

Abstract
PURPOSE
Children with pediatric gliomas harboring a BRAF V600E mutation have poor outcomes with current chemoradiotherapy strategies. Our aim was to study the role of targeted BRAF inhibition in these tumors.
PATIENTS AND METHODS
We collected clinical, imaging, molecular, and outcome information from patients with BRAF V600E–mutated glioma treated with BRAF inhibition across 29 centers from multiple countries.
RESULTS
Sixty-seven patients were treated with BRAF inhibition (pediatric low-grade gliomas [PLGGs], n = 56; pediatric high-grade gliomas [PHGGs], n = 11) for up to 5.6 years. Objective responses were observed in 80% of PLGGs, compared with 28% observed with conventional chemotherapy (P < .001). These responses were rapid (median, 4 months) and sustained in 86% of tumors up to 5 years while receiving therapy. After discontinuation of BRAF inhibition, 76.5% (13 of 17) of patients with PLGG experienced rapid progression (median, 2.3 months). However, upon rechallenge with BRAF inhibition, 90% achieved an objective response. Poor prognostic factors in conventional therapies, such as concomitant homozygous deletion of CDKN2A, were not associated with lack of response to BRAF inhibition. In contrast, only 36% of those with PHGG responded to BRAF inhibition, with all but one tumor progressing within 18 months. In PLGG, responses translated to 3-year progression-free survival of 49.6% (95% CI, 35.3% to 69.5%) versus 29.8% (95% CI, 20% to 44.4%) for BRAF inhibition versus chemotherapy, respectively (P = .02).
CONCLUSION
Use of BRAF inhibition results in robust and durable responses in BRAF V600E–mutated PLGG. Prospective studies are required to determine long-term survival and functional outcomes with BRAF inhibitor therapy in childhood gliomas.
INTRODUCTION
Pediatric low-grade gliomas (PLGGs) are the most common brain tumors in children.1 Over the last 10 years, the molecular landscape of this heterogeneous group of tumors has been characterized, identifying alterations in the RAS/mitogen-activated protein kinase (MAPK) pathway in a majority of tumors.2-6 Recent data suggest that specific alterations result in different clinical behaviors and prognoses in PLGG.
CONTEXT
Key Objective
To describe patient outcomes and kinetics of tumor response to targeted BRAF V600E inhibition in patients with pediatric glioma.
Knowledge Generated
Objective response was observed in 80% of patients with low-grade gliomas, with progression-free survival (PFS) superior to that seen with chemotherapy. These responses occurred rapidly and were sustained while patients were receiving treatment. However, upon stopping, rapid progression occurred. Patients who were rechallenged with BRAF V600E inhibition responded again with tumor reduction back to their baseline. In contrast, BRAF V600E high-grade gliomas experienced progression, even when initial tumor shrinkage was observed, and patient outcomes were poor.
Relevance
High response rates to targeted inhibition (partial or complete response, 53%; minor response, 27%) were observed, with PFS at 3 years of 49.6% for BRAF V600E–mutated pediatric low-grade gliomas. This was associated with favorable short-term outcomes. Future prospective clinical trials are required to address long-term management strategies and outcomes in these patients.
BRAF V600E is the second most common mutation observed in PLGG, responsible for 15% to 20% of cases and occurring in most pathologic subtypes.3,7 Among the common PLGG molecular subtypes, including NF1-mutated and KIAA1549-BRAF–fused subtypes, PLGGs harboring a BRAF V600E mutation have the poorest survival, especially when co-occurring with homozygous CDKN2A deletion.3,8-10 BRAF V600E–mutated PLGGs also have a higher propensity toward malignant transformation.8,9 Although the role of CDKN2A deletion in overall outcome and transformation is well described, it is unclear whether this alteration affects response to therapy.
The standard of care for patients with unresectable and/or progressive PLGG consists of chemotherapy, with radiotherapy generally reserved for progressive cases. Current chemotherapy strategies achieve tumor control in 40% to 50% of patients.11-14 Radiotherapy achieves excellent tumor control, approaching 80%, but is accompanied by an increased risk of secondary malignancies, ischemic events, and cognitive adverse effects.15,16 Overall, the long-term outcomes of PLGG with such therapies are favorable, with overall survival (OS) > 90% at 20 years.14
In contrast, in BRAF V600E–mutated PLGG, tumor control is achieved in fewer than 30% of patients using standard therapies, including radiotherapy and chemotherapy, suggesting a different clinical-biologic group of cancers.9 These data suggest that a novel approach is required for childhood BRAF V600E–mutated PLGG. In addition, pediatric high-grade glioma (PHGG) remains one of the most challenging childhood cancers. Irrespective of treatment strategy, including surgery, radiotherapy, and chemotherapy, prognosis remains dismal, with survival of only 10% at 5 years.17 Primary and secondary transformed BRAF V600E–mutated PHGGs exhibit similar grim outcomes with current chemoradiotherapy approaches.8
BRAF V600E mutation is a commonly observed somatic event in colon, melanoma, and other adult cancers, which led to the development of targeted therapies aimed at BRAF V600E inhibition. This has changed the landscape of melanoma treatment, with improvement in survival but with eventual resistance to this monotherapy.18-21 Several case reports have also described encouraging responses in patients with BRAF V600E–mutated PLGG.22-24 However, clinical experience in the use of BRAF inhibition is lacking in these patients.
To address this urgent need, we assembled clinical and molecular data from a large cohort of patients treated with BRAF inhibitor monotherapy from multiple leading neurooncology centers. Our results provide new insights into the impact of BRAF inhibition on this high-risk group of patients.
PATIENTS AND METHODS
We collected and reviewed clinical and molecular data from pediatric BRAF V600E–mutated gliomas treated with BRAF inhibition (dabrafenib or vemurafenib) from 29 different institutions around the world. All samples and clinical annotations were collected and analyzed after approval by and in accordance with research institutional review boards at participating institutions and at the Hospital for Sick Children. Patients age < 25 years at diagnosis treated with BRAF inhibition outside of a clinical trial and with follow-up of at least 6 months, with clinical and radiologic information available for review at an institutional level, were eligible. Data collection was performed at each site, and centralized review of radiology and/or pathology was not performed unless requested by the treating physician. All patients had a BRAF V600E mutation confirmed by either immunohistochemistry or sequencing.25 CDKN2A status was tested by single-nucleotide polymorphism array, multiplex ligation-dependent probe amplification, or droplet digital polymerase chain reaction in patients for whom tissue was available.
Response was evaluated according to reduction in tumor size, as measured by the product of two dimensions on T2 or fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) for low-grade gliomas (modified Response Assessment in Neuro-Oncology criteria for PLGG); measurements on T1 postcontrast MRI were used for high-grade glioms.26,27 Minor response (MR) was defined as reduction in tumor size between 25% and 49%, partial response (PR) as reduction in tumor size between 50% and 99%, and complete response (CR) as disappearance of disease on FLAIR or T2 imaging. Progressive disease (PD) was defined as either any tumor growth or clinical deterioration, as judged by treating physician, requiring change in management. Objective response (OR) was defined as reduction in tumor size of ≥ 25%, and overall response rate (ORR) was determined by combining PR and CR.28,29 Median time to best response was evaluated in patients with available imaging at 2 to 3 months and 6 months after starting treatment.
Adverse events were recorded when requiring dose reduction and/or discontinuation of BRAF inhibitors. Additionally, we analyzed and compared the PLGG cohort with a previous published cohort of patients with a BRAF V600E mutation treated with chemotherapy (SickKids, n = 29; international institutions, n = 36).9 Previous chemotherapy regimens included the common regimens of carboplatin with vincristine, vinblastine, and temozolomide.11-13,30,31 Detailed clinical characteristics and demographics of this cohort are presented in the Data Supplemental.
Statistical Analyses
Descriptive analysis was summarized. Progression-free survival (PFS) was analyzed using the Kaplan-Meier method, and differences in PFS and OS between chemotherapy and BRAF inhibition cohorts were tested using the log-rank test. R software (version 3.5.1; https://www.r-project.org) was used for all statistical analyses (packages used, ggplot and lattice). Fisher’s exact test was used to determine differences in response between BRAF inhibition and chemotherapy; P < .05 was considered statistically significant.
RESULTS
Overall, our cohort included 67 patients. Of those, 56 with BRAF V600E–mutated PLGG and 11 with BRAF V600E–mutated PHGG were treated with BRAF inhibition. The Results for these two cohorts will be described separately in the following sections.
Outcomes of PLGGs
Demographic and clinical characteristics of the patients with PLGG are listed in Table 1. Median age at diagnosis was 4.5 years (range, 0.1-22.3 years), and treatment with BRAF inhibition was initiated at a median age of 8.2 years. The most common histopathologic diagnosis in BRAF V600E–mutated PLGG was pilocytic astrocytoma followed by ganglioglioma, with midline tumors representing 83% of cases. Within the PLGG cohort, 15% of tested patients had a concomitant CDKN2A deletion, and H3.3 K27M mutation was also observed concomitantly with BRAF V600E in one patient with brainstem PLGG.
TABLE 1.
Clinical and Tumor Characteristics of the BRAF Inhibition Cohort
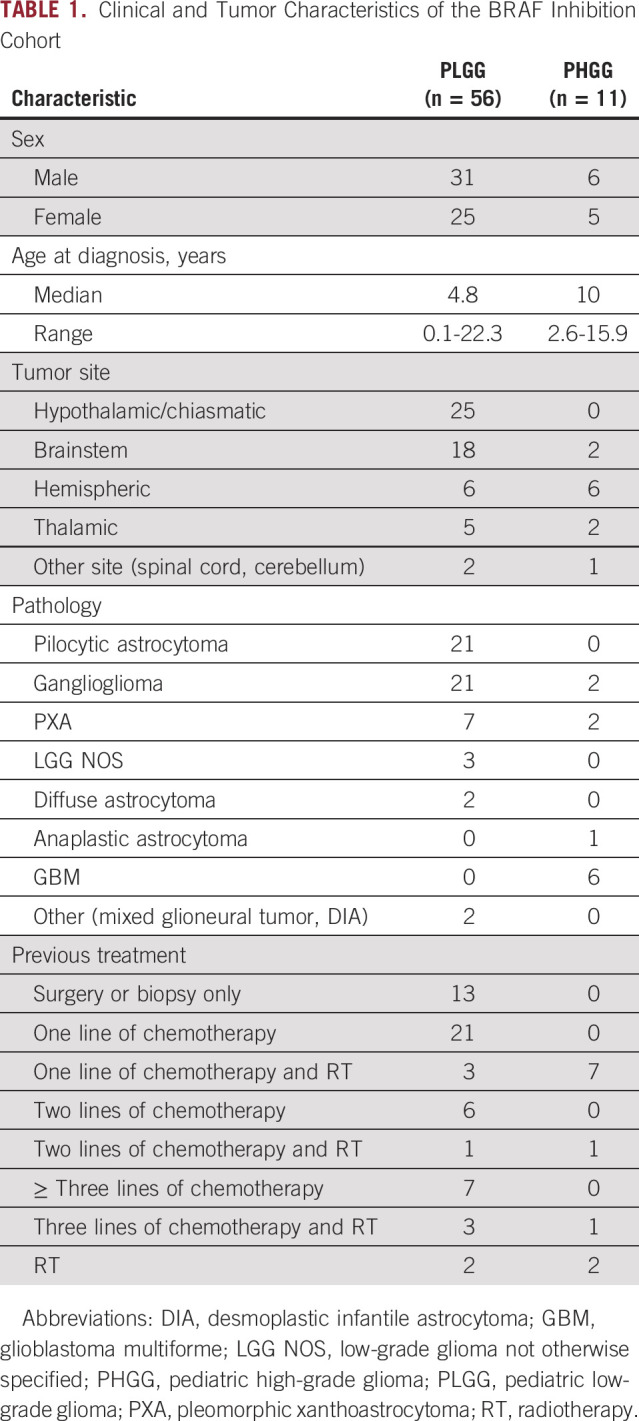
A majority of patients (76,8%) had received at least one previous line of therapy (chemotherapy, n = 34; chemotherapy and radiotherapy, n = 7; radiotherapy only, n = 2). The remaining 23.2% of patients were treated with BRAF inhibition after partial resection (six of 13) or diagnostic biopsy (seven of 13) as first-line treatment. All patients had measurable disease at start of therapy.
Treatment was generally well tolerated, with 23% of patients needing dose reductions or temporary discontinuation of the drugs because of adverse effects (mainly skin toxicity; n = 11). Only three patients (5%) stopped medication because of severe adverse events (skin toxicity with rash, n = 1; hepatotoxicity, n = 1; benign melanotic lesions after 15 months of treatment that resolved upon discontinuation, n = 1).
BRAF V600E–mutated PLGG responds to BRAF inhibition.
OR was observed in 80% of tumors (PR [n = 28 of 56] or CR [n = 2 of 56], 53%; MR [n = 15 of 56], 27%; Fig 1A). These responses were observed equally among pathologic subtypes (Data Supplement). Average time to response (MR, PR, or CR) was 4 months in assessable patients (n = 48 of 56; Fig 1B), and 94% had achieved best response by 6 months of therapy.
FIG 1.
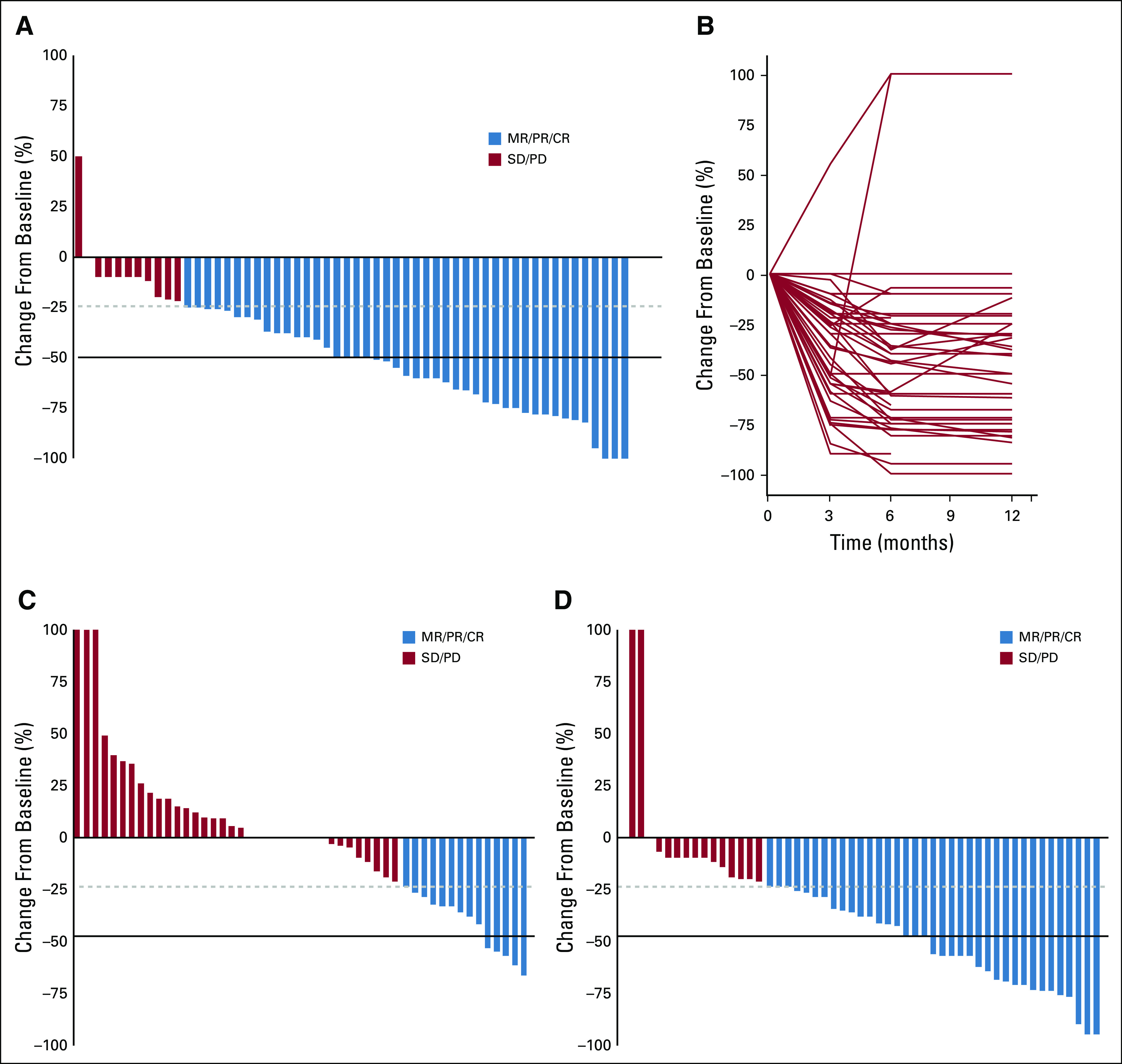
Response of BRAF V600E–mutated gliomas to BRAF inhibition. (A) Waterfall plot of best response in pediatric low-grade gliomas (PLGGs) to BRAF inhibition as measured by the products of perpendicular measures in T2 or fluid-attenuated inversion recovery magnetic resonance imaging. (B) Spider plot revealing time to response of PLGGs to BRAF inhibition. (C) Waterfall plot of response at 6 months in PLGGs treated with (C) chemotherapy or (D) BRAF inhibition. CR, complete response; MR, minor response; PD, progressive disease; PR, partial response; SD, stable disease.
To compare the response of PLGG to BRAF inhibition versus chemotherapy, we used a large control cohort (n = 65) of BRAF V600E PLGGs from our PLGG taskforce database. Response was assessed for patients with available imaging at 6 months in both cohorts (BRAF inhibition, n = 52 v chemotherapy, n = 50). Whereas chemotherapy resulted in OR (reduction in tumor size ≥ 25%) and ORR (reduction in tumor size ≥ 50%) of 28% and 10%, BRAF inhibition resulted in OR and ORR of 71% and 42%, respectively (P < .001; Fig 1C).
Most PLGGs had sustained response to BRAF inhibition, with median treatment time of 17.4 months (range, 6-61 months; Fig 2). PD was observed in only eight (14.2%) of 56 tumors during therapy. Six of those had initial OR, with median time to progression of 9.4 months, suggesting acquired resistance. Of these, five of eight patients received combination treatment at progression with BRAF inhibition and MEK inhibition, with no further progression at a median follow-up of 8 months (range 3-14 months) after starting the combination.
FIG 2.

Swimmer plot for BRAF V600E–mutated gliomas treated with BRAF inhibition. Legend describes parameters of response, progression, and ongoing therapy. CR, complete response; MR, minor response; PD, progressive disease; PR, partial response; SD, stable disease.
Seventeen patients stopped BRAF inhibition due to patient/physician decision (n = 14) or toxicity (n = 3). Of those, 13 (76.5%) experienced rapid progression (median time to progression, 2.3 months; range, 0.3-20.8 months). Importantly, nine of these 13 patients were rechallenged with BRAF inhibition, either alone (n = 8) or in combination with MEK inhibition (n = 1), with OR seen in all patients with PLGG except one, who required dose reduction because of toxicity. This patient eventually experienced further tumor progression and died (Fig 2).
Molecular markers of response to BRAF inhibition.
CDKN2A deletion has been previously shown to be a marker of poor prognosis in BRAF V600E–mutated PLGG. To assess whether CDKN2A homozygous deletion predicts response to BRAF inhibition, CDKN2A status was determined in 33 PLGGs, five of which harbored a homozygous deletion (15.1%). All CDKN2A-deleted tumors responded, with best response ranging from 21% to 100%. Furthermore, no difference in response rates or PFS between CDKN2A-deleted and balanced BRAF V600E–mutated PLGGs was observed (Data Supplement).
One brainstem tumor was reported to present concomitantly with an H3 K27M mutation. This tumor progressed after discontinuation of therapy, and the patient now has stable disease with a combination of BRAF inhibition and MEK inhibition (for 9 months), with overall follow-up of 22.8 months from treatment initiation.
Favorable survival for patients with BRAF V600E–mutated PLGG who continue receiving BRAF inhibition.
To determine whether treatment discontinuation affects survival, we compared PFS for patients who continued versus discontinued BRAF inhibition after at least 1 year of treatment. Of the 28 patients who had been receiving continuous treatment for > 10 months, PFS at 2 years was 81.6% (95% CI, 65% to 100%), with no further progression up to 5 years of therapy (Fig 3B). In contrast, PFS for those with PLGG in whom therapy was discontinued after a minimum of 10 months was 26.7% (95% CI, 11.5% to 61.7%) at 1 year after cessation of therapy (Fig 3C).
FIG 3.
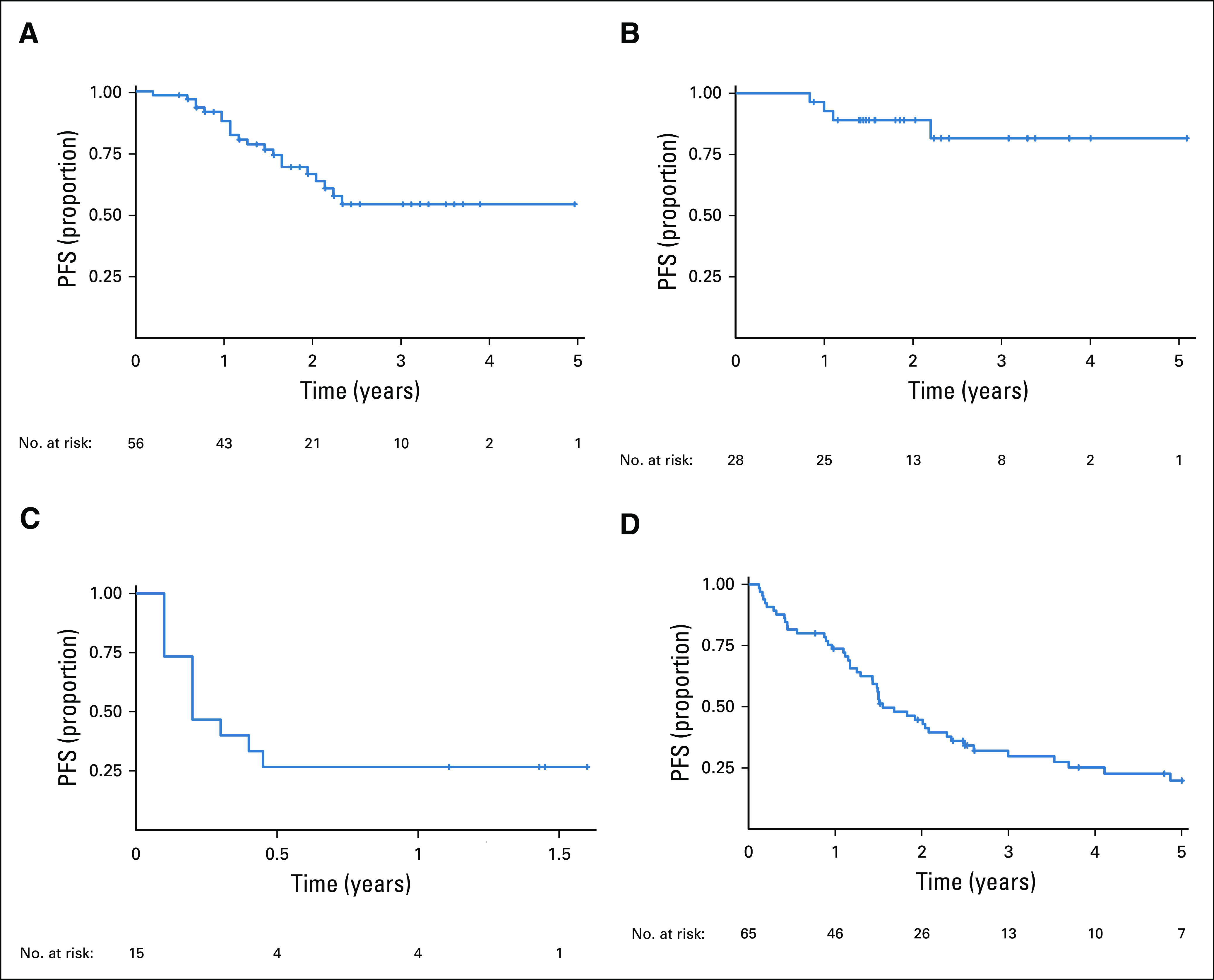
Survival plot for BRAF V600E–mutated pediatric low-grade gliomas (PLGGs). Progression-free survival (PFS) for (A) 56 patients with PLGG treated with BRAF inhibition, (B) those in whom BRAF inhibition therapy was continued for ≥ 10 months, and (C) those in whom BRAF inhibition was discontinued after ≥ 10 months. (D) PFS for patients with BRAF V600E–mutated PLGG treated with chemotherapy.
We then compared these data with survival for patients receiving chemotherapy. BRAF V600E–mutated PLGG responded poorly to chemotherapy and continued to progress, resulting in PFS of 20% (95% CI, 11.2% to 35.1%) at 5 years (Fig 3D). In contrast, patients treated with BRAF inhibition had significantly better PFS of 49.6% at 3 years (95% CI, 35.3% to 69.5%), with a potential plateau afterward (log-rank test P = .02). OS was not different between the groups, although longer follow-up time is required (Data Supplement).
Outcomes of PHGGs Treated With BRAF Inhibition
Because BRAF V600E–mutated PLGG tends to transform to PHGG, we collected information on 11 patients with PHGG treated with BRAF inhibition. Median age at diagnosis was 10.6 years (range, 2.6-15.9 years), and treatment with BRAF inhibition was initiated at a median age of 12.3 years. The most common pathologic subtype and tumor location were glioblastoma multiforme (n = 6 of 11) and supratentorial location, respectively. Although responses were also seen, with best ORR of 36% (Fig 4A), most PHGGs progressed rapidly, and all but one patient (with epithelioid glioblastoma multiforme) experienced progression within 18 months (range, 1.4-16.3 months; Fig 4B). H3.3 K27M mutations were also observed in three patients with PHGG (thalamic, n = 2; diffuse intrinsic pontine glioma, n = 1). Despite initial responses, with stabilization of disease (Data Supplement), these patients eventually experienced PD, at a median time of 10 months (range, 3.7-16.2 months), and all died as a result of their disease (Fig 4B).
FIG 4.

Outcomes of BRAF V600E–mutated pediatric high-grade gliomas (PHGGs) treated with BRAF inhibition. (A) Waterfall plot of best response, (B) swimmer plot, and (C) Kaplan-Meier plot for progression-free survival (PFS) comparing children with pediatric low-grade glioma (PLGG) and PHGG. CR, complete response; MR, minor response; PD, progressive disease; PR, partial response; SD, stable disease.
Therefore, tumor grade was a strong predictor of poor outcome in pediatric patients with BRAF V600E–mutated gliomas receiving BRAF inhibition. PFS at 1 year was 27% (95% CI, 10.4% to 71.6%) versus 86.4% (95% CI, 77.5% to 96.3%) for PHGG and PLGG, respectively (P < .001; Fig 4C).
DISCUSSION
Here we report, to our knowledge, the largest cohort to date of pediatric BRAF V600E–mutated gliomas treated with BRAF inhibition. Our data show encouraging initial responses and sustained disease control in BRAF V600E–mutated PLGG using this targeted therapy approach. We also uncovered several key concepts critical to clinicians in the optimal management of these childhood cancers.
Despite the majority of our cohort comprising patients with relapsed disease treated with BRAF inhibition, the responses of BRAF V600E–mutated PLGG to BRAF inhibition were remarkable. Overall ORs were observed in 80% of PLGGs, with more than half showing ≥ 50% reduction in tumor size. Notably, these responses were usually achieved within 3 months, contrasting the slow responses and mostly stabilization observed with chemotherapy and/or radiotherapy approaches (Fig 1). However, it is important to note that BRAF inhibition leads to CR in a minority of tumors. Whether a heterogeneous cell population with different tumor content of mutated cells is responsible for these heterogeneous responses or only cycling cells are sensitive to BRAF inhibition is still to be clarified.
Because BRAF inhibition is currently reserved for recurrent tumors, our data suggest that earlier treatment with BRAF inhibition has the potential to lead to prevention of morbidities and sequelae in patients with PLGG. Improvement of visual status, seizure control, and other neurologic dysfunctions has been reported in the literature.22,24 Although we did not collect these data, it is likely that the dramatic cytoreduction seen across this cohort would be associated with clinical and neurologic improvements. These clinical improvements are uncommon in most PLGGs treated with conventional chemoradiotherapy and therefore should be better characterized in prospective trials with BRAF inhibition moving forward.
Unfortunately, for PHGG, the initial response observed in 36% of tumors was transient. All but one patient with PHGG experienced tumor progression within 18 months. Although these data are limited, our observation suggests that resistance in PHGG occurs in most cases, and new strategies beyond BRAF inhibitor monotherapy are needed.
Previous reports have identified both CDKN2A deletion and H3.3 K27M mutation as poor prognostic markers in PLGGs with concomitant BRAF V600E mutation.8,9 Although still preliminary, our data suggest that CDKN2A-deleted tumors still respond to BRAF inhibition in a manner similar to that of other BRAF V600E–mutated tumors (Data Supplement). It would be important to closely monitor these patients to determine whether BRAF inhibition can change the natural history of these aggressive tumors. Similarly, the initial tumor stabilization observed in four recurrent gliomas harboring both H3 K27M and BRAF V600E mutations is notable, but whether outcome is different in these cancers is yet to be determined. Additional studies in larger cohorts are required to verify these outcomes.
Eight patients with BRAF V600E–mutated PLGG experienced progression while receiving therapy. Of these, five were started on combination therapy with BRAF inhibition and MEK inhibition. Combination treatment resulted in stable disease; however, no further reduction in tumor size was seen. Resistance to BRAF inhibition is well described in melanoma because of additional mutations in other genes in the RAS/MAPK pathway.20 Combinatorial therapies have, in this context, shown reresponse rates of 42.3%.32 Acquiring tissue from these patients with resistant disease will be critical in understanding this process in those with pediatric glioma.33
One of the most important findings of this study was the behavior of BRAF V600E–mutated PLGG when BRAF inhibition therapy was discontinued. As observed in other gliomas, such as subependymal giant cell astrocytoma (SEGA), in which stopping mammalian target of rapamycin (mTOR) inhibitors results in inevitable tumor regrowth, in our PLGG cohort, 76.5% of tumors had rapid regrowth after BRAF inhibition was stopped.34 Importantly, 90% of PLGGs that progressed after BRAF inhibition was discontinued responded if rechallenged with BRAF inhibition alone or combined with MEK inhibition. Although these data are encouraging, they raise several issues regarding interpretation of the efficacy of BRAF inhibition in BRAF V600E–mutated PLGG. First, chronic long-term use of BRAF inhibition may be needed, as is commonly done with mTOR inhibitors in patients with tuberous sclerosis and SEGA. If so, studies of long-term adverse effects and potential developmental issues unique to children are required. Second, although our data reveal relatively durable responses to BRAF inhibition in a large fraction of patients with PLGG, discontinuing therapy may have a major effect on interpretation of the data and management of these patients (Fig 3).
This study has the classic limitations of a retrospectively collected cohort. These include the retrospective nature, as well as the lack of consistent starting and stopping rules, central imaging, and pathologic and molecular review. Nevertheless, the strikingly consistent and clear responses observed in BRAF V600E–mutated PLGG suggest that BRAF inhibition represents a highly feasible and efficacious treatment option regardless of pathologic subtype or imaging characteristics.
In summary, the data presented in this study demonstrate high rates of initial response as well as sustained response of most BRAF V600E–mutated PLGGs to BRAF inhibition. This effect may be lost if BRAF inhibition is discontinued, and inhibition is less effective in BRAF V600E–mutated PHGG. Although these findings support the potential for incorporation of BRAF inhibition into the management of BRAF V600E–mutated PLGG, the long-term efficacy and potential risks of this continuous therapy need to be fully explored. Additional studies are required to better understand the mechanisms of resistance, use of upfront BRAF inhibition, and potential use of BRAF inhibition with radiotherapy, chemotherapy, or other targeted combinations. Finally, understanding the role of concomitant alterations observed in BRAF V600E–mutated pediatric glial tumors will help in designing new approaches for these high-risk tumors.
Presented at the 18th International Symposium on Pediatric Neuro-Oncology, Denver, CO, June 29-July 3, 2018, and at the Pediatric Neuro-Oncology Basic and Translational Research Conference, San Francisco, CA, May 3-4, 2019.
SUPPORT
Supported by Canadian Cancer Society Grant No. 702296; by Genome Canada; by Ontario Genomics Institute Grant No. OGI-121; by A Kid’s Brain Tumor Cure/PLGA Foundation; by the LivWise Foundation; by the Brain Child Foundation; by Canadian Institutes for Health Research Grant No. 159805; by the Elmaglachli Family Foundation; by the Mckeddie Family Foundation; by Memorial Sloan Kettering Cancer Center Grant No. P30 CA008748 (M. Karajannis, G.K., I.J.D.); in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology; in part by National Cancer Institute Cancer Center Core Grant No. P30-CA008748; by the Garron Family Cancer Centre with funds from the SickKids Foundation; by the Garron Family Chair in Childhood Cancer Research at the Hospital for Sick Children (U.T., E.B.); by the Meagan’s Walk Fellowship in Pediatric Neuro-Oncology (L.N., M.Z.); and by the Garron Family Cancer Center Fellowship and Restracomp (M.Z.).
AUTHOR CONTRIBUTIONS
Conception and design: Liana Nobre, Michal Zapotocky, Cynthia Hawkins, Uri Tabori
Financial support: Cynthia Hawkins and Uri Tabori
Provision of study material or patients: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Vijay Ramaswamy
Honoraria: AstraZeneca
Miriam Bornhorst
Consulting or Advisory Role: AstraZeneca/MedImmune
Daniel R. Boue
Stock and Other Ownership Interests: Vertex Pharmaceuticals, Intuitive Surgical, Illumina
Adela Canete
Consulting or Advisory Role: EUSA Pharma, Bayer
Speakers’ Bureau: EUSA PHarma
Research Funding: EUSA Pharma (Inst)
Travel, Accommodations, Expenses: EUSA Pharma
Ira J. Dunkel
Consulting or Advisory Role: Bayer, Apexigen, Celgene, Roche/Genentech, AstraZeneca
Research Funding: Bristol-Myers Squibb (Inst), Genentech (Inst), Novartis (Inst)
Karen Gauvain
Employment: Iqvia Biotech
Consulting or Advisory Role: Bayer, Axiom Health Care Sciences
Jordan R. Hansford
Consulting or Advisory Role: Bayer
Sarah G. Injac
Research Funding: Takeda
Matthias Karajannis
Consulting or Advisory Role: Bayer, Recursion Pharma
Research Funding: Novartis
Travel, Accommodations, Expenses: Bayer
Uncompensated Relationships: Debiopharm (Inst)
Open Payments Link: https://openpaymentsdata.cms.gov/physician/710370/summary
Normand Laperriere
Honoraria: Merck/Schering Plough
Consulting or Advisory Role: AbbVie
Alvaro Lassaletta
Consulting or Advisory Role: Shire, Jazz Pharmaceuticals, Roche
Travel, Accommodations, Expenses: Shire, Gilead Sciences
Roger Packer
Honoraria: Novartis
Consulting or Advisory Role: Novartis, AstraZeneca
Jaroslav Sterba
Research Funding: Roche/Genentech (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Derek S. Tsang
Other Relationship: Varian Medical Systems (Inst), Mevion Medical Systems (Inst), Hitachi (Inst), RaySearch Laboratories (Inst), IBA (Inst), ProTom (Inst)
Cornelis M. van Tilburg
Consulting or Advisory Role: Novartis, Bayer
Olaf Witt
Consulting or Advisory Role: Novartis, AstraZeneca, Janssen Research & Development, Bristol-Myers Squibb, Roche, Bayer
Eric Bouffet
Research Funding: Roche (Inst), Bristol-Myers Squibb (Inst)
Cynthia Hawkins
Consulting or Advisory Role: Bayer
Patents, Royalties, Other Intellectual Property: IP for low-grade glioma and sarcoma fusion panels as well as medulloblastoma subgrouping panel
No other potential conflicts of interest were reported.
REFERENCES
- 1.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro-oncol. 2014;16(suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones DT, Hutter B, Jäger N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48:273–282. doi: 10.1038/ng.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergthold G, Bandopadhayay P, Hoshida Y, et al. Expression profiles of 151 pediatric low-grade gliomas reveal molecular differences associated with location and histological subtype. Neuro-oncol. 2015;17:1486–1496. doi: 10.1093/neuonc/nov045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33:1015–1022. doi: 10.1200/JCO.2014.58.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lassaletta A, Zapotocky M, Mistry M, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. 2017;35:2934–2941. doi: 10.1200/JCO.2016.71.8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahiya S, Haydon DH, Alvarado D, et al. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125:901–910. doi: 10.1007/s00401-013-1120-y. [DOI] [PubMed] [Google Scholar]
- 11.Packer RJ, Ater J, Allen J, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747–754. doi: 10.3171/jns.1997.86.5.0747. [DOI] [PubMed] [Google Scholar]
- 12.Lassaletta A, Scheinemann K, Zelcer SM, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: A Canadian Pediatric Brain Tumor Consortium study. J Clin Oncol. 2016;34:3537–3543. doi: 10.1200/JCO.2016.68.1585. [DOI] [PubMed] [Google Scholar]
- 13.Massimino M, Spreafico F, Cefalo G, et al. High response rate to cisplatin/etoposide regimen in childhood low-grade glioma. J Clin Oncol. 2002;20:4209–4216. doi: 10.1200/JCO.2002.08.087. [DOI] [PubMed] [Google Scholar]
- 14.Krishnatry R, Zhukova N, Guerreiro Stucklin AS, et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: A population-based study. Cancer. 2016;122:1261–1269. doi: 10.1002/cncr.29907. [DOI] [PubMed] [Google Scholar]
- 15.Merchant TE, Kun LE, Wu S, et al. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598–3604. doi: 10.1200/JCO.2008.20.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cherlow JM, Shaw DWW, Margraf LR, et al. Conformal radiation therapy for pediatric patients with low-grade glioma: Results from the Children’s Oncology Group phase 2 study ACNS0221. Int J Radiat Oncol Biol Phys. 2019;103:861–868. doi: 10.1016/j.ijrobp.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakacki RI, Cohen KJ, Buxton A, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro-oncol. 2016;18:1442–1450. doi: 10.1093/neuonc/now038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu H, Liu S, Zhang G, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas [published correction appears in Nature. 565:E4, 2019] Nature. 2017;550:133–136. doi: 10.1038/nature24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriceau G, Hugo W, Hong A, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–256. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lassaletta A, Guerreiro Stucklin A, Ramaswamy V, et al. Profound clinical and radiological response to BRAF inhibition in a 2-month-old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr Blood Cancer. 2016;63:2038–2041. doi: 10.1002/pbc.26086. [DOI] [PubMed] [Google Scholar]
- 23.van Tilburg CM, Selt F, Sahm F, et al. Response in a child with a BRAF V600E mutated desmoplastic infantile astrocytoma upon retreatment with vemurafenib. Pediatr Blood Cancer. 2018;65:XXXX. doi: 10.1002/pbc.26893. [DOI] [PubMed] [Google Scholar]
- 24.Bavle A, Jones J, Lin FY, et al. Dramatic clinical and radiographic response to BRAF inhibition in a patient with progressive disseminated optic pathway glioma refractory to MEK inhibition. Pediatr Hematol Oncol. 2017;34:254–259. doi: 10.1080/08880018.2017.1360971. [DOI] [PubMed] [Google Scholar]
- 25.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): Assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12:583–593. doi: 10.1016/S1470-2045(11)70057-2. [DOI] [PubMed] [Google Scholar]
- 27.Chukwueke UN, Wen PY. Use of the Response Assessment in Neuro-Oncology (RANO) criteria in clinical trials and clinical practice. CNS Oncol. 2019;8:CNS28. doi: 10.2217/cns-2018-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warren KE, Poussaint TY, Vezina G, et al. Challenges with defining response to antitumor agents in pediatric neuro-oncology: A report from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Pediatr Blood Cancer. 2013;60:1397–1401. doi: 10.1002/pbc.24562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gnekow AK, SIOP Brain Tumor Subcommittee. International Society of Pediatric Oncology Recommendations of the Brain Tumor Subcommittee for the reporting of trials. Med Pediatr Oncol. 1995;24:104–108. doi: 10.1002/mpo.2950240209. [DOI] [PubMed] [Google Scholar]
- 30.Chintagumpala M, Eckel SP, Krailo M, et al. A pilot study using carboplatin, vincristine, and temozolomide in children with progressive/symptomatic low-grade glioma: A Children’s Oncology Group study. Neuro-oncol. 2015;17:1132–1138. doi: 10.1093/neuonc/nov057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ater JL, Xia C, Mazewski CM, et al. Nonrandomized comparison of neurofibromatosis type 1 and non-neurofibromatosis type 1 children who received carboplatin and vincristine for progressive low-grade glioma: A report from the Children’s Oncology Group. Cancer. 2016;122:1928–1936. doi: 10.1002/cncr.29987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valpione S, Carlino MS, Mangana J, et al. Rechallenge with BRAF-directed treatment in metastatic melanoma: A multi-institutional retrospective study [published correction appears in Eur J Cancer. 93:158, 2018] Eur J Cancer. 2018;91:116–124. doi: 10.1016/j.ejca.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Yao Z, Jonsson P, et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAFV600E-mutant brain tumor. Cancer Discov. 2018;8:1130–1141. doi: 10.1158/2159-8290.CD-17-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]