

Abstract
Immune checkpoint inhibitors have emerged as a revolutionary treatment option for patients with various types of malignancy. Although these agents afford a significant improvement in outcomes for melanoma and other previously untreatable malignancies, their novel mechanism of action may predispose patients to immune‐related adverse effects (irAEs). In the tumour neoantigen environment, these irAEs are due to the activation of the immune system by the blockade of suppressive checkpoints, leading to increases in T‐cell activation and proliferation. IrAEs have been reported in almost any organ and at any point in time, even months to years after discontinuation of therapy. Certain populations with distinct physiological changes, genetic risk factors, and specific antigen exposures may be more highly predisposed to develop irAEs. This review discusses the incidence and mechanisms of irAEs and the relationship between host factors and irAE occurrence.
Keywords: cytotoxic T‐lymphocyte antigen‐4, immune checkpoint inhibitors, immune‐related adverse effects, immunotherapy, programmed cell death 1, programmed cell death ligand 1
1. INTRODUCTION
1.1. Scope of toxicity related to immunomodulatory therapies
Immune checkpoint inhibitors (ICIs) are a novel form of cancer treatment that unleash host immune responses to attack the neoplasm. There are currently 3 United States Food and Drug Administration‐approved targets for ICIs: cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) inhibitors; programmed cell death 1 (PD‐1) inhibitors; and programmed cell death 1 (PD‐L1; the ligand for PD‐1) inhibitors. Currently, ICIs can be used as monotherapy for many malignancies, or in a multitude of combinations. 1 , 2 CTLA‐4 inhibitors can be used in combination with PD‐1 inhibitors for disease states such as melanoma. 3 ICIs can also be combined with chemotherapy for malignancies such as small cell lung cancer, non‐small cell lung cancer (NSCLC), and triple negative breast cancer. 4 , 5 , 6 Most recently, ICIs have been approved in combination with oral tyrosine kinase inhibitors for the treatment of renal cell carcinoma and endometrial cancer. 7 , 8
ICIs were discovered after a breakthrough finding that T‐cell immune responses are controlled through immune checkpoints that are physiologically expressed to downregulate T‐cell activation (Figure 1) and prevent immune‐mediated damage to self. 9 These agents are monoclonal antibodies targeted at increasing T‐cell activation by blocking the above checkpoint molecules. 10 While both pathways have been shown to mediate control of CD4+ and CD8+ T‐cell responses, CTLA‐4 displays more dominant regulation of CD4+ T‐cells within the tumour microenvironment and these agents were the first ICIs to be developed as anticancer agents. 10 , 11 The clinical utility of blocking the PD‐1/PD‐L1 pathway was discovered years later in 2010 when utilized against solid tumours in a phase I trial. 12 , 13 During clinical trial phases, new terms were used for these medications in order to describe the response to therapy, such as immune‐related response criteria and immune‐related progression‐free survival, which allowed the possibility of tumour growth radiographically before shrinkage. 14 Along with this new efficacy criterion another new term came about: immune‐related adverse events (irAEs). 10 Although the mechanisms of irAEs are diverse and poorly understood at this time, they are in broad terms thought to be caused by breaks in peripheral tolerance outside of the tumour microenvironment, resulting in activation of the immune system towards self‐peptides and other nontumour antigens (Figure 2a,b) that have the potential to affect almost any organ system given the holistic dysregulation imposed (Figure 2c). The most common irAEs are generally mild to moderate, and manifest as skin, gut, endocrine, lung, neurological and musculoskeletal toxicities. 15 These adverse effects can occur at any time and, although more likely to occur with early doses, chronic toxicities can persist months to years after cessation of the ICI. Following the introduction of the CTLA‐4 inhibitor ipilimumab, PD‐1 inhibitors (nivolumab, pembrolizumab and cemiplimab) and PD‐L1‐inhibitors (atezolizumab, avelumab and durvalumab) were licensed. Although milder in frequency, and in some cases severity, these agents also displayed irAEs due to their independent but similar effect on T‐cell activation. 16 As with CTLA‐4 signalling, PD‐1 binding inhibits T‐cell proliferation and decreases T‐cell activation. Although in the literature, the expression of PD‐1 on T‐cells has been purported to mark an immune exhaustion phenotype due to overstimulation or a reduction of T‐cell activation, this is overly simplistic and imprecise as PD‐1 has other functions and it is only 1 of several markers that contribute. 17 , 18 However, the blockade of PD‐1 signalling is sufficient to increase T‐cell production and proliferation. 19 PD‐L1 is expressed on professional antigen presenting cells and other immune and nonimmune cells, including tumour cells. The specific blockage of PD‐L1, as opposed to targeting PD‐1, will block the PD‐1:PD‐L1 interaction only, preserving the interaction with the alternate PD‐1 ligand, PD‐L2. It has been proposed that this narrowed therapeutic effect decreases toxicity and preserves self‐tolerance to some degree, although the toxicity profiles of anti‐PD‐1 and anti‐PD‐L1 are similar overall. 19
FIGURE 1.
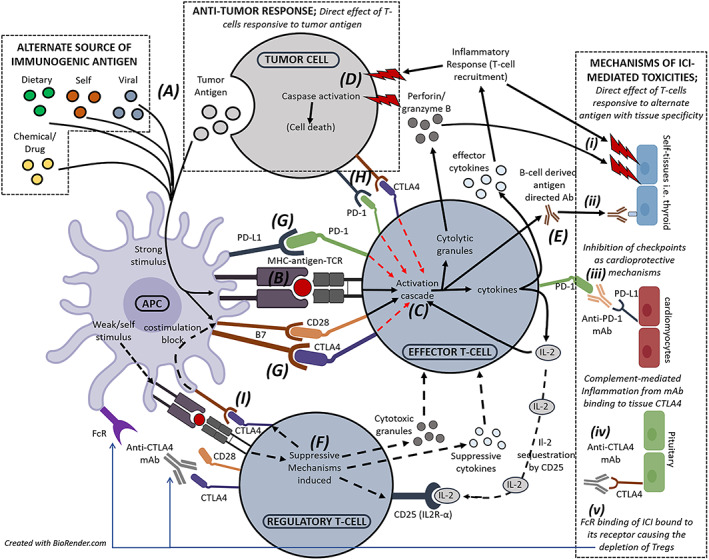
Cellular interplay and mechanisms targeted by immune checkpoint inhibitor (ICI) therapy that drive antitumour response to tumour antigens and ICI toxicities to alternate antigens with a range of tissue specificities. For effector T‐cell activation (solid black lines), immunogenic antigens (A) form a stable immunological synapse with corresponding major histocompatibility complex and T‐cell receptor (TCR; B). In the presence of costimulatory signals including B7‐CD28 ligation, the T‐cell activation cascade ensues (C) driving secretion of interleukin (IL)‐2, inflammatory cytokines and cytolytic granules that instigate immune cell recruitment and target cell death (D and i). Antigen‐directed antibodies may also be released by B‐cells in response to T‐cell signalling (E and ii). Alternatively, weak or continued antigenic stimulus may promote regulatory T‐cell (Treg) activity (F), inducing multiple suppressive mechanisms (black dotted lines) to repress or dampen effector T‐cell activity, including the release of cytotoxic granules and suppressive cytokines, expression of the high affinity IL‐2R to sequester IL‐2 in the local environment, and expression of coinhibitory receptors to block antigen presenting cell (APC) costimulation of effector T‐cells. T‐cells express coinhibitory checkpoint receptors (i.e. programmed cell death 1 [PD‐1], cytotoxic T‐lymphocyte antigen 4 [CTLA4]) that upon ligation may outweigh costimulatory signals and inhibit the T‐cell activation cascade (red dotted lines). Tumour cells use these pathways as mechanisms of immune evasion. Therapeutic ICIs block these interactions that exist between APC and T‐cells (G), between T‐cells and tumour cells (H), and between APC and Tregs (I), driving effector T‐cell activation. While tumour cells may be targeted in response to tumour antigens, peptide antigens from alternate sources may be more immunogenic under the dysregulated environment created by ICI therapy, driving similar T‐cell activation that mediate toxicities directed towards self‐tissues. This may be driven by T‐cells with distinct or shared specificities, or alternatively memory T‐cells cross reactive to both tumour and alternative antigens. Alternatively, inhibition of checkpoint receptors may give rise to toxicities through (iii) preventing the immune‐protective ligation of checkpoint ligands on stressed tissues as described for cardiomyocytes, (iv) instigating complement‐mediated inflammation when ICI bind to checkpoint receptors expressed on tissues as described for the thyroid, or (v) when CTLA4 monoclonal antibody (mAb) that is blocking the dominant expression of CTLA4 on Tregs is recognised by FcR on APC, internalising and depleting the suppressive Treg population
FIGURE 2.
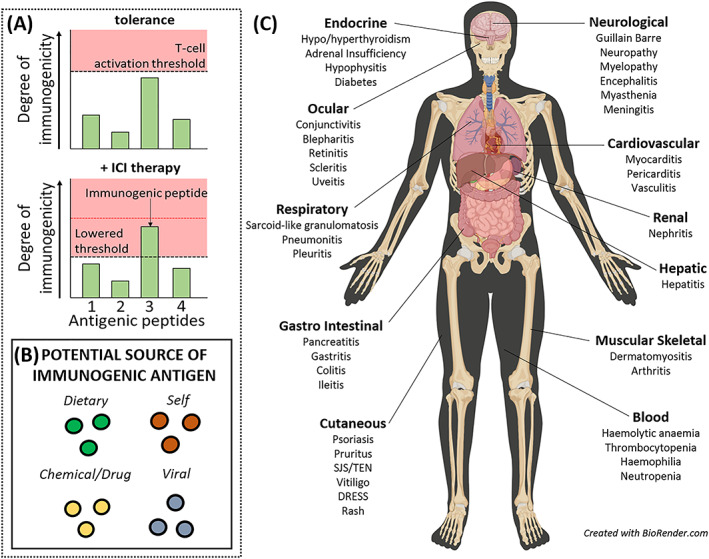
Modulation of immune activation thresholds by immune checkpoint inhibitor (ICI) admin drives onset of varied toxicities. (A) ICI therapy disrupts homeostatic tolerance, lowering immune activation thresholds and effectively increasing the immunogenicity of antigens. Thus, an effector response may ensue to an antigen that is normally tolerated. (B) the antigens responsible for driving individual reactions may be from varied sources which may have tissue‐specific localities/properties. (C) Toxicities affecting varied organs have been observed. DRESS, drug reaction with eosinophilia and systemic symptoms; SJS/TEN, Stevens–Johnson syndrome/toxic epidermal necrolysis
1.2. Specific drug issues related to use of individual and combination ICI therapy
CTLA‐4 and PD‐1 binding to their respective ligands both lead to downregulation of T‐cell activation, although their breadth of cellular expression and timing of inhibition differs. CTLA‐4 expression is limited to T‐cells, whereas PD‐1 is expressed on activated T‐cells, B‐cells and myeloid cells. 12 The CTLA‐4 ligands CD80 and CD86, which are additionally and competitively sought by the costimulatory receptor CD28, are also only expressed on professional antigen presenting cells, while PD‐L1 and PD‐L2 are more widely expressed. Early mechanistic work highlighted that PD‐1 ligation blocks T‐cell receptor (TCR)‐induced PI3K phosphorylation early in the T‐cell signalling cascade following stable immunological synapse formation. However, CTLA‐4 blocked this cascade further downstream and thus was proposed to have less capacity to hinder T‐cell activation given a lack of influence upon more membrane proximal signalling pathways. Indeed, Parry et al. found that 90% of associated T‐cell activation transcripts were diminished by PD‐1, compared to 67% in response to CTLA‐4 ligation. 20 However, the locality of T‐cells and level of expression of individual pathways means this is likely an overly simplistic view for the regulatory dominance of individual pathways. Nonetheless, in vitro T‐cell activation platforms utilizing antigen‐inexperienced healthy donors has shown that both pathways are able to regulate both the nontumour antigen‐specific priming of naïve T‐cells and the activation of pre‐existing memory T‐cells. 21
PD‐1:PD‐L1 interactions preserve the PD‐1:PD‐L2 interaction, which may explain the lower incidence of irAEs with PD‐L1 inhibitors reported in some studies. 19 , 22 In 2 reviews, PD‐1 inhibitors had higher rates of observed pneumonitis compared to PD‐L1 inhibitors, whereas PD‐L1 inhibitors trended towards higher rates of hypothyroidism. 23 , 24 Overall, the difference in toxicities between PD‐1 and PD‐L1 inhibitors is minimal, approximately 70% in both, compared to the substantially higher irAE incidences observed with CTLA‐4 inhibitors. 25 All grade irAEs occur in approximately 90% of patients treated with CTLA‐4 inhibitors. 26 The mechanisms that drive the variability in toxicity incidence remain undefined, but may include Fc‐dependent recognition of bound ICI on immunosuppressive regulatory T‐cells (Tregs), which have higher expression of CTLA‐4, leading to their depletion (Figure 1, v). 27 IrAEs generally occur within the first 3–6 months when using CTLA‐4 or PD‐1/PD‐L1 inhibitors. 25 CTLA‐4 inhibitor toxicities directly correlate with dose and can occur at any time during treatment, but most commonly manifest after the third or fourth dose. 28 In a phase II trial of patients receiving ipilimumab for metastatic melanoma, all grade, severe, treatment limiting, and fatal irAEs were higher in patients receiving 10 mg/kg compared to those who received 3 mg/kg. 29 Combination ICI therapy is utilized in various malignancies for synergistic benefit, but as the mechanisms of efficacy mirror the mechanisms of toxicities, this is often accompanied by a similar enhancement of toxicity onset rates. In a phase III trial in patients with advanced melanoma receiving combination nivolumab/ipilimumab therapy, grade 3–4 irAEs occurred in 59% of patients, compared to 21% in nivolumab therapy and 28% in ipilimumab therapy. 30 Similar results were also observed in another trial utilizing combination ICI therapy, and this combination is now approved in melanoma, renal cell carcinoma, mismatch‐repair deficient colorectal cancer, and most recently was granted priority review by the Food and Drug Administration for the treatment of advanced NSCLC. 31 , 32
1.3. Types of toxicities
1.3.1. Skin toxicity
Multiple studies have shown that skin toxicity presents as the first and most common irAE with CTLA‐4, PD‐1 or PD‐L1 inhibitors. 33 , 34 Skin associated irAEs generally present as maculopapular rashes or sometimes pruritus even in the absence of rash, with a higher incidence in CTLA‐4 inhibitors. 35 All grade dermatological toxicity has been reported in approximately 30–40% of patients receiving PD‐1 or PD‐L1 inhibitors and approximately 50% of patients receiving CTLA‐4 inhibitors. 36 The onset of symptoms occurs at a median of 5 weeks for PD‐1 and PD‐L1 inhibitors and 3.6 weeks with CTLA‐4 inhibitors. 33 , 34 Despite the high frequency of mild skin reactions these are fortunately not typically treatment‐limiting and can be managed symptomatically. Most commonly, the clinical presentation of rashes is a maculopapular eruption, which affects the trunk and arms. However, a wide spectrum of types and severities of skin reactions have been described including lichenoid reactions, psoriaform reactions, vitiligo, and less commonly severe events with defined cytotoxic T‐cell mediated aetiologies such as drug reaction with eosinophilia and systemic symptoms and Stevens–Johnson syndrome/toxic epidermal necrolysis. The clinical appearance of the rash overall has poor correlation with the histopathology of other severe grade rashes and are more typically histopathologically defined as clinically mild. 37 The response of these rashes to treatment may give some clue as to mechanisms as mild rashes may respond to topical steroids and antihistamines whereas pruritus without rash appears to respond better to drugs targeting a neurological mechanism of itch such as gabapentin. Overall, the presence of vitiligo and rash, more than pruritus alone correlated with superior outcomes in metastatic melanoma patients receiving PD‐1 inhibition. 38 Similar observations, where early onset rash is an indicator for successful tumour response and better overall survival, have also been noted for a range of other cancers including NSCLC and metastatic renal cell carcinoma. 39 , 40
1.3.2. Gastrointestinal toxicities
Onset of immune‐mediated colitis is usually characterized by diarrhoea and less commonly abdominal pain, bloody or mucosal stool, or ileus. Symptoms manifest most often 6–8 weeks after starting treatment. 41 While both CTLA‐4 inhibitors and PD‐1/PD‐L1 inhibitors can cause colitis, the reported incidence is greater in patients receiving CTLA‐4 inhibitors or combination PD‐1 and CTLA‐4 inhibitors. 42 The frequency and severity of gastrointestinal (GI) toxicity is also known to be dose‐dependent, particularly for CTLA‐4 inhibitors. The frequency of GI toxicity in patients receiving ipilimumab 3 mg/kg (all grade colitis 32.4% and grade 3–5 2.8%) was lower compared to patients receiving ipilimumab 10 mg/kg (all‐grade colitis 39.4% and grade 3–4 15.5%). 43 Patients may also experience exocrine pancreatitis, and mucositis less commonly.
1.3.3. Hepatic toxicities
ICI‐induced hepatic toxicity, commonly referred to as autoimmune hepatitis, has been observed in as many as 17% of patients. 44 While the incidence varies in clinical trials, that for autoimmune hepatitis appears much lower with monotherapy compared to a synergistic effect of combination CTLA‐4 and PD‐1 inhibition. 45 CTLA‐4 and PD‐1 inhibitor monotherapy had observed autoimmune hepatitis occurrences in 1–7 and 1–6% respectively, compared to approximately 30% with combination ICI. 46 In trials, the median onset of transaminase elevation is around 6–14 weeks after initiation of therapy. 45 , 46 , 47 , 48 Although generally asymptomatic, when evaluating for autoimmune hepatitis, it is pertinent to exclude other drug causes, environmental factors, and viral hepatitis. 46 While ICI‐induced hepatitis described to date has been described to lack the typical antibody response observed in traditional autoimmune hepatitis, and there are slight differences between the anatomical targeting of reaction within the liver, both conditions are associated with influx of largely CD8+ liver‐infiltrating T‐cells highly indicative of a similar role for cytotoxic T‐cell mediated tissue‐damage. 49
1.3.4. Endocrine toxicities
Endocrine abnormalities are another common adverse effect of ICIs. In 1 meta‐analysis, the incidence of endocrine abnormalities with exposure to CTLA‐4 inhibitors or PD‐1/PD‐L1 inhibitors was 12.9% overall. 50 Hypothyroidism, which may be proceeded by transient hyperthyroidism, has been observed more frequently with PD‐1 inhibitors compared to CTLA‐4 inhibitors, with 1 trial displaying an incidence of 18.8% compared to 2%, respectively. 44 However, hypophysitis occurs more commonly with CTLA‐4 inhibitors (13%) compared to PD‐1 inhibitors (0.6%). 51 Hypophysitis is more common at higher doses of ipilimumab, with an incidence of 17% with 10 mg/kg dosing, compared to ≤10% with 3 mg/kg dosing. 15 With CTLA‐4 inhibitors, it is proposed that a type II hypersensitivity occurs due to the CTLA‐4 antibody binding to the cognate antigen expressed on pituitary glands, which then activates complement and leads to the breakdown of the gland (Figure 1, iv). 52 Adrenal insufficiency and type 1 diabetes may also occur. Principally linked with inhibition of PD‐1 or PD‐L1, onset of type I diabetes is also described in 1% of ICI‐treated patients, with risk of associated, life‐threatening diabetic ketoacidosis. Interestingly, the defined HLA‐DR4‐DQ8 and DR3‐DQ2‐risk alleles for development of type I diabetes in general population were highly observed in ICI‐treated patients who developed type I diabetes post treatment, with 84% carrying at least 1 risk allele. 53 Many affected patients also presented with typical disease‐specific antibodies, largely directed towards glutamic acid decarboxylase, 1 of the 4 known islet antigens. While suggestive of an ICI‐driven CD4 + T‐cell mediated response, and supported by the accumulation of insulin‐specific CD4+ T‐cells in the pancreatic lymph node of PD‐1 deficient NOD mice, in a single patient from whom biopsy was possible, an increased population of CD8+ T‐cells compared to CD4+ are described in the pancreatic islets suggesting an important CD8+ cytotoxic role in destructive response. 54 , 55
1.3.5. Pulmonary toxicities
Pneumonitis is the most commonly manifested lung toxicity in patients receiving immune checkpoint inhibitors. 15 In a pooled analysis of 19 trials of PD‐1 or PD‐L1 inhibitors, PD‐1 inhibitors were found to have significantly higher rates of all grade pneumonitis compared to PD‐L1 inhibitors (3.6 vs 1.3%). Incidence of grade 3–4 pneumonitis was also higher with PD‐1 inhibitors compared to PD‐L1 inhibitors (1.1 vs 0.4%). 56 Overall, the incidence of all grade pneumonitis across ICIs is <5% when used as single agents. 57 Combination therapy of PD‐1/PD‐L1 and CTLA‐4 inhibitors carries a higher risk of pulmonary toxicity, with an incidence of 6.88% for all grade pneumonitis and 1.88% incidence for grade 3–4. 58 While validated risk factors for pulmonary are currently unknown, there is thought that characteristics such as sex, age, smoking history and baseline lung disease may predispose patients. 59 Studies have observed that patients with baseline interstitial lung disease have increased risk, as well as tumour type, such as lung and renal cell carcinoma, may lead to an increased incidence of pulmonary toxicity. 60 , 61 In a 2017 study of affected tissue from 2 PD‐1 inhibited cancer patients who subsequently developed pulmonary toxicities, activated CD8+ T‐cells were found at the site of the inflammatory lesion. Moreover, overlapping TCR repertoires were observed between the cancer and pulmonary tissues which were not as distinct in the lymph nodes or in peripheral circulation suggesting shared antigen specificity drives antitumour and irAE response. 62 These data correlate with those that show that ICIs drive diversification of the TCR repertoire, which probably expands overall reactivity to shared auto‐antigens but indeed potentially other exogenous antigenic sources as previously described within. 63 Given the specificity of TCR‐antigen interactions with individual HLA, these data motion that HLA‐risk alleles may form important genotype correlations with distinct reactions upon future, widespread screening.
1.3.6. Neurological toxicities
Neurological toxicities related to ICIs are relatively rare, with an overall occurrence rate of <6% across drug classes. However, combination therapy is associated with up to a 12% risk of neurological manifestations in 1 series. 64 Most documented events occur with nonspecific symptoms such as a headache, but can include potentially irreversible toxicities with long‐term morbidity such as autoimmune encephalitis, myasthenia gravis, peripheral sensorimotor neuropathies (e.g. Guillain–Barre syndrome) and posterior reversible encephalopathy syndrome. 65 In a recent pharmacovigilance study examining neurological toxicities in association with ICI therapy, the development of myasthenia gravis was associated with ~20% mortality rate, occurred early (median onset 29 days), and frequently occurred in conjunction with myocarditis. 66 In the same study, the development of all other neurological toxicities occurred later (median onset 61–80 days) and carried a lower mortality rate of 6–12%. 66 Upon sampling cerebrospinal fluid from a single patient with ICI‐induced fluid‐attenuated inversion recovery hyperintensities and gadolinium enhancing lesions, both CD4 and CD8+ T‐cell populations were identified but with different TCR to those of the patients melanoma suggestive on a nontumour antigen driven T‐cell mediated aetiology. 67
1.3.7. Cardiovascular toxicities
Although the precise mechanism of cardiotoxic irAEs is not fully understood, preclinical data suggest that CTLA‐4 and PD‐1/PD‐L1 regulate and limit both the magnitude and duration of T‐cell responses in the heart. 68 In murine models, CTLA‐4 blockade led to fatal autoimmune myocarditis mediated by CD8 + T‐cells, while the blockage of PD‐1 in mice resulted in dilated cardiomyopathy secondary to anti‐cardiac troponin autoantibody‐mediated myocardial injury (Figure 1, iii). 69 , 70 , 71 Increases in PD‐L1 have also been shown to have a protective influence in the setting of T‐cell mediated inflammation. 72 Although the incidence of cardiovascular toxicities reported in phase I‐III trials for anti‐CTLA‐4 and anti‐PD‐1 agents was low, pooled case reports have demonstrated an incidence of myocarditis up to 2.4% with combination ICI therapy. 73 Fulminant and fatal cases of myocarditis may additionally occur. ICI‐associated myocarditis is characterized by electrocardiographic disturbances and can occur concomitantly with myocarditis and myasthenia gravis. 74 , 75 T cells may be targeting the antigen shared by tumour and striated muscle, or the same TCR may be targeting a tumour antigen and a different muscle antigen. 76 In a recent pharmacovigilance study, myocarditis occurred with a median time to onset of 30 days, but as early as after the first dose of treatment. This same study reported a 50% mortality rate in patients developing ICI‐related myocarditis. 75
1.3.8. Renal toxicities
In an analysis of all published phase II and III clinical trials, 1 review found an overall incidence of acute kidney injury (AKI) of 2.2% across all agents, with grade 3 or 4 AKI having an incidence of 0.6%. 77 This review also found that patients were at a higher risk of developing AKI with combination therapy (4.9%) when compared to monotherapy with ipilimumab (2.0%), nivolumab (1.9%) or pembrolizumab (1.4%). 77 In patients receiving CTLA‐4 inhibitors, the median onset of renal toxicity was 6–12 weeks, compared to 6–12 months with PD‐1 inhibitors. 36 , 78 The disparity of onset between agents is an important distinction to note when discussing therapies. When CTLA‐4 is inhibited, Tregs and other suppressive mechanisms may be lost (Figure 1, v), leading to an uncontrolled activation of autoreactive T cells, which can migrate and infiltrate the kidney. 78 As a protective mechanism, PD‐L1 can be upregulated in renal cells which then bind to PD‐1 to suppress T‐cell activation and prevent proliferation and damage of kidney tissue. When PD‐1 is blocked, this process no longer occurs, and T cells can proliferate and injure kidney tissue. 79 PD‐L1 expression is critical for mitigating inflammatory responses throughout many tissues, such as in the kidney. 80
1.3.9. Mechanisms of toxicity
ICI efficacy relates to the enhanced activation of T cells responsive to tumour antigen, though as discussed, the same mechanisms are thought to drive the associated toxicities, albeit in response to nontumour antigens. While self‐antigens are implicated, it is possible that antigens from other sources, such as viruses and coadministered drugs also drive ICI‐treatment associated adverse responses. These antigens are associated with the development of tissue‐specific toxicities with distinct similarities to those seen with ICI therapy, and may be similarly seen as immunogenic in the checkpoint dysregulated environment (Figure 2a,b). Indeed, reports are now published that describe a high incidence of sulfasalazine‐ or vemurafenib‐induced cutaneous drug reactive T‐lymphocyte associated hypersensitivity reactions in patients previously administered ICIs. 81 , 82 As early reports, detailed mechanistic studies have not yet extracted and validated the drug specificity of T‐cells from these patients, but severe T‐cell mediated reactions to sulfonamides that lead to self‐tissue damage are well‐documented in their own right. Although ICI toxicities occur in many patients without pre‐existing autoimmune disease or antigen exposure and patients with underlying autoimmune disease will necessarily exacerbate their underlying disease, these studies highlight the potential that ICI could enhance susceptibility to both autoimmune and allergic disease as sulfasalazine was administered in this cohort to treat ICI‐induced inflammatory arthritis. This is a deeply worrying concept for the prediction of adverse events due to the enormous variability in antigen exposure between patients. This complexity alongside a requirement for the presence of many additional host and genetic risk factors that are required for the presentation of a particular antigen i.e. HLA risk alleles and corresponding TCR, will further define the relationships between ICI efficacy and toxicity.
1.4. Host predisposition
1.4.1. Autoimmune diseases
The mechanism of irAEs resulting from activation of T‐cells against host tissues closely resembles that of autoimmune diseases, leading to fears that this could lead to: (i) unacceptable immune activation in patients with pre‐existing autoimmune disease; and (ii) autoimmune onset in patients without prior disease. 83 Because of this concern, most clinical trials excluded patients with a history of autoimmune disease. A retrospective study assessing patients with pre‐existing autoimmune disorders treated with ipilimumab for advanced melanoma showed that 27% had an exacerbation of their autoimmune disease while 33% experienced a conventional irAE. All but 1 event resolved with corticosteroids, where a patient with psoriasis died from complications resulting from colitis. The overall response rate of 20% with ipilimumab did not differ from the response rate seen in clinical trials. 84 Another study of patients with pre‐existing autoimmune diseases treated with a PD‐1 or PD‐L1 inhibitor showed that 30% of patients experienced an exacerbation of their autoimmune disease, while 29% experienced a conventional irAE. This study noted that no patients with a gastrointestinal or neurological autoimmune disease experienced an exacerbation, and that 52% of patients with rheumatological disorders experienced low grade flares. More patients who experienced conventional irAEs had to discontinue therapy (8%) compared to those who experienced autoimmune disease flares (4%). The overall response rate of 33% was consistent with response rates seen in clinical trials. 85 Although not observed in the previous studies, there are case reports of patients with pre‐existing myasthenia gravis experiencing flares after the initiation of an ICI. 86 These reviews suggest that while checkpoint inhibitors may increase autoimmune disease exacerbations, they may be just as likely to be associated with de novo disease in those who would not have otherwise been predisposed. Furthermore, flares in patients with underlying immune disease can be managed and overall tumour response rates do not appear to be affected.
1.4.2. Microbiome
The human microbiome is the collective microbial community that lives symbiotically in the body and the gut represents the internal organ with the largest presence of microbes. The human microbiome is driven by host genomic and ecological factors including host immunogenomic background. In the gut, 98% of the microbes are represented by Firmicutes, Bacteroides, Proteobacteria, and Actinobacteria. Disruption by external factors can cause a change in the composition of the gut microbiome called gut dysbiosis. 87 The greatest external factor in microbiome disruption is thought to be from broad‐spectrum antibiotics. 88 While historically it was known that the microbiota plays a role in immune tolerance against opportunistic infections, recent evidence has shown the microbiota has an even broader than anticipated role with effects on innate and adaptive immunity. 89 The largest source of lymphocytes in the human body is found in the gut lamina propria and is associated with a careful balance of various players in adaptive immunity, such as T‐helper cells and Tregs. Autoimmune colitis has been associated with a disruption of this balance. 90 Studies have also shown the importance of the microbiome in relation to immune checkpoint inhibitor‐related toxicity. Studies have demonstrated that supplementation of certain gut microbiota such as Bacteroides fragilis and Burkholderia cepacia resulted in improvement of toxicity in preclinical models of mice treated with CTLA‐4 inhibitors. 91 In melanoma patients treated with CTLA‐4 inhibitors, those who were colitis‐free were noted to have an abundance of Bacteroides and an increase in the expression of genetic pathways involved in polyamine transport and vitamin B synthesis. 92 To further characterize the effects of the microbiome on toxicity, 2 studies have demonstrated that patients treated with CTLA‐4 inhibitors who had a higher abundance of Faecalibacterium prausnitzii and lower levels of Bacteroides had higher rates of colitis. 93 , 94 Aside from certain microbiota, 1 study demonstrated an increased incidence of colitis after ICIs in patients with increased expression of the costimulatory checkpoint molecule inducible costimulator on effector CD4+ T‐cells in addition to decreased Tregs and low circulating inflammatory proteins (interleukin [IL]‐6, IL‐8 and sCD25). These differences in expression are thought to be mediated by differences in the composition of the individual patient's gut microbiome. 93 Efficacy has also been affected by microbiome composition. One study of melanoma patients demonstrated an increase in response to PD‐1 inhibitors in patients with a microbiome with higher levels of Bifidobacterium longum, Collinsella aerofaciens and Enterococcus faecium. 95 Thus, immunotherapy efficacy and toxicity are both affected by the balance of microbiota in the gut.
1.5. Biomarkers for toxicity
The use of quantitative imaging biomarkers has provided promising opportunities to help predict both efficacy and toxicity risk associated with ICI use. 96 Genome‐wide association studies have identified genetic variants predictive for protection or risk in relation to autoimmunity. The mechanism of ICIs probably represents a complex interplay between the type of ICI used, the tumour, the host and other ecological factors. Given the strong association between host genetics and early immune responses including the development of the microbiome, it is very likely that germline genetic variations could also impact irAEs. 97 Variants in the major histocompatibility complex locus are a strong predictor for a wide spectrum of immunologically mediated diseases; however, the relationship between these experiments of nature and the experiments of man that ICIs create is unclear at this time. Evidence supports that not all patients carrying traditional HLA class I or II risk factors in the presence of an underlying autoimmune illness will present de novo or flare on ICIs. It may be that the complex environment of neoantigens that the tumour creates in combination with ICIs may be permissive for patients to develop autoimmune and other immune‐mediated diseases in the absence of traditional immunogenomic risk factors. 98
In 2 patients who developed fulminant myocarditis after ipilimumab and nivolumab, an analysis of T‐cells infiltrating the myocardium, skeletal muscle and tumour revealed common clones of T‐cells in muscle and tumour. The tumours (in this case melanoma) expressed muscle‐specific antigens, such as desmin and troponin. 76 Troponin is a protein present in cardiomyocytes that is released into the blood in response to cardiac damage. 99 In 1 systematic review, while nonspecific, elevations in troponin were noted in 98% of irAE cardiotoxicity cases. 100 This discovery led to the theory that an epitope shared by tumour and healthy tissue contributed to the development of myocarditis. 76 Autoimmune‐related effects may be due to epitope spreading related to immunotherapy‐induced inflammation and tumour lysis. Ipilimumab can induce epitope spreading which can prompt autoimmunity against healthy tissue. 101
Circulating cytokines have been observed as a marker for toxicity. A study of melanoma patients receiving combination PD‐1 and CTLA‐4 inhibitor therapy examined peripheral blood in pre‐, early‐, mid‐ and late‐treatment phases. Of the 51 patients enrolled, 24 (48%) experienced severe toxicity. Eleven cytokines were associated with severe toxicity (granulocyte colony‐stimulating factor, granulocyte‐macrophage colony‐stimulating factor, fractalkine, fibroblast growth factor 2, interferon α2, IL12p70, IL1a, IL1B, IL1RA, IL2, IL13). 102 Another study of patients receiving ICI monotherapy and combination therapy showed that baseline expression of IL‐17, CXCL10 and TGFb1 was associated with the development of irAEs. 103 A retrospective trial of all patients treated with checkpoint inhibitors for advanced cancers demonstrated that elevations in sCD40L, PDGF‐A and PDGF‐B prior to initiation of therapy were associated with an increased risk of developing an irAE. The biomarker, sCD40L, has been shown to be elevated in patients with advanced malignancies, but was not associated with checkpoint inhibitor toxicity previously. 104 While these data highlight cytokines as markers for development of irAE, there remains little consensus correlation between studies. This may relate to different focus criteria of each study; however, it may be that different cytokine biomarkers exist for different cancers and suggests a panel approach, rather than an individual cytokine of interest, would be more predictive.
Recently, a new study has identified the potential of autoimmune antibodies in the development of irAEs by screening autoantibodies in the plasma of ICI‐treated patients. 105 Anti‐GNAL and anti‐ITM2B autoantibodies were associated with the development of hypophysitis. Increases in anti‐CD74 autoantibody was associated with the development of pneumonitis. The study also determined a potential role of the anti‐GNAL autoantibody as a predictive and treatment‐related biomarker for the development of hypophysitis. Anti‐ITM2B has a potential role as an on‐treatment biomarker for hypophysitis. Lastly, elevations in anti‐CD74 may represent a predictive and treatment‐related biomarker for the development of pneumonitis. 105
In clinical practice, the use of biomarker monitoring is currently being explored for prediction and earlier diagnosis of irAEs. In many cases, the recommendation of which biomarker should be tested depends on the specific organ that may be affected. For example, serial measurements of troponin may suggest evidence of myocarditis and at least some groups advocate for surveillance of troponin in high‐risk patients (for example, those given combination ICI). 106 In patients where the onset of irAEs is considered likely, current recommendations include the collection of troponin, C‐reactive protein, autoimmune antibodies such as antinuclear body, rheumatoid factor, anticitrullinating protein body and erythrocyte sedimentation rate. 47 In addition, serial troponin has been used as a marker for early detection of cardiotoxicity in patients with NSCLC, although this theory is still being explored further. 107
While several biomarkers have been studied and associated with an increased risk of irAEs, this is still an evolving field in which new data continue to surface as these agents are utilized in more diverse patient populations. The utilization of validated biomarkers as both predictive and serial determinants of irAEs will help manage ICI therapy.
The emerging knowledge of mechanisms and the relationship of biomarkers with ICI therapeutic response will shed important light for optimizing treatment. A recent analysis observed a relationship between confirmed response and the development of irAEs. This analysis found the odds ratio (OR) of developing an irAE based on responders (OR 5.38) compared to nonresponders (OR 3.77). 108 With this correlation, and if confirmed to efficacy, this could lead to the clinical question of the use of corticosteroids to decrease the extent of irAEs. Although thought to dampen immune responses, this analysis observed no difference on duration of response in patients who developed irAEs. However, recent data have suggested that use of high‐dose steroids, particularly at the start of treatment, may be associated with inferior outcomes. 109 , 110
2. DISCUSSION
Checkpoint inhibitors have revolutionized the treatment of many cancers. While these agents have produced impressive and durable responses, irAEs remain a common occurrence. Due to nonspecific activation of the immune system, these agents have the potential to affect almost any organ system. 15 Most mechanisms of irAEs are inclusive of effects on effector T‐cells, Tregs and tissues that express these receptors/ligands. IrAEs have the potential to occur at any time during or after cessation of therapy and long‐term follow‐up is required for patients who have received ICIs. Patients treated with CTLA‐4 inhibitors had a higher occurrence of all‐grade irAEs when compared to those treated with PD‐1 or PD‐L1 inhibitors. Skin and GI toxicities are the most common across all agents and are typically manageable with standard treatment algorithms. Patients with baseline autoimmune illnesses have been shown to be at an increased risk for irAEs due to the nonspecific activation of T‐cells, mimicking their autoimmune disease, although as new data become available, this statement may not be as simple. While this patient population experienced higher rates of all grade irAEs, there was not a significant increase in grades 3–5 irAEs, and most were manageable with steroid treatment. 111 Adverse effects on the microbiome, such as those that lead to low diversity have been correlated with effects on ICI efficacy and an increase in toxicity. 112 The lack of certain microbiota such as Bacteroides, and an increase in inducible costimulator expression, have been linked to the development of colitis. Certain biomarkers have also been linked to development of ICI toxicity. While these data are nascent, certain elevated biomarkers such as various interleukins and sCD40L have been observed with increases in toxicity. There is currently limited information on what the genetic risk factors are that predispose to irAEs; however, these will be important to help elucidate mechanisms, risk stratification and toxicity pre‐emption and prevention.
2.1. Future directions and unmet needs
ICIs are promising agents that have transformed the treatment of melanoma and a number of other tumours. Although the short‐term toxicities have been now relatively well described, the long‐term effects on the host remain unclear. An understanding of the mechanisms of toxicities and risk stratification and early diagnosis in patient populations will be essential to managing the risk benefit ratio of treatment and developing the most targeted strategies for treating irAEs that do not compromise tumour outcomes. As more patients are treated with ICI therapy, a more comprehensive efficacy and toxicity clinical picture will be developed. There are currently clinical trials underway aimed at unstudied patient populations, including those with pre‐existing autoimmune and inflammatory diseases and patients on antibiotics. While the mechanism of toxicity is of great interest, specific risk stratification has not yet been developed. New agents are on the horizon as well, with inhibition of novel checkpoints unleashing natural killer (NK) cells. NKG2A is an intracytoplasmic tyrosine‐based inhibitory motif‐bearing receptor expressed on both T and NK cells. NKG2A blockage was shown to enhance antitumour immunity meditated by both NK and CD8+ T‐cells. 113 Killer immunoglobulin‐like receptors have been known to regulate NK‐cell activation. 114 The inhibition of killer immunoglobulin‐like receptors has demonstrated an increase in NK activity with a trend towards an increase in response rates when combined with PD‐1 inhibition in head and neck cancer patients (NCT101714739). 115 For the future, knowledge of how these drugs might benefit patients and which specific patient populations, both alone and in combination with other ICIs, will be important.
2.2. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
COMPETING INTERESTS
Dr Phillips is codirector of IIID Pty Ltd that holds a patent for HLA‐B*57:01 testing for abacavir hypersensitivity; she holds a provisional patent for HLA‐A*32:01 testing for vancomycin hypersensitivity.
Douglas B. Johnson is on advisory boards for Array Biopharma, BMS, Merck, Novartis, and research funding from BMS and Incyte.
Javid J. Moslehi is on advisory boards for Novartis, Pfizer, BMS, GSK, Takeda, Nektar, AstraZeneca, Intrexon and Regeneron.
No other authors on this paper have any actual or potential conflicts of interests pertaining to the information in this review article.
CONTRIBUTORS
All authors on this paper contributed to both the research, writing and review of this article. Each author carries an expertise in an area of this review and was able to significantly impact the quality of this review.
ACKNOWLEDGEMENTS
Dr Phillips receives funding from: National Institutes of Health (1P50GM115305‐01, R21AI139021, and 1 R01 HG010863‐01) and the National Health and Medical Research Council of Australia. D.B.J., E.J.P. and J.M.B. receive funding from NCI 1R01CA227481.
Mangan BL, McAlister RK, Balko JM, et al. Evolving insights into the mechanisms of toxicity associated with immune checkpoint inhibitor therapy. Br J Clin Pharmacol. 2020;86:1778–1789. 10.1111/bcp.14433
REFERENCES
- 1. Mok TSK, Wu YL, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): a randomised, open‐label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819‐1830. [DOI] [PubMed] [Google Scholar]
- 2. Eggermont AM, Chiarion‐Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high‐risk stage III melanoma (EORTC 18071): a randomised, double‐blind, phase 3 trial. Lancet Oncol. 2015;16(5):522‐530. [DOI] [PubMed] [Google Scholar]
- 3. Hodi FS, Chiarion‐Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4‐year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19(11):1480‐1492. [DOI] [PubMed] [Google Scholar]
- 4. Paz‐Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non‐small‐cell lung cancer. N Engl J Med. 2018;379(21):2040‐2051. [DOI] [PubMed] [Google Scholar]
- 5. Horn L, Mansfield AS, Szczęsna A, et al. First‐line Atezolizumab plus chemotherapy in extensive‐stage small‐cell lung cancer. N Engl J Med. 2018;379(23):2220‐2229. [DOI] [PubMed] [Google Scholar]
- 6. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379(22):2108‐2121. [DOI] [PubMed] [Google Scholar]
- 7. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus Sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2019;380(12):1116‐1127. [DOI] [PubMed] [Google Scholar]
- 8. Makker V, Rasco D, Vogelzang NJ, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open‐label, single‐arm, phase 2 trial. Lancet Oncol. 2019;20(5):711‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoos A. Development of immuno‐oncology drugs ‐ from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15(4):235‐247. [DOI] [PubMed] [Google Scholar]
- 11. Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti‐CTLA‐4 and anti‐PD‐1 checkpoint blockade. Cell. 2017;170:1120‐33.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26(1):677‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167‐3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune‐related response criteria. Clin Cancer Res. 2009;15(23):7412‐7420. [DOI] [PubMed] [Google Scholar]
- 15. Puzanov I, Diab A, Abdallah K, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of cancer (SITC) toxicity management working group. J Immunother Cancer. 2017;5(1):1‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toi Y, Sugawara S, Kawashima Y, et al. Association of Immune‐Related Adverse Events with clinical benefit in patients with advanced non‐small‐cell lung cancer treated with Nivolumab. Oncologist. 2018;23(11):1358‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492‐499. [DOI] [PubMed] [Google Scholar]
- 18. Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchbinder EI, Desai A. CTLA‐4 and PD‐1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA‐4 and PD‐1 receptors inhibit T‐cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543‐9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gibson A, Faulkner L, Lichtenfels M, et al. The effect of inhibitory signals on the priming of drug Hapten‐specific T cells that express distinct Vbeta receptors. J Immunol. 2017;199(4):1223‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ott PA, Hodi FS, Robert C. CTLA‐4 and PD‐1/PD‐L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19(19):5300‐5309. [DOI] [PubMed] [Google Scholar]
- 23. Huang YF, Xie WJ, Fan HY, Du J. Comparative safety of PD‐1/PD‐L1 inhibitors for cancer patients: systematic review and network meta‐analysis. Front Oncol. 2019;9:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pillai RN, Behera M, Owonikoko TK, et al. Comparison of the toxicity profile of PD‐1 versus PD‐L1 inhibitors in non‐small cell lung cancer: a systematic analysis of the literature. Cancer. 2018;124(2):271‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Michot JM, Bigenwald C, Champiat S, et al. Immune‐related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139‐148. [DOI] [PubMed] [Google Scholar]
- 26. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simpson TR, Li F, Montalvo‐Ortiz W, et al. Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J Exp Med. 2013;210(9):1695‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18(6):733‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ascierto PA, Del Vecchio M, Robert C, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double‐blind, multicentre, phase 3 trial. Lancet Oncol. 2017;18:611‐622. [DOI] [PubMed] [Google Scholar]
- 30. Wolchok JD, Chiarion‐Sileni V, Gonzalez R, et al. Overall survival with combined Nivolumab and Ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hellmann MD, Paz‐Ares L, Bernabe Caro R, et al. Nivolumab plus Ipilimumab in advanced non‐small‐cell lung cancer. N Engl J Med. 2019;381(21):2020‐2031. [DOI] [PubMed] [Google Scholar]
- 33. Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of Nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35(7):785‐792. [DOI] [PubMed] [Google Scholar]
- 34. Weber JS, Kahler KC, Hauschild A. Management of immune‐related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691‐2697. [DOI] [PubMed] [Google Scholar]
- 35. Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19(3):345‐361. [DOI] [PubMed] [Google Scholar]
- 36. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune‐related toxicities. Transl Lung Cancer Res. 2015;4(5):560‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lacouture M, Sibaud V. Toxic side effects of targeted therapies and immunotherapies affecting the skin, Oral mucosa, hair, and nails. Am J Clin Dermatol. 2018;19(S1):31‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quach HT, Dewan AK, Davis EJ, et al. Association of Anti‐Programmed Cell Death 1 cutaneous toxic effects with outcomes in patients with advanced melanoma. JAMA Oncol. 2019;5(6):906‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verzoni E, Cartenì G, Cortesi E, et al. Real‐world efficacy and safety of nivolumab in previously‐treated metastatic renal cell carcinoma, and association between immune‐related adverse events and survival: the Italian expanded access program. J Immunother Cancer. 2019;7(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teraoka S, Fujimoto D, Morimoto T, et al. Early immune‐related adverse events and association with outcome in advanced non‐small cell lung cancer patients treated with Nivolumab: a prospective cohort study. J Thorac Oncol. 2017;12(12):1798‐1805. [DOI] [PubMed] [Google Scholar]
- 41. Friedman CF, Proverbs‐Singh TA, Postow MA. Treatment of the immune‐related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2(10):1346‐1353. [DOI] [PubMed] [Google Scholar]
- 42. Gupta A, De Felice KM, Loftus EV Jr, Khanna S. Systematic review: colitis associated with anti‐CTLA‐4 therapy. Aliment Pharmacol Ther. 2015;42(4):406‐417. [DOI] [PubMed] [Google Scholar]
- 43. Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double‐blind, multicentre, phase 2, dose‐ranging study. Lancet Oncol. 2010;11(2):155‐164. [DOI] [PubMed] [Google Scholar]
- 44. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521‐2532. [DOI] [PubMed] [Google Scholar]
- 45. Reynolds K, Thomas M, Dougan M. Diagnosis and Management of Hepatitis in patients on checkpoint blockade. Oncologist. 2018;23(9):991‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016;44:51‐60. [DOI] [PubMed] [Google Scholar]
- 47. Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of Immune‐Related Adverse Events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2018;36:1714‐1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim KW, Ramaiya NH, Krajewski KM, et al. Ipilimumab associated hepatitis: imaging and clinicopathologic findings. Invest New Drugs. 2013;31(4):1071‐1077. [DOI] [PubMed] [Google Scholar]
- 49. Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug‐induced liver injury. Mod Pathol. 2018;31(6):965‐973. [DOI] [PubMed] [Google Scholar]
- 50. Villa NM, Farahmand A, Du L, et al. Endocrinopathies with use of cancer immunotherapies. Clin Endocrinol (Oxf). 2018;88(2):327‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cukier P, Santini FC, Scaranti M, Hoff AO. Endocrine side effects of cancer immunotherapy. Endocr Relat Cancer. 2017;24:T331‐t47. [DOI] [PubMed] [Google Scholar]
- 52. Laurent S, Queirolo P, Boero S, et al. The engagement of CTLA‐4 on primary melanoma cell lines induces antibody‐dependent cellular cytotoxicity and TNF‐alpha production. J Transl Med. 2013;11(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, Michels AW. Immune checkpoint inhibitor‐induced type 1 diabetes: a systematic review and meta‐analysis. Diabet Med. 2019;36(9):1075‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoneda S, Imagawa A, Hosokawa Y, et al. T‐lymphocyte infiltration to islets in the pancreas of a patient who developed type 1 diabetes after administration of immune checkpoint inhibitors. Diabetes Care. 2019;42:e116‐e118. [DOI] [PubMed] [Google Scholar]
- 55. Martinov, Spanier JA, Pauken KE, Fife BT. PD‐1 pathway‐mediated regulation of islet‐specific CD4(+) T cell subsets in autoimmune diabetes. Immunoendocrinology (Houst). 2016;3:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khunger M, Rakshit S, Pasupuleti V, et al. Incidence of pneumonitis with use of programmed death 1 and programmed death‐ligand 1 inhibitors in non‐small cell lung cancer: a systematic review and meta‐analysis of trials. Chest. 2017;152(2):271‐281. [DOI] [PubMed] [Google Scholar]
- 57. Naidoo J, Wang X, Woo KM, et al. Pneumonitis in patients treated with anti‐programmed Death‐1/programmed death ligand 1 therapy. J Clin Oncol. 2017;35(7):709‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu J, Hong D, Zhang X, Lu X, Miao J. PD‐1 inhibitors increase the incidence and risk of pneumonitis in cancer patients in a dose‐independent manner: a meta‐analysis. Sci Rep. 2017;7(1):1‐12, 44173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang H, Guo X, Zhou J, et al. Clinical diagnosis and treatment of immune checkpoint inhibitor‐associated pneumonitis. Thorac Cancer. 2020;11(1):191‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cho JY, Kim J, Lee JS, et al. Characteristics, incidence, and risk factors of immune checkpoint inhibitor‐related pneumonitis in patients with non‐small cell lung cancer. Lung Cancer. 2018;125:150‐156. [DOI] [PubMed] [Google Scholar]
- 61. Ma K, Lu Y, Jiang S, Tang J, Li X, Zhang Y. The relative risk and incidence of immune checkpoint inhibitors related pneumonitis in patients with advanced cancer: a meta‐analysis. Front Pharmacol. 2018;9:1‐12, 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Läubli H, Koelzer VH, Matter MS, et al. The T cell repertoire in tumors overlaps with pulmonary inflammatory lesions in patients treated with checkpoint inhibitors. Oncoimmunology. 2018;7(2):1‐7, e1386362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Oh DY, Cham J, Zhang L, et al. Immune toxicities Elicted by CTLA‐4 blockade in cancer patients are associated with early diversification of the T‐cell repertoire. Cancer Res. 2017;77(6):1322‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cuzzubbo S, Javeri F, Tissier M, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. 2017;73:1‐8. [DOI] [PubMed] [Google Scholar]
- 65. Hottinger AF. Neurologic complications of immune checkpoint inhibitors. Curr Opin Neurol. 2016;29(6):806‐812. [DOI] [PubMed] [Google Scholar]
- 66. Johnson DB, Manouchehri A, Haugh AM, et al. Neurologic toxicity associated with immune checkpoint inhibitors: a pharmacovigilance study. J Immunother Cancer. 2019;7(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dubey D, David WS, Reynolds KL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing Spectrum. Ann Neurol. 2020;87(5):659‐669. [DOI] [PubMed] [Google Scholar]
- 68. Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun. 2013;45:90‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Love VA, Grabie N, Duramad P, Stavrakis G, Sharpe A, Lichtman A. CTLA‐4 ablation and interleukin‐12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ Res. 2007;101(3):248‐257. [DOI] [PubMed] [Google Scholar]
- 70. Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD‐1 receptor‐deficient mice. Science. 2001;291(5502):319‐322. [DOI] [PubMed] [Google Scholar]
- 71. Okazaki T, Tanaka Y, Nishio R, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD‐1‐deficient mice. Nat Med. 2003;9(12):1477‐1483. [DOI] [PubMed] [Google Scholar]
- 72. Grabie N, Gotsman I, DaCosta R, et al. Endothelial programmed death‐1 ligand 1 (PD‐L1) regulates CD8+ T‐cell mediated injury in the heart. Circulation. 2007;116(18):2062‐2071. [DOI] [PubMed] [Google Scholar]
- 73. Mahmood SS, Fradley MG, Cohen JV, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J am Coll Cardiol. 2018;71(16):1755‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Moslehi JJ, Salem JE, Sosman JA, Lebrun‐Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor‐associated myocarditis. Lancet. 2018;391(10124):933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Salem JE, Manouchehri A, Moey M, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;19(12):1579‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cortazar FB, Marrone KA, Troxell ML, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90(3):638‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wanchoo R, Karam S, Uppal NN, et al. Adverse renal effects of immune checkpoint inhibitors: a narrative review. Am J Nephrol. 2017;45(2):160‐169. [DOI] [PubMed] [Google Scholar]
- 79. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD‐1 pathway in the immune response. Am J Transplant. 2012;12(10):2575‐2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Menke J, Lucas JA, Zeller GC, et al. Programmed death 1 ligand (PD‐L) 1 and PD‐L2 limit autoimmune kidney disease: distinct roles. J Immunol. 2007;179(11):7466‐7477. [DOI] [PubMed] [Google Scholar]
- 81. Ford M, Sahbudin I, Filer A, Steven N, Fisher BA. High proportion of drug hypersensitivity reactions to sulfasalazine following its use in anti‐PD‐1‐associated inflammatory arthritis. Rheumatology (Oxford). 2018;57(12):2244‐2246. [DOI] [PubMed] [Google Scholar]
- 82. Uhara H, Kiyohara Y, Tsuda A, Takata M, Yamazaki N. Characteristics of adverse drug reactions in a vemurafenib early post‐marketing phase vigilance study in Japan. Clin Transl Oncol. 2018;20(2):169‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123(11):1904‐1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Johnson DB, Sullivan RJ, Ott PA, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol. 2016;2(2):234‐240. [DOI] [PubMed] [Google Scholar]
- 85. Menzies AM, Johnson DB, Ramanujam S, et al. Anti‐PD‐1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol. 2017;28(2):368‐376. [DOI] [PubMed] [Google Scholar]
- 86. Amy Kamien AK. Cheruppolil Santhosh‐Kumar. Reactivation of Myasthenia Gravis Secondary to Nivolumab: Case Report and Literature Review Journal of Hematology Oncology Pharmacy. 2019;9:24‐29. [Google Scholar]
- 87. Baiden‐Amissah REM, Tuyaerts S. Contribution of aging, obesity, and microbiota on tumor immunotherapy efficacy and toxicity. Int J Mol Sci. 2019;20(14):1‐12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kho ZY, Lal SK. The human gut microbiome ‐ a potential controller of wellness and disease. Front Microbiol. 2018;9:1‐23, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535(7610):75‐84. [DOI] [PubMed] [Google Scholar]
- 90. Sethi V, Vitiello GA, Saxena D, Miller G, Dudeja V. The role of the microbiome in immunologic development and its implication for pancreatic cancer immunotherapy. Gastroenterology. 2019;156:2097‐115.e2. [DOI] [PubMed] [Google Scholar]
- 91. Vetizou M, Pitt JM, Daillere R, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science. 2015;350:1079‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dubin K, Callahan MK, Ren B, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint‐blockade‐induced colitis. Nat Commun. 2016;7(1):1‐8, 10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chaput N, Lepage P, Coutzac C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2019;30(12):1368‐1379. [DOI] [PubMed] [Google Scholar]
- 94. Frankel AE, Coughlin LA, Kim J, et al. Metagenomic shotgun sequencing and unbiased Metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. 2017;19(10):848‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Trebeschi S, Drago SG, Birkbak NJ, et al. Predicting response to cancer immunotherapy using non‐invasive Radiomic biomarkers. Ann Oncol. 2019;30(6):998‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Khan Z, Hammer C, Guardino E, Chandler GS, Albert ML. Mechanisms of immune‐related adverse events associated with immune checkpoint blockade: using germline genetics to develop a personalized approach. Genome Med. 2019;11(1):1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gutierrez‐Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases ‐ connecting risk alleles with molecular traits of the immune system. Nat Rev Genet. 2016;17(3):160‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mahajan VS, Jarolim P. How to interpret elevated cardiac troponin levels. Circulation. 2011;124(21):2350‐2354. [DOI] [PubMed] [Google Scholar]
- 100. Pradhan R, Nautiyal A, Singh S. Diagnosis of immune checkpoint inhibitor‐associated myocarditis: a systematic review. Int J Cardiol. 2019;296:113‐121. [DOI] [PubMed] [Google Scholar]
- 101. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles' heel of cancer immunotherapy? Nat Med. 2017;23(5):540‐547. [DOI] [PubMed] [Google Scholar]
- 102. Lim SY, Lee JH, Gide TN, et al. Circulating cytokines predict immune‐related toxicity in melanoma patients receiving anti‐PD‐1‐based immunotherapy. Clin Cancer Res. 2019;25(5):1557‐1563. [DOI] [PubMed] [Google Scholar]
- 103. Meghan Mooradian XG, Donald P. Lawrence, Justine Vanesa Cohan, Tatyana Sharova, Genevieve Marie Boland, Towia a Libermann, Ryan K. Sullivan. Predictive plasma proteomic biomarkers of immunotherapy toxicity in patients (pts) with metastatic melanoma (MM). J Clin Oncol. 2018;36. [Google Scholar]
- 104. Meti NEK, Colmegna I, Fritzler MJ, et al. Elevated sCD40L as a predictive biomarker of immune‐related adverse events in patients receiving immune checkpoint inhibitors [abstract]. Arthritis Rheumatol. 2018. [Google Scholar]
- 105. Tahir SA, Gao J, Miura Y, et al. Autoimmune antibodies correlate with immune checkpoint therapy‐induced toxicities. Proc Natl Acad Sci U S a. 2019;116(44):22246‐22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hu JR, Florido R, Lipson EJ, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. 2019;115(5):854‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sarocchi M, Grossi F, Arboscello E, et al. Serial troponin for early detection of Nivolumab cardiotoxicity in advanced non‐small cell lung cancer patients. Oncologist. 2018;23(8):936‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Maher VE, Fernandes LL, Weinstock C, et al. Analysis of the association between adverse events and outcome in patients receiving a programmed death protein 1 or programmed death ligand 1 antibody. J Clin Oncol. 2019;37(30):2730‐2737. [DOI] [PubMed] [Google Scholar]
- 109. Arbour KC, Mezquita L, Long N, et al. Impact of baseline steroids on efficacy of programmed cell Death‐1 and programmed death‐ligand 1 blockade in patients with non‐small‐cell lung cancer. J Clin Oncol. 2018;36(28):2872‐2878. [DOI] [PubMed] [Google Scholar]
- 110. Faje AT, Lawrence D, Flaherty K, et al. High‐dose glucocorticoids for the treatment of ipilimumab‐induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer. 2018;124(18):3706‐3714. [DOI] [PubMed] [Google Scholar]
- 111. Cortellini A, Buti S, Santini D, et al. Clinical outcomes of patients with advanced cancer and pre‐existing autoimmune diseases treated with anti‐programmed Death‐1 immunotherapy: a real‐world transverse study. Oncologist. 2019;24:e327‐e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Andre P, Denis C, Soulas C, et al. Anti‐NKG2A mAb is a checkpoint inhibitor that promotes anti‐tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731‐43.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Delgado DC, Hank JA, Kolesar J, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18‐IL2 immunotherapy. Cancer Res. 2010;70(23):9554‐9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bi J, Tian Z. NK cell dysfunction and checkpoint immunotherapy. Front Immunol. 2019;10:1‐10, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]