cell Differentiation Potential. PLOS ONE. 2012;7(11):e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maryu G, Matsuda M, Aoki K. Multiplexed Fluorescence Imaging of ERK and Akt Activities and Cell-cycle Progression. Cell Struct Funct. 2016;41(2):81–92. [DOI] [PubMed] [Google Scholar]
- 22.Spinosa P, Humphries B, Lewin Mejia D, Buschhaus J, Linderman J, Luker G, et al. Tunable responsiveness in cell signaling. Science Signaling. 2019;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. High-sensitivity measurements of multiple kinase activities in live single cells. Cell. 2014;157(7):1724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aka JA, Lin S-X. Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PloS one. 2012;7(2):e31532–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Yu C, Gao X, Welte T, Muscarella A, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haley HR, Shen N, Qyli T, Buschhaus JM, Pirone ME, Luker KE, et al. , editors. Enhanced Bone Metastases in Skeletally Immature Mice Tomography; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh AJ, Poole KM, Duvall CL, Skala MC. Ex vivo optical metabolic measurements from cultured tissue reflect in vivo tissue status. Journal of biomedical optics. 2012;17(11):116015-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnér ESJ, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. European Journal of Biochemistry. 2000;267(20):6102–9. [DOI] [PubMed] [Google Scholar]
- 29.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis Marcia C, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature Cell Biology. 2014;16:992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nature chemical biology. 2014;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustacich D, Powis G. Thioredoxin reductase. The Biochemical journal. 2000;346 Pt 1(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 32.Scalcon V, Bindoli A, Rigobello M. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radical Biology and Medicine. 2018;127. [DOI] [PubMed] [Google Scholar]
- 33.Zhang D, Xu Z, Yuan J, Zhao Y-X, Qiao Z-Y, Gao Y-J, et al. Synthesis and Molecular Recognition Studies on Small-Molecule Inhibitors for Thioredoxin Reductase. Journal of Medicinal Chemistry. 2014;57(19):8132–9. [DOI] [PubMed] [Google Scholar]
- 34.Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018;28(1):69–86.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarosz EL, Chang C-H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018;18(1):e14–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nature Reviews Drug Discovery. 2019;18(9):669–88. [DOI] [PubMed] [Google Scholar]
- 37.Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. Journal of Experimental & Clinical Cancer Research. 2019;38(1):403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sosa MS, Avivar-Valderas A, Bragado P, Wen H-C, Aguirre-Ghiso JA. ERK1/2 and p38α/β signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(18):5850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Wang L, Zou C, Xia Y, Qin S, Keller E, et al. Tumor microenvironment promotes prostate cancer cell dissemination via the Akt/mTOR pathway. Oncotarget. 2018;9(10):9206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang XHF, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent Bone Metastasis in Breast Cancer Tied to Src-Dependent Survival Signals. Cancer Cell. 2009;16(1):67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, Jücker M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cellular Signalling. 2011;23(12):1952–60. [DOI] [PubMed] [Google Scholar]
- 42.Jabbarzadeh Kaboli P, Salimian F, Aghapour S, Xiang S, Zhao Q, Li M, et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer - A comprehensive review from chemotherapy to immunotherapy. Pharmacological research. 2020;156:104806. [DOI] [PubMed] [Google Scholar]
- 43.Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2(11):1080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding W, Shanafelt TD, Lesnick CE, Erlichman C, Leis JF, Secreto C, et al. Akt inhibitor MK2206 selectively targets CLL B-cell receptor induced cytokines, mobilizes lymphocytes and synergizes with bendamustine to induce CLL apoptosis. British journal of haematology. 2014;164(1):146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue G, Zippelius A, Wicki A, Mandalà M, Tang F, Massi D, et al. Integrated Akt/PKB Signaling in Immunomodulation and Its Potential Role in Cancer Immunotherapy. JNCI: Journal of the National Cancer Institute. 2015;107(7). [DOI] [PubMed] [Google Scholar]
- 46.Tripathy D, Chien AJ, Hylton N, Buxton MB, Ewing CA, Wallace AM, et al. Adaptively randomized trial of neoadjuvant chemotherapy with or without the Akt inhibitor MK-2206: Graduation results from the I-SPY 2 Trial. Journal of Clinical Oncology. 2015;33(15_suppl):524-.25584001 [Google Scholar]
- 47.Heinz S, Freyberger A, Lawrenz B, Schladt L, Schmuck G, Ellinger-Ziegelbauer H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Scientific reports. 2017;7:45465-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thakur S, Daley B, Gaskins K, Vasko VV, Boufraqech M, Patel D, et al. Metformin Targets Mitochondrial Glycerophosphate Dehydrogenase to Control Rate of Oxidative Phosphorylation and Growth of Thyroid Cancer In Vitro and In Vivo. Clinical Cancer Research. 2018;24(16):4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell communication and signaling : CCS. 2016;14(1):20-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plava J, Cihova M, Burikova M, Matuskova M, Kucerova L, Miklikova S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Molecular cancer. 2019;18(1):67-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallabhaneni KC, Penfornis P, Dhule S, Guillonneau F, Adams KV, Yuan Mo Y, et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget; Vol 6, No 7 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong W, Tong Y, Li Y, Yuan J, Hu S, Hu T, et al. Mesenchymal stem cells in inflammatory microenvironment potently promote metastatic growth of cholangiocarcinoma via activating Akt/NF-κB signaling by paracrine CCL5. Oncotarget. 2017;8(43):73693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Özdemir B, Sflomos G, Brisken C. The challenges of modeling hormone receptor-positive breast cancer in mice. Endocrine-Related Cancer. 2018;25:ERC-18. [DOI] [PubMed] [Google Scholar]
- 54.Ottewell PD, Wang N, Brown HK, Reeves KJ, Fowles CA, Croucher PI, et al. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin Cancer Res. 2014;20(11):2922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer cell. 2011;20(6):701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang XHF, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis dormancy in estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(23):6389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogba N, Manning NG, Bliesner BS, Ambler SK, Haughian JM, Pinto MP, et al. Luminal breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells. Breast Cancer Research. 2014;16(6):489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holen I, Walker M, Nutter F, Fowles A, Evans CA, Eaton CL, et al. Oestrogen receptor positive breast cancer metastasis to bone: inhibition by targeting the bone microenvironment in vivo. Clinical & Experimental Metastasis. 2016;33(3):211–24. [DOI] [PubMed] [Google Scholar]
- 59.Buschhaus JM, Luker KE, Luker GD. A Facile, In Vitro 384-Well Plate System to Model Disseminated Tumor Cells in the Bone Marrow Microenvironment In: Lacorazza HD, editor. Cellular Quiescence: Methods and Protocols. New York, NY: Springer New York; 2018. p. 201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavnar S, Xiao A, Gibbons A, Rickelmann A, Neely T, Luker K, et al. Imaging sensitivity of quiescent cancer cells to metabolic perturbations in bone marrow spheroids. Tomography. 2016;2(2):146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cavnar S, Salomonsson E, Luker K, Luker G, Takayama S. Transfer, imaging, and analysis plate for facile handling of 384 hanging drop 3D tissue spheroids. J Lab Autom. 2014;19(2):208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phansalkar N, More S, Sabale A, Joshi MS. Adaptive local thresholding for detection of nuclei in diversity stained cytology images. 2011 International Conference on Communications and Signal Processing 2011:218–20. [Google Scholar]
- 63.Eckley SS, Buschhaus JM, Humphries BA, Robison TH, Luker KE, Luker GD. Short-term environmental conditioning generates cellular memory that enhances tumorigenic potential of triple-negative breast cancer cells. Tomography. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Molecular Cancer Therapeutics. 2010;9(7):1956. [DOI] [PubMed] [Google Scholar]
- 65.Ma G, He J, Yu Y, Xu Y, Yu X, Martinez J, et al. Tamoxifen inhibits ER-negative breast cancer cell invasion and metastasis by accelerating Twist1 degradation. Int J Biol Sci. 2015;11(5):618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.