

Abstract
Mycobacterium avium, one of the closest relatives of Mycobacterium tuberculosis (MTB), offers an advantage in studying MTB because of its tuberculosis-like effect in humans and host immune tolerance. This study examined the antimycobacterial action of ursolic acid and its regulation in macrophages during infection. Colonyforming units of the bacteria were determined in the cell lysate of macrophages and in the supernatant. The effect of ursolic acid on macrophages during infection was determined by analyzing the phosphorylation of the mitogen-activated protein kinase signaling pathway and the concentrations of tumor necrosis factor-α, interleukin-1β, interleukin-6, and nitrite. The colony-forming units analysis demonstrated that ursolic acid reduced the presence of Mycobacterium avium both intracellularly (in macrophages) and extracellularly. It decreased the levels of tumor necrosis factor- α and interleukin-6 but increased the concentrations of interleukin-1β and nitrite during infection. It also inhibited the phosphorylation of ERK1/2 but phosphorylated the C-Jun N-terminal kinase signaling pathway. The antimycobacterial effect of ursolic acid correlated with its ability to regulate the activation of macrophages. This dual ability made the ursolic acid-related elimination of the mycobacteria more effective.
Key words: ursolic acid, Mycobacterium avium, MAPK, pro-inflammatory cytokines, macrophages
Introduction
Mycobacterium tuberculosis (MTB) causes an infectious disease leading to death in adults worldwide.1 Principally, the success of MTB as a causative agent of tuberculosis (TB) is based on three characteristics: the regulation of macrophages after phagocytosis to prevent apoptosis or destruction, the formation of granulomas in which the intracellular bacillus can survive and replicate, and conversion into a stage of dormancy that makes MTB resistant to drug treatment and host defense.2 MTB that has been phagocytosed by macrophages becomes less susceptible to elimination with antituberculosis drugs compared to extracellular MTB. This mechanism induces a high probability of reinfection. On the basis of this background, it is necessary to develop a new compound that not only is able to kill the mycobacteria but also can regulate the host’s immune system to increase the ability of antituberculosis drugs to eliminate MTB, primarily intracellular MTB.
UA (3β-hydroxy-urs-12-ene-28-oic acid) is a pentacyclic triterpenoid that is widely known to have various pharmacological effects. This secondary metabolite can be found in apple (Malus domestica) fruit peel, rosemary (Rosmarinus officinalis) leaves, thyme (Thymus vulgaris) leaves, and the waxy layer of many edible fruits.3,4 Jyoti et al.5 reported that there is a reduction of mycolic acid concentration in the MTB H37Ra strain when it is treated with ursolic acid (UA). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of a sensitive MTB strain treated with UA revealed morphological abnormalities and lysis of cells.5,6 Combination studies of UA and antituberculosis drugs (isoniazid, ethambutol, streptomycin, and rifampicin) also showed a synergistic effect in killing MTB.7
Mitogen-activated protein kinase (MAPK) provides an essential signaling pathway in eukaryotic cells. It is responsible for complex cellular programs like proliferation, differentiation, and apoptosis. Three MAPK families have been characterized, namely, extracellular-signal-regulated kinase (ERK) or classical MAPK, C-Jun Nterminal kinase (JNK) or stress-activated kinase (SAPK), and p38 kinase. It has been reported that UA inhibited p38 and increased the activity of the ERK signaling pathway in Huh-7 cells.8 UA is also reportedly able to induce apoptosis via JNK in leukemic cells.9 On the basis of these facts, UA is a strong candidate for further investigation.
This study examined the effect of UA as an antimycobacterial agent and its regulation in macrophages during infection. Mycobacterium avium was used as the infectious agent in this study because its characteristics of intracellular activity and macrophage modulation function are similar to those caused by MTB. The study hypothesized that the decrease in the number of mycobacteria in macrophages was related to UA’s ability to regulate the MAPK signaling pathway and proinflammatory cytokines during mycobacterial infection. In addition, the bactericidal effect shown by UA affects factors that prevent mycobacteria from being recognized by macrophage surface receptors. To test these hypotheses, researchers chose to examine the impact of UA treatment in the concentration of cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and nitrite produced by infected macrophages and correlate it with the ability of UA to kill the bacteria, as assessed by the number of colony-forming units (CFU) found in the experiment. For further insight, we determined the activation of the MAPK pathway in terms of multiple targets, such as p38, ERK, and JNK, in infected macrophages that were treated with UA.
Materials and methods
Materials
Female C57BL/6 mice 6–8 weeks of age were obtained from the Preclinical Research Facility (University of Leicester, UK) and were used as a source for bonemarrow- derived macrophages (BMDM). UA (purity >98% and endotoxin free) was isolated and obtained by Tokyo Chemical Industry (Tokyo, Japan). Dimethyl sulfoxide (DMSO) of cell culture grade for use as a diluent and tetramethylbenzidine for use as a substrate for enzyme-linked immunosorbent assay (ELISA) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Isolation and culture of murine bone-marrow-derived macrophages
BMDM were cultured using Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), HEPES buffer, MEM amino acid, sodium pyruvate solution, and L-glutamine (complete DMEM; Thermo Fisher Scientific, Waltham, MA, USA). Twenty-four-well tissue culture plates were initially seeded with 5.8.105 cells/well and incubated at 37°C in a 5% CO2 humidified air chamber for 24h. The following day, the cells were supplemented with additional fresh complete DMEM containing 10 ng/mL GM-CSF. The medium was changed every 3 days, and the cultures were incubated for 10 days. On the day before infection (day 9), the medium was replaced with complete DMEM without granulocyte macrophage colony-stimulating factor (GM-CSF), and the adherent cells were ready for infection on day 10.
In vitro infection of murine macrophages with Mycobacterium avium
Mycobacterium avium 724 with a density of 2.11.109 CFU/mL was used as the infection agent and was prepared from Cooper’s Lab (Department of Respiratory Sciences, College of Life Sciences, University of Leicester, UK). Macrophages in 24-well plates were infected with a suspension of Mycobacterium avium at a multiplicity of infection (MOI) of 8:1. Three models of the experiment were made in this study. Model 1, as control, was treated with different concentrations of UA without infection of Mycobacterium avium and incubated for 18h. Model 2 was treated with Mycobacterium avium and UA at the same time and incubated for 18h. Model 3, as a pretreatment model, was pretreated with Mycobacterium avium for 3h, followed by with UA for 15h. All models were incubated at 37°C in a 5% CO2 humidified air chamber. After 18h, the supernatants were collected for analysis of cytokines and nitrite.
Measurement of proinflammatory cytokine production
TNF-α, IL-1β, and IL-6 produced by murine macrophages in the supernatants from all three models were measured using an ELISA kit purchased from Biolegend (San Diego, CA, USA). ELISAs were performed according to the protocol given by the manufacturer. Tetramethylbenzidine was used as a visualizing reagent. ELISA plates were read for absorbance at 450 nm using an LT-4500 microplate reader and were analyzed using the LT-com data analysis software. The concentration of cytokines was analyzed by interpolating from the sample absorbance to the standard curve.
Determination of nitrite
Nitrite (NO2 −) concentration was determined by using Griess reagent, which is composed of 1% sulfanilamide, 0.1% N-(1- naphthyl)ethylenediamine, and 2.5% phosphoric acid. In a flat-bottom 96-well microtiter plate, 100 μL of Griess reagent was added to 100 μL of each sample, and incubation for 10 min at room temperature followed. The absorbance was read at 550 nm. The concentration of NO2 − was analyzed by interpolating from the sample absorbance to the standard curve.
Determination of colony-forming units (CFU)
After 18h of incubation, the supernatant in previous studies was collected for analysis of TNF-α, IL-6, IL-1β, and nitrite. The macrophages were then incubated with lysis buffer (0.05% Tween 80). Cell lysate and supernatant were used to determine CFU by making a 10. serial dilution. For each serial dilution, a total of 100 μL was plated on Middlebrook 7H9 agar media (Sigma- Aldrich, St. Louis, MO, USA), followed by incubation for 15 days at 37°C. After 15 days of incubation, the number of CFU was then determined by counting colonies that grew on the surface of the media while paying attention to the dilution factors used.
Preparation of cell lysates
BMDMs prepared as already described and cultured in 24-well tissue culture plates were treated with Mycobacterium avium (MOI 8:1) and UA at a concentration of 50 μg/mL. As controls, Mycobacterium avium and UA were also incubated in the same plates. Nuclear protein was extracted at two time points (15 and 30 min). After treatment, macrophages were washed with icecold phosphate-buffered saline, and the cellular extract was harvested using RIPA buffer (Cell Signaling Technology, Danvers, MA). After the addition of 400 μL of lysis buffer per well, the macrophages were incubated on ice for 5 min, scraped, and centrifuged for 10 min at 16,000.g at 4°C. The supernatant was collected, and the concentration of protein in the cell lysate was determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA; USA) according to the manufacturer’s instructions.
MAPK multitarget sandwich ELISA
Protein from cell lysates was analyzed for the activation of ERK1/2, p38, and JNK using the PathScan MAPKinase Multi- Target Sandwich ELISA kit purchased from Cell Signaling Technology (Danvers, MA, USA). The kit provides analysis of phospho- p44/42 MAPK (Thr202/Tyr204), phospho- p38 (Thr180/Tyr182), total protein of MEK1, phospho-MEK1/2 (Ser217/221), total protein of JNK, and phospho-JNK (Thr183/Tyr185). The test procedure used was according to the manufacturer’s instructions.
Statistical analysis
Intergroup differences in treatment effects were probed with one-way analysis of variance, using Tukey’s post hoc test for multiple comparisons. The significance of intergroup differences for other parameters was determined using two-way analysis of variance and Bonferroni’s post hoc test. All analyses and graphs were carried out using Prism software (version 8.0 GraphPad Software, La Jolla, CA, USA). All experiments were performed at least three separate times with data obtained in duplicate wells in each experiment.
Results
Effect of UA on proinflammatory cytokine production and nitrite concentration and its bactericidal activity during infection of murine macrophages with Mycobacterium avium
The optimal concentration of UA for the experiment was determined by the cytotoxicity assay in macrophages. It was started with the in vitro minimum inhibitory concentrations reported from the previous experiment.7 It was found that 100 μg/mL was toxic to cells at 24h of treatment. Cell viability was under 65% after 24h of treatment with 100 μg/mL. Therefore, the concentrations chosen for use ranged from 25 to 100 μg/mL. Initial screening of the housekeeping genes expression also demonstrated that UA did not affect the expression of glyceraldehyde-3-phosphate dehydrogenase (GADPH) and β-actin in noninfected macrophages, but interestingly, it decreased the expression of those two housekeeping genes in Mycobacterium avium-infected macrophages (data not shown).
Three research models had been made to investigate the production of TNF-α, IL- 6, IL-1β, and NO (analyzed as nitrite) due to the administration of UA to macrophages infected with Mycobacterium avium. These models also illustrated how UA could kill Mycobacterium avium that infected macrophages. Model 1 incubated UA with noninfected macrophages, whereas model 2 incubated UA with macrophages infected with Mycobacterium avium.
UA was seen to reduce the concentration of TNF-α, IL-6, and nitrite weakly in noninfected macrophages and did not affect the production of IL-1β (model 1). The presence of Mycobacterium avium infection in macrophages could drastically increase the production of all three cytokines and nitrites when compared to controls. Conversely, the administration of UA to macrophages infected with Mycobacterium avium (model 2) could reduce the production of all three cytokines and nitrites, with the lowest decrease in UA concentration of 50 μg/mL (Figure 1). The results of CFU model 2 also showed the absence of Mycobacterium avium colonies both in cell lysate and in supernatant, beginning at a UA concentration of 50 μg/mL (Figure 2).
Afterward, the study was conducted using a pretreatment model (Model 3). In this model, macrophages were initially incubated with Mycobacterium avium for 3h, followed by incubation of macrophages with UA for 15h. After 18 h of incubation, the macrophages were lysed to determine the CFU of Mycobacterium avium.
On the basis of Figure 3, pretreatment of macrophages with Mycobacterium avium showed a significant increase in concentration, which was evident compared to the control after 18h of incubation. Interestingly, after treatment with UA, TNF-α and IL-6 concentrations decreased significantly, starting at UA with the lowest concentrations of 25 μg/mL for TNF-α and 50 μg/mL for IL-6. Conversely, the levels of IL-1β and nitrite increased in the experimental group with a UA concentration of 50 μg/mL. The CFU results in model 3 (Figure 4) showed that cell lysate had a higher CFU value of Mycobacterium avium than the supernatant had. The CFU value decreased significantly in the experimental group, with UA concentrations of 50 μg/mL, when compared to the control.
Figure 1.
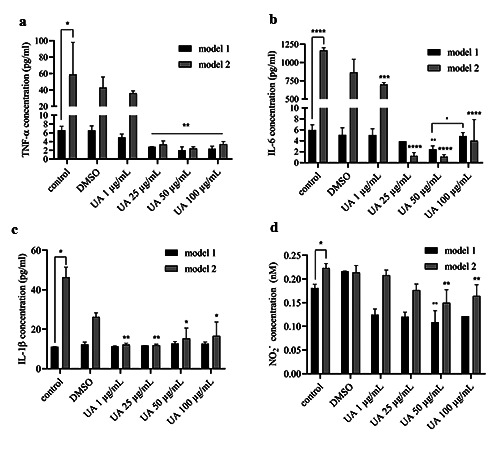
The concentrations of TNF-α (a), IL-6 (b), IL-1β (c), and NO2− (d) after UA treatment on noninfected macrophages (model 1) and macrophages infected with Mycobacterium avium (model 2). The results are described as mean of ± SD, each performed in triplicate; ****p<0.0001; ***p< .001; **p<0.01; *p<0.05.
Figure 2.
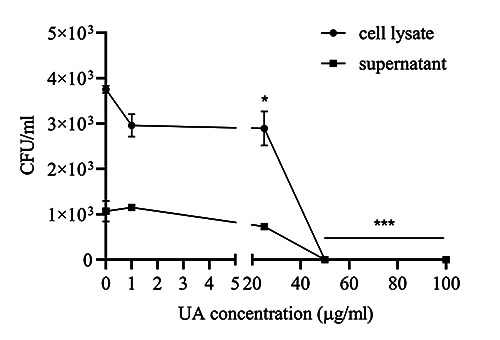
Colony-forming units (CFU) of Mycobacterium avium in model 2. The results are described as mean of ± SD, with each experiment performed in triplicate; ***p<0.001.
The study was continued by analyzing the signaling pathways in macrophages that were influenced by UA and were responsible for the production of cytokines and nitrites. This had been analyzed also in previous studies. The data generated from this study will actively support how UA regulates the MAPK signaling pathway in macrophages and how UA influences the production of cytokines and nitrites as observed in previous studies.
Well plates containing macrophages were incubated with Mycobacterium avium (MOI 8:1) and UA concentration of 50 μg/mL. Nuclear extraction of macrophages was carried out in 15 and 30 min, and analysis was carried out using sandwich ELISA. The results of the investigation were presented as absorbance, which is directly proportional to the amount of protein produced.
UA given to Mycobacterium aviuminfected macrophages was found to phosphorylate SAPK/JNK on Thr182/185 but did not phosphorylate ERK1/2 (p44/42) on Thr202/Tyr204 and MKK1/2 on Ser217/211. As a control, the UA was seen to phosphorylate MKK1/2, ERK1/2, and SAPK/JNK. Mycobacterium avium itself was only able to phosphorylate MKK1/2 and ERK1/2 (Figure 5).
Discussion
Interaction between mycobacteria and the immune system begins with phagocytosis of mycobacteria by macrophages and activates the immune response by regulating the production of cytokines and chemokines. Differences in surface lipids, sugars, and protein compositions in MTB and mycobacteria strains may affect the innate immune response.10,11 TLR signaling has been implicated in the host response to mycobacteria, as well as MAPK activation following stimulation with the mycobacterial component. MTB can activate macrophages through TLR2, stimulate ERK1/2 phosphorylation, and increase the production of proinflammatory cytokines such as TNF-α, IL-6, IL-1, and NO.12,13 Infection of macrophages with Mycobacterium avium will also stimulate TLR2 and induce the production of TNF-α, IL-1, and NO.14,15 Li et al16 showed that Mycobacterium avium and MTB share significant homology of polyketides, which were transported to the surface of mycobacteria and are associated with modulation of the host and immune response to infection, impacting on cytokine production by infected macrophages. Palucci et al11 also identified PE-PGRS 33 (a protein that belongs to the PE-PGRS family and has been linked to the virulence of MTB) in Mycobacterium avium, which also contributes in MTB entry into macrophages through the interaction with TLR2. On the basis of these similarities, we choose Mycobacterium avium as an infection agent for studying the host immune response affected by UA.
Figure 3.
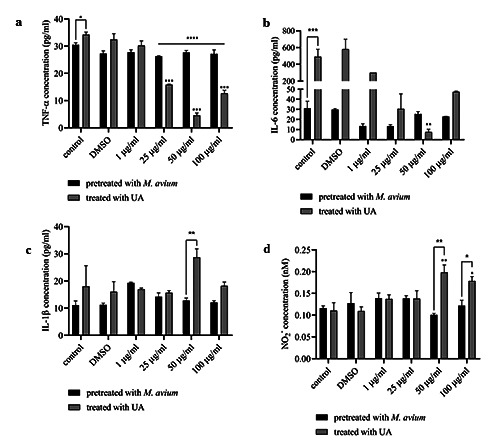
The concentrations of TNF-α (a), IL-6 (b), IL-1β (c), and NO2− (d) in model 3. The results are described as mean of ± SD, with each performed in triplicate. The results are described as mean of ± SD, with each performed in triplicate: ****p<0.0001; ***p<0.001; **p<0.01; * p<0.05.
Figure 4.
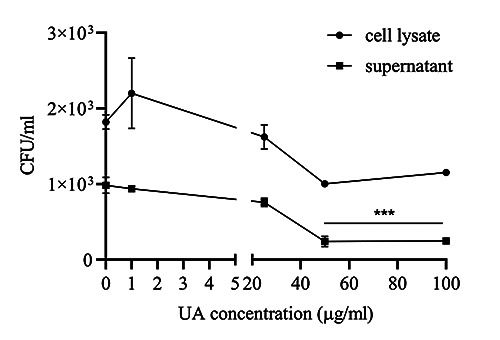
Colony-forming units (CFU) of Mycobacterium avium in model 3. The results are described as mean of ± SD, with each experiment performed in triplicate; ***p<0.001. Effect of UA on phosphorylation of ERK1/2, P38, and SAPK/JNK during infection of murine macrophages with Mycobacterium avium.
ERK1/2 and p38 activation occurred after 30–60 min of Mycobacterium avium infection, and activation is needed in the production of TNF-α and regulation of the inducible nitric oxide synthase (iNOS) genes that are responsible for NO production. 14,15 Some studies also reported that MTB induces the production of TNF-α, IL- 6, and IL-1β via P38 and ERK1/2.17,18 UA was shown to inhibit the phosphorylation of ERK1/2 in macrophages infected with Mycobacterium avium, so it is supposed that this inhibition decreases the production of cytokines and nitrites. UA reportedly severely affected the cell wall peptidoglycan, and there were signs of cell wall damage distinguished in the H37Ra and H37Rv sensitive strain of MTB by using SEM and TEM.5,6 This phenomenon is suspected to inhibit the interaction of Mycobacterium avium and macrophages from activating the signaling pathway that is responsible for the inhibition of the cytokine production.
It was observed that UA was able to increase the phosphorylation of the SAPK/JNK signaling pathways in macrophages infected with Mycobacterium avium. The final target of JNK is the activation of the c-Jun transcription factor, which is responsible for regulating the expression of proapoptotic or antiapoptotic genes, Bax (Bcl2-related X protein), and Bcl-2 (B cell lymphoma 2).19,20 When activated, JNK will phosphorylate the Ser63/Ser73 amino acid residues in part N of the c-Jun terminal so that c-Jun will be activated and increase its transcriptional activity.21 These data are supported by the decreasing expression of housekeeping genes, which indicates a decrease of macrophage viability.
The results of the research showed that UA could inhibit TNF-α production in macrophages infected with Mycobacterium avium. It will cause an unstable granuloma structure and macrophage necrosis. This phenomenon will cause MTB to pass out of the cell and become extracellular, coming into contact with the antituberculosis drugs so that they can work optimally in eliminating MTB.22 Decreasing the concentration of IL-6 induced by UA on Mycobacterium avium-infected macrophages also becomes vital because it will increase the sensitivity of macrophages to interferon (IFN)-γ. These cytokines play a role in accelerating cell mortality by increasing caspase-dependent DNA cutting and the formation of vesicle apoptosis, as well as increasing the release of lactate dehydrogenase in a caspase- dependent manner.23
The presence of IL-1β can control severe MTB infections, inflammation of the lungs, and necrosis.24 The experimental results here indicated that UA could increase the production of IL-1β, especially in model 3. IL-1β is known to suppress the replication of intracellular MTB via IL-1R receptors and MyD88. In addition, IL-1β can directly kill MTB in macrophages and influence the recruitment of other antimicrobial effector molecules and can activate caspase 3 via extrinsic pathways.25
NO also plays a vital role in bacteriostatic and bactericidal processes as part of the defense mechanism against MTB infection. Inflammatory stimulation due to mycobacterial infection can increase NO release through the upregulation of iNOS in macrophages. NO will be converted into a highly reactive nitrogen species such as NO2 − to encourage MTB mortality. A reactive oxygen intermediate such as superoxide (O2 −) and hydroxyl radical (OH−) can form a reactive conjugate with NO2 − and increase the production of peroxynitrite (ONOO−) to kill MTB in phagosomes.13 UA is found to increase nitrite production in macrophages infected with Mycobacterium avium. This effect is vital to eliminating intracellular MTB.
Conclusions
On the basis of the results of this and other studies that have been carried out and supported by their findings, it can be concluded that UA has antimycobacterial activity and prevents the pathogen from activating the MAPK pathway. This mechanism decreases the production of proinflammatory cytokines such as TNF-α, IL-6, IL-1β, and nitrite. The ability of UA to inhibit phosphorylation of ERK1/2 and P38 signaling pathways also enables the reduction of the production of these proinflammatory cytokines and nitrites induced by mycobacteria. Conversely, the increase in the activation of the SAPK/JNK pathway by UA is suspected of activating the apoptotic process in mycobacteria-infected macrophages, which will also be beneficial for the process of eliminating the mycobacteria. This study provides experimental evidence on the application of UA and indicates that UA may be a candidate for the treatment of TB and of infection caused by Mycobacterium avium in the future.
Figure 5.
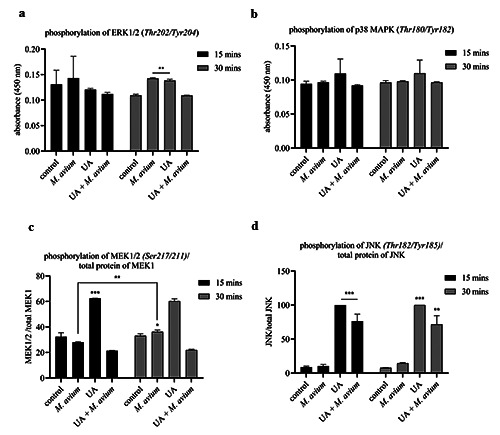
Nuclear protein absorbance in the experimental group in 15 and 30 min, consisting of (a)ERK1/2 MAPK phosphorylation, (b)phosphorylation of p38 MAPK, (c)relative phosphorylation of MEK1/2 to the total protein of MEK1, and (d)relative phosphorylation of JNK to the total protein of JNK. The results are described as mean of ±SD, with each performed in triplicate; ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05.
Acknowledgements
This research was supported by Directorate General of Resources for Science Technology and Higher Education of the Republic of Indonesia.
Funding Statement
Funding: This research was supported by
References
- 1.World Health Organization. Global tuberculosis report 2018. World Health Organization; 2019. Available from https://www.who.int/tb/publications/global_report/en/ Accessed: May 2019. [Google Scholar]
- 2.Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol Rev 2012;36:514–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szakiel A, Pączkowski C, Pensec F, Bertsch C. Fruit cuticular waxes as a source of biologically active triterpenoids. Phytochem Rev 2012;11:263–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jager S, Trojan H, Kopp T, et al. Pentacyclic triterpene distribution in various plants - rich sources for a new group of multi-potent plant extracts. Molecules 2009;14:2016–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jyoti MdA, Zerin T, Kim T-H, et al. In vitro effect of ursolic acid on the inhibition of Mycobacterium tuberculosis and its cell wall mycolic acid. Pulm Pharmacol Ther 2015;33:17–24. [DOI] [PubMed] [Google Scholar]
- 6.Pitaloka DAE, Damayanti S, Artarini AA, Sukandar EY. Molecular docking, dynamics simulation, and scanning electron microscopy (SEM) Examination of Clinically Isolated Mycobacterium tuberculosis by Ursolic Acid: A pentacyclic triterpenes. Indones J Chem 2019;19: 328-36. [Google Scholar]
- 7.Pitaloka DAE, Sukandar EY. In vitro study of ursolic acid combination firstline antituberculosis drugs against drugsensitive and drug-resistant strains of Mycobacterium tuberculosis. Asian J Pharm Clin Res 2017;10:216-8. [Google Scholar]
- 8.Lee K-C, Chen Y-L, Lin P-Y, Chuang W-L. Ursolic Acid-Induced Apoptosis via Regulation of the PI3K/Akt and MAPK Signaling Pathways in Huh-7 Cells. Molecules 2018;23:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armstrong JA, Hart PD. Response of Cultured Macrophages to Mycobacterium tuberculosis, with Observations on Fusion of Lysosomes with Phagosomes. J Exp Med 1971; 134:713–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med 1995;181:257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palucci I, Camassa S, Cascioferro A, et al. PE_PGRS33 Contributes to Mycobacterium tuberculosis Entry in Macrophages through Interaction with TLR2. PloS One 2016;11:1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T, Lafuse WP, Zwilling BS. Regulation of Toll-Like Receptor 2 Expression by Macrophages Following Mycobacterium avium Infection. J Immunol 2000;165:6308–13. [DOI] [PubMed] [Google Scholar]
- 13.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 2000;97:8841–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan ED, Morris KR, Belisle JT, et al. Induction of inducible nitric oxide synthase- NO* by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-kappaB signaling pathways. Infect Immun 2001;69:2001–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhattacharyya A, Pathak S, Kundu M, Basu J. Mitogen-activated protein kinases regulate Mycobacterium avium-induced tumor necrosis factoralpha release from macrophages. FEMS Immunol Med Microbiol 2002;34:73–80. [DOI] [PubMed] [Google Scholar]
- 16.Li YJ, Danelishvili L, Wagner D, et al. Identification of virulence determinants of Mycobacterium avium that impact on the ability to resist host killing mechanisms. J Med Microbiol 2010;59:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulthard LR, White DE, Jones DL, et al. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol Med 2009;15:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HM, Shin DM, Jo EK. Mycobacterium tuberculosis induces the production of tumor necrosis factor- α, interleukin-6, and CXCL8 in pulmonary epithelial cells through reactive oxygen species-dependent mitogenactivated protein kinase activation. J Bacteriol Virol 2009;39:1–10. [Google Scholar]
- 19.Zhang P, Miller BS, Rosenzweig SA, Bhat NR. Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J Neurosci Res 1996;46:114–21. [DOI] [PubMed] [Google Scholar]
- 20.Bos JL, Dansen TB, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002;419: 316-321. [DOI] [PubMed] [Google Scholar]
- 21.Bogoyevitch MA, Kobe B. Uses for JNK: the Many and Varied Substrates of the c-Jun N-Terminal Kinases. Microbiol Mol Biol Rev 2006;70:1061–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson CM, Jung J-Y, Nau GJ. Interferon-γ, Tumor Necrosis Factor, and Interleukin-18 Cooperate to Control Growth of Mycobacterium tuberculosis in Human Macrophages. Cytokine 2012;60:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Kornfeld H. Interferon-γ Regulates the Death of M. tuberculosis- Infected Macrophages. J Cell Death 2010;3:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourigault M-L, Segueni N, Rose S, et al. Relative contribution of IL-1α, IL-1β and TNF to the host response to Mycobacterium tuberculosis and attenuated M. bovis BCG. Immun Inflamm Dis 2013;1:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jayaraman P, Sada-Ovalle I, Nishimura T, et al. IL-1β Promotes Antimicrobial Immunity in Macrophages by Regulating TNFR Signaling and caspase- 3 activation. J Immunol 2013; 190:4196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]