

Abstract
Sialidosis is an autosomal recessive lysosomal storage disease, belonging to the glycoproteinoses. The disease is caused by deficiency of the sialic acid-cleaving enzyme, sialidase 1 or neuraminidase 1 (NEU1). Patients with sialidosis are classified based on the age of onset and severity of the clinical symptoms into type I (normomorphic) and type II (dysmorphic). Patient-derived skin fibroblasts from both disease types were reprogrammed using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit. iPSCs were characterized for pluripotency, three germ-layer differentiation, normal karyotype and absence of viral components. These cell lines represent a valuable resource to model sialidosis and to screen for therapeutics.
1. Resource table
| Unique stem cell lines identifier and STJUDEi001-A | 1. Set up an account at https://hpscreg.eu/about/naming-tool | ||||
| 2. The system generates and guarantees a unique name based on: researcher’s institution; type of cell line type (iPSC/hESC); additional clone from patient or subclone of a line already present in the database. | |||||
| 3. Include all the unique cell lines name generated HERE. For example: | |||||
| STJUDEi001-A | Male | African American | c.649G > A/ 644T > C | Sialidosis type I | |
| STJUDEi002-A | Male | Hispanic | c.1109A > G/ c.1109A > G | Sialidosis type II | |
| STJUDEi003-A | Female | White | c.1195_1200dup/c.679G > A | Sialidosis type I | |
| Alternative names of stem cell lines | |||||
| Institution | St. Jude Children’s Research Hospital | ||||
| Contact information of distributor | Alessandra d’Azzo (sandra.dazzo@stjude.org) | ||||
| Type of cell lines | iPSCs | ||||
| Origin | Human | ||||
| Cell Source | Fibroblasts | ||||
| Clonality | Clonal | ||||
| Method of reprogramming | Sendai virus (CytoTune™-iPS 2.0 Sendai Reprogramming Kit) | ||||
| Multiline rationale | Disease types, non-isogenic cell lines | ||||
| Gene modification | Yes | ||||
| Type of modification | Spontaneous Mutation | ||||
| Associated disease | Sialidosis (OMIM: #256550) | ||||
| Gene/locus | NEU1 (OMIM: 608272) | ||||
| Mutations: | |||||
| c.649G > A/644 T > C (sialidosis type I patient/STJUDEi001-A) | |||||
| c.1109A > G/c.1109A > G (sialidosis type II patient/STJUDEi002-A) | |||||
| c.1195_1200dup/c.679G > A (sialidosis type I patient/STJUDEi003-A) | |||||
| Method of modification | N/A | ||||
| Name of transgene or resistance | N/A | ||||
| Inducible/constitutive system | N/A | ||||
| Date archived/stock date | |||||
| Cell line repository/bank | |||||
| Ethical approval | Fibroblasts from sialidosis patients type I and II were obtained from the Pediatric Undiagnosed Diseases Program, National Human Genome Research Institute/NIH (Bethesda MD, USA),); from G. Visser (Department of Metabolic Diseases, Wilhelmina Children’s Hospital, University Medical Centre Utrecht, Utrecht, The Netherlands) and from Dr. D. Wenger (Division of Medical Genetics, Jefferson Medical College, Philadelphia, PA, USA). Original consent was obtained by the clinicians from the patient or a family member and the study was approved by the ethics committees of the three institutions.IRB number: 19-0191 | ||||
1.1. Resource utility
We describe the generation of the first human iPSCs reprogrammed from patient-derived type I and type II sialidosis fibroblasts. These iPSCs will serve as versatile tool to understand NEU1 function in undifferentiated and differentiated cells, including neurons, and for drug development.
2. Resource details
NEU1 is a lysosomal exoglycosidase whose main function is to catalyse the cleavage of sialic acids from sialylated glycoconjugates. Owing to this function, the enzyme is involved in numerous, basic physiological processes (d’Azzo et al., 2015; Bonten et al., 1996). NEU1 is catalytically active and stable in lysosomes only when in complex with the glycosidase β-galactosidase (β-GAL) and the carboxypeptidase protective protein/cathepsin A (PPCA) (Bonten et al., 2014). Mutations in the NEU1 gene are the primary cause of sialidosis, an autosomal recessive glycoprotein storage disease. Deficiency/defective NEU1 activity results in lysosomal accumulation of sialylated glycoproteins, which are the natural substrates of the enzyme in vivo. Patients with sialidosis are classified into types I and II, depending on the age of onset and severity of their clinical symptoms. Type I sialidosis, also known as cherry-red spot myoclonus syndrome, is the mild non-neuropathic form of the disease. Symptoms occur in the second decade of life and primarily consist of progressive loss of vision, myoclonus epilepsy and ataxia (d’Azzo et al., 2015). Sialidosis type II is the more severe, neuropathic form of the disease, comprising three subtypes: congenital or hydropic with onset in utero, infantile with onset between birth and 1 year of age, and juvenile with onset after the second year of life (d’Azzo et al., 2015). Patients with type II have a severe systemic disease and develop, among other symptoms, dysmorphic features and severe mental retardation. The penetrance and degree of severity of the symptoms in these patients correlate with the type of NEU1 mutations involved and, in turn, the levels of residual enzyme activity. Most disease-causing mutations result in single amino acid substitutions and are present either in homozygosity or compound heterozygosity. Skin fibroblasts from sialidosis patients carrying NEU1 mutations C.649G > A/644 T > C, c.1195_1200dup/c.679G > A (Schene et al., 2016) and C.1109A > G/C.1109A > G (Bonten et al., 2000) were chosen as representative of sialidosis type I or type II respectively (see Table 1). The generation of iPSCs from patients’ fibroblasts was carried out using the non-integrating Sendai virus vectors encoding the four Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) (Fig. 1A and Table 1). Generated iPSC lines were free of exogenous reprogramming vectors, as confirmed by RT-PCR (Fig. 1B and Table 1). Their pluripotency was examined by immunocytochemistry and flow cytometry, which confirmed the expression of OCT4, SSEA-3 and SSEA-4 (Fig. 1C and Table 1). Pluripotency was further proven by differentiation of the iPSCs into into ectodermal, endodermal and mesodermal germ layers using the STEM-diff™ Trilineage Differentiation kit. Concomitant expression of PAX6 and Nestin (ectoderm), Brachury and NCAM/CXCR4 (mesoderm), and SOX17 and CXCR4/FOXA2 (endoderm) confirmed the identity of the differentiated cells (Fig. 1D, E and Table 1). Karyotype analyses of the iPSCs demonstrated that no abnormal chromosomal changes occurred during the reprogramming process (Fig. 1F and Table 1). NEU1 gene mutations in the parental fibroblasts and the respective iPSCs were confirmed by Sanger sequencing (Supp. Fig. 1). Comparison of short tandem repeats (STR) between the parental fibroblasts and the derived iPSCs showed that they share identical haplotypes. The established iPSCs from sialidosis types I and II could be used to model this disease and to test novel therapeutics.
Table 1.
Characterization and validation.
| Classification | Test | Result | Data |
|---|---|---|---|
| Morphology | Photography | Normal morphology | Fig. 1 panel A |
| Phenotype | Qualitative analysis (Immunocytochemistry) | Expression of pluripotency markers: Oct4 | Fig. 1 panel B |
| Quantitative analysis (Flow cytometry) |
SSEA-4: 99% SSEA-3: 78% (LS)
SSEA-4: 98% SSEA-3: 62% (GV) |
Fig. 1 panel B | |
| Genotype | Karyotype (G-banding) and resolution |
46XY, 46XY
Resolution 375-525 |
Fig. 1 panel D |
| Identity | Microsatellite PCR (mPCR) OR STR analysis | not performed | N/A |
| Tested 16 sites, all matched | Supplementary Fig. 1 and full analysis data were submitted | ||
| Mutation analysis (IF APPLICABLE) | Sequencing | Heterozygous (LS)/homozygous (GV), type of mutation | Fig. 1 panel D |
| Southern Blot OR WGS | not performed | N/A | |
| Microbiology and virology | Mycoplasma | Mycoplasma testing luminescence. All negative | Supplementary Fig. 1D |
| Differentiation potential | Embryoid body formation and Directed differentiation (3 germ layer) | Expression of Sox17 (Endo), Brachyury (Meso), and Otx2 (Ecto) | Fig. 1 panel E and F |
| Donor screening (OPTIONAL) | HIV 1 + 2 Hepatitis B, Hepatitis C | N/A | |
| Genotype additional info (OPTIONAL) | Blood group genotyping | N/A | |
| HLA tissue typing | N/A |
Fig. 1.
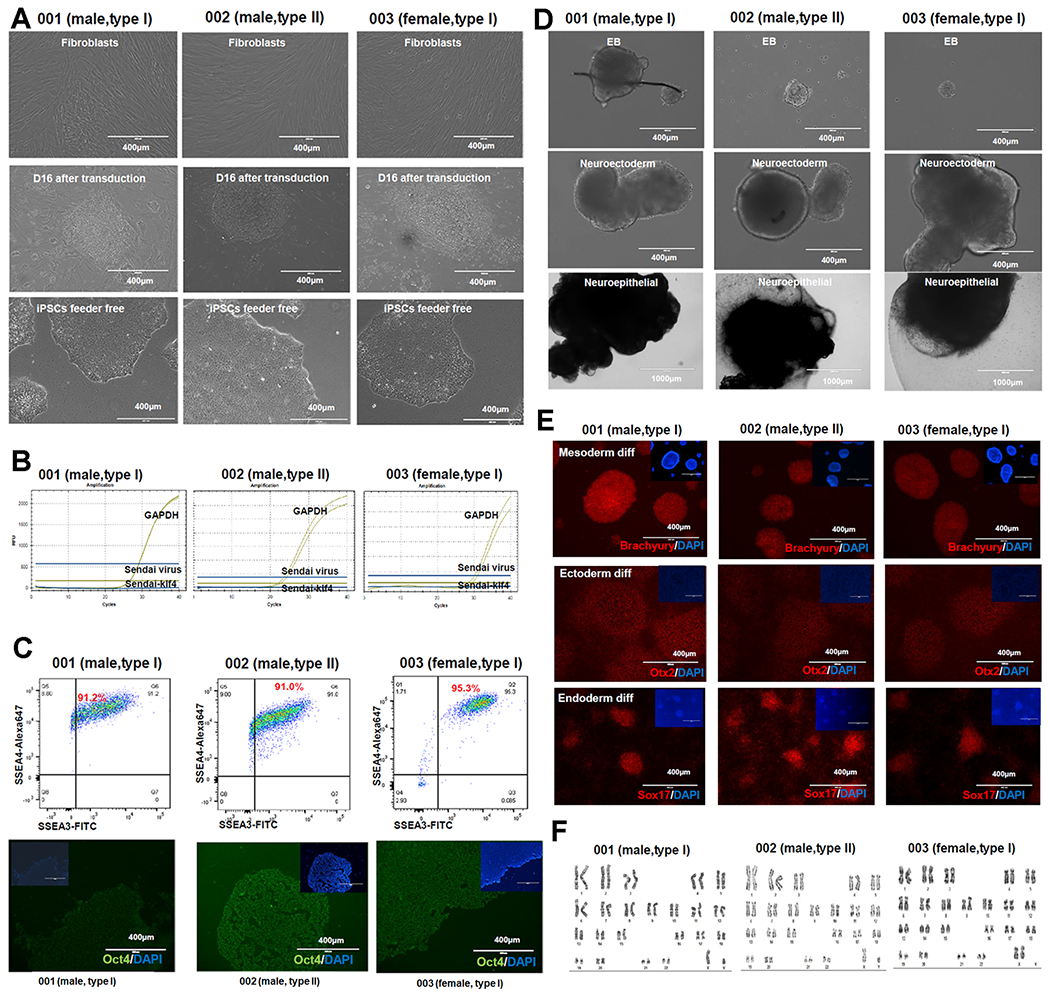
Characterization of generated sialidosis-patient derived iPS cells.
3. Materials and methods
3.1. Fibroblast cell reprogramming
Fibroblasts from sialidosis patients type I and II were obtained from the Pediatric Undiagnosed Diseases Program, National Human Genome Research Institute/NIH (Bethesda MD, USA),); from G. Visser (Department of Metabolic Diseases, Wilhelmina Children’s Hospital, University Medical Centre Utrecht, Utrecht, The Netherlands) and from Dr. D. Wenger (Division of Medical Genetics, Jefferson Medical College, Philadelphia, PA, USA). Original consent was obtained by the clinicians from the patient or a family member and the study was approved by the ethics committees of the three institutions. Fibroblasts were plated in 6-well plates at 2 × 105 cells per well. After 48 h of culture at 37 °C/5% CO2, fibroblasts were transformed using the CytoTune™-iPS 2.0 Sendai reprogramming Kit (Invitrogen) following the manufacturer’s instructions. On day 6 post viral transduction fibroblasts were re-plated on mouse embryonic fibroblast feeder (MEF) plates at 2 × 104–1 × 105 cells per well. iPSC colonies were manually picked and transferred to feeder-free plates and cultured to passage 5 for characterization. Culture media (mTeSR) was changed daily.
3.2. Immunofluorescence (IF)
iPSCs were cultured on 12-well culture plates for IF analysis. Cells were fixed with 4% Paraformaldehyde for 10 min at room temperature, permeabilized in 0.2%Triton™-X-100 (Sigma) for 10 min, blocked in 20% Goat Serum (Life Technologies) for 60 min. Cells were then incubated with primary antibodies for 2 h, followed by secondary antibodies for 2 h at room temperature (Table 2). Subsequently, nuclei were stained with DAPI (VectorLabs) and images were captured with EVOS fluorescence microscope (Invitrogen).
Table 2.
Reagents details.
| Antibodies used for immunocytochemistry/flow-citometry | |||
|---|---|---|---|
| Antibody | Dilution | Company Cat # and RRID | |
|
| |||
| Pluripotency Markers | Rabbit anti-OCT4 | 1:1000 | Thermo Fisher Scientific Cat# A13998 |
| Mouse anti-SSEA4-Alexa647 | 1:50 | Thermo Fisher Scientific Cat# SSEA421 | |
| Mouse anti-SSEA3-FITC | 1:50 | BD Cat# 560236 | |
| Differentiation Markers | Anti-human Otx2 NL557-conjugated Goat IgG | 1:1000 | R&D Systems Cat# SC022 |
| Anti-human Brachyury NL557-conjugated Goat IgG | 1:1000 | R&D Systems Cat# SC022 | |
| Anti-human SOX17 NL637-conjugated Goat IgG | 1:1000 | R&D Systems Cat# SC022 | |
| Secondary antibodies | Goat anti-Rabbit Alexa488 | 1:1000 | Thermo Fisher Scientific Cat# A-11034 |
| Primers | |||
|
| |||
| Target | Forward/Reverse primer (5′-3′) | ||
|
| |||
| Sendai virus detection | SeV | TaqMan probe Mr04269880_mr | |
| Sendai virus detection | SeV/Klf4 | TaqMan probe Mr04421256_mr | |
| House-Keeping Genes (qPCR) | GAPDH | VIC/TAMRA probe 4310884E | |
3.3. Confirmation of pluripotency and Flow cytometry (FACS) analysis
Flow cytometry analysis with antibodies to SSEA3/SSEA4 confirmed the pluripotency of the established iPSCs. Single cell dissociated iPSCs were filtered and incubated with Fc-Block to prevent non-specific binding. Cells were incubated with anti-SSEA3-FITC and anti-SSEA4-Alexa647 antibodies (Table 2) for 30 min on ice, protected from light. Samples were run by the Flow Cytometry Core of St. Jude Children’s Research Hospital. Data were analysed with the FlowJo software.
3.4. Differentiation
Three-germ-layer differentiation was performed according to the manufacturer’s protocol (STEMdiff™ Trilineage Differentiation Kit, STEMCELLTechnologies). To validate expression of each germ layer differentiation, immunofluorescence assays were performed with anti-OTX2 (Ectoderm), anti-BRACHURY (Mesoderm), and anti-SOX17 (Endoderm) antibodies (Table 2). Subsequently, nuclei were stained with DAPI (VectorLabs) and images were captured with EVOS fluorescence microscope (Invitrogen).
3.5. Detection of Sendai virus genome and transgenes
After 10 passages, iPSC lines were tested for elimination of Sendai virus. Total RNA was extracted and transcribed into cDNA using the Zymo Research Direct-zol RNA MiniPrep Plus (Zymo Research). RT-PCR was performed using the listed primers according to the manufacturer’s instructions (Table 2). Positive control RNA was derived from cells harvested immediately after Sendai virus transduction.
3.6. Karyotype analysis
We performed G-banding karyotyping analysis at passage 5 after confirmation of viral clearance. The number of metaphases that we analyzed was 30. G-band karyotyping analyses were performed by the Cytogenetic Shared Resource laboratory at St. Jude Children’s Research Hospital.
3.7. Mycoplasma detection
Absence of mycoplasma contamination was confirmed by analysis with the MycoAlert mycoplasma detection kit (Lonza), following the procedure outlined by the manufacturer.
Supplementary Material
Acknowledgements
A.d’A. holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy. This work was supported, in part, by NIH grants GM104981, the Assisi Foundation of Memphis, the American Lebanese Syrian Associated Charities (ALSAC). We thank the Cytogenetic Shared Resource and the Flow Cytometry and Cell Sorting Shared Resource for karyotyping and FACS analyses.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scr.2020.101836.
References
- d’Azzo A, Machado E, Annunziata L, 2015. Pathogenesis, emerging therapeutic targets and Treatment in Sialidosis. Exp. Opin. Orphan Drugs 3, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonten EJ, van der Spoel A, Fornerod M, Grosveld G, d’Azzo A, 1996. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 10 (31), 56–69. [DOI] [PubMed] [Google Scholar]
- Bonten EJ, Annunziata I, d’Azzo A, 2014. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci. 71, 2017–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schene IF, Kalinina Ayuso V, de Sain-van der Velden M, van Gassen KL, Cuppen I, van Hasselt PM, Visser G, 2016. Pitfalls in diagnosing neuraminidase deficiency: psychosomatics and normal sialic acid excretion. JIMD Rep. 25, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R, Zammarchi E, d’Azzo A, 2000. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet 9 (27), 2715–2725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.