

Abstract
Photo-controlled affinity reagents seek to provide modular spatiotemporal control of bioactivity by conferring photo-switchability of function on an affinity reagent scaffold. Here we used Rosetta-based computational methods to screen for sites on the Fynomer affinity reagent structure for attachment of photoswitchable cross-linkers. Both established UV-based cross-linkers (azobenzene-iodoacetamide (IAC)) and an azonium-based efficient red light switchable cross-linker, piperazino-tetra-ortho-methoxy azobenzene (PIP), were then tested experimentally. Several sites compatible with Fynomer function were identified, including sites showing rapid (<10s) red light (633 nm) modulation of function. While a range of overall target binding affinities were observed, the degree of photo-switchability of Fynomer function was generally small (<2-fold). Computational models suggest that local flexibility limits the degree of switching seen in these designs.
Keywords: azobenzene, azonium, photopharmacology, fynomer, optogenetic
Graphical Abstract
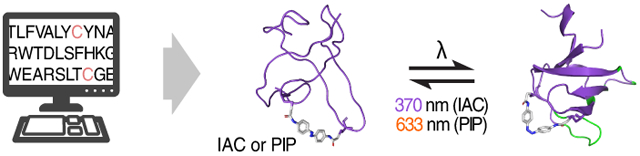
A light touch on proteins. With the advent of photopharmacology, a modular, generalizable strategy to confer photoswitchability to biomolecules is crucial. We present a strategy (see picture) where computational methods identified potential cross-linking sites on the Fynomer protein scaffold for azobenzene-based cross-linkers, IAC and PIP. Testing these sites experimentally showed that IAC and PIP isomerization by light could be used to control Fynomer activity.
Introduction
Photo-control of bioactive molecules can permit remote control of biological processes with unprecedented spatiotemporal precision.[1] Photo-controlled biomolecules can serve as probes for studying complex living systems and also have clinical potential as photopharmaceuticals.[2] While several promising photopharmaceuticals have been developed via modification of small molecule drugs with photoswitchable moieties,[2b] in practice, development of a photopharmaceutical for each new target requires extensive optimization.[3] Furthermore, the delivery of light into tissue is limited by wavelength, where red and near-infrared wavelengths have the deepest penetration.[4] A requirement for long wavelength photoswitching adds additional restrictions on the chemical nature of the photoswitchable moiety, increasing the difficulty of small molecule photopharmaceutical design.
Photoswitchable affinity reagents offer a potential generalizable photopharmaceutical design strategy. Numerous protein affinity reagent scaffolds have been developed that can readily be evolved to recognize diverse targets with high affinity.[5] We and others have shown that such scaffolds can be made photoswitchable via the introduction of azobenzene-based cross-linkers.[6] Isomerization of the azobenzene cross-linker by light can distort the affinity reagent structure, thereby coupling light and protein activity (Figure 1). The modularity of the approach means that target affinity and photoswitching characteristics (e.g. wavelength dependence, thermal relaxation rate) can be independently optimized. In previous work, we showed that we could confer photo-switchability on a Fynomer affinity reagent that inhibits the activity of the serine protease, chymase, by attaching the UV-switchable azobenzene cross-linker BSBCA or the red light switchable cross-linker TOM (Scheme I).[6a]
Figure 1.
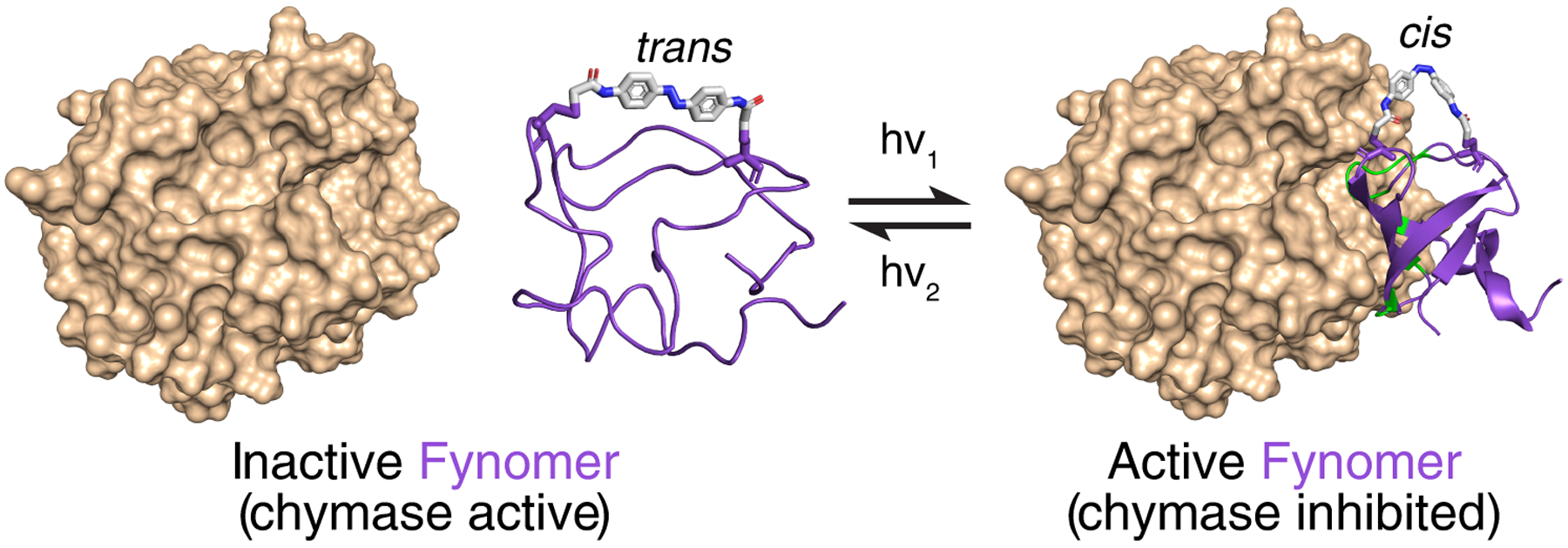
Photo-modulation of Fynomer (purple) structure by an intramolecular photoswitchable cross-linker (shown as sticks) can affect the binding of the Fynomer to its target (chymase, brown).
Scheme 1.
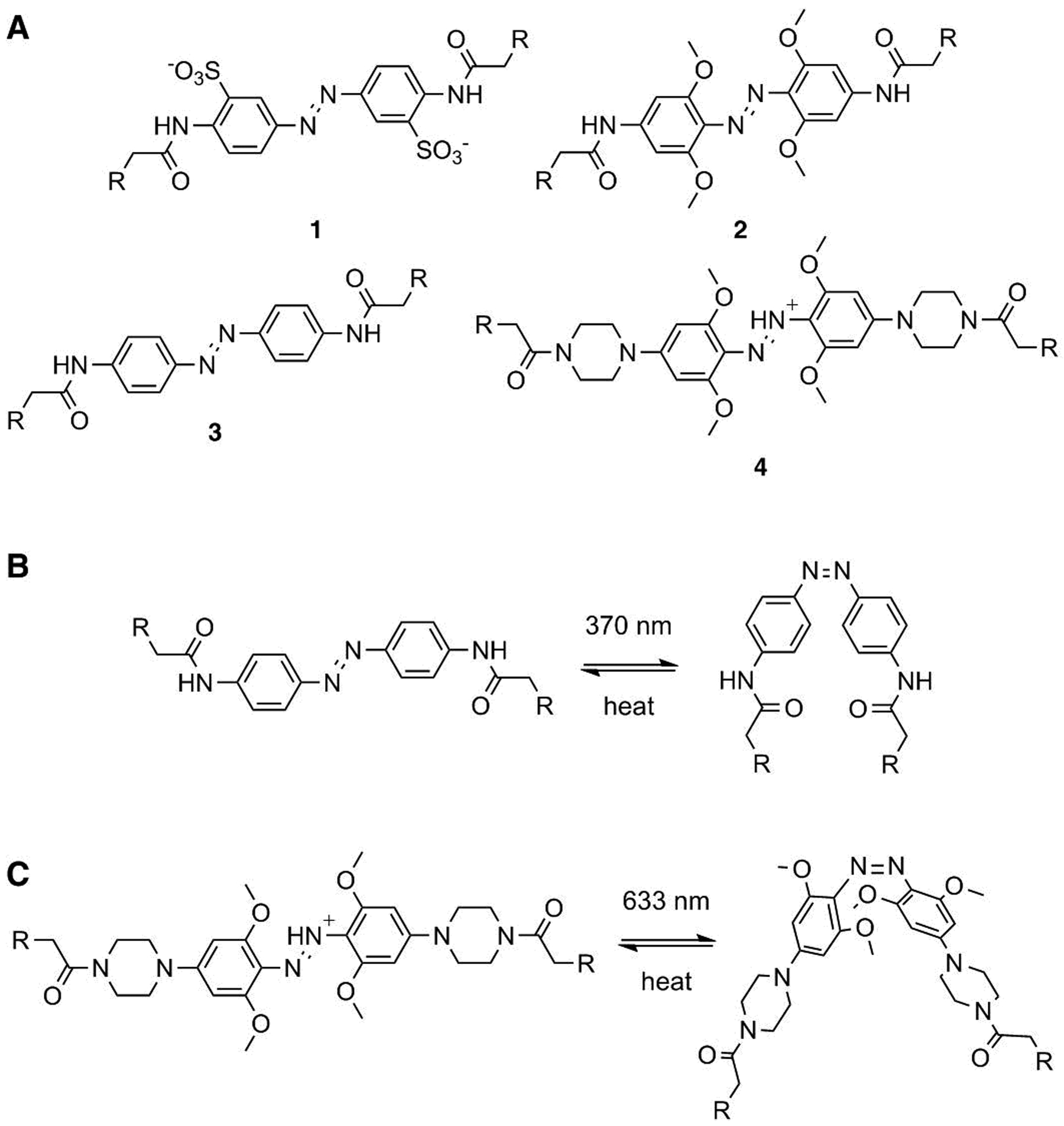
(A) Azobenzene-based cross-linkers used in previous work, BSBCA (1) and TOM (2), and in this study, IAC (3) and PIP (4). (B) The trans-to-cis isomerization of IAC (3) is induced by 370 nm light and relaxation to trans occurs thermally. (C) The trans-to-cis isomerization of the azonium-ion based PIP cross-linker (4) is induced by 633 nm light and relaxation to trans occurs thermally.
Although the structure and activity of Fynomers could be successfully controlled with azobenzenes and light, these first generation photoswitchable Fynomers had a number of limitations. First, the affinity of the cross-linked Fynomers for the target chymase was significantly lower, even in the active state, than the affinity of the unmodified Fynomer. Second, the degree of switching of affinity between the cis-state and trans-state of both the BSBCA-Fynomer and TOM-Fynomer was low ~(2-fold). Third, the efficiency of red light switching of the TOM-Fynomer was very low since the absorption coefficient of TOM in the red region is small, [7] and thermal relaxation is very slow (>10h).[6a, 7]
In the present study, we used computational methods to identify alternative cross-linking sites on the Fynomer scaffold in an effort to address the first and second of these limitations. The decreased affinity of the cross-linked Fynomers for chymase, even in their active states, may be a consequence of the cross-linker location on the Fynomer causing a steric clash with a chymase loop in the chymase-bound state. We reasoned that different cross-linker locations should relieve this clash and also may be expected to show different degrees of coupling between cross-linker isomerization and protein folding/unfolding. We explored cross-linking with the established UV-light cross-linker, IAC, analogous to BSBCA but with higher Cys reactivity,[8] and also with the azonium ion based red-light cross-linker, PIP (Scheme 1). The azonium form of PIP has a high absorption coefficient (ε630 = ~20,000 M−1cm−1)[9] in the red region of the spectrum and an intrinsic thermal relaxation time of seconds[9–10] making it more suitable for red light photoswitching than TOM under physiological conditions.[10]
Overall 9 different cross-linking sites were identified using our computational design approach[11] (one site L3CL29C was identified previously), the appropriate Cys mutations were introduced, and the Fynomer proteins expressed and purified. Of these, some could be cross-linked easily in high yield whereas others gave mixtures of un-crosslinked, correctly cross-linked, and doubly reacted species under standard cross-linking conditions (see Experimental section). With the UV-cross-linker IAC, five cross-linked Fynomer mutants were purified and characterized. Amongst these IAC-Fynomers, a range of behaviours was seen. One mutant (N11CT44C) was a substantially stronger inhibitor of chymase activity than the BSBCA-Fynomer characterized previously, however, the degree of switching observed was similar. With the red light cross-linker PIP, seven different cross-linked Fynomer mutants were purified and characterized. Of these, some showed enhanced inhibition of chymase activity under red light, others showed chymase inhibition but no difference under red light, and one mutant (L3CL29C) showed reverse behaviour with decreased inhibition under red light. This is the first example of azonium-ion modified biomolecules having photoswitchable activity.
Results
Computational models
For all the azobenzene derivatives studied here, dark-adaptation quantitatively produces the trans isomeric form, whereas irradiation produces a photostationary state substantially enriched for the cis isomer.[12] To maintain an affinity reagent in its inactive form until activation is desired, and to maximize the degree of activation, cross-linking sites were chosen to be compatible with cis isomeric forms of the cross-linkers.[6a] Trans isomers were intended to promote unfolding of the Fynomer structure by adopting end-to-end distances too long to be accommodated by the folded Fynomer structure.[6a, 13]
Using a computational protocol we developed previously,[11] we used the RosettaMatch algorithm to query all pairs of residue positions in the Fynomer-chymase complex structure (PDB: 4AG1) to find suitable cysteine substitutions that could sterically accommodate the attachment of an azobenzene derivative in the cis conformation. We chose two derivatives, IAC, an established UV-switchable cross-linker[14] and PIP, an azonium-ion based cross-linker that can be switched efficiently with red light.[10] The generation of conformational ensembles of each of these azobenzene derivatives for use in the RosettaMatch algorithm is described in the Experimental section.
Five different cis-IAC cross-linked models are shown in Figure 2. The range of end-to-end distances of the cis-PIP cross-linkers includes larger distances than for cis-IAC,[15] however, models indicated it could also be accommodated at the same set of sites, especially if local backbone and side-chain movements occurred (see SI and Discussion section).
Figure 2.
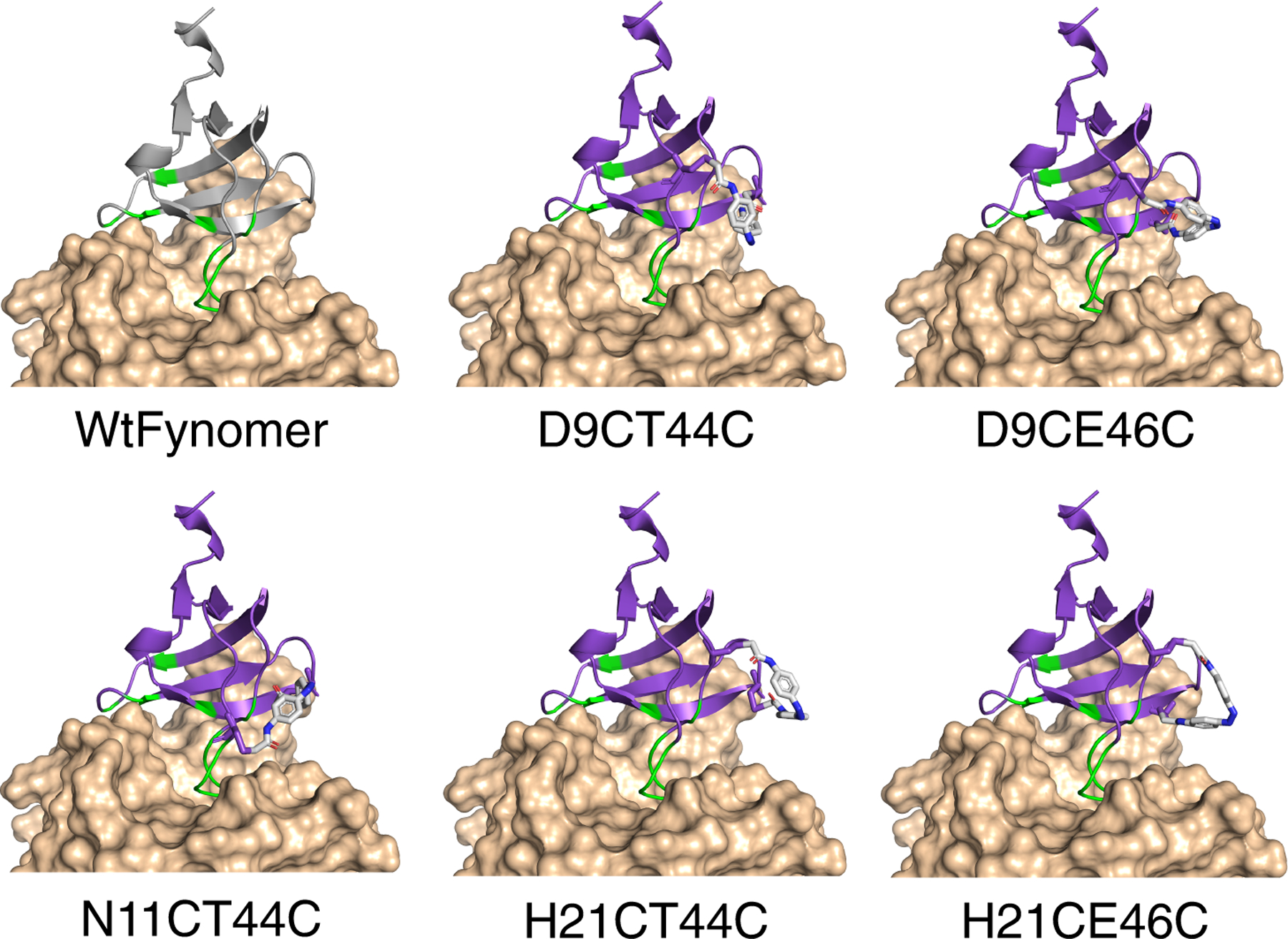
Structure of the parent Fynomer (PDB: 4AG1, gray) and computational models of cis-IAC-cross-linked Fynomers (purple) bound to chymase (beige). Binding occurs via modified RT- and n-src-loops (green).
Fynomer purification and cross-linking with IAC
Based on the computationally selected cross-linking points, dual-cysteine mutants of Fynomer were constructed from the chymase-binding Fynomer variant 4C-E4 (see Experimental section)(Table I).[16] These were then moved into the pET system for expression in E. coli. After purification, Fynomer mutants were cross-linked with IAC under denaturing conditions with guanidine (Gdn) (see Experimental section). The relative yield of the cross-linked product versus un-cross-linked or doubly reacted protein was assessed by MALDI or ESI-MS (see Supplementary Figure 1). Of the 10 Fynomer mutants expressed, 5 Fynomers (D9CT44C, D9CE46C, N11CT44C, H21CT44C, and H21CE46C) were efficiently cross-linked and resulted in the singly-cross-linked protein as the major product. L3CL29C was cross-linked previously.[6a] The reactions of the remaining 4 Fynomer mutants resulted in a mixture of uncross-linked, correctly cross-linked, and/or doubly-cross-linked Fynomer.
Table 1.
Fynomer cross-linker attachment sites tested
| Mutant | Cross-link point | Res. apart | Half-life (IAC) [min] | Half-life (PIP) [sec] | Res. flanking 1st Cys | Res. flanking 2nd Cys |
|---|---|---|---|---|---|---|
| D9CT44C | D9C + T44C | 34 | 42.2 | — | YCY | TCG |
| D9CE46C | D9C + E46C | 36 | 10.4 | 2.1 | YCY | GCT |
| N11CH21C | N11C + H21C | 9 | — | — | YCA | FCK |
| N11CT44C | N11C + T44C | 33 | 11.8 | 3.5 | YCA | TCG |
| H21CK25C | H21C + K25C | 3 | — | — | FCK | ECF |
| H21CT43C | H21C + T43C | 21 | — | 3.0 | FCK | LCT |
| H21CT44C | H21C + T44C | 22 | 14.6 | 4.4[a] | FCK | TCG |
| H21CE46C | H21C + E46C | 24 | 16.4 | 8.6 | FCK | GCT |
| K25CT43C | K25C + T43C | 21 | — | 6.8 | ECF | LCT |
| L3CL29C[b] | L3C + L29C | 25 | — | 130 | TCF | ICE |
Includes uncross-linked protein.
Previously characterized with BSBCA.[6a]
Photoswitching was confirmed and half-lives for thermal cis-to-trans relaxation were measured using UV-VIS spectroscopy (Table 1, Supplementary Figure 2). Fynomer mutants D9CE46C, N11CT44C, H21CT44C, and H21CE46C showed half-lives similar to IAC alone (20.1 min). For D9CT44C, the half-life was substantially longer (42.2 min). We hypothesize that aggregation of D9CT44C in solution in the dark-adapted state could be slowing trans-to-cis isomerization (vide infra).
Chymase inhibition assay
The parent Fynomer used here binds chymase at the active site and inhibits chymase activity.[16] To test the activity of cross-linked Fynomer mutants we used a colorimetric assay for chymase activity. In this assay, chymase catalyzed hydrolysis of N-succinyl-Ala-Ala-Pro-Phe p-nitroanilide produces p-nitroaniline, which can be quantified by UV-Vis spectroscopy. The addition of a light-activated inhibitor, i.e., a cross-linked Fynomer, decreases chymase activity under light conditions (370 nm for IAC, 633 nm for PIP) compared to dark conditions.
Fynomer mutants cross-linked with IAC (D9CT44C, D9CE46C, N11CT44C, H21CT44C, H21CE46C) exhibited light-activated inhibition of chymase activity albeit with varying degrees of photoswitching and apparent IC50 values (Figure 3). N11CT44C showed the lowest IC50 in both light (26 ± 7 nM) and dark (36 ± 11 nM) conditions. The light (72 ± 2 nM) and dark (121 ± 14 nM) activity of H21CE46C was on par with the previously reported BSBCA-Fynomer.[6a] H21CT44C gave higher IC50 values in both light (243 ± 55 nM) and dark (398 ± 94 nM) conditions. D9CT44C showed weak inhibitory activity in the dark and enhanced inhibition upon UV-irradiation (Supplementary Figure 3). However, this mutant exhibited visibly aggregated in the dark over time, thus increased activity upon irradiation likely reflects photoisomerization-induced monomerization. When D9CT44C was pre-incubated with Gdn just prior to the enzyme assay to prevent aggregation, results were more consistent to other cross-linked Fynomers (see Supplementary Figure 3). In contrast, D9CE46C showed very weak inhibition under both light and dark conditions without any apparent tendency to aggregate.
Figure 3.
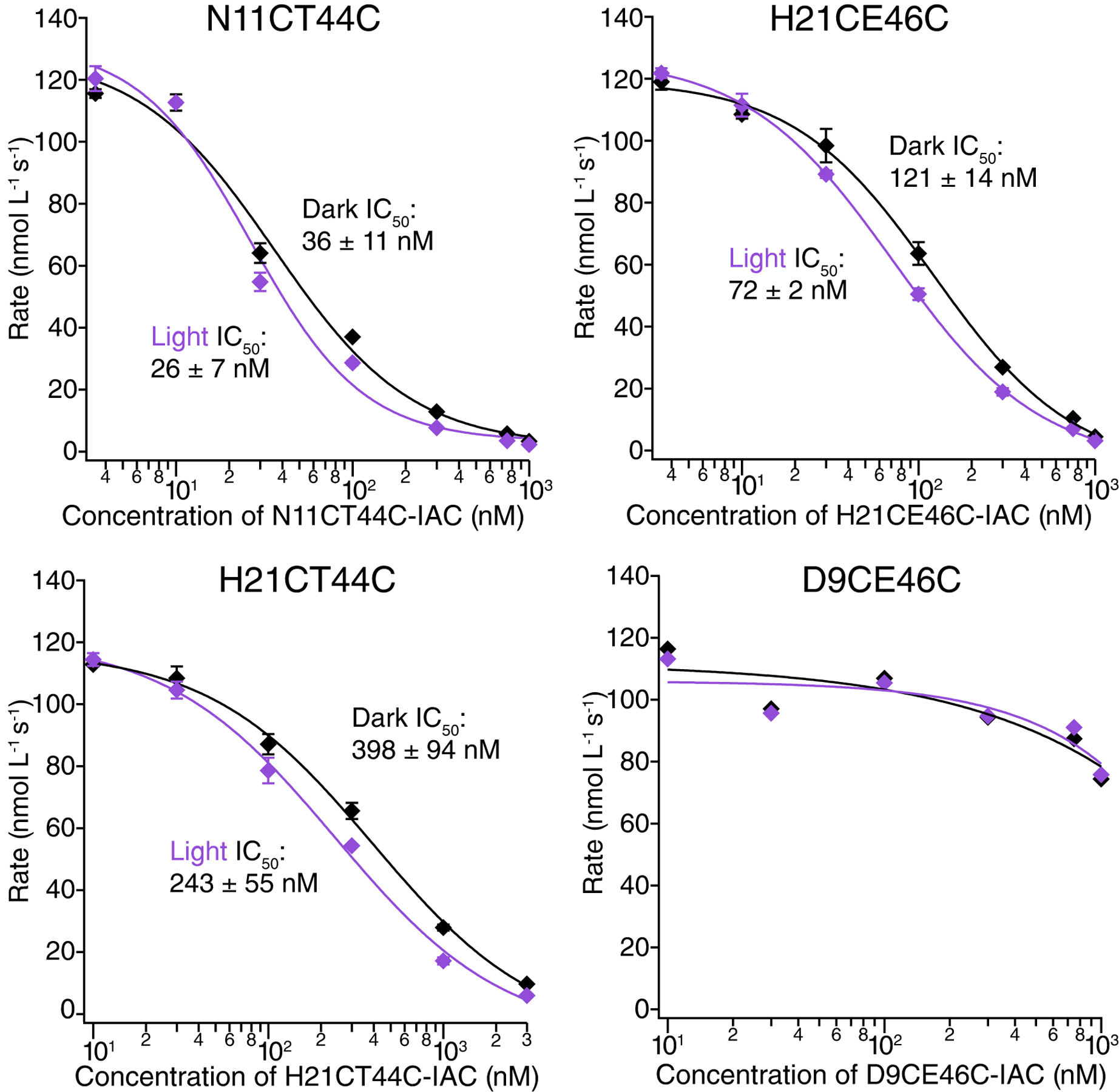
Chymase inhibition assay data for IAC-cross-linked N11CT44C, H21CT44C, H21CE46C, and D9CE46C in dark (trans-state, black) and 370 nm light (cis-state, purple) conditions.
Red-light switching PIP-Fynomers
Red-shifted photopharmaceuticals are particularly sought after because red light penetrates tissues more deeply than shorter wavelengths. For localized activation of an affinity reagent, rapid trans-to-cis isomerization with low intensity red or near-IR light is desirable, with rapid (~seconds) thermal relaxation to avoid activated molecules being carried outside the area by the circulatory system.[4b] Azonium ions meet these criteria, and the PIP switch is readily synthesized.[10]
We found that D9CE46C, N11CT44C, H21CT43C, H21CT44C, H21CE46C, K25CT43C, and L3CL29C mutants could be readily cross-linked with PIP (Table 1). Most Fynomer mutants cross-linked with PIP exhibited half-lives comparable to PIP alone (7.3 s) with the exception of Fynomer mutant L3CL29C (Fig. 4, Supplementary Fig. 4). The lifetime of cross-linked L3CL29C was ~15-fold longer than the life-time of PIP alone. The longer half-life did not appear to be due to aggregation since the value observed was independent of concentration.
Figure 4.
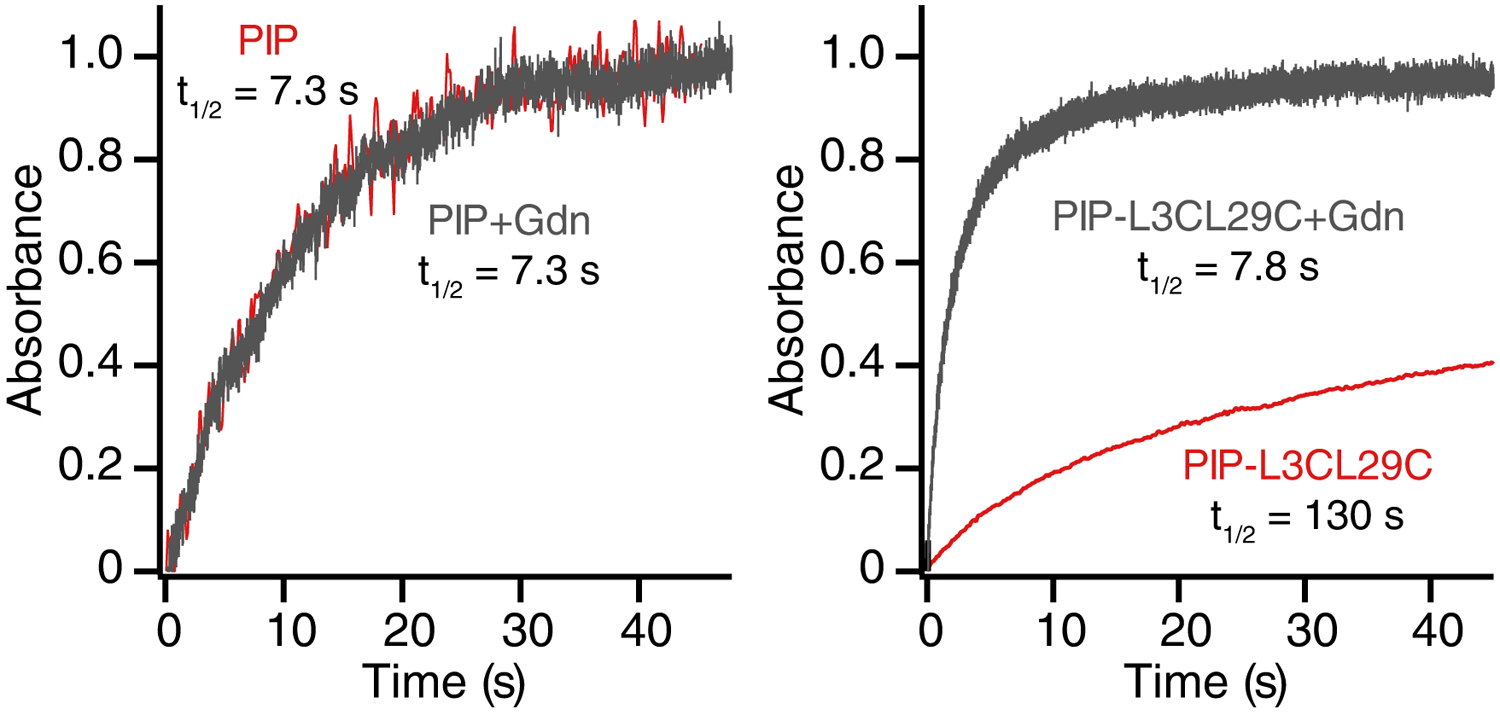
Time-course of cis-to-trans relaxation for PIP alone (left) and PIP-cross-linked L3CL27C (right), either with Gdn (gray) or without Gdn (red). Assays were performed in 10 mM sodium phosphate (pH 7.0).
To determine if the folded protein structure of L3CL29C was impacting the half-life of PIP, the half-life was measured in denaturing conditions (6M Gdn) (Figure 4). Under these conditions, the half-life (7.8 s) became comparable to the half-life of PIP alone (7.3 s), indicating that the folded structure of PIP-L3CL29C causes the longer half-life.
Models of PIP-cross-linked Fynomers showed that PIP lies over neutral or acidic patches on the folded Fynomer structure in all cases with the exception of L3CL29C. In the L3CL29C case, the local environment around PIP is positively charged (Fig. 5). We hypothesize that the local positively charged environment could be destabilizing the cationic azonium ion, promoting dissociation to the neutral azo species, thereby increasing the half-life of PIP.[10]
Figure 5.
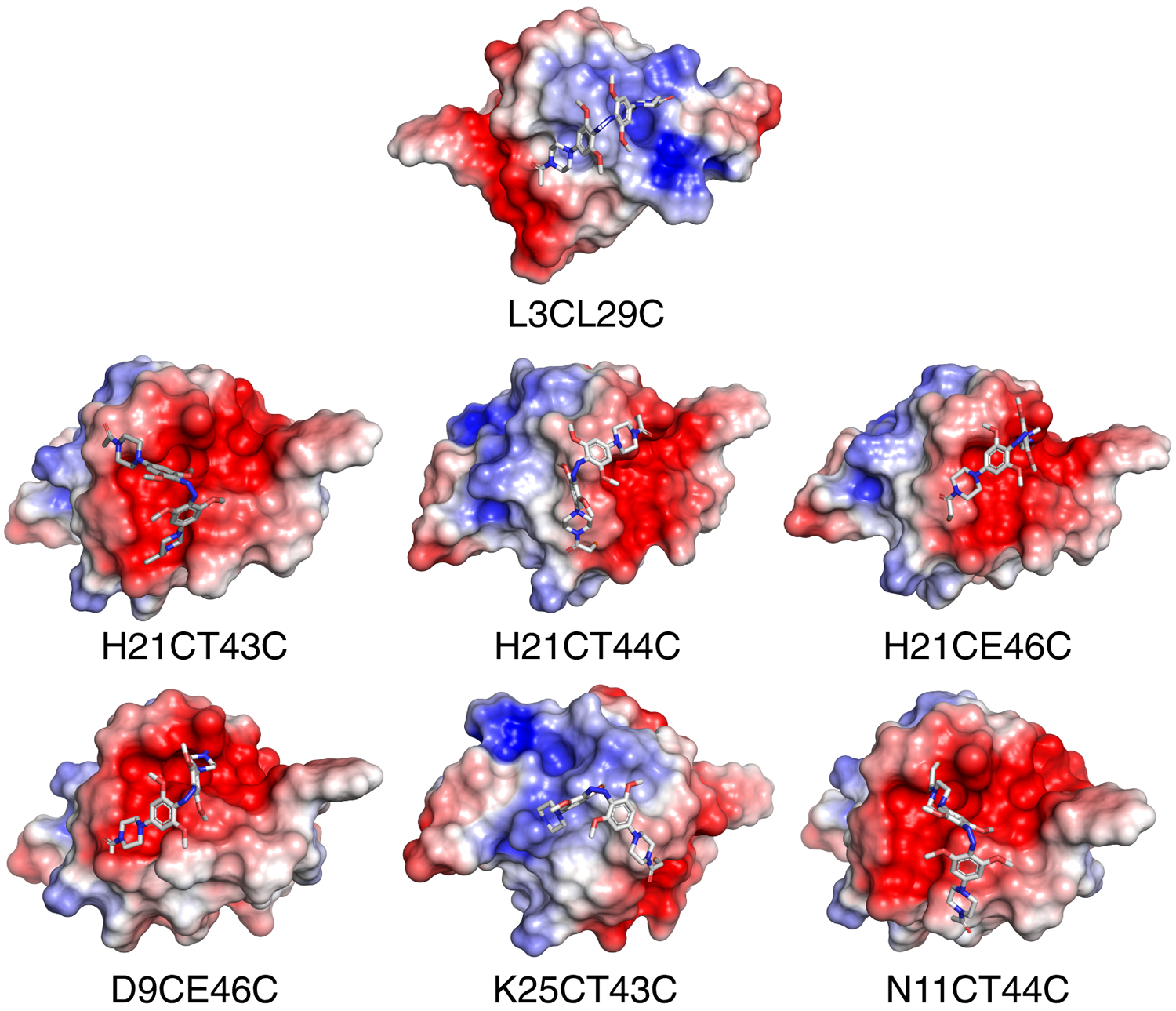
Electrostatic-potential maps of PIP-cross-linked Fynomers in the folded state. Areas of negative electrostatic potential are red and blue shows positive areas on the Fynomer surface. PIP in the cis-state is shown in sticks.
Chymase assays for red-light PIP-Fynomers
Because of the rapid relaxation of PIP-cross-linked species in the dark, end point assays were performed as opposed to reaction rate determinations (see Experimental section). Samples were either continuously irradiated with red light (633 nm, 90 mW/cm2) or kept in the dark, and the extent of substrate hydrolysis was measured after a fixed time interval. Red light alone had no effect on the assay. When PIP-Fynomers were tested using this assay, an array of behaviours was found. Mutants D9CE46C and K25CT43C did not exhibit any light-dependent change in inhibition (Figure 6). While D9CE46C was a relatively weak inhibitor (IC50 approx. 70 nM), K25CT43C was almost comparable to wild-type Fynomer (IC50 ~10 nM). Mutants N11CT44C, H21CT43C, and H21CE46C showed some difference in chymase inhibition between light and dark at specific concentrations (e.g. 10 nM for H21CT43C and H21CE46C, and 50 nM for N11CT44C). PIP-L3CL29C was unusual because it showed a light-dependent decrease in inhibition at high concentrations (100 nM). As noted above, at the L3CL29C cross-linker site, steric clashes can occur between the cross-linker and chymase. Neither cis or trans isomers of PIP-L3CL29C are particularly good inhibitors, as was observed for BSBCA-L3CL29C previously.[6a] Perhaps the cis form of PIP-L3CL29C produces more severe steric clashes with the chymase loop than the trans form accounting for the reversed sense of photo-modulation.
Figure 6.
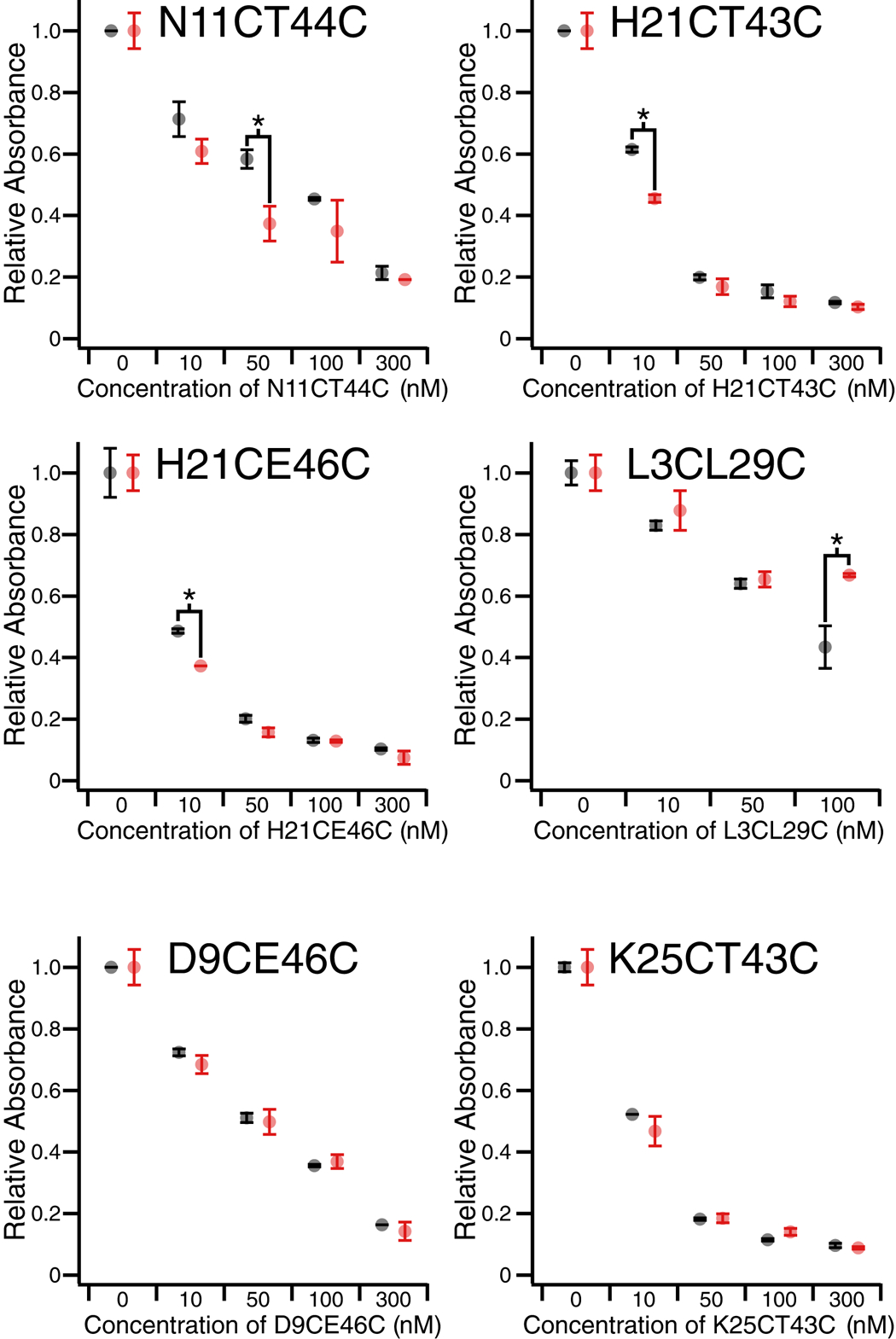
End point assay chymase inhibition data for PIP-cross-linked N11CT44C, H21CT43C, H21CE46C, L3CL29C, D9CE46C, K25CT43C in dark (trans-state, black) and 633 nm light conditions (cis-state, red). Absorbance corresponds to the amount of substrate turned over during a 5 min incubation with chymase and Fynomer. Significant differences between inhibition in the dark compared to the light are marked with an asterisk (*). Error bars show standard deviation from 2 replicates.
Discussion
Computational design led to the identification of a number of attachment sites for the cross-linkers IAC and PIP on the Fynomer structure. In total, nine new Fynomers were predicted to be amenable to cross-linker attachment, in addition to the L3CL29C mutant studied previously. Of these, five were successfully cross-linked with IAC and seven were cross-linked with PIP and purified (Table I).
Cross-linking was carried out under denaturing conditions so that reaction between Cys residues on the Fynomer and the iodo- or chloroacetamide groups on the cross-linkers occurred from the unfolded ensemble of Fynomer structures. We hypothesize that the distance between the cysteines and the local environment around the cysteines affects the efficiency of the cross-linking reaction. For example, cross-linking of the two mutants with the shortest sequence separation (N11CH21C, H21CK25C) proved most problematic (Table I). Fynomer mutants containing a Cys with a flanking Gly residue generally gave good yields of correctly cross-linked products, perhaps due to decreased steric hindrance.
The UV-irradiated forms of the five successfully cross-linked IAC-Fynomer mutants (D9CT44C, D9CE46C, N11CT44C, H21CT44C, and H21CE46C) were capable of inhibiting chymase to varying degrees (Figure 3) The strongest inhibitor was N11CT44C (IC50 in light: 26 ± 7 nM and dark: 36 ± 11 nM) whereas the weakest was D9CE46C (IC50 >1000 nM). Weak inhibition was also seen with D9CT44C but appeared to be due to self-association of the cross-linked Fynomer in that case. This range of IC50 values suggests that modification of a Fynomer with a cross-linker can impact activity significantly (~100 fold-range vs. an unmodified Fynomer (see SI and[6a]). Interestingly, models of both weakly-binding designs (D9CT44C and D9CE46C) required small but significant changes in the backbone conformation (sampled via Rosetta FastRelax) of the bound-state structure of the Fynomer (Supplementary Fig. 8), indicating that a high degree of cis-cross-linker compatibility with the native backbone structure may be necessary for tight binding of cross-linked variants. Since the modelled cis-IAC-D9CE46C structure was only slightly distorted from the unmodified Fynomer-chymase complex (Supplementary Fig. 8), this may imply that local distortion can in some cases have significant effects on target binding affinity. Alternatively, cross-linking at certain sites may introduce substantial changes in Fynomer dynamics, and thereby activity,[17] a feature that would not be captured in these models.
Despite improvements in cis-state affinity for the target compared to first generation cross-linked Fynomers (i.e. for N11CT44C), the degree of switching observed was similar to that seen previously for the L3CL29C mutant with BSBCA or TOM.[6a] In an effort to understand this, we used a computational approach to simulate cis-to-trans isomerization of the cross-linker. The model of the cis-IAC ligand cross-linked to N11CT44C was “mutated” to its trans isomer, and the same constraints between the cysteine sidechains and ligand that were applied during the geometric matching and optimization stages were also applied to the resulting conformation. The Rosetta FastRelax protocol was then used to sample backbone and sidechain movements in cross-link-neighbouring and binding loop regions to simultaneously optimize geometries of cross-linker/Cys connections and the energy of the protein. This simulation was conducted with the Fynomer domain both bound and unbound to the chymase domain in order to remove steric hindrances that may constrain any sampling of the loop conformation. The presence of the chymase domain had little impact on the results. Figure 7 shows the resulting structure of a low-energy conformation for trans IAC-linked N11CT44C overlaid with cis-IAC-linked N11CT44C. It appears that a collection of small loop backbone movements and Cys side-chain rotamer changes can accommodate the trans IAC cross-linker without a major shift in the overall Fynomer structure or in the binding interface of the complex. The observed small increase in the calculated Rosetta energy of the complex in the trans state (Fig. 7 and Supplementary Fig. 9), indicating slight destabilization, is compatible with the experimentally observed minor decrease in binding affinity of this variant under light compared to dark conditions (Fig. 3). For the L3CL29C mutant, a previous explicit chain model analysis suggested that trans isomers could be accommodated by a disengagement of the N-terminal strand, also without a major shift in the overall Fynomer structure.[17] Thus, a local structural distortion may not result in a substantial change in Fynomer activity depending on the conformational features of the system. Additionally, protein dynamics and/or solvation properties of the photoswitch, which are not modeled in our current approach, are also expected to be altered upon cross-linking and irradiation. Design success may be improved by further development of methodology to identify cross-linking sites where local distortion is more costly to the overall fold, or allosteric sites which are conformationally coupled to the functional regions of the protein.[18]
Figure 7.
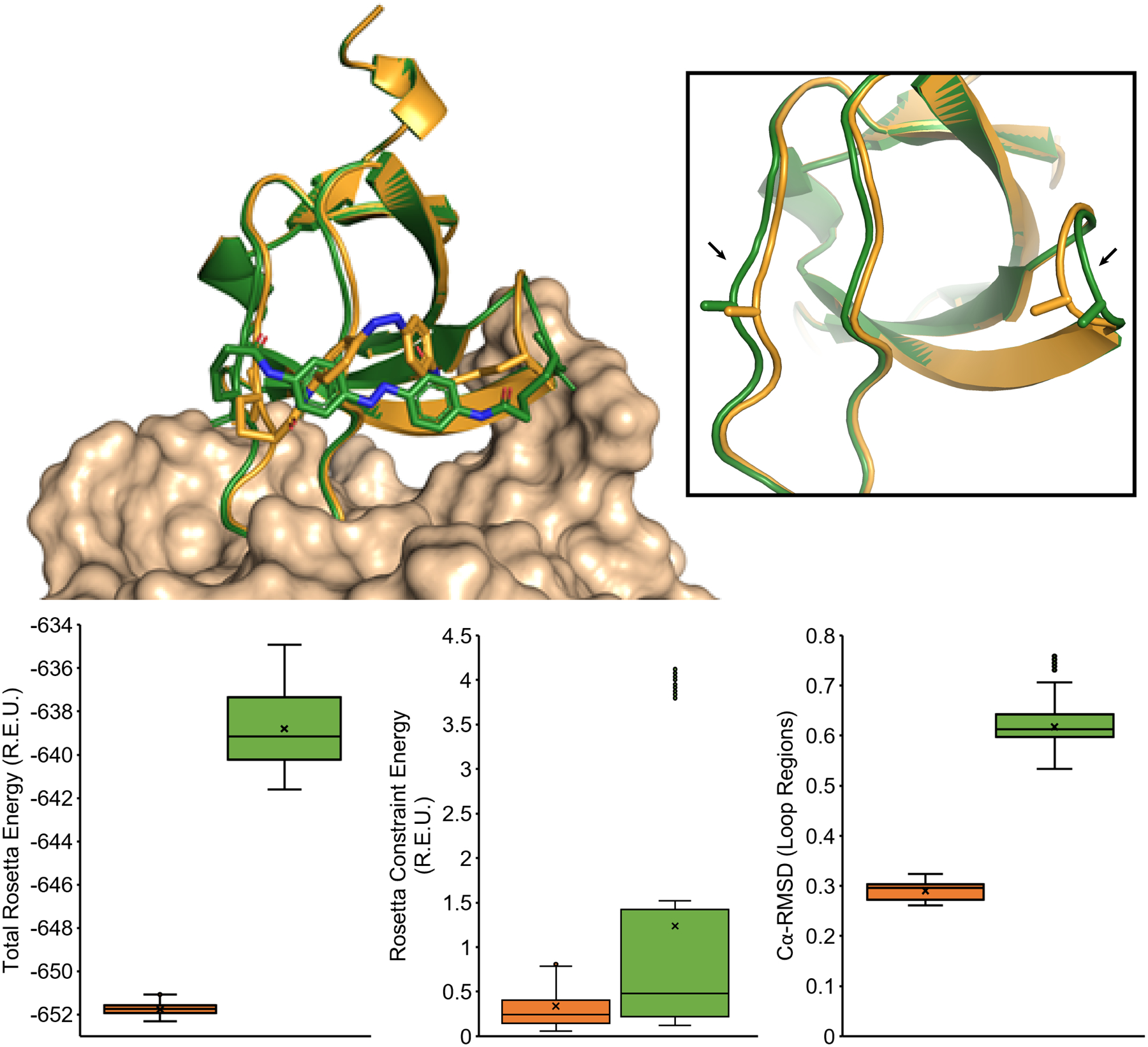
Models of N11CT44C cross-linked with IAC in the cis-state (orange) and trans-state (green), where the insert highlights movements in the loop regions (arrows). The box plots below compare distributions of Total Rosetta Energy, Rosetta Constraint Energy, and Cα-RMSD from 500 FastRelax simulation trajectories of N11CT44C in the cis (orange, left) and trans (green, right) states, which show a small destabilization in the trans conformation but similarly low constraint scores and small changes in binding loop Cα-RMSD distributions. The models illustrate how small changes in overall Fynomer structure and loop residues (6–23, 41–45) can accommodate IAC isomerization with local flexibility.
To address the third limitation noted in the first generation photoswitchable Fynomers, i.e. that red light switching was very slow, we applied the recently described azonium ion based PIP photoswitch.[9–10] We found that PIP could be successfully cross-liked at seven sites on the Fynomer scaffold. At most cross-linking sites, thermal relaxation of the PIP attached to the Fynomer occurred in a few seconds, similar to the half-life observed for PIP alone. At one site, PIP-L3CL29C, the half-life was substantially longer (~100s), perhaps due to the site having cationic character. By extension, it may be possible to affect the half-life of the azonium ion by making point mutations on the surface of the Fynomer. As for the IAC-Fynomers, a range of apparent affinities for the target chymase was observed with some (e.g. PIP-H21CE46C) comparable to the unmodified Fynomer. The cross-linking sites that produced the highest affinity were different for PIP-Fynomers and IAC-Fynomers, likely as a consequence of the different conformational and distance preferences of the two cross-linkers. A range of switching behaviours was again observed; in general photoswitchable changes in activity were small, but comparable to those seen with IAC, BSBCA, and TOM.[6a] This finding, combined with the superior optical features (wavelength and thermal relaxation rate) of PIP-modified affinity reagents make them well-suited for further development. Perhaps a wider screen of cross-linking sites on this or other affinity reagent scaffolds,[6b] coupled with computational design to maximize protein structural consequences of cross-linker isomerization will produce effective affinity-reagent based red-light switchable photopharmaceuticals.
Experimental Section
Computational generation of ligand ensembles
The IAC and PIP derivatives of azobenzene (Scheme 1) were built in Avogadro[19] using a crystal structure of the cis-form of azobenzene (CCDC ID AZBENC01) as a starting conformation. Internal structural geometries were optimized using the Auto Optimize tool with default settings while the cis-azobenzene core was kept static. Bond angles and torsions were manually set to values representing the highest probable conformation according to histograms of angles and torsions found in fragments from the CCDC with matching atom connectivity. After converting the models from Avogadro to Rosetta ligand parameter files, specific torsion angles (see Supplementary Figure 6) were sampled discretely using the Rosetta generate_ligens.linuxiccrelease executable to generate ligand ensemble files (for further details see SI).
Computational search for suitable cross-linking sites
The crystal structure of the 4C-E4 Fynomer, derived from the human Fyn SH3 domain, bound to human chymase (PDB ID 4AG1) was initially optimized using the Rosetta FastRelax[20] algorithm with flexible backbone and sidechain degrees of freedom and coordinate constraints. The lowest energy structure from 100 individual applications of the protocol was used as the starting conformation for subsequent computational cross-linking searches.
The Rosetta Match protocol[21] was used to scan the Fynomer scaffold for pairs of cysteine substitutions capable of binding to a cis-form azobenzene derivative and was followed by binding geometry optimization, and repacking and minimization of nearby residue sidechains using Rosetta EnzRepackMinimize module.[22] No sequence changes (apart from the two cysteines) were permitted but rotamer optimization of sidechains spatially proximal to the cross-linker was allowed in the Rosetta packing simulations. Promising designs were selected by requiring the constraint energy (CST energy, a penalty term for cross-linking geometries that vary from ideal values) to be less than 5.0 Rosetta energy units (R.E.U.). From this set of designs, differing in the placement of the dye and cysteine residues, the lowest total energy designs were chosen for further experimental characterization.
While the above design protocol was performed for each dye individually, we experimentally tested both dyes, IAC and PIP, with all identified cysteine pairs. For generating models of non-cognate dye-cysteine combinations (IAC-D9CT44C, IAC-D9CE46C, PIP-H21CT43C, PIP-K25CT43C), we first performed a sweep of increasing tolerance for parameters defining crosslinking compatibility in the RosettaMatch algorithm to place the dye in reasonable proximity of the cysteine residues, without altering the backbone conformation of the Fynomer. Starting with these conformations, a Rosetta FastRelax in which all cysteine-neighboring loop regions were allowed to sample backbone and sidechain dihedrals was used to optimize the dye cross-linking geometry (CST energy < 5.0 R.E.U.).
The PIP-cross-linked model of mutant L3CL29C could not be optimized (i.e. CST energy 5.0) via either of the EnzRepackMinimize- or FastRelax-based protocols described above due to clashes between the azobenzene derivative and a static loop of chymase. Therefore, the Fynomer was removed from its chymase-bound context and the binding geometry between the cysteine pair and PIP ligand were optimized via the EnzRepackMinimize protocol as described previously. RosettaScripts and example commandlines used for simulations are described in SI.
Simulation of trans-isomerization and evaluation of loop conformation
The low-energy, lowest-constraint-score model of the cis-IAC ligand cross-linked to N11CT44C, generated by the Rosetta EnzRepackMinimize optimization step described previously, was used as the starting conformation for subsequent trans-isomerization simulations. During the simulation, the cis-IAC ligand was “mutated” to its trans isomer, which was generated in the same fashion as the cis ligand ensemble. The same constraints between the cysteine sidechains and ligand that were applied during the matching and optimization stages were also applied to the resulting conformation. The Rosetta FastRelax protocol was then used to sample backbone and sidechain movements in cross-link-neighbouring loop regions in order to achieve a constraint energy of less than 5.0, where the weighting factor for the constraint energy was increased incrementally until a trans-cross-linked decoy with a constraint energy of less than 5.0 could be found. At each of these weighting factors, starting from 1, the trans-isomerization simulation protocol was applied 100 times, and the lowest energy decoy with a sub-5.0 constraint energy was chosen as the trans-model. This simulation was conducted with the Fynomer domain both bound and unbound to the chymase domain in order to remove steric hindrances that may constrain any sampling of the loop conformation.
Construction of paired Cys mutants
A synthetic gene sequence of chymase binding Fynomer variant 4C-E4[16] was cloned into a custom p3-phagmid.[23] The coding sequence of Fyn-SH3 variant cloned into the phagemid is shown below (partial ORF).
| GATTATAAAGATGATGATGATAAAGGCGGATCCGTCACCCTGTTTGTTGCGCTGTATGAC | |||||||||||||||||||
| D | Y | K | D | D | D | D | K | G | G | S | V | T | L | F | V | A | L | Y | D |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||||||||
| TACAATGCCACGCGCTGGACAGACCTGAGCTTCCACAAAGGCGAAAAGTTCCAGATCCTG | |||||||||||||||||||
| Y | N | A | T | R | W | T | D | L | S | F | H | K | G | E | K | F | Q | I | L |
| 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 |
| GAATTCGGTCCGGGAGATTGGTGGGAGGCTCGTTCTCTGACCACGGGTGAAACCGGTTAC | |||||||||||||||||||
| E | F | G | P | G | D | W | W | E | A | R | S | L | T | T | G | E | T | G | Y |
| 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | 48 | 49 |
| ATCCCAAGCAATTACGTGGCACCGGTAGACTCCATTGGGAGCTCTGGAGAC | |||||||||||||||||||
| I | P | S | N | Y | V | A | P | V | D | S | I | G | S | S | G | D | |||
| 50 | 51 | 52 | 53 | 54 | 55 | 56 | 57 | 58 | 59 | 60 | 61 | ||||||||
The oligonucleotides shown in the table below were used to create pairs of Cys mutations using standard oligonucleotide based site-directed mutagenesis protocols.[24] Two oligonucleotides were used for each mutation pair positions separated by >=30 bp sequence. A single oligonucleotide was used to mutate both positions if separated by <30bp. Several clones from each mutation reaction were verified by Sanger sequencing to obtain Cys-pair mutant clones. Cys mutant Fynomer gene were moved from the p3 vector to a pET24b expression vector using HiFi assembly (New England Biolabs)
| Fynomer | Oligonucleotide 1 | Oligonucleotide 2 |
|---|---|---|
| D9CT44C | TTTGTTGCGCTGTATTGCTACAATGCCACGCGC | GCTCGTTCTCTGACCTGCGGTGAAACCGGTTAC |
| D9CE46C | TTTGTTGCGCTGTATTGCTACAATGCCACGCGC | TCTCTGACCACGGGTTGCACCGGTTACATCCCA |
| N11CH21C | GCGCTGTATGACTACTGCGCCACGCGCTGGACA | ACAGACCTGAGCTTCTGCAAAGGCGAAAAGTTC |
| N11CT44C | GCGCTGTATGACTACTGCGCCACGCGCTGGACA | GCTCGTTCTCTGACCTGCGGTGAAACCGGTTAC |
| H21CK25C | ACAGACCTGAGCTTCTGCAAAGGTGAATGCTTCCAGATCCTGGAA | |
| H21CT43C | ACAGACCTGAGCTTCTGCAAAGGCGAAAAGTTC | GAGGCTCGTTCTCTGTGCACGGGTGAAACCGGT |
| H21CT44C | ACAGACCTGAGCTTCTGCAAAGGCGAAAAGTTC | GCTCGTTCTCTGACCTGCGGTGAAACCGGTTAC |
| H21CE46C | ACAGACCTGAGCTTCTGCAAAGGCGAAAAGTTC | TCTCTGACCACGGGTTGCACCGGTTACATCCCA |
| K25CT43C | TTCCACAAAGGCGAATGCTTCCAGATCCTGGAA | GAGGCTCGTTCTCTGTGCACGGGTGAAACCGGT |
| L3CL29C | GGCGGATCCGTCACCTGCTTTGTTGCGCTGTAT | GAAAAGTTCCAGATCTGCGAATTCGGTCCGGGA |
Protein expression and purification
For expression of each Fynomer mutant, the corresponding pET24b vector was transformed into chemically competent E. coli (BL21*DE3) cells using the heat shock method and plated on agar/lysogeny broth (LB) plates containing 50 μg/mL kanamycin. A colony was used for inoculating 25 mL of LB culture media on the following day. The culture was grown overnight at 37°C in the presence of 50 μg/mL kanamycin. The overnight culture was transferred to 1 L LB containing the same concentration of kanamycin and incubated at 37°C on a shaker (180 rpm) until an optical density (OD) of 0.6 was reached. Protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 750 μM. The induced culture was then incubated and shaken for 4 hours at 37°C. The culture was centrifuged at 4000 rpm for 30 minutes and the harvested cell pellets were resuspended in 30 mL of 100 mM phosphate buffer 6 M Gdn solution (pH=8.0, containing 10 mM imidazole) and was stored at −20°C. For purification, the cell lysis mixture was thawed for an hour in a water bath at room temperature. The cell lysis mixture was sonicated for 10 minutes (10 seconds on and 20 seconds off cycle) at ~12 W on ice. Cell debris was removed by centrifugation at 12000 rpm for an hour. The crude supernatant was filtered through a 0.45 μm syringe filter and then applied to a Ni-NTA column which was pre-equilibrated with the same buffer. Once loaded with the His-tagged protein, the Ni-NTA resin was washed with 15 mL of the same buffer. The protein was eluted by lowering the pH to 4.5 using a buffer of 6 M Gdn, 100 mM sodium phosphate, 100 mM sodium acetate. The expression yield was found to be 5 mg per liter of culture on average.
Cross-linking of Fynomers with azobenzene cross-linkers
The absorbance of mutant Fynomer stock solution (elution buffer: pH 4.5, 6M Gdn, 100 mM phosphate, 100 mM acetate) was measured at 280 nm to determine protein concentration. Molar extinction coefficients for wildtype and mutant Fynomers were obtained from the ProtParam online tool: 22,460 M−1cm−1. The pH of the 40 μM mutant Fynomer solution was adjusted to a pH near 8.5 using 1M NaOH. The protein was then treated with 2 equivalents of tris(2-carboxyethyl)phosphine (TCEP) and incubated at room temperature for at least 10 minutes to ensure the cysteine thiols were fully reduced. The protein was concentrated using centrifugal concentrator filters (Millipore Sigma, 3K MWCO). Solution of 400 μM of IAC and PIP (synthesized as described previously)[10, 14] were prepared in anhydrous DMSO (Sigma).
To cross-link a Fynomer with IAC, 1.5 equivalents of IAC were added to the reduced protein and the reaction mixture was shaken at room temperature (~ 22°C) for 2 hours. The mass of the protein was measured by MALDI after 2 hours and if uncross-linked protein remained, 2 equivalents of TCEP and 1.5 equivalents of IAC were added, and the reaction was continued for another 12 hours.
To cross-link a Fynomer with PIP, 1.5 equivalents of PIP were added to the reduced protein and the reaction mixture was shaken at 37–50°C for 6 hours. The mass of the protein was measured by MALDI after 6 hours and if uncross-linked protein remained, another 2 equivalents of TCEP and 1.5 equivalents of PIP were added and the reaction was continued for another 12 hours.
When the reaction was completed, the cross-linked protein was dialyzed using a membrane with 3.5K MWCO in 10 mM sodium phosphate buffer. The cross-linked protein was concentrated using centrifugal concentrator filters. The product was analyzed with ESI or MALDI-TOF mass spectrometry. If impurities were detected (ex. uncross-linked protein or protein with two cross-linker), the reaction mixture was further purified by reverse-phase HPLC. This was performed with a C18 semi-preparatory column (Zorbax, RxC18) using 0.1% (v/v) TFA water and 0.1% TFA (v/v) acetonitrile as solvents with a linear gradient of 5% to 70% acetonitrile over the course of 25 minutes (Flow rate was 1.3 mL / min). The eluate was monitored using a dual wavelength Waters detector at 280 nm (chromophore: protein and azobenzene) and 360 nm (only azobenzene). The cross-linked Fynomers eluted at ~42% acetonitrile. The eluate was then lyophilized and reconstituted in 50 mM sodium phosphate pH 8.0. Some samples of purified cross-linked Fynomer did not dissolve in sodium phosphate buffer. In this case, the cross-linked Fynomer was dissolved in sodium phosphate buffer containing 6 M Gdn pH 8.0.
UV-Visible spectroscopy:
UV-Visible spectra were obtained using a Perkin-Elmer Lambda 35 spectrophotometer or a diode array UV–Vis spectrophotometer (Ocean Optics Inc., USB4000). Temperature was maintained at 22°C for all measurements (Quantum Northwest), using 10 mm or 1.5 mm quartz cuvettes (Hellma Analytics).
To measure spectra of IAC-cross-linked Fynomers, samples in 10 mM sodium phosphate pH 7.0 were dark-adapted by heating at 42°C for 30 minutes in the dark. Irradiation of the sample (at 90° to the light source and detector used for the absorbance measurements) was carried out using a 370 nm LED for 1 minute. (897-LZ440U610 LedEngin LED, San Jose, CA, USA, operating at 68 mW/cm2) For PIP-cross-linked Fynomers with a thermal relaxation half-life at pH 7 of several seconds, only dark-adapted spectra were obtained.
To measure the thermal relaxation rates for IAC-cross-linked Fynomers, samples were periodically scanned after irradiation. For PIP-cross-linked Fynomers, the sample was irradiated with a red LED for 30 seconds. (LedEngin LZ4-40R200-0000, 700 mA, 635 nm, 90 mW/cm2) and then absorbance changes were measured using a photomultiplier tube (PMT) as described previously.[4b]
Chymase inhibition assays
The chymase activity assay was based on measurement of the production of p-nitroaniline which absorbs at 400 nm. Chymase and the substrate peptide were purchased from Sigma (CS1140 and S7388). Activity was tested with two different assay methods since the thermal relaxation half-life of IAC after irradiation with light at 25 ℃ in pH 8.0 buffer was ~15 minutes while PIP was ~7 seconds. For the PIP-cross-linked proteins, it was necessary to always irradiate with light. To ensure that both assays gave comparable results, IAC-N11CT44C was tested using both assays (see Supplementary Figure 7).
Chymase inhibition assays for IAC-cross-linked Fynomer mutants
A stock solution of the substrate peptide was prepared in DMSO. The concentration was calculated from the absorbance at 315 nm. (ε315 = 14000 M−1cm−1).[25] To prepare the dark-adapted IAC-Fynomers, the cross-linked protein solution was heated at 42°C for 30 minutes in the dark, converting the azobenzene to its trans form. The sample was then cooled to room temperature in the dark. The concentration of IAC-Fynomer was calculated from its absorbance at around 360 nm (ε363 = 24000 M−1cm−1).[26] The substrate peptide stock solution was diluted to 750 μM with 50 mM sodium phosphate buffer pH 8. Chymase (Sigma, CS1140, ~9 μM) was diluted 25-fold with the Assay buffer (Sigma, CS1140).
The following operations were then carried out under dim red light in a dark room. A Fynomer/chymase solution was prepared as follows: 0.4% (v/v) chymase, 26.35% (v/v) Assay buffer, 0.04% (v/v) Triton X-100 and an IAC-cross-linked Fynomer stock solution diluted with sodium phosphate buffer were mixed so that the cross-linked Fynomer would have the desired final concentration.
For the assay, 75 μL of diluted substrate peptide solution and 75 μL of IAC-Fynomer/chymase solution (cross-linked Fynomer mutant, chymase, Triton X-100 with sodium phosphate and assay buffers) were mixed in a 1 cm microcuvette (Sigma, BR759235), and then immediately placed in the Lambda 35 instrument equipped with a moving cuvette rack. Absorbance at 400 nm was measured every 20 seconds for 5 minutes while inside a temperature-controlled jacket at 22°C. Measurements were made two or three times in each dark-adapted and light-adapted sample. Mutant IAC-H21CT44C was not tested because it contained 30% uncross-linked species
Once mixed with the substrate solution, the final concentration of the substrate peptide was 375 μM, and the nominal Chymase concentration was ~18 nM.
For light-adapted IAC-Fynomers, the Fynomer/chymase solution was irradiated with 370 nm LED light for 30 seconds while inside a temperature-controlled holder at 22°C. The solution was mixed by inversion. The solution was then quickly mixed with the substrate and the measurement was initiated.
Rate data as a function of Fynomer concentration was fitted to a standard 4-parameter equation to calculate the IC50.
Enzyme Assay for PIP-cross-linked Fynomer mutants:
In this assay, 75 μL of diluted substrate peptide solution, 15 μL of chymase solution, and 60 μL of PIP-Fynomer solution (crosslinked Fynomer mutants, the enzyme buffer) were mixed in a 96 well plate (Thermo Scientific, Nunc MicroWell 96-Well Microplates, 243656) at room temperature. After 5 minutes, a quench solution (>6 M Guanidine HCl, 50 mM sodium phosphate buffer, pH 8.0) was added to the reaction. When all samples were quenched, the absorbance at 400 nm of all samples were measured using a microplate reader (BMG LABTECH, CLARIOstar).
For dark-adapted PIP-Fynomers, the concentration of Fynomer was calculated by measuring absorbance at the maximum near 550 nm using pH ~2 buffer in order to convert all the PIP to its azonium ion form (ε~550 = 47000 M−1cm−1). The PIP cross-linked Fynomer solution was diluted to 750 nM with an enzyme buffer (0.02% (v/v) Triton X-100, 50 mM sodium phosphate buffer, pH 8.0). When the Fynomer stock solution contained guanidine HCl to dissolve the cross-linked Fynomer PIP-L3CL29C, the guanidine final concentration of the reaction solution at the time of assay (150 μL solution in a well plate) was 0.4 M or less. A control experiment without Fynomer showed that this concentration did not inhibit chymase.
The substrate peptide stock solution was diluted to 750 μM with enzyme buffer. Chymase (Sigma, CS1140, ~9 μM) was diluted 50-fold with assay buffer (Sigma, CS1140).
The following operations were done under a dim red light in a dark room. Enzyme buffer and a 750 nM PIP-cross-linked Fynomer stock solution were mixed so that the cross-linked Fynomers would have the desired final concentration. A 60 μL volume of this Fynomer solution was then mixed with 15 μL of a diluted Chymase solution (Fynomer/Chymase solution). After that 75 μL of diluted substrate peptide solution was added quickly to the Fynomer/Chymase solution and incubated at room temperature (~ 22°C). After 5 minutes, 150 μL of a quench solution (>6 M Guanidine HCl, 50 mM sodium phosphate buffer, pH 8.0) was added. Once mixed with the substrate solution, the final concentration of the substrate peptide was 375 μM, and the nominal chymase concentration was ~18 nM, i.e. the same concentrations as used for the IAC-Fynomer assay.
For light-adapted PIP-Fynomers, the Fynomer/chymase solution was irradiated with a 633 nm LED light for 30 seconds at room temperature. The LED was kept ~8 cm above the 96-well plate to keep the PIP-Fynomer in its cis enriched form. The diluted substrate peptide solution was added quickly to the solution and incubated for 5 minutes.
To ensure that UV irradiation or red light irradiation had no effect on the enzyme or any other component of the system, this assay was performed with and without irradiation in the absence of a cross-linked Fynomer. Absorbance at 400 nm from both sets of experiments did not differ significantly. Wild-type Fynomer was also tested using the same assay method to ensure that there was no influence from light irradiation.
Supplementary Material
Acknowledgements
We would like to thank Dr. Amir Babalhavaeji for advice and students in the 2018 CHM379 biomolecular chemistry lab class (Dept. of Chemistry, University of Toronto) for assistance.
References
- [1].a) Szymanski W, Beierle JM, Kistemaker HA, Velema WA, Feringa BL, Chem. Rev 2013, 113, 6114–6178; [DOI] [PubMed] [Google Scholar]; b) Bamberg E, Gartner W, Trauner D, Chem. Rev 2018, 118, 10627–10628. [DOI] [PubMed] [Google Scholar]
- [2].a) Broichhagen J, Frank JA, Trauner D, Acc. Chem. Res 2015, 48, 1947–1960; [DOI] [PubMed] [Google Scholar]; b) Hull K, Morstein J, Trauner D, Chem. Rev 2018, 118, 10710–10747; [DOI] [PubMed] [Google Scholar]; c) Lerch MM, Hansen MJ, van Dam GM, Szymanski W, Feringa BL, Angew. Chem. Int. Ed. Engl 2016, 55, 10978–10999. [DOI] [PubMed] [Google Scholar]
- [3].a) Rastogi SK, Zhao Z, Barrett SL, Shelton SD, Zafferani M, Anderson HE, Blumenthal MO, Jones LR, Wang L, Li X, Streu CN, Du L, Brittain WJ, Eur. J. Med. Chem 2018, 143, 1–7; [DOI] [PubMed] [Google Scholar]; b) Szymanski W, Ourailidou ME, Velema WA, Dekker FJ, Feringa BL, Chemistry 2015, 21, 16517–16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Dong M, Babalhavaeji A, Samanta S, Beharry AA, Woolley GA, Acc. Chem. Res 2015, 48, 2662–2670; [DOI] [PubMed] [Google Scholar]; b) Dong M, Babalhavaeji A, Collins CV, Jarrah K, Sadovski O, Dai Q, Woolley GA, J Am Chem Soc 2017, 139, 13483–13486. [DOI] [PubMed] [Google Scholar]
- [5].a) Azhar A, Ahmad E, Zia Q, Rauf MA, Owais M, Ashraf GM, Int. J. Biol. Macromol 2017, 102, 630–641; [DOI] [PubMed] [Google Scholar]; b) Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA, Nat. Biotechnol 1997, 15, 772–777; [DOI] [PubMed] [Google Scholar]; c) k A, Annu. Rev. Pharmacol. Toxicol 2015, 55, 489–511. [DOI] [PubMed] [Google Scholar]
- [6].a) Babalhavaeji A, Woolley GA, Chem. Commun 2018, 54, 1591–1594; [DOI] [PubMed] [Google Scholar]; b) Heu W, Choi JM, Kyeong HH, Choi Y, Kim HY, Kim HS, Angew. Chem. Int. Ed. Engl 2018, 57, 10859–10863. [DOI] [PubMed] [Google Scholar]
- [7].Samanta S, Beharry AA, Sadovski O, McCormick TM, Babalhavaeji A, Tropepe V, Woolley GA, J. Am. Chem. Soc 2013, 135, 9777–9784. [DOI] [PubMed] [Google Scholar]
- [8].Ali AM, Forbes MW, Woolley GA, Chembiochem 2015, 16, 1757–1763. [DOI] [PubMed] [Google Scholar]
- [9].Dong M, Babalhavaeji A, Hansen MJ, Kalman L, Woolley GA, Chem. Commun 2015, 51, 12981 – 12984. [DOI] [PubMed] [Google Scholar]
- [10].Samanta S, Babalhavaeji A, Dong MX, Woolley GA, Angew. Chem. Int. Ed. Engl 2013, 52, 14127–14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blacklock KM, Yachnin BJ, Woolley GA, Khare SD, J. Am. Chem. Soc 2018, 140, 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beharry AA, Woolley GA, Chem. Soc. Rev 2011, 40, 4422–4437. [DOI] [PubMed] [Google Scholar]
- [13].Zhang F, Zarrine-Afsar A, Al-Abdul-Wahid MS, Prosser RS, Davidson AR, Woolley GA, J. Am. Chem. Soc 2009, 131, 2283–2289. [DOI] [PubMed] [Google Scholar]
- [14].Kumita JR, Smart OS, Woolley GA, Proc. Natl. Acad. Sci. USA 2000, 97, 3803–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Beharry AA, Sadovski O, Woolley GA, Org. Biomol. Chem 2008, 6, 4323–4332; [DOI] [PubMed] [Google Scholar]; b) Woolley GA, Acc. Chem. Res 2005, 38, 486–493. [DOI] [PubMed] [Google Scholar]
- [16].Schlatter D, Brack S, Banner DW, Batey S, Benz J, Bertschinger J, Huber W, Joseph C, Rufer A, van der Klooster A, Weber M, Grabulovski D, Hennig M, MAbs 2012, 4, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beharry AA, Chen T, Al-Abdul-Wahid MS, Samanta S, Davidov K, Sadovski O, Ali AM, Chen SB, Prosser RS, Chan HS, Woolley GA, Biochemistry 2012, 51, 6421–6431. [DOI] [PubMed] [Google Scholar]
- [18].Dokholyan NV, Chem. Rev 2016, 116, 6463–6487. [DOI] [PubMed] [Google Scholar]
- [19].Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR, J. Cheminform. 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Tyka MD, Keedy DA, Andre I, Dimaio F, Song Y, Richardson DC, Richardson JS, Baker D, J. Mol. Biol 2011, 405, 607–618; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Misura KM, Baker D, Proteins 2005, 59, 15–29. [DOI] [PubMed] [Google Scholar]
- [21].Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Rothlisberger D, Baker D, Protein Sci. 2006, 15, 2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D, PLoS One 2011, 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reis JM, Xu X, McDonald S, Woloschuk RM, Jaikaran ASI, Vizeacoumar FS, Woolley GA, Uppalapati M, ACS Synth. Biol 2018, 7, 2355–2364. [DOI] [PubMed] [Google Scholar]
- [24].a) Kunkel TA, Roberts JD, Zakour RA, Methods Enzymol. 1987, 154, 367–382; [DOI] [PubMed] [Google Scholar]; b) Tonikian R, Zhang Y, Boone C, Sidhu SS, Nat. Protoc 2007, 2, 1368–1386. [DOI] [PubMed] [Google Scholar]
- [25].Bru R, Walde P, Eur. J. Biochem 1991, 199, 95–103. [DOI] [PubMed] [Google Scholar]
- [26].Borisenko V, Woolley GA, J. Photochem. Photobiol. A. Chem 2005, 173, 21–28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.