

Abstract
mRNA levels are determined by the balance between mRNA synthesis and decay. Protein factors that mediate both processes, including the 5'-3' exonuclease Xrn1, are responsible for a cross-talk between the two processes that buffers steady-state mRNA levels. However, the roles of these proteins in transcription remain elusive and controversial. Applying native elongating transcript sequencing (NET-seq) to yeast cells, we show that Xrn1 functions mainly as a transcriptional activator and that its disruption manifests as a reduction of RNA polymerase II (Pol II) occupancy downstream of transcription start sites. By combining our sequencing data and mathematical modeling of transcription, we found that Xrn1 modulates transcription initiation and elongation of its target genes. Furthermore, Pol II occupancy markedly increased near cleavage and polyadenylation sites in xrn1Δ cells, whereas its activity decreased, a characteristic feature of backtracked Pol II. We also provide indirect evidence that Xrn1 is involved in transcription termination downstream of polyadenylation sites. We noted that two additional decay factors, Dhh1 and Lsm1, seem to function similarly to Xrn1 in transcription, perhaps as a complex, and that the decay factors Ccr4 and Rpb4 also perturb transcription in other ways. Interestingly, the decay factors could differentiate between SAGA- and TFIID-dominated promoters. These two classes of genes responded differently to XRN1 deletion in mRNA synthesis and were differentially regulated by mRNA decay pathways, raising the possibility that one distinction between these two gene classes lies in the mechanisms that balance mRNA synthesis with mRNA decay.
Keywords: mRNA buffering, native elongating transcript sequencing (NET-seq), transcription regulation, transcriptional profiling, yeast, mRNA decay, Xrn1, transcription factor, transcriptomics, gene regulation
Steady-state mRNA levels are determined by the balance between synthesis and decay rates. Once thought to function separately, recent studies have discovered that these two processes are linked. In previous work we showed that the major cytoplasmic yeast mRNA degradation pathway, consisting of the decapping enzyme Dcp1/2, the decapping activator Pat1/Lsm1-7, the helicase Dhh1, and the 5'-3' exonuclease Xrn1, shuttles between the cytoplasm and the nucleus to participate in both processes. Notably, the elements of this pathway were found to degrade most mRNAs in the cytoplasm while stimulating transcription in the nucleus. The proteins Dcp2, Lsm1, and Xrn1 were further shown to bind chromatin, probably as a complex, and to stimulate transcription initiation and elongation (1). We also uncovered a connection between how Xrn1 functions in transcription and mRNA decay by revealing the correlation between the effects of Xrn1 disruption on mRNA synthesis and decay in the nucleus and cytoplasm, respectively (1, 2). We subsequently ranked genes according to their responsiveness to Xrn1 disruption in optimally proliferating yeast cells; the most responsive were dubbed the “Xrn1 synthegradon” and consisted of genes whose transcription and decay rates exhibited the highest sensitivity to Xrn1 disruption (2). This group is highly enriched with genes required for cell growth and proliferation, including genes encoding ribosome biogenesis and translation factors.
“Classic” mRNA decay factors are not the only bridges between transcription and mRNA decay. For example, Rpb4 and Rpb7, two canonical RNA polymerase II (Pol II) subunits, and the Ccr4-NOT complex function in both processes (3–10), and even promoters are capable of regulating mRNA decay (11, 12). Hence the cross-talk between mRNA synthesis and decay is complex and involves an interplay between canonical transcription and degradation factors. Although the links are clear, the mechanism mediating mRNA buffering remains enigmatic and controversial. Some publications have proposed a simple feedback mechanism involving a repressor (13, 14), although others have suggested that components of the mRNA decay machinery function directly in transcription (1, 2). In fact, the former articles proposed that the deletion of Xrn1 leads to transcription activation, whereas the latter group asserted the opposite.
The realization of the critical role of mRNA buffering requires changes in the approaches used to analyze transcription. In the past, mRNA levels were regarded as a good proxy for transcription, and prior studies have relied upon changes in these levels to infer alterations in transcription. As an example, earlier work classified genes as SAGA- or TFIID-dominated based on measured changes in mRNA levels after inactivation of central components of the SAGA (mainly Spt3) or TFIID (mainly Taf1) complexes (15, 16). However, it was recently reported that virtually all promoters recruit both the SAGA and TFIID complexes and subsequent transcriptional profiling experiments demonstrated that mutations in either complex result in widespread defective transcription (17–19). Nonetheless, the disruption of most components of either complex did not lead to decreases in the levels of most mRNAs due to feedback mechanisms that involve mRNA decay (17–19). It is now clear that mRNA levels are not simply determined by two unrelated processes of mRNA synthesis and decay; rather, each of these processes affects the other by a hitherto elusive mechanism.
Following transcription initiation, many metazoan genes undergo a regulatory step termed promoter-proximal pausing (reviewed recently in Refs. 20 and 21). Specifically, after transcribing 30–120 nucleotides downstream of transcription start sites (TSS), Pol II pauses; its release into productive elongation requires the activity of specific factors, including TFIIS. Following its release, Pol II interacts with additional elongation factors that modulate its processivity. It was recently reported that the release of mammalian Pol II from a paused state in the promoter-proximal region is a key step in the regulation of transcription, both generally (22) and in response to environmental stress (22, 23). Interestingly, Pol II recruitment rate was proposed to have only a marginal impact on overall transcription rates (23). Conversely, common wisdom posits that promoter-proximal pausing does not play a major role in budding yeast as it is less prominent than in metazoans (24). However, there is evidence that Pol II in Saccharomyces cerevisiae accumulate downstream of TSS (25), although this phenomenon and its contribution to transcriptional regulation has been little studied. In contrast with the inconclusive nature of 5' pausing, a conspicuous Pol II pausing event does occur at polyadenylation sites (PAS) (e.g. Ref. 26). It is plausible that this pausing is required to provide the necessary time for the assembly of polyadenylation (PA) machinery, but gaps in the mechanistic understanding of this pausing event persist. Nevertheless, it is clear that factors of the PA pathway affect transcription termination events that occur downstream.
To probe the effects of mRNA decay factors (DFs) on transcription, we employed native elongating transcript sequencing (NET-seq), an experimental protocol which assays Pol II occupancy at single nucleotide resolution. This technique sequences nascent RNA strands attached to actively engaged Pol II (27) and maps the 3' ends of nascent RNAs to yield the positions of Pol II active sites. Therefore, unlike RNA-seq, NET-seq data are not confounded by mRNA decay rates and give the precise locations of bound Pol II. Additionally, noncoding RNAs (ncRNAs) are frequently difficult to detect using RNA-seq because of their low transcript stabilities but are easily identified using NET-seq, permitting more thorough investigations of additional classes of transcripts. In contrast to other transcription profiling methods such as Genomic Run-On (GRO) (28) and its high-resolution cousin Biotin Genomic Run-On (BioGRO) (29, 30), which assay only actively elongating Pol II, NET-seq can report both elongating Pol II as well as arrested Pol II (25). As a result, run-on methods and NET-seq are particularly informative when used in combination and can reveal information about Pol II processivity and pausing.
In light of the poorly understood functions of DFs in transcription, we applied NET-seq to obtain Pol II occupancy profiles in various DF deletion strains to facilitate study of the roles of DFs in transcription. In addition to effects on initiation, Xrn1 and our other studied DFs seem to affect transcription primarily via elongation changes that are plausibly attributable to modified Pol II pausing and/or backtracking (31).
These effects primarily manifest in the ends of genes, occurring ∼100 bp downstream of TSS in the 5' end and from ∼75 bp upstream of PAS until the PAS. Similar changes in Pol II occupancy were identified in the 5', but not 3', ends of ncRNAs, implicating DFs in the regulation of the early stages of ncRNA transcription. Furthermore, deletion of XRN1 affected Pol II elongation efficiency in a manner consistent with reduced Pol II processivity. We additionally employed a recently developed mathematical model (32) to infer changes in spatial transcriptional dynamics. This methodologically novel model uses our metagene profiles to estimate baseline values for relative initiation and elongation rates, while offering a framework to systematically vary unknown parameters. This allowed us to perform in silico experiments suggesting that Xrn1 is required for efficient initiation of its target genes. In contrast to the most affected genes, NET-seq signals increase in response to Xrn1 disruption in a small repertoire of so-called “repressed” genes. Interestingly, these genes displayed demonstrably different 5' and 3' occupancy patterns upon DF deletion. Given these observed differences and comparisons with external data, we propose that the considered DFs modulate transcription of a subset of genes, perhaps as a complex, via the regulation of pausing and/or backtracking during the early and late stages of transcription.
Results
Deletions of mRNA decay factors lead to overall decreases in pol II occupancy
Previously we reported that Xrn1 binds to promoters and gene bodies and directly stimulates transcription initiation and elongation (1). Shortly thereafter, Sun et al. (14) reported that the deletion of XRN1 leads to the up-regulation of transcription, implying that Xrn1 represses transcription. To resolve this discrepancy and gain insight into the mechanisms linking transcription and mRNA decay, we used NET-seq to compare Pol II occupancy in WT strains and those carrying a deletion of XRN1 (xrn1Δ). Because NET-seq does not directly capture global changes, we began by normalizing across runs by selecting sets of housekeeping genes based on prior GRO and cDTA experiments (see “Materials and methods”). As GRO and cDTA measure mRNA synthesis rates, we are assuming that genes who display the smallest changes upon DF deletion according to these assays will similarly not experience much disruption to their normal Pol II occupancy as measured by NET-seq. After normalizing, overall Pol II occupancy in xrn1Δ cells generally decreased (Fig. 1A), indicating the down-regulation of transcription. That Pol II occupancy is negatively affected by XRN1 deletion is consistent with Xrn1's proposed role as a transcriptional activator (1, 2). To provide confidence in our results, both the WT and xrn1Δ experiments were performed in replicate, producing similar NET-seq patterns (Fig. S1). We further studied additional mutant strains, each carrying a single deletion of CCR4, DHH1, or LSM1 and observed decreases in Pol II occupancy in each respective knockout, although not as strongly as in xrn1Δ cells (Fig. S2A). These results support roles for the encoded proteins as transcriptional stimulators. It is valuable to note that our findings are robust to the choice of normalization, as using other common approaches (e.g. all genes as references, total counts, and re-scaling such that genes have the same numbers of reads) produces nearly identical results (Fig. S3). We also wish to emphasize that regardless of normalization, we are able to infer relative changes in transcription dynamics from the respective NET-seq profiles, described below.
Figure 1.
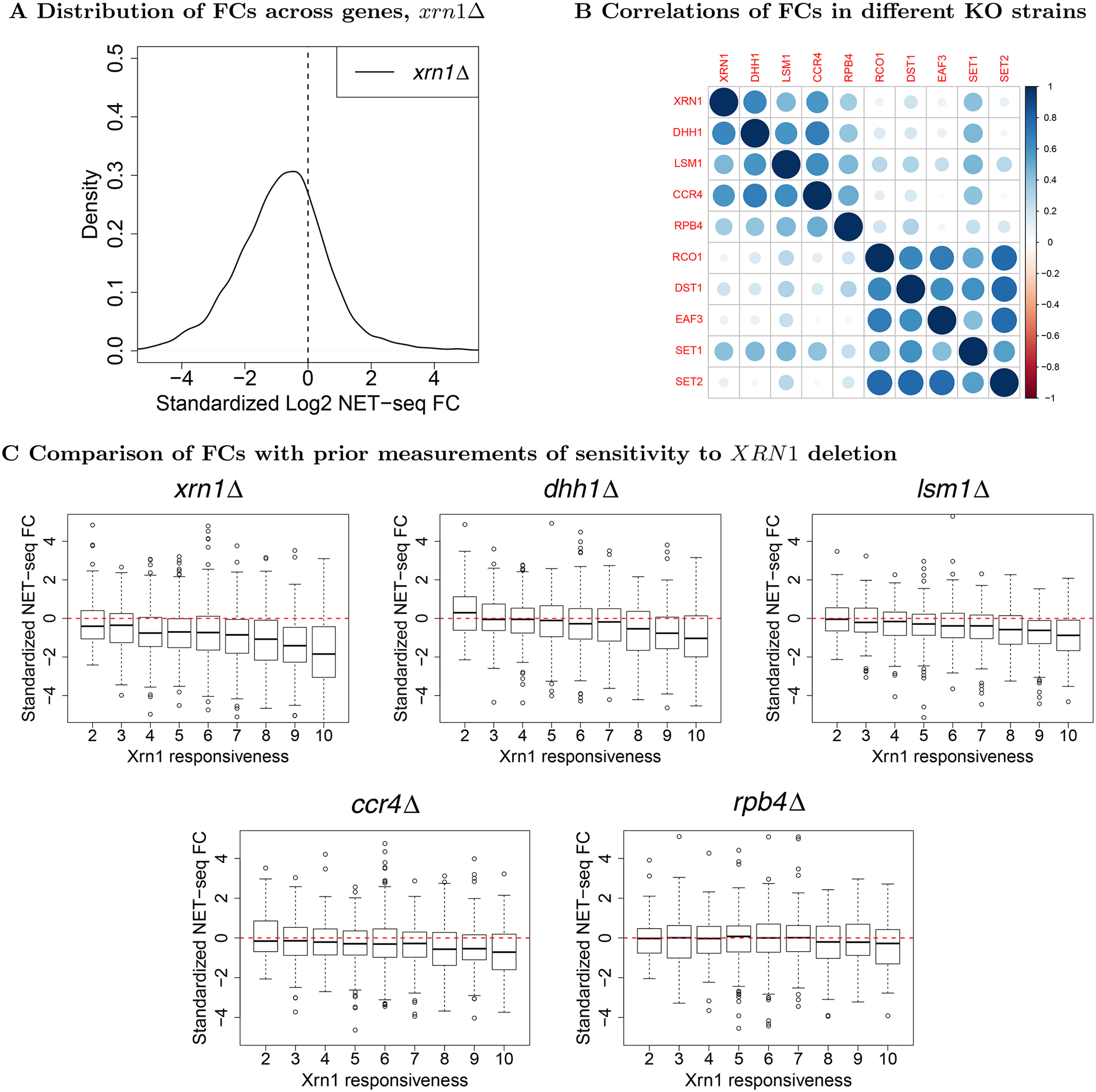
Fold changes in NET-seq Pol II occupancy. A, we aggregated NET-seq reads within annotated gene boundaries (TSS to PAS) and applied DESeq2 (33) to estimate standardized fold changes (FC) in each gene's normalized signal with respect to the WT. Both xrn1Δ and WT were done in two replicates. Normalization across runs was performed by selecting sets of housekeeping genes (see text and “Materials and methods”). B, the visualized correlation matrix for standardized fold changes in NET-seq reads in genes. Each entry corresponds to the Spearman correlation between the fold change with respect to the WT in NET-seq reads in annotated genes. xrn1Δ, dhh1Δ, lsm1Δ, ccr4Δ, and rpb4Δ come from the experiments associated with this paper, whereas the rest come from Ref. 25. Fold changes were estimated using DESeq2 (33) with both experiments analyzed simultaneously. C, genes were stratified using previously obtained measures of Xrn1 responsiveness, an aggregated measure of the sensitivity of synthesis and decay rates to Xrn1 deletion as measured in (1). A value of 2 indicates the lowest sensitivity and 10 the highest. Standardized NET-seq fold changes from our experiments were then plotted for genes falling into each responsiveness classification.
In earlier work, we found that Xrn1, Lsm1, and Dcp2 produced highly similar ChIP-exo profiles (1), raising the possibility that they function in transcription as a complex. Analogously, we sought to characterize the extent to which deletion strains induced similar genome-wide transcriptional responses using NET-seq. We constructed a correlation matrix of gene Pol II fold changes (Fig. 1B), uncovering strong similarities in transcriptomic response to deleted DFs. As an external control, we compared the correlations between our NET-seq data and previously generated NET-seq profiles from strains carrying respective deletions of EAF3, RCO1, and SET2, genes encoding a set of proteins, which are known to act in concert and are required for proper function of the Rpd3S H4 deacetylation complex (25). Strains carrying deletions in each of DST1 and SET1, encoding proteins that regulate Pol II release from backtracking and promoter directionality (25), were also included. Fold changes among these non-DF strains correlated well (ρ ∼ 0.75−0.8) as expected due to their shared functions. In contrast, correlations between these non-DFs and our studied DFs were typically much lower with the exception of SET1 (Fig. 1B). Although these experiments were done in the same laboratory using the same protocols and reagents (see “Materials and methods”), we still interpret this disparity cautiously because these data were collected by different investigators at different times. More relevant is that the correlations among our DFs were comparable with those from the other set of mutants, supporting the possibility that they act together in a complex, as has been proposed for Lsm1 and Xrn1 (1), or at least through similar mechanisms. Rpb4 is a protein that functions in both mRNA synthesis and decay (3, 7, 9, 34). To examine whether Rpb4 function is indeed related to those of our studied DFs, we performed NET-seq on an rpb4Δ strain and compared it to our other samples. As suspected, the rpb4Δ NET-seq profile correlated well with all considered DFs (ρ ∼ 0.5) (Fig. 1B). This suggests that Rpb4 functions similarly to the studied DFs in linking mRNA synthesis and decay, consistent with its known interactions with both Pol II (3) and the scaffold of the mRNA decay complex, Pat1 (7), as well as its distinct functions in transcription and in the major cytoplasmic mRNA decay (5, 7, 8, 10, 34). We therefore included the rpb4Δ strain in our subsequent analysis.
To examine whether Xrn1 or Rpb4 are required for the overall processivity of Pol II, we compared NET-seq signals with previously reported GRO signals (1, 35). GRO results are sensitive to backtracking because the RNA 3' end of backtracked Pol II is displaced from the active site and transcription elongation cannot proceed in vitro. The log2 ratio between GRO signal and Pol II occupancy detected by NET-seq (henceforth the elongation efficiency) is substantially compromised in the xrn1Δ strain (Fig. S2B), suggesting that Xrn1 mediates proper elongation of Pol II. Compared with xrn1Δ, the rpb4Δ strain displayed a smaller decrease in efficiency. We do not suggest that Rpb4 is not important for transcription, as was demonstrated previously by other means (see Introduction); rather, we conclude that the overall effects of RPB4 deletion identified using GRO are similarly reflected in the NET-seq data. Indeed, as we show later, Rpb4 does impact Pol II activity in 5' and 3' ends of genes. To further probe the impact of Xrn1 and Rpb4 on elongation, we compared fold changes in elongation efficiency to gene length. Consistent with the above, fold changes in xrn1Δ were more negative than those in rpb4Δ for genes of all lengths, but both deletion strains showed that longer genes tended to see larger reductions in elongation efficiency (Fig. S4). These findings suggest Xrn1 and Rpb4 are important for efficient Pol II elongation in a manner that becomes more essential for longer genes (1, 2, 36).
We previously rated genes according to the sensitivity of their mRNA synthesis and decay rates to XRN1 deletion. mRNAs whose synthesis and decay were highly responsive to Xrn1 disruption were named the Xrn1 synthegradon, whereas those least affected were dubbed the “Xrn1 anti-synthegradon” (2). We compared the changes in NET-seq signals as a function of these ratings, finding that higher sensitivity is strongly correlated with larger decreases in Pol II occupancy in xrn1Δ, dhh1Δ, and lsm1Δ; a weaker pattern was apparent for ccr4Δ and none for rpb4Δ (Fig. 1C). Our findings support earlier classification of genes into the Xrn1 synthegradon using GRO (2) and additionally demonstrate that transcription of the same genes is also activated by Dhh1, Lsm1, and Ccr4. Rpb4's role in transcription is unrelated to this classification, most likely because it affects the transcription of most, if not all, genes (9).
To understand whether particular classes of genes are more affected by DF deletions, we ranked genes according to their fold changes in total Pol II occupancies. Genes whose Pol II counts decreased or increased significantly were called “up-regulated” (normally their transcription is induced by the concerned DFs) or “down-regulated” genes (normally their transcription is repressed by the concerned DFs), respectively. Briefly, we found that the most affected genes in xrn1Δ strains are those that are required for cell proliferation under optimal conditions when glycolysis is the main producer of ATP (and aerobic metabolism is partially repressed). For example, deletion of XRN1 results in reduced transcription of ribosomal protein (RP) and ribosome biogenesis (RiBi) (Fig. S5, B and C), and increased transcription of aerobic metabolic genes (cellular respiration, mitochondria, ATP synthesis and transport, and cytochromes) (Fig. S5, A and C). Highly similar classes of affected genes were identified among Dhh1-up-regulated genes in dhh1Δ (results not shown). Because the deletion of XRN1 results in lower Pol II levels in RP and RiBi genes, but increased levels in aerobic metabolic genes, it seems that Xrn1 is involved in the balance between building cell mass and metabolism. It is possible that the deletion strains experience stress. Although we regard this possibility as unlikely because we failed to detect abnormal levels of “classical” stress-induced genes, we nevertheless compared our results to those obtained by juxtaposing transcription rates before and after the depletion of Xrn1 for 30 min (37). We identified highly similar patterns among their enrichments (Fig. S5, C and D), indicating that the balance between building cell mass and metabolism was similarly affected even after using a different technique. These commonalities help provide additional credibility to our general findings via NET-seq.
Deletion of mRNA decay factors affects Pol II occupancy in both ends of transcription units of protein coding genes
A notable feature of NET-seq is that it can capture arrested Pol II in addition to those that are productively elongating. Although this may complicate direct estimation of transcription rates, it allows for a more refined interrogation of changes in Pol II processivity. For instance, the role of the elongation factor TFIIS (Dst1) in facilitating the release of backtracked Pol II was studied using NET-seq (25). In the same vein, we examined whether our deleted genes affect Pol II distributions across genes by constructing metagene densities (see “Materials and methods”). We first observed that WT samples displayed a ramp-like accumulation of reads ∼100 bp downstream of TSS, in agreement with previous results (25). Remarkably, TSS-proximal densities decreased strongly in xrn1Δ and dhh1Δ. This was observed for transcripts of length greater than 1100 bp (Fig. 2, A and B), whose metagene analysis involved no compression (see Fig. 2, legend), as well as an aggregate comparison that included all transcripts regardless of length (Fig. S6). ccr4Δ and rpb4Δ exhibited even sharper Pol II occupancy profiles in these regions that also resided closer to TSS than those present in WT samples (Fig. 2C). Pol II additionally pauses at the sites where Pol II transcripts are cleaved and post-transcriptionally polyadenylated, henceforth denoted PAS (38–43). We observed a trend of higher densities near PAS across all mutants, with a particularly pronounced increase in xrn1Δ and dhh1Δ, and smaller changes apparent in the remaining mutants (Fig. 2). Together, these results indicate that deletions of XRN1 and other DFs contribute to the altered Pol II occupancy ∼100 bp downstream of TSS. The high degree of similarity among the measured responses of xrn1Δ, dhh1Δ, and lsm1Δ (as well as ccr4Δ with rpb4Δ) provides confidence in these results. Moreover, we note that the 5' and 3' changes in Pol II occupancy are seemingly unrelated to growth rate given the lack of correlation between these changes (Fig. 2) and growth rates (Fig. S7). For example, both xrn1Δ and rpb4Δ cells grow more slowly than WT but display quite different metagenes. Likewise, lsm1Δ, dhh1Δ, and ccr4Δ grow similarly but present notably distinct average Pol II profiles. Conversely, ccr4Δ and rpb4Δ cells grow at different rates yet possess similar metagenes.
Figure 2.

Comparison of normalized full-body metagenes. Normalized reads were aggregated from 200 bp before TSS to 200 bp after PAS. The −200:500 with respect to TSS and −500:200 with respect to PAS were kept fixed and the remaining parts of genes were re-scaled to 500 bp. To avoid compression, only genes at least 1100 bp long were included. Fig. S6 shows that similar profiles were obtained when all genes were analyzed and re-scaled to lengths of 1000 bp. Finally, the read counts corresponding to the new “metapositions” were averaged to yield a picture of Pol II occupancy along whole gene bodies. Different panels show comparisons between WT and the indicated deletion strains.
Pol II initiation and elongation at the 5' ends of protein-coding genes
To examine the most apparent changes, we focused on the respective ends of transcription units, beginning with the 5' end. We generated metagene profiles for reads adjacent to TSS (Fig. 3A and Fig. S8) and relied on mathematical modeling (see below) to propose that the deletion of any one of these mRNA decay factors results in defective transcription initiation in addition to previous findings that XRN1 deletion leads to defective elongation (1, 44). We computed additional Pol II metagenes after stratifying genes into those that were up- or down-regulated by Xrn1 (Fig. 3A). As expected, genes strongly up-regulated by Xrn1 (FC < −2) underwent notable reductions in 5' Pol II occupancy in xrn1Δ strains. Differences between these gene classes were also apparent in WT cells; genes up-regulated by Xrn1 are highly transcribed and exhibited relatively higher Pol II levels with steeper slopes in 5' regions. In contrast, those that are down-regulated did not (Fig. 3A), suggesting that XRN1 deletion differentially affects genes based on their normal transcriptional patterns, although the observed differences may be related to transcription levels. Up- and down-regulated genes were subsequently determined for the other deletion strains using the same criteria (Fig. S8). The resulting profiles from lsm1Δ and dhh1Δ cells displayed occupancy changes similar to those of xrn1Δ, suggesting that Xrn1, Lsm1, and Dhh1 function similarly (Fig. 3A and Fig. S8, A and B). On the other hand, changes in ccr4Δ and rpb4Δ profiles exhibited different patterns (Fig. S8, C and D). Up-regulated genes displayed reductions, whereas 5' peaks in down-regulated genes increased and exceeded levels in the WT for both mutants.
Figure 3.
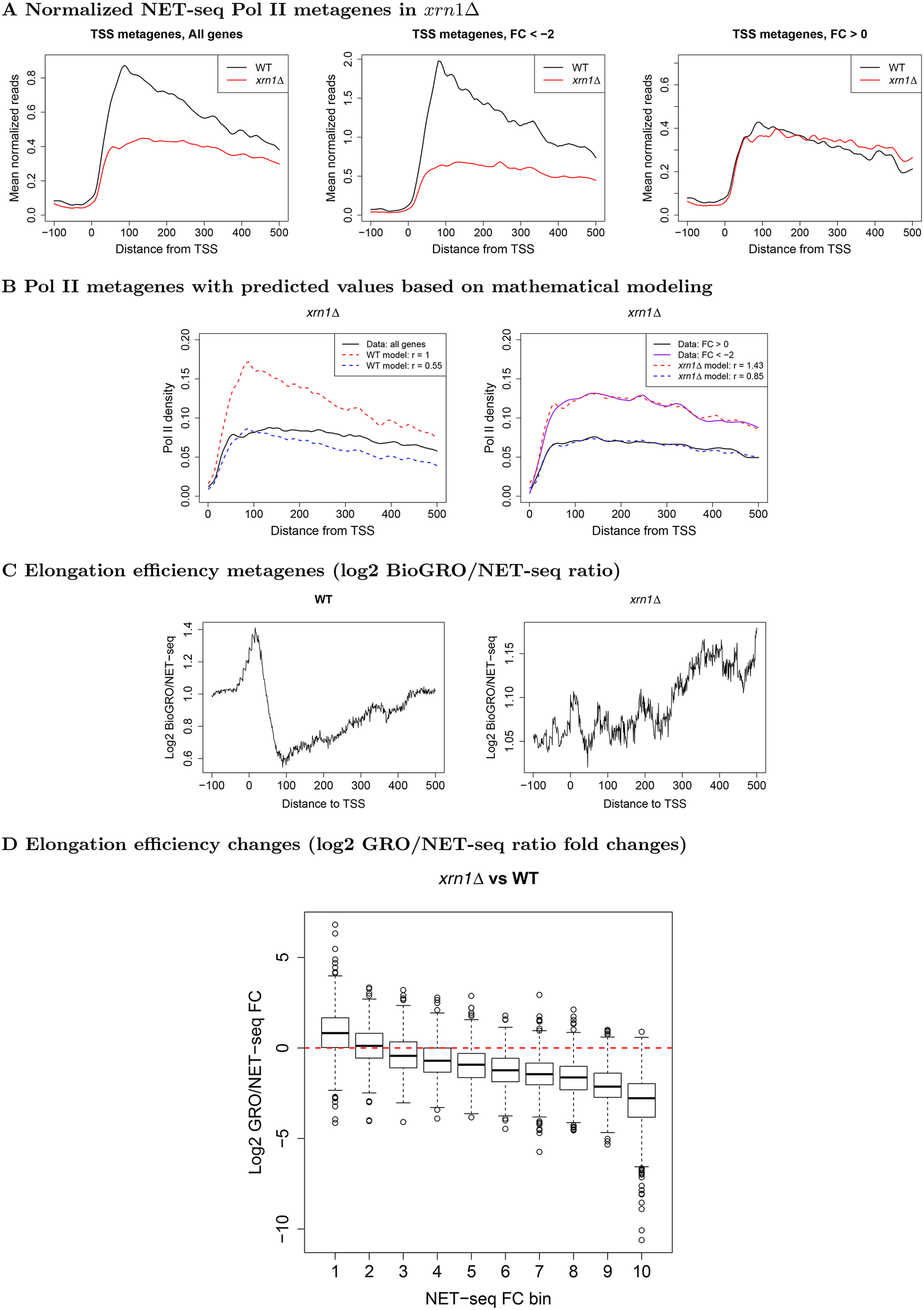
Metagene profiles near TSS in WT and xrn1Δ for Pol II and BioGRO/NET-seq ratios. A, we extracted NET-seq reads (–100:500 relative to TSS), normalized, and averaged them. Genes were separated into those which are stimulated (FC < 2) or repressed by Xrn1 (FC > 0). Note that the axes are on different scales to facilitate comparison of profile shapes within each panel. B, we applied a mathematical model (see “Materials and methods”) to investigate how initiation and elongation rates affect metagenes. Elongation rates for WT and mutant metagenes were estimated and initiation rates (r) were varied to find the best fits. L, varying initiation rates while using only the estimated WT elongation rates; R, varying initiation rates while using the estimated elongation rates from the xrn1Δ metagene. See Fig. S8 for other mutants. C, we extracted BioGRO and NET-seq values in the −100:500 region with respect to the TSS for all genes. For each gene, we smoothed the BioGRO and NET-seq profiles and took the log2 of their ratios. We then averaged over all genes to yield elongation efficiency metagenes. The profile shapes should be compared rather than the raw values due to potential differences in the scales of the BioGRO data. D, comparison of fold changes in elongation efficiency as a function of fold changes in NET-seq Pol II occupancy in xrn1Δ cells in the first 500 bp downstream of TSS. Genes were sorted into bins such that those in bin 1 had the largest reductions in occupancy, whereas those in bin 10 had slight increases. See “Materials and methods” for more details.
We sought to bolster our hypothesis of reduced elongation rates in xrn1Δ by looking for signs of increased pausing or backtracking. As indicated earlier, only Pol II that can incorporate nucleotides in vitro are captured by run-on methods, Pol II, which enter backtracked configurations, or cannot incorporate nucleotides by otherwise undefined mechanisms, are not mapped by GRO or BioGRO. In contrast, NET-seq is indifferent to the in vitro activity of Pol II and can identify these Pol II molecules; hence, Pol II processivity can be evaluated by comparing data from these two assays. To investigate transcriptional activity per unit Pol II, the elongation efficiency, in WT and xrn1Δ strains, we compared published BioGRO data (29, 30) and our NET-seq data. This is analogous to the analysis in Fig. S2B but with spatial resolution of Pol II activity. We focused on the two regions that demonstrated strong responses to Xrn1 deletion, the 5' and 3' ends, the latter of which is discussed in the subsequent section. In WT cells, we observed high elongation efficiency extending from TSS until ∼30 bp post-TSS, followed by a drop until ∼100 bp post-TSS (Fig. 3C). We thus propose that WT Pol II backtracks and/or pauses more often as it approaches ∼100 bp beyond TSS. In cells lacking Xrn1, this initial high elongation efficiency region vanishes (Fig. 3C), suggesting dysregulation of these processes.
The accumulation of NET-seq reads at ∼100 bp downstream of TSS could represent a controlled Pol II pausing phenomenon akin to what has been described for many metazoan genes (see Introduction). Alternatively, the trademark buildup of Pol II near TSS may simply be the result of unbalanced initiation and 5' elongation rates. To investigate the plausibility of the latter scenario, we employed a recent mathematical model (32) that models particles moving along a one-dimensional path (Fig. 4). We first used our computed metagene profiles to estimate reference initiation and site-specific elongation rates and then examined the results of perturbing these parameters. This allowed us to perform in silico experiments to separate the contributions of initiation and elongation rate changes and to infer the contributions of the studied DFs to elongation dynamics. Although Monte Carlo models of transcription have been considered in a handful of prior studies (45–49), to the best of our knowledge this is the first attempt to apply a model that rigorously and flexibly handles both spatial heterogeneity in elongation rates and the mutual interference of co-localized Pol II. Furthermore, it permits us to obtain analytical solutions from input parameters, increasing the precision of our analysis. Based on this model, we found that the observed WT Pol II metagene was indeed consistent with slower 5' elongation compared with initiation (Fig. 3B). Thus, a controlled pausing event is not necessary to reproduce the observed profiles. Of course, our simulation does not conclusively rule out such a possibility; nevertheless, given the absence of supporting data in this work or the wider literature, we propose that imbalanced rates of transcription initiation and elongation constitute the major cause of 5' Pol II accumulation in WT cells. Given the computed transcription elongation efficiency profiles (Fig. 3C), we additionally propose that the gradual decrease in Pol II processivity as Pol II approaches the 100-bp position exacerbates the imbalance between rates of initiation and elongation.
Figure 4.

Schematic of mathematical model. The key parameters are α, β, λ, and ℓ, corresponding to the initiation, termination, and profile of elongation rates, respectively, plus the width of the polymerase. Note that λi gives the site-specific elongation rates, and a polymerase cannot move when blocked by another. These quantities can be estimated from the Pol II occupancy profiles produced using our NET-seq data, which are denoted as ρi. In particular, up to a common constant multiplicative factor J, λi ∼ J × [ρi(1 – ρi)]−1, α ∼ J × [1 – ℓ ρ1]−1, and β ∼ J × [ρL]−1. See “Materials and methods” or Ref. 32 for additional details.
We next performed a similar analysis on mutant metagenes. To test whether the changes between mutant and WT profiles could be replicated by solely modulating initiation, we fixed elongation rates to the values inferred for the WT (see “Materials and methods”) and varied initiation rates over a range of values in the simulation model. This procedure produced similar simulated profiles to those observed for xrn1Δ and lsm1Δ, suggesting that deletion of these genes compromised transcription initiation. However, the observed NET-seq profiles were notably flatter than the simulated ones (Fig. 3B and Fig. S8B), indicating that defects in elongation in the mutants should also be considered. For the dhh1Δ, ccr4Δ, and rpb4Δ mutants, simulated profiles using WT elongation rates were unable to recapitulate the appearance of pronounced peaks slightly upstream of 100 bp (Fig. S8, A, C, and D). This indicates that, for these strains, our observed metagene profiles cannot be explained by simple changes in the overall balance between initiation and elongation. Hence it is likely that more complex kinetics are involved in which the ∼100 bp location may serve as a transition point. This notion is supported by the clear differences in behavior observed at the 30- and 100-bp positions post-TSS in the WT and xrn1Δ elongation efficiency (BioGRO/NET-seq) profiles (Fig. 3C). In summary, although the differences between heights of 5' peaks in WT and mutant strains can be explained by reduced initiation rates, the differences in profile shapes cannot be totally accounted for by manipulating this single quantity. Hence transcription elongation is affected both before and after the 100-bp mark in xrn1Δ cells (Fig. 3). We therefore propose that initiation rate reduction is a major consequence of XRN1 and LSM1 deletion with additional decreases also occurring in elongation rates. Furthermore, although the respective deletions of DHH1, CCR4, and RPB4 also reduce initiation rates, they have additional targeted effects on elongation rates in the first 100 bp of genes that differ from those of Xrn1 and Lsm1.
Pol II elongation can be hampered by Pol II collisions. Thus, the net change in elongation efficiency due to XRN1 deletion consists of a balance between the competing effects of increased pausing or backtracking (reducing efficiency) and reduced interference between Pol II molecules due to lower initiation rates (reducing collisions and increasing efficiency). To further explore the joint effects on initiation and elongation changes in xrn1Δ cells, we looked at log2 fold changes in the elongation efficiency (GRO/NET-seq ratio; full description under “Materials and methods”) as a function of changes in the initiation rate. As our simulations indicated that the initiation rate is the dominant contributor to Pol II occupancy just downstream of TSS, we used the log2 fold changes in NET-seq reads in the first 500 bp of genes as a proxy for initiation rate changes. After binning genes based on these occupancy fold changes, we found a clear trend in which genes with less severe initiation defects displayed larger reductions in their computed elongation efficiencies (Fig. 3D). Hence the observed trend comports with our hypothesis that Xrn1 mediates productive elongation across the genome. In particular, bins 7-9 correspond to genes with the most similar 5' NET-seq occupancies in both xrn1Δ and WT cells, so they represent our best estimate of changes in elongation without the confounding effect of initiation; we find a clear reduction in efficiency for all three bins (Fig. 3D).
Deletion of mRNA decay factors affects NET-seq profiles near PAS
WT Pol II pauses at PAS, probably to provide time for the PA mechanism to function (50). In the xrn1Δ, dhh1Δ, and lsm1Δ strains, abnormally high spikes were observed in this region (Fig. 5 and Fig. S9A), perhaps due to enhanced pausing in the absence of these DFs (see “Discussion”). Interestingly, mutant strains displayed abnormally high accumulations of reads beginning ∼75 bp upstream of PAS and lasting until PAS, suggesting that these factors are involved in Pol II processivity as it approaches PAS (Figs. 2 and 5A). Downstream of these PAS, NET-seq reads accumulated due to transcription that continues beyond PAS before reaching transcription termination sites (38). Atypically low NET-seq reads were observed downstream of PAS in the xrn1Δ mutant strains, suggesting that less Pol II could be released from a paused state in the absence of Xrn1. Taken together, these results favor a model whereby Xrn1, Dhh1, and Lsm1 are required for Pol II processivity as Pol II approaches PAS and are involved in the release of Pol II from PAS (alternative interpretations are considered under “Discussion”). In the rpb4Δ and ccr4Δ strains, we detected accumulations of reads upstream and downstream of PAS, but the actual PAS peaks were comparable with those in the WT (Fig. S9B). Separation into genes up- and down-regulated by Xrn1 revealed 3' occupancy patterns unlike those in 5' ends. Whereas up-regulated genes (FC < −2) had displayed large reductions in 5' Pol II levels (Fig. 3A), 3' occupancy demonstrated relatively little sensitivity to the presence of Xrn1 (Fig. 5A). Genes down-regulated by Xrn1 (FC > 0) displayed nearly the opposite behavior, as 5' occupancy was insensitive to XRN1 deletion, whereas 3' pausing greatly increased (Figs. 3A and 5A). Deletions of CCR4 and RPB4 had smaller effects on peaks at PAS, although general increases were present in down-regulated genes (Fig. S9B).
Figure 5.
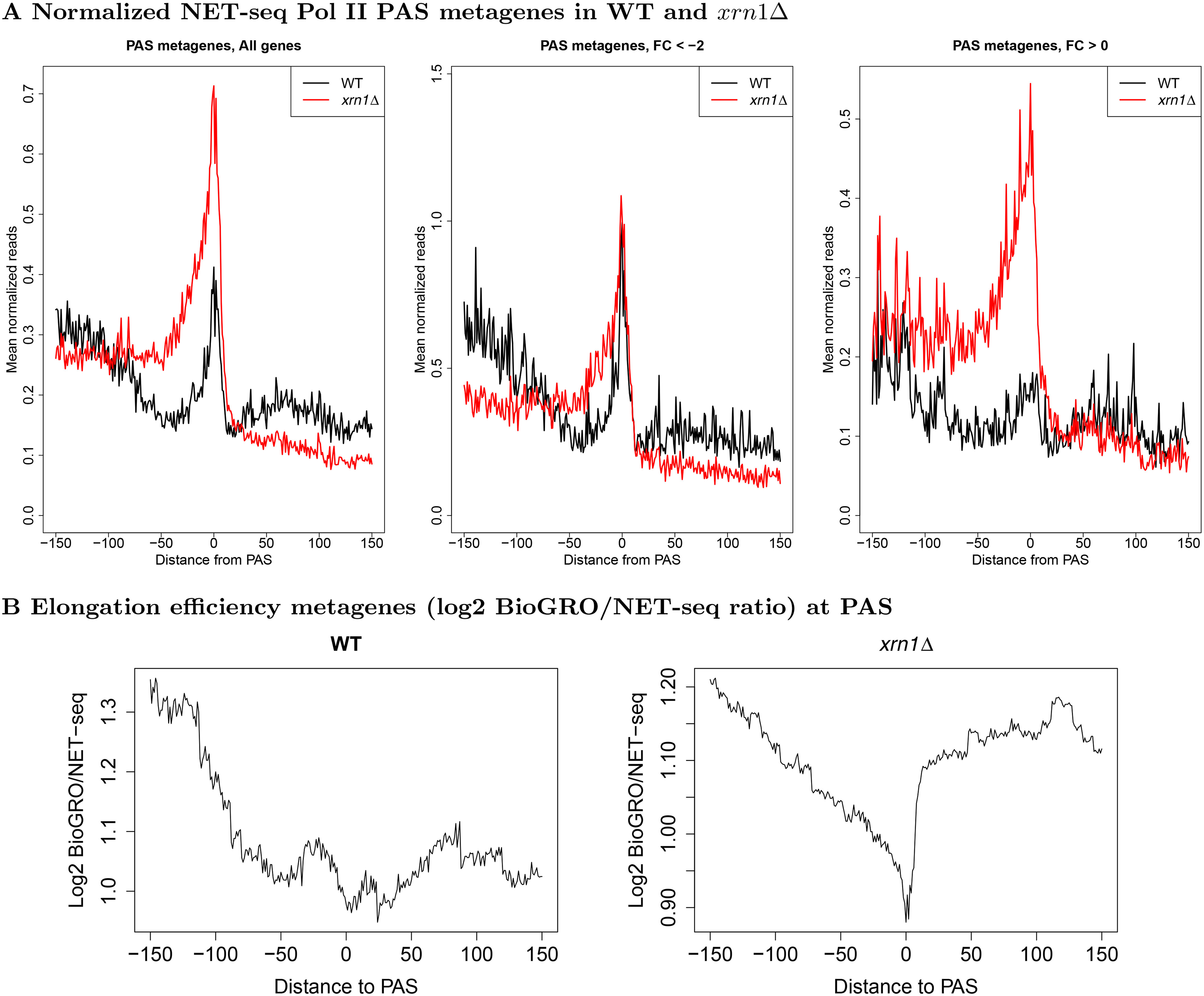
Metagene profiles near PAS in WT and xrn1Δ for Pol II and BioGRO/NET-seq ratios. A, we extracted NET-seq reads (−150:150 relative to PAS), normalized, and averaged them. Genes were separated into those that are stimulated (FC < 2) or repressed by Xrn1 (FC > 0). Note that the axes are on different scales to facilitate comparison of profile shapes within each panel. B, we extracted BioGRO and NET-seq values in the −150:150 region with respect to the PAS for all genes. For each gene, we smoothed the BioGRO and NET-seq profiles and took the log2 of their ratios. We then averaged over all genes to yield elongation efficiency metagenes. The profile shapes should be compared rather than the raw values due to potential differences in scales of the BioGRO data.
Our metagene analysis showed that xrn1Δ and dhh1Δ cells accumulate abnormally small numbers of reads in 5' regions and unusually high numbers of reads in 3' regions (∼-100 until PAS). Because these analyses aggregated reads across all genes, it was unclear whether profile changes were driven by widespread behavior or simply a small number of highly impacted genes. To address this issue more directly, we determined Pol II 5'/3' ratios for each mutant (Fig. S10). Indeed, Pol II 5'/3' ratios were substantially lower in xrn1Δ compared with WT strains, and the overall shift in the distribution of these ratios suggests that the changes in the 5' and 3' metagene profiles are not confined to a small number of genes. Interestingly, the dhh1Δ strain exhibited a similar pattern of Pol II 5'/3' ratios, and additional modest decreases in Pol II 5'/3' ratios were observed in the remaining mutants.
Much like before, we generated BioGRO/NET-seq ratio profiles near PAS (Fig. 5B). We found that WT Pol II elongation efficiency decreased as it moved toward PAS, finally bottoming out near the PAS. Meanwhile, xrn1Δ cells displayed a smooth efficiency reduction culminating in a sharp valley at the PAS. Thus, Pol II that accumulate upstream of PAS are relatively inactive, both in WT but notably more so in xrn1Δ cells, possibly in a backtracked configuration or one that is incompatible with in vitro polymerization. Collectively, the most pronounced effects of DFs in transcription are at the ends of genes where Pol II processivity decreases in the mutant strains, potentially relating to increased Pol II pausing or backtracking. Moreover, the differences between DF deletion-induced responses indicate additional defects in transcription initiation. These differences cannot be simply attributed to growth rates because lsm1Δ cells proliferate comparably to dhh1Δ and ccr4Δ (Fig. S7), despite different effects of the deletions of LSM1, CCR4, and DHH1 on metagene profiles (Fig. S8).
Transcription termination (downstream of PAS) seems to be affected by deletion of the studied mRNA decay factors
Transcription termination, which occurs downstream of PAS, is allosterically modulated by the PA mechanism (50, 51). Because we found that our studied DFs function in the PA process, we examined whether transcription termination is also affected by the deletion of DFs. Direct analysis of changes in termination using NET-seq is challenging because it does not identify transcription termination efficiently, probably because there are multiple termination events (27). Therefore, we examined the effect of DFs on this process indirectly by taking advantage of the capacity of NET-seq to report Pol II pausing due to collisions of two convergently transcribed Pol II molecules. After defining the midpoint between convergent genes as the halfway point between the ends of paired 3' UTRs, we found that the NET-seq signal in WT cells decreases gradually as a function of distance from the 3' ends, consistent with a gradual termination post-PAS, occuring before the midpoints. In contrast, mutant strains displayed accumulations of Pol II near the midpoints of convergent gene pairs as evidenced by midpoint peaks (Fig. 6A). Our results are reminiscent of the previous demonstration of the Pol II buildup between convergent genes in strains lacking Elc1, a protein that aids in the removal of stalled Pol II (52). This raises an alternative explanation in which the studied DFs stimulate the degradation of colliding Pol II. To verify that the accumulation between convergent gene pairs was truly a result of Pol II collisions, we stratified genes based on the distances between their respective PAS. This analysis demonstrated that Pol II occupancy between such pairs gradually decreased as a function of the distance from the midpoints (Fig. S11), suggesting that as the distance between respective PAS increases, Pol II has more opportunities to terminate in both WT and mutant strains. This is consistent with a model wherein Pol II normally terminates within a window 100-200 bp downstream of PAS in WT strain, but instead continues to transcribe further downstream in mutant counterpart because of less efficient termination. As a point of reference, we also performed this analysis for divergent pairs, finding only the expected differences due to reduced Pol II occupancy downstream of TSS (Fig. 6B).
Figure 6.
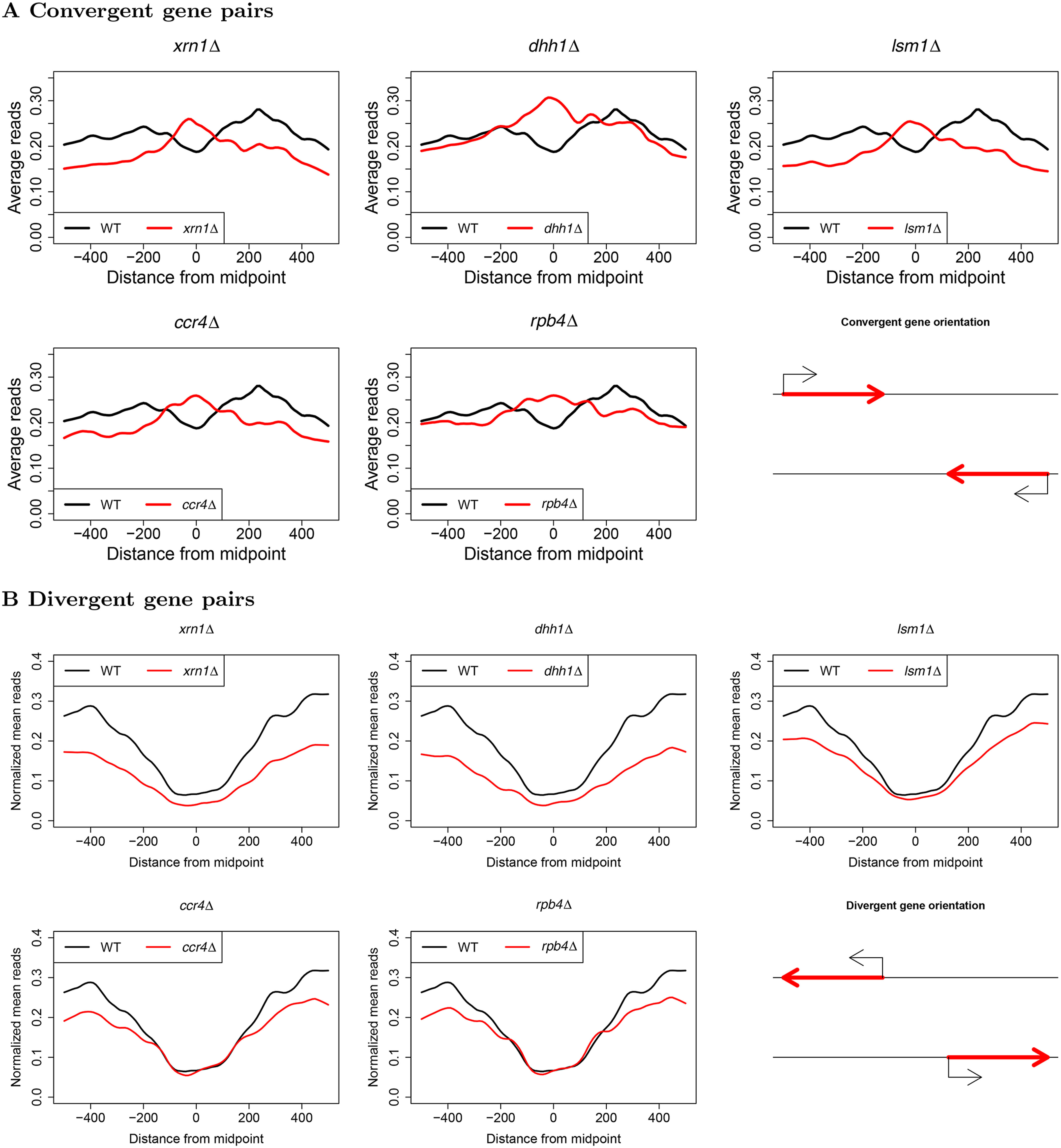
Metagene analysis with respect to midpoints between genes for convergent and divergent gene pairs. Convergent and divergent gene pairs were determined by the lengths between their PAS (convergent) and TSS (divergent). Midpoints between genes were defined as the halfway point between these respective features, and gene distances were computed as the difference between the annotated features on the negative and positive strand, respectively. Normalized NET-seq reads were then extracted for sites within 500 bp of gene midpoints and subsequently averaged to produce the metagene profiles.
Transcription in noncoding regions is also compromised by DF deletions
NET-seq provides an opportunity to monitor the production of unstable transcripts because it quantifies bound Pol II rather than mature RNAs and is little affected by RNA stability. Consequently, we investigated the effect of DF deletions on the transcription of ncRNAs by considering the changes in Pol II occupancy at chromosomal loci encoding cryptic unstable transcripts (CUTs), Nrd1-unterminated transcripts (NUTs), stable unannotated transcripts (SUTs), and Xrn1-sensitive unstable transcripts (XUTs) (53–57). Computation of NET-seq fold changes across gene bodies demonstrated global reductions in Pol II occupancy in xrn1Δ, dhh1Δ, and rpb4Δ (Fig. S12). To identify potential distributional changes in Pol II occupancy, we computed metagene densities (Fig. S13). These showed changes resembling those of protein-coding genes at TSS but little effect in the 3' regions. To determine whether ncRNAs could play cis-regulatory roles in transcription, we examined the relationship between sense and antisense Pol II occupancies for annotated transcripts, finding essentially no correlation both in WT and deletion strains (Fig. S14A). Thus, there seems to be no obvious global relationship between coding and ncRNA genes, consistent with what has been reported previously (58, 59), and any mild correlations among divergent pairs are plausibly attributable to the common chromatin environment of nearby divergent promoters (57, 58). In summary, DFs are involved in regulating the early stages of ncRNA transcription. However, we could not find any indication that these effects modulate the transcription of protein-coding genes.
DFs differentiate between SAGA- and TFIID-dominated genes
Early studies classified genes as SAGA- or TFIID-dominated, according to measured changes in their mRNA levels after inactivation of central components of the SAGA (mainly Spt3) and TFIID (mainly Taf1) complexes (15, 16). However, having focused only on mRNA levels, analysis such as these are unable to disentangle the contributions of mRNA synthesis and decay. Indeed, by examining transcription directly, recent studies demonstrated that all promoters recruit both SAGA and TFIID (17–19); mutations in either complex resulted in defective transcription, but in most cases mRNA levels were unaffected due to feedback mechanisms that involve mRNA decay (17–19). As our studied DFs have been implicated in buffering, we considered the possibility that they differentially affect the two groups of genes by comparing the respective fold changes in Pol II levels after DF deletions (Fig. 7A). As expected, we found decreases in Pol II occupancy throughout TFIID genes due to XRN1 deletion. However, median Pol II occupancy in “SAGA-dominated” genes remained unaffected in xrn1Δ, lsm1Δ, and rpb4Δ strains and even slightly increased in ccr4Δ and dhh1Δ strains (Fig. 7A), and similar gaps were identified at either end of genes (Fig. 7B).
Figure 7.
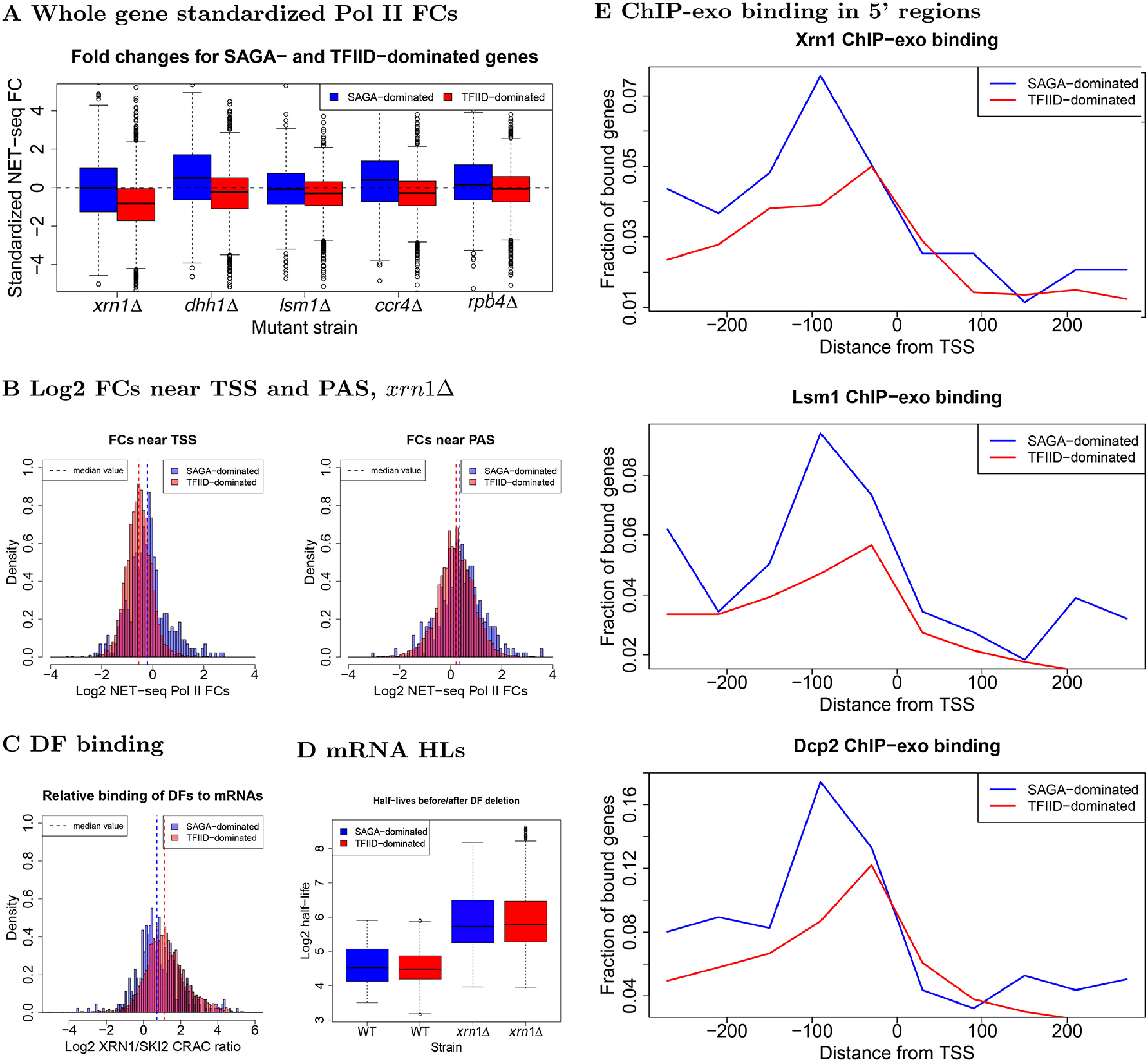
Comparison of transcription, decay, and protein binding for SAGA- and TFIID-dominated genes. A, fold changes, computed as described in the legend to Fig. 1A, for SAGA- or TFIID-dominated genes, as indicated. B, histograms of log2 NET-seq Pol II FCs for regions near TSS (−100:500) and PAS (−150:150) in xrn1Δ. C, we summed Xrn1 and Ski2 CRAC data (60) mapped to each gene and took the log2 ratio of mapped reads for each DF in each gene. D, comparison of mRNA half-lives before and after XRN1 deletion (2). E, reads were binned into windows of 60 bp starting 300 bp upstream and extending 300 bp downstream of TSS. The proportion of bins having more than 10 recorded reads was then computed across the genome and plotted (1). Due to coverage differences across the Xrn1, Lsm1, and Dcp2 experiments, only the qualitative behavior between different panels should compared. Within panels, the fractions may be compared without worry.
The discrepancy between measured mRNA levels and transcription rates suggests that an important aspect of the division between SAGA- and TFIID-dominated genes may lie in the regulation of transcript decay, possibly mediated by DFs (see above). To explore this possibility, we analyzed publicly available UV cross-linking and analysis of cDNA (CRAC) data for DFs representing binding of the 3'-5' (Ski2) and the 5'-3' (Xrn1) cytoplasmic mRNA decay factors to mRNAs (60). We found that each DF bound to both classes of genes (Fig. 7C), suggesting they play a role in the decay of transcripts regardless of their annotation as SAGA- or TFIID-dominated. However, both Ski2 and Xrn1 bind more frequently to SAGA-dominated genes than TFIID-dominated genes even after accounting for transcript length and steady-state mRNA levels (∼1.86 and ∼1.47 ×, respectively). That binding of Ski2 occurs at a higher comparative rate between classes than Xrn1 suggests that the decay of SAGA-dominated transcripts is more dependent on the exosome than the Xrn1-led 5'-3' pathway. At any rate, SAGA- and TFIID-dominated genes are distinguished by Xrn1 and Ski2 binding. However, despite the higher binding rates for both proteins, a comparison of the half-lives of mRNAs from SAGA- and TFIID-dominated genes based on previously published data (2) failed to detect a discernible difference (Fig. 7D).
Inspired by the different effects of DFs on SAGA and TFIID genes, we explored the DNA-binding patterns of three decay factors by analyzing previously generated ChIP-exo data for Dcp2, Lsm1, and Xrn1 (1) after stratifying genes according to their classic SAGA/TFIID labels. We found that all three proteins bind to SAGA-dominated genes at higher frequencies than TFIID-dominated genes (Fig. 7E), consistent with the higher transcription levels of SAGA-dominated as compared with TFIID-dominated genes (Figs. S15A and S16A, see scales). Moreover, these proteins tend to bind further upstream of TSS for SAGA-dominated genes (peak ∼90 bp) compared with TFIID-dominated genes (∼30 bp). Given the recent finding that SAGA localizes further upstream than TFIID and binds more frequently in SAGA-dominated than TFIID-dominated promoters (17), it is possible that the interactions of Dcp2, Lsm1, and Xrn1 with promoters are influenced by the positions of bound SAGA and TFIID complexes as well as their binding frequencies (see “Discussion”). In summary, transcription in SAGA- and TFIID-dominated genes depends on both the SAGA and TFIID complexes, and their mRNA products have comparable half-lives in WT and in xrn1Δ strains alike. However, SAGA- and TFIID-dominated genes differ by (i) the effect that the studied DFs have on their transcription (Fig. 7A), (ii) chromatin binding features of Xrn1, Lsm1, and Dcp2 (Fig. 7E), and (iii) the binding of Xrn1 and Ski2 to their mRNAs (Fig. 7C). Thus, these results highlight the capacity of the studied DFs to differentiate between SAGA- and TFIID-dominated genes.
Discussion
In recent years, interest in understanding the cross-talk between mRNA synthesis and decay has grown. Under optimal proliferation conditions, various mRNA decay factors are involved in mRNA “buffering,” a feedback mechanism that minimizes changes in mRNA levels. In this coupling, reductions in either mRNA synthesis or decay are associated with compensatory reductions in the other process, resulting in relatively consistent concentrations of mRNAs (Ref. 61 and references therein). Whereas the functions of DFs in cytoplasmic mRNA decay are well-characterized, we currently know very little about their roles in transcription, motivating this work. For instance, although Xrn1 was identified as an effector of buffering, its mode of action in transcription has remained controversial (see Introduction). Using NET-seq, we found that the deletion of XRN1 generally resulted in the down-regulation of transcription (Fig. 1A) and notably reduced the elongation efficiency of Pol II (BioGRO/NET-seq, Figs. 3C and 5B). These results, combined with ChIP-exo results showing that Xrn1 (as well as Dcp2 and Lsm1) binds promoters (Fig. 7E and Ref. 1), are consistent with a role for Xrn1 as a stimulator (1, 2) rather than repressor (14) of transcription. As a transcriptional activator, Xrn1 primarily targets genes required for proliferation under optimal conditions, when cells are dependent mainly on fermentation (Fig. S5, B and C). These GO terms are similar to those that characterize the Xrn1 synthegradon group identified by a GRO-based analysis (2). We also found that the absence of Xrn1 results in increased Pol II levels of a minor population of relatively lowly expressed genes that mainly encode proteins related to aerobic metabolism (Fig. S5, A and C). Given earlier findings of direct binding of Xrn1 and Lsm1 to affected genes (1), and our finding that these proteins also bind promoters of both stimulated and repressed genes (results not shown), we suspect that this effect is direct. Thus, Xrn1 seems to function in the balance between building cell mass and metabolism and to permit efficient proliferation when fermentation is preferred. Interestingly, Xrn1 is regulated by Snf1-activated phosphorylation (62) and XRN1 interacts genetically with TOR2 (62). Snf1 and Tor2 are kinases that function in a similar balance. Our work also uncovered an underlying function by which Xrn1 targets transcription initiation and Pol II processivity, possibly via pausing and backtracking.
Recent studies have reported that the release of promoter-proximal–paused Pol II is a crucial component of the regulation of transcription under both optimal (22) and stress conditions in metazoans (22, 23) (see Introduction). Although not as severe as in Schizosaccharomyces pombe or metazoans, promoter proximal Pol II accumulation has been reported in S. cerevisiae (17, 25, 64) but the underlying cause is uncharacterized (24). This early build-up of Pol II has been implicated as a “checkpoint” of Pol II elongation regulated by the CTD kinase Kin28 (65). Moreover, depletion of sirtuin proteins (Hst3 and Hst4) increases 5' proximal accumulation (64). Thus, although the correspondence to mammalian Pol II pausing remains unclear, it seems that Pol II is subject to promoter-proximal regulation in S. cerevisiae as well. Here we show that Xrn1 and other yeast DFs also affect Pol II accumulation roughly 100 bp post-TSS in genes that they up-regulate. We also note reductions in initiation rates for many genes across mutants and demonstrate via mathematical modeling that both initiation and elongation rate reductions are necessary to recapitulate observed Pol II profiles. Furthermore, we showed that although the initiation rate changes appear to be restricted to a subset of genes, the elongation rate changes are more ubiquitous, as metagenes of both activated and repressed genes displayed flatter Pol II profiles in mutants than the WT (Fig. 3, A and B, and Fig. S8). Recently, Pol II was shown to frequently backtrack in promoter-proximal regions of human genes, with TFIIS-stimulated RNA cleavage helping to release Pol II from pause sites (22, 23). High elongation efficiency (BioGRO to NET-seq ratio) was observed in WT cells for the first ∼30 bp downstream of TSS (roughly coincident with capping), followed by a gradual drop until around 100 bp downstream (Fig. 3C). Given that NET-seq captures all bound Pol II whereas BioGRO only detects those that are productively elongating in vitro, this suggests that Pol II enters a state that is incompatible with in vitro elongation, such as a backtracked configuration, with increasing frequency as it approaches the ∼100 bp mark post-TSS, as was found in mammalian cells (22, 23). In the absence of Xrn1, this pattern is disrupted (Fig. 3C), suggesting that normal regulation is compromised. DFs have previously been implicated to function in the context of backtracking. For example, Ccr4 has been shown to physically interact with TFIIS and to directly modulate backtracking in conjunction with TFIIS (66, 67). Likewise, Dhh1, Pat1, and Lsm1 interact with TFIIS both physically and genetically (63, 66, 68–73). Recently, Xrn1 was shown to increase TFIIS recruitment to Pol II (44), consistent with the role we assign to Xrn1 as a regulator of backtracking.
The effects of Xrn1 on transcription initiation and elongation are interconnected. Specifically, genes whose transcription initiation is little affected by XRN1 deletion are highly affected in elongation and vice versa (Fig. 3D). We interpreted this observation to indicate that the decrease in initiation decreases Pol II collisions and consequently increases processivity (the underlying rationale is discussed under “Results”). Alternatively, the observed behavior could suggest a mechanistic link between Xrn1's roles in the regulation of initiation and elongation. For example, it is possible that Xrn1 exists in two configurations, one that stimulates initiation and one that prevents backtracking. When adopting the latter configuration, Xrn1 is not involved in initiation and instead binds Pol II transcripts early in transcription to enhance processivity by repressing backtracking. For any given transcription cycle, Xrn1 can exist in one of the two configurations, so by looking at samples consisting of many cells or genes with multiple Pol II, we can learn about the relative frequency with which it adopts each configuration, perhaps corresponding to the variable elongation efficiencies that characterize the bins shown in Fig. 3D.
All these observations provide outside credibility to our proposals. Why, then, do we detect differences in the impact of different DFs on 5' Pol II accumulation? For example, whereas the deletion of CCR4 led to enhanced Pol II accumulation (Fig. S8C), the deletion of XRN1 led to the obliteration of Pol II accumulation at this position (Fig. 3A). The simplest explanation is that different DFs differentially affect the initiation/elongation ratio by targeting initiation, elongation/backtracking, or both as demonstrated using our mathematical model. This is consistent with the recent report that Xrn1 and Ccr4 differentially regulate Pol II elongation; namely, whereas deletion of XRN1 led to increased TFIIS-Pol II interaction, that of CCR4 had the opposite effect (44). Nonetheless, the exact mechanism remains to be determined. These results not only provide insight into the manner in which Xrn1 affects transcription, but they also highlight the need to understand the balance between initiation and elongation rates as a possible regulatory feature in yeast transcription.
WT Pol II pauses at PAS, probably to provide time for assembly of the full complement of the PA complex and/or to permit time to execute the process (50). During PA, the nascent RNA is cleaved, giving rise to a 5' long RNA that is the substrate of poly(A) polymerase and a 3' short RNA whose 3' end is in the Pol II active site. Downstream of these PAS, NET-seq reads accumulate due to transcription that continues beyond PAS before reaching the transcription termination sites (38). However, not much is known about pausing of Pol II at PAS and what triggers its release. If, during the NET-seq protocol, the 5' long RNA is pulled down by Pol II, it would be sequenced together with the 3' short RNA and thus contribute to the NET-seq signal at PAS. Moreover, if Pol II continues to polymerize beyond PAS after RNA cleavage and without releasing the 5' long RNA, it might further confound our results. Specifically, pulling down this Pol II would map both the growing RNA whose 3' end is in the Pol II active site and the 5' long RNA to provide a signal at PAS. Thus, PAS reads do not always report the exact location of Pol II. In the xrn1Δ, dhh1Δ, and lsm1Δ strains, abnormally high spikes were observed in the PAS region (Figs. 5A and S9A). We propose the following three possible interpretations to explain the observed effects of DF deletion: (i) longer Pol II pausing; (ii) abnormally slow cleavage and polyadenylation rates combined with dragging of the 5' long RNA with Pol II as it continues to transcribe past PAS; and (iii) increased cleavage at the major PAS at the expense of alternative PAS. Interestingly, mutant strains displayed abnormally high accumulations of reads beginning ∼75 bp upstream of PAS and lasting until PAS. This supports a model whereby Pol II processivity is compromised already upstream of PAS. Importantly, atypically low NET-seq reads were observed downstream of PAS in the xrn1Δ mutant strains (Fig. 5A), suggesting that less Pol II could be released from a paused state in the absence of Xrn1. Taken together, our results favor a model whereby Xrn1, Dhh1, and Lsm1 are required for Pol II processivity as Pol II approaches PAS, whereas Xrn1 is involved in the release of Pol II from PAS. One possibility is that Xrn1 sterically impedes backward movement of Pol II (i.e. backtracking), as was proposed for Ccr4 (66, 67). Accordingly, in xrn1Δ and the other mutant strains, backward motion of Pol II is not repressed and therefore backtracking is enhanced, burdening the TFIIS-stimulated RNA cleavage process and leading to an accumulation of reads upstream of and at PAS. It is also possible that the concerned DFs are involved in selecting alternative PAS. Interestingly, Rpb4 was reported to contribute to proper co-transcriptional 3'-end processing (74). Although our data highlight a unique feature of PAS and the region immediately upstream of them, more work is required to pinpoint the exact regulatory mechanisms governing transcription in these regions. It hence remains possible that unique chromatin architecture and DNA/RNA sequences combined with the recruitment of PA factors, TFIIS, and some DFs in this region are involved in regulating proper Pol II elongation rate at PAS.
Transcription termination occurs downstream of PAS (43). Cleavage and polyadenylation factors are presumed to act in the Pol II release step of transcription termination by either allosterically modifying the properties of the transcription elongation complex or acting via the torpedo mechanism (for a recent review, see Ref. 75). Our studied DFs, which are involved in cleavage and polyadenylation, further appear to affect termination as well. This was determined indirectly because NET-seq does not identify transcription termination efficiently (27), probably because, for any single gene, termination does not occur at a single locus. Correspondingly, we found that the NET-seq signal decreases gradually as a function of distance from the 3' ends, consistent with a gradual termination post-PAS. In contrast, the signals in several of the mutant strains, most notably rpb4Δ and ccr4Δ, increased relative to the WT. We interpreted these results to indicate that transcription termination post-PAS is less efficient in our mutant cells, thus increasing the probability that two opposing Pol II would collide.
We also studied the effects of DF deletions on noncoding transcription. As in the case of coding genes, overall transcription of noncoding transcripts is compromised upon DF deletion. Moreover, changes in Pol II occupancy in noncoding regions largely mirrored those in coding regions, particularly for NUT genes (compare Fig. 2 and Fig. S13), which suggests that initiation and early elongation is affected by the studied DFs similarly in both coding and noncoding regions. However, changes in noncoding 3'-proximal pausing of Pol II did not match what was observed for coding genes. First, we note that 3' pausing in ncRNA regions is less apparent than in their coding counterparts, perhaps implying that they are regulated differently. Indeed, production of the 3' ends of NUTs is controlled by a unique mechanism (54). Perhaps a key distinction between transcription of coding and noncoding genes lies in the mechanisms controlling their respective terminations. Analyzing the effects of DFs at PAS of noncoding transcripts revealed that unlike in coding transcripts, deletions of the studied DFs did not lead to increased 3' Pol II accumulation, implying that DFs interact differently with the PA machinery of coding and noncoding regions. Additionally, the lack of correlations between sense and antisense transcription for multiple ncRNA types and the insignificant effect of DF deletion on these correlations (results not shown) belies the possibility that regulation of antisense ncRNAs is the preferred mechanism by which DFs modulate coding transcription. Moreover, we found that genes that are highly sensitive to XRN1 deletion (i.e. the Xrn1 synthegradon) are less likely to have convergent antisense transcripts of any type (results not shown), suggesting that convergent ncRNAs are not the direct mechanism that mediates the effect of Xrn1 on coding regions.
Our data highlight additional distinctions between TFIID- and SAGA-dominated genes. First, we found that deletion of XRN1, and to a lesser extent our other studied DFs, affects transcription of TFIID-dependent genes more than SAGA-dominated genes (Fig. 7, A and B). Second, the binding of Xrn1, Lsm1, and Dcp2 to promoters occurs at different positions between the two classes, with SAGA-dominated genes having binding sites located further upstream of the TSS than TFIID-dominated genes (Fig. 7E). Third, we found that both Xrn1 and Ski2 bind SAGA-dominated gene transcripts more than TFIID-dominated gene transcripts; moreover, Xrn1 and Ski2 exhibit different preferences to the two classes of mRNAs (Fig. 7C). On the other hand, both the SAGA and TFIID complexes regulate transcription of nearly all genes (17–19), and their respective transcripts have comparable half-lives (Fig. 7D). Cumulative results, shown here and published by others, suggest that the two classes are each characterized by distinct chromatin structure and different transcriptional plasticity (76–78). Based on the results discussed here, we propose that the two classes of genes differ in the buffering mechanisms controlling their mRNA levels, involving a linkage between RNA and chromatin binding features of some of the factors examined in this paper. In particular, we suggest that deletions of TAF1 and SPT3 compromise the cross-talk between mRNA synthesis and decay for TFIID- and SAGA-dominated genes, respectively, leading to decreases in steady-state mRNA levels.
Previously, we classified genes based on their sensitivity to XRN1 deletion (2). Genes whose synthesis and transcript stability were highly sensitive to this deletion were labeled the Xrn1 sythegradon. Here we found that our xrn1Δ NET-seq data are consistent with our prior classifications, as fold changes in Pol II occupancy nicely correlated with previously assigned responsiveness values (Fig. 1C, “xrn1Δ” panel) (2). Our results further demonstrate that these scores agree well with NET-seq fold changes of lsm1Δ, dhh1Δ, somewhat with ccr4Δ, but not with rpb4Δ (Fig. 1C). This suggests that Lsm1 and Dhh1 function in the link between mRNA synthesis and decay similarly to Xrn1, perhaps as a complex. In general, our analysis differentiate between two types of DFs. Type I comprises Xrn1, Lsm1, and Dhh1, the deletion of any one reduces 5' Pol II occupancy and elongation rates of DF-stimulated genes but enhances Pol II pausing at PAS. Type II includes Ccr4 and Rpb4, whose deletions inhibit the release of Pol II from positions roughly 100 bp post-TSS or enhance elongation rates downstream of these locations (Fig. S8, C and D). This distinction is further underlined by the aforementioned difference in the correlations between knockout-associated changes in NET-seq signals and Xrn1 responsiveness (Fig. 1C). Nevertheless, in contrast with the lack of correlation between DFs and the genes of the Rpd3S H4 deacetylation complex, we observed positive correlations among all studied DFs (Fig. 1B). We hence propose that they act similarly at the global level, akin to our previous suggestions for Xrn1, Lsm1, and Dcp2 (1).
In aggregate, our results point toward roles for Xrn1 and other DFs in facilitating the efficient elongation of Pol II early and, more clearly, late during transcription, potentially via control of pausing and backtracking. The identified functions of DFs further highlight the key roles of these processes in the regulation of transcription. In recent years, promoter-proximal pausing has been a focal point in the study of transcription of many metazoans; whether Pol II pausing plays a comparable key role at PAS and in transcription termination of S. cerevisiae and other organisms remains to be examined.
Materials and methods
Yeast strains, growth conditions, RNA extraction, and library preparation
Yeast strains were derivatives of 4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0). Deletion of the studied genes was done using standard PCR-based methods by replacing open reading frames with the Kan resistance gene. Epitope-tagged Rpb3 (C-terminal 3× FLAG) was expressed from its endogenous locus. Cell growth and library preparation were done in Churchman's laboratory as described previously (25). Briefly, 1 liter of log phase culture in rich medium (YPD) (2% Bacto Peptone, 1% yeast extract (Difco Laboratories), 2% dextrose) was harvested at 1 × 107 cells/ml by filtration and flash frozen by plunging into liquid nitrogen. Frozen cells were lysed cryogenically via six cycles of pulverization using a mixer mill 400 (RETSCH). Affinity purification of Rpb3-(FLAG)x3-containing Pol II, RNA extraction, and library preparation were done as described previously (25).
Data collection and pre-processing
We used the NET-seq protocol to measure the number of RNA polymerase II (Pol II) bound to DNA, which are engaged in mRNA synthesis; experiments were performed as detailed in Ref. 27, generating single-end reads. We consider the bound Pol II levels of 4973 genes in six genotypes comprising one control (in duplicate) and five mutant knockouts (xrn1Δ in duplicate). TSS and PAS annotations for ORFs were taken from Ref. 79. The data were pre-processed using cutadapt and prinseq (80, 81), and mapping was done via TopHat (82) with unique reads retained.
Identification and interpretation of differentially transcribed genes
The normalization used throughout the manuscript was performed by selecting genes whose productive transcription as measured by cDTA (ccr4Δ, dhh1Δ, lsm1Δ) and GRO (rpb4Δ and xrn1Δ) was least perturbed (log2 FC between −0.25 and 0.25) by DF deletion (13, 28), as these are the genes for which we expect the least disruption in transcriptional processes. The accession numbers for the corresponding datasets are provided under “Data availability.” Each strain was then separately normalized to WT samples by using the normalization procedure of DESeq2 restricted to the appropriate sets of genes (33). This process yields a size factor for each strain by which all read counts in that strain should be divided. Although not used in the main text, we also performed analysis with other normalization procedures to confirm our results were not due to procedural choices. TSS metagenes under each of the respective normalizations are displayed in Fig. S3. Normalization 1 is as described above, whereas normalization 2 is the same except for the fact that all ORFs are used as references rather than the subset that showed small changes in GRO or cDTA. Normalization 3 was a form of total counts normalization, as reads in a given sample were divided by the total number of NET-seq reads mapping to ORFs in that sample. Each sample was then multiplied by a factor of 2.5 million to approximate the total number of reads in samples normalized by the other procedures. Finally, in normalization 4, we divided the counts in each ORF profile by the total number of reads in that ORF. These profiles were then averaged across all ORFs within the sample and rescaled by a global factor (500) to roughly match the total number of reads in the samples when normalized by other methods.
Standardized fold changes of Pol II levels in genes and ncRNAs were computed using DESeq2 (33); these values account for the heteroskedastic mean variance trend for gene Pol II counts. Gene ontology (GO) enrichment analysis among genes were performed using GOrilla in single-ranked list mode (83, 84), where genes were ranked by their p values and sign of log2 fold changes to prevent mixing of over- and underexpressed genes. Transcriptional efficiencies were computed by applying a log2 transformation after taking the ratio of GRO to NET-seq reads (for whole genes) or BioGRO to NET-seq reads (for profiles). When fold changes of these quantities are presented, the difference between mutant and WT values were taken. Mathematically, the log2 fold changes in transcriptional efficiency for a mutant with respect to the WT can be expressed as Equation 1.
| (Eq. 1) |
For Fig. 3D, these ratios were computed for the xrn1Δ mutant. Genes were then grouped into deciles based on their log2 fold changes in the total number of NET-seq reads in the first 500 bp downstream of TSS. Deciles were ordered such that bin 1 contains genes with the largest reductions in occupancy.
Metagene construction
Full-transcript metagene densities for protein-coding genes of length at least 1100 nt were generated by fixing the regions located −200:500 with respect to TSS and −500:200 with respect to PAS and re-scaling the remainder of each transcript to a length of 500 nt. We then averaged reads across these re-scaled genes to create metagene profiles (Fig. 2). This allowed us to preserve the important spatial context near TSS and PAS without compressing the reads. We also generated metagenes for all genes and ncRNAs by re-scaling each transcript to a length of 1000 nt. We then aggregated reads across genes and applied LOWESS smoothing to create metagene profiles (Fig. S6 and Fig. S13). The metagene profiles of TSS-adjacent and PAS-adjacent sites instead display the average number of normalized reads in defined regions. Specifically, reads no more than 100 nt upstream and 500 nt downstream of annotated TSS were incorporated for 5' metagenes, while all reads within 150 nt upstream and 150 nt downstream of PAS were aggregated to give characteristic profiles for Pol II distributions near PAS (Fig. 5A and Fig. S9). We computed elongation efficiency metagenes in the WT and xrn1Δ strains by taking BioGRO reads in the same regions described above and smoothing the profiles for each gene. We then took the log2 of the ratio of the smoothed BioGRO and NET-seq profiles at each location of the defined regions for each gene and averaged these profiles across all genes. Any described smoothing was done via LOWESS smoothing (85). Hence, for sample s, gene g, and location i, we define the elongation efficiency as,
| (Eq. 2) |
Where and refer to the BioGRO and NET-seq profile values for gene g in sample s. These values are then averaged to produce the metagene plots of Figs. 3C and 5B.
Application of mathematical model
Our mathematical analysis of initiation and elongation rates is based on the recently obtained analytical solutions (32) to the Totally Asymmetric Simple Exclusion Process (TASEP), which we applied to model the behavior of Pol II in transcription. Denoting the NET-seq Pol II metagene for a given sample at position x as ρ(x), site-specific elongation rates λ(x) were approximated as λ(x) ∼ 1/(ρ(x) × (1 − ρ(x))) after appropriate re-scaling of ρ(x) to lie in the interval (0,1). The 1/(ρ(x) term of the approximation arises because the Pol II occupancy at a location should be inversely proportional to the speed at which Pol II are moving forward at that site. The 1/(1-ρ(x)) term accounts for the fact that the ability to progress is affected by whether or not other Pol II are already present at that location. Ideally, the re-scaling would be given by the number of cells in each sample, such that ρ(x) is the probability of finding a Pol II at position x. As these numbers were unknown, a range of re-scalings were tried and the results remained robust over the range of choices. It is worth noting that the actual initiation and elongation rates are not identifiable without the exact re-scaling; however, the ratio of initiation and elongation rates is, permitting our analysis. In our analysis, ρ(x) is obtained from our NET-seq metagene profiles and then λ, α, and β are inferred using the analytical solutions to the model in the low-density regime (32). The relevant equations are presented in the caption of Fig. 4.
External data sets
External data sets are available as follows: Remaining NET-seq, GSE25107 (25, 87); CRAC, GSE46742 (60, 88); RNA-seq FCs, GSE107841 (89, 90); GRO, GSE29519 and GSE57467 (1, 35, 91, 92); cDTA, E-MTAB-1525 (14, 93); BioGRO, GSE58859 (29, 30, 94); ChIP-exo, GSE44312 (1, 95); ORF annotations, GSE49026 (79, 96); mRNA half-lives accompany (2).
Data availability
NET-seq data for WT, xrn1Δ, dhh1Δ, lsm1Δ, ccr4Δ, and rpb4Δ cells were generated as part of this project and are available at ArrayExpression (accession number E-MTAB-8749) (86). Custom analysis scripts are available at: https://github.com/jrfischer/NET-seq_DF_KOs.
Note added in proof
Perturbation of Pol II occupancy across the genome in both the 5' and 3' ends of gene bodies shortly after depletion of Xrn1 has recently been posted in bioRxiv. This work determined Pol II occupancy by means different from reported here, Rpb1 CRAC, see Ref. 97.
Supplementary Material
Acknowledgments
We thank Sebastián Chávez, José E. Pérez-Ortín, and Antonio Jordán-Pla for comments on our results and use of BioGRO data. We also thank Dan D. Erdmann-Pham for discussion regarding application of the computational model used for simulations.
This article contains supporting information.
Author contributions—J. F. and J. d. I. data curation; J. F. software; J. F. formal analysis; J. F. and M. C. writing-original draft; Y. S. S. and N. Y. supervision; Y. S. S. and M. C. funding acquisition; Y. S. S. methodology; L. S. C. resources; M. C. conceptualization; M. C. investigation; M. C. project administration.
Funding and additional information—This work was supported in part by Israel Science Foundation Grant 1472/15, National Institutes of Health Training Grants T32-HG000047 and R01-GM094402, and a Packard Fellowship for Science and Engineering. N. Y. and Y. S. S. are Chan Zuckerberg Biohub Investigators. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare no conflicts of interest.
- Pol II
- RNA polymerase II
- TSS
- transcription start site
- PAS
- polyadenylation site
- PA
- polyadenylation
- DF
- decay factor
- NET-seq
- native elongating transcript sequencing
- ncRNA
- noncoding RNA
- RP
- ribosomal protein
- nt
- nucleotide
- TF
- transcription factor
- cDTA
- comparative dynamic transcriptome analysis.
References
- 1. Haimovich G., Medina D. A., Causse S. Z., Garber M., Millán-Zambrano G., Barkai O., Chávez S., Pérez-Ortín J. E., Darzacq X., and Choder M. (2013) Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell 153, 1000–1011 10.1016/j.cell.2013.05.012 [DOI] [PubMed] [Google Scholar]
- 2. Medina D. A., Jordán-Pla A., Millán-Zambrano G., Chávez S., Choder M., and Pérez-Ortín J. E. (2014) Cytoplasmic 5′-3′ exonuclease Xrn1p is also a genome-wide transcription factor in yeast. Front. Genet. 5, 1 10.3389/fgene.2014.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choder M. (2004) Rpb4 and Rpb7: subunits of RNA polymerase II and beyond. Trends Biochem. Sci 29, 674–681 10.1016/j.tibs.2004.10.007 [DOI] [PubMed] [Google Scholar]
- 4. Collart M. A. (2016) The CCR4-NOT complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 7, 438–454 10.1002/wrna.1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goler-Baron V., Selitrennik M., Barkai O., Haimovich G., Lotan R., and Choder M. (2008) Transcription in the nucleus and mRNA decay in the cytoplasm are coupled processes. Genes Dev. 22, 2022–2027 10.1101/gad.473608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta I., Villanyi Z., Kassem S., Hughes C., Panasenko L. L., Steinmetz L. M., and Collart M. A. (2016) Translational capacity of a cell is determined during transcription elongation via the CCR4-NOT complex. Cell Rep. 15, 1782–1794 10.1016/j.celrep.2016.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lotan R., Bar-On V. G., Harel-Sharvit L., Duek L., Melamed D., and Choder M. (2005) The RNA polymerase II subunit Rpb4p mediates decay of a specific class of mRNAs. Genes Dev. 19, 3004–3016 10.1101/gad.353205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lotan R., Goler-Baron V., Duek L., Haimovich G., and Choder M. (2007) The Rpb7p subunit of yeast RNA polymerase II plays roles in the two major cytoplasmic mRNA decay mechanisms. J. Cell Biol. 178, 1133–1143 10.1083/jcb.200701165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schulz D., Pirkl N., Lehmann E., and Cramer P. (2014) Rpb4 subunit functions mainly in mRNA synthesis by RNA polymerase II. J. Biol. Chem. 289, 17446–17452 10.1074/jbc.M114.568014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shalem O., Groisman B., Choder M., Dahan O., and Pilpel Y. (2011) Transcriptome kinetics is governed by a genome-wide coupling of mRNA production and degradation: a role for RNA Pol II. PLoS Genet. 7, e1002273 10.1371/journal.pgen.1002273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bregman A., Avraham-Kelbert M., Barkai O., Duek L., Guterman A., and Choder M. (2011) Promoter elements regulate cytoplasmic mRNA decay. Cell 147, 1473–1483 10.1016/j.cell.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 12. Trcek T., Larson D. R., Moldón A., Query C. C., and Singer R. H. (2011) Single-molecule mRNA decay measurements reveal promoter-regulated mRNA stability in yeast. Cell 147, 1484–1497 10.1016/j.cell.2011.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun M., Schwalb B., Schulz D., Pirkl N., Etzold S., Lariviere L., Maier K. C., Seizl M., Tresch A., and Cramer P. (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 22, 1350–1359 10.1101/gr.130161.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun M., Schwalb B., Pirkl N., Maier K. C., Schenk A., Failmezger H., Tresch A., and Cramer P. (2013) Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52, 52–62 10.1016/j.molcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 15. Basehoar A. D., Zanton S. J., and Pugh B. F. (2004) Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709 10.1016/S0092-8674(04)00205-3 [DOI] [PubMed] [Google Scholar]
- 16. Huisinga K. L., and Pugh B. F. (2004) A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13, 573–585 10.1016/S1097-2765(04)00087-5 [DOI] [PubMed] [Google Scholar]
- 17. Baptista T., Grünberg S., Minoungou N., Koster M. J., Timmers H. M., Hahn S., Devys D., and Tora L. (2017) SAGA is a general cofactor for RNA polymerase II transcription. Mol. Cell 68, 130–143 10.1016/j.molcel.2017.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonnet J., Wang C. Y., Baptista T., Vincent S. D., Hsiao W. C., Stierle M., Kao C. F., Tora L., and Devys D. (2014) The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 28, 1999–2012 10.1101/gad.250225.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Warfield L., Ramachandran S., Baptista T., Devys D., Tora L., and Hahn S. (2017) Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol. Cell 68, 118–129 10.1016/j.molcel.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen F. X., Smith E. R., and Shilatifard A. (2018) Born to run: control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 19, 464–478 10.1038/s41580-018-0010-5 [DOI] [PubMed] [Google Scholar]
- 21. Wissink E., Vihervaara A., Tippens N., and Lis J. (2019) Nascent RNA analyses: tracking transcription and its regulation. Nat. Rev. Genet. 20, 705–723 10.1038/s41576-019-0159-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheridan R. M., Fong N., D'Alessandro A., and Bentley D. L. (2019) Widespread backtracking by RNA Pol II is a major effector of gene activation, 5' pause release, termination, and transcription elongation rate. Mol. Cell 73, 107–118 10.1016/j.molcel.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartman C. R., Hamagami N., Keller C. A., Giardine B., Hardison R. C., Blobel G. A., and Raj A. (2019) Transcriptional burst initiation and polymerase pause release are key control points of transcriptional regulation. Mol. Cell 73, 519–532 10.1016/j.molcel.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adelman K., and Lis J. T. (2012) Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 10.1038/nrg3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Churchman L. S., and Weissman J. S. (2011) Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mayer A., Landry H. M., and Churchman L. S. (2017) Pause & Go: from the discovery of RNA polymerase pausing to its functional implications. Curr. Opin. Cell Biol. 46, 72–80 10.1016/j.ceb.2017.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Churchman L. S., and Weissman J. S. (2012) Native elongating transcript sequencing (NET-seq). Curr. Protoc. Mol. Biol. 98, 144.1–14.4.17 10.1002/0471142727.mb0414s98 [DOI] [PubMed] [Google Scholar]
- 28. García-Martínez J., Aranda A., and Pérez-Ortín J. E. (2004) Genomic run-on evaluates transcription rates for all yeast genes and identifies gene regulatory mechanisms. Mol. Cell 15, 303–313 10.1016/j.molcel.2004.06.004 [DOI] [PubMed] [Google Scholar]
- 29. Jordán-Pla A., Gupta I., de Miguel-Jiménez L., Steinmetz L. M., Chávez S., Pelechano V., and Pérez-Ortín J. E. (2015) Chromatin-dependent regulation of RNA polymerases II and III activity throughout the transcription cycle. Nucleic Acids Res. 43, 787–802 10.1093/nar/gku1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jordán-Pla A., Miguel A., Serna E., Pelechano V., and Pérez-Ortín J. E. (2016) A high-resolution method for the analysis of nascent transcription in yeast. Methods Mol. Biol. Biotin-Genomic Run-On (Bio-GRO): 1361, 125–139 10.1007/978-1-4939-3079-1_8 [DOI] [PubMed] [Google Scholar]
- 31. Gómez-Herreros F., de Miguel-Jiménez L., Millán-Zambrano G., Peñate X., Delgado-Ramos L., Muñoz-Centeno M. C., and Chávez S. (2012) One step back before moving forward: regulation of transcription elongation by arrest and backtracking. FEBS Lett. 586, 2820–2825 10.1016/j.febslet.2012.07.030 [DOI] [PubMed] [Google Scholar]
- 32. Erdmann-Pham D. D., Dao Duc K., and Song Y. S. (2020) The key parameters that govern translation efficiency. Cell Syst. 10, 183–192 10.1016/j.cels.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duek L., Barkai O., Elran R., Adawi I., and Choder M. (2018) Dissociation of Rpb4 from RNA polymerase II is important for yeast functionality. PLoS ONE 13, e0206161 10.1371/journal.pone.0206161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. García-Martínez J., Delgado-Ramos L., Ayala G., Pelechano V., Medina D. A., Carrasco F., González R., Andrés-León E., Steinmetz L., Warringer J., Chávez S., and Pérez-Ortín J. E. (2016) The cellular growth rate controls overall mRNA turnover, and modulates either transcription or degradation rates of particular gene regulons. Nucleic Acids Res. 44, 3643–3658 10.1093/nar/gkv1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verma-Gaur J., Rao S. N., Taya T., and Sadhale P. (2008) Genome-wide recruitment analysis of Rpb4, a subunit of polymerase II in Saccharomyces cerevisiae, reveals its involvement in transcription elongation. Eukaryot. Cell 7, 1009–1018 10.1128/EC.00057-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blasco-Moreno B., de Campos-Mata L., Böttcher R., García-Martínez J., Jungfleisch J., Nedialkova D. D., Chattopadhyay S., Gas M. E., Oliva B., Pérez-Ortín J., Leidel S. A., Choder M., and Díez J. (2019) The exonuclease Xrn1 activated transcription and translation of mRNAs encoding membrane proteins. Nat. Commun. 10, 1298 10.1038/s41467-019-09199-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bentley D. L. (2014) Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 10.1038/nrg3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hyman L. E., and Moore C. L. (1993) Termination and pausing of RNA polymerase II downstream of yeast polyadenylation sites. Mol. Cell Biol. 13, 5159–5167 10.1128/mcb.13.9.5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kazerouninia A., Ngo B., and Martinson H. G. (2010) Poly (A) signal-dependent degradation of unprocessed nascent transcripts accompanies poly(A) signal-dependent transcriptional pausing in vitro. RNA 16, 197–210 10.1261/rna.1622010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuehner J. N., Pearson E. L., and Moore C. (2011) Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol. 12, 283–294 10.1038/nrm3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larson D. R., Zenklusen D., Wu B., Chao J. A., and Singer R. H. (2011) Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332, 475–478 10.1126/science.1202142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mischo H. E., and Proudfoot N. J. (2013) Disengaging polymerase: terminating RNA polymerase II transcription in budding yeast. Biochim. Biophys. Acta 1829, 174–185 10.1016/j.bbagrm.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Begley V., Corzo D., Jordán-Pla A., Cuevas-Bermúdez A., de Miguel-Jiménez L., Pérez-Aguado D., Machuca-Ostos M., Navarro F., Chávez M. J., Pérez-Ortín J. E., and Chávez S. (2019) The mRNA degradation factor Xrn1 regulates transcription elongation in parallel to Ccr4. Nucleic Acids Res. 47, 9524–9541 10.1093/nar/gkz660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Darzacq X., Shav-Tal Y., De Turris V., Brody Y., Shenoy S. M., Phair R. D., and Singer R. H. (2007) In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 14, 796–806 10.1038/nsmb1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ehrensberger A. H., Kelly G. P., and Svejstrup J. Q. (2013) Mechanistic interpretation of promoter proximal peaks and RNAPII density maps. Cell 154, 713–715 10.1016/j.cell.2013.07.032 [DOI] [PubMed] [Google Scholar]
- 47. Grosso A. R., de Almeida S. F., Braga J., and Carmo-Fonseca M. (2012) Dynamic transitions in RNA polymerase II density profiles during transcription termination. Genome Res. 22, 1447–1456 10.1101/gr.138057.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jonkers I., Kwak H., and Lis J. T. (2014) Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 3, e02407 10.7554/eLife.02407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Le Martelot G., Canella D., Symul L., Migliavacca E., Gilardi F., Liechti R., Martin O., Harshman K., Delorenzi M., Desvergne B., Herr W., Deplancke B., Schibler U., Rougemont J., Guex N., CycliX Consortium, et al. (2012) Genome-wide RNA polymerase II profiles and RNA accumulation reveal kinetics of transcription and associated epigenetic changes during diurnal cycles. PLos Biol. 10, e1001442 10.1371/journal.pbio.1001442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian B., and Manley J. L. (2017) Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 18, 18–30 10.1038/nrm.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Richard P., and Manley J. L. (2009) Transcription termination by nuclear RNA polymerases. Genes Dev. 23, 1247–1269 10.1101/gad.1792809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hobson D. J., Wei W., Steinmetz L. M., and Svejstrup J. Q. (2012) RNA polymerase II collision interrupts convergent transcription. Mol. Cell 48, 365–374 10.1016/j.molcel.2012.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bumgarner S. L., Dowell R. D., Grisafi P., Gifford D. K., and Fink G. R. (2009) Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc. Natl. Acad. Sci. U.S.A. 106, 18321–18326 10.1073/pnas.0909641106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schulz D., Schwalb B., Kiesel A., Baejen C., Torkler P., Gagneur J., Soeding J., and Cramer P. (2013) Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155, 1075–1087 10.1016/j.cell.2013.10.024 [DOI] [PubMed] [Google Scholar]
- 55. Tisseur M., Kwapisz M., and Morillon A. (2011) Pervasive transcription-lessons from yeast. Biochimie (Paris) 93, 1889–1896 10.1016/j.biochi.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 56. van Dijk E. L., Chen C. L., d'Aubenton-Carafa Y., Gourvennec S., Kwapisz M., Roche V., Bertrand C., Silvain M., Legoix-Né P., Loeillet S., Nicolas A., Thermes C., and Morillon A. (2011) XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475, 114–117 10.1038/nature10118 [DOI] [PubMed] [Google Scholar]
- 57. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., and Steinmetz L. M. (2009) Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037 10.1038/nature07728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murray S. C., Haenni S., Howe F. S., Fischl H., Chocian K., Nair A., and Mellor J. (2015) Sense and antisense transcription are associated with distinct chromatin architectures across genes. Nucleic Acids Res. 43, 7823–7837 10.1093/nar/gkv666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wery M., Gautier C., Descrimes M., Yoda M., Vennin-Rendos H., Migeot V., Gautheret D., Hermand D., and Morillon A. (2018) Native elongating transcript sequencing reveals global anti-correlation between sense and antisense nascent transcription in fission yeast. RNA 24, 196–208 10.1261/rna.063446.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tuck A. C., and Tollervey D. (2013) A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154, 996–1009 10.1016/j.cell.2013.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pérez-Ortín J. E., Tordera V., and Chávez S. (2019) Homeostasis in the Central Dogma of Molecular Biology: the importance of mRNA instability. RNA Biol. 16, 1659–1666 10.1080/15476286.2019.1655352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Braun K. A., and Young E. T. (2014) Coupling mRNA synthesis and decay. Mol. Cell Biol. 34, 4078–4087 10.1128/MCB.00535-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Costanzo M., VanderSluis B., Koch E. N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S. D., Pelechano V., Styles E. B., Billmann M., van Leeuwen J., van Dyk N., et al. (2016) A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 10.1126/science.aaf1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Feldman J. L., and Peterson C. L. (2019) Yeast sirtuin family members maintain transcription homeostasis to ensure geNome Stability. Cell Rep. 27, 2978–2989 10.1016/j.celrep.2019.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rodríguez-Molina J. B., Tseng S. C., Simonett S. P., Taunton J., and Ansari A. Z. (2016) Engineered covalent inactivation of TFIIH-kinase reveals an elongation checkpoint and results in widespread mRNA stabilization. Mol. Cell 63, 433–444 10.1016/j.molcel.2016.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dutta A., Babbarwal V., Fu J., Brunke-Reese D., Libert D. M., Willis J., and Reese J. C. (2015) Ccr4-Not and TFIIS function cooperatively to rescue arrested RNA polymerase II. Mol. Cell Biol. 35, 1915–1925 10.1128/MCB.00044-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kruk J. A., Dutta A., Fu J., Gilmour D. S., and Reese J. C. (2011) The multifunctional Ccr4-Not complex directly promotes transcription elongation. Genes Dev. 25, 581–593 10.1101/gad.2020911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Collins S. R., Kemmeren P., Zhao X. C., Greenblatt J. F., Spencer F., Holstege F. C., Weissman J. S., and Krogan N. J. (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 6, 439–450 10.1074/mcp.M600381-MCP200 [DOI] [PubMed] [Google Scholar]
- 69. Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E. D., Sevier C. S., Ding H., Koh J. L. Y., Toufighi K., Mostafavi S., Prinz J., St. Onge R. P., VanderSluis B., Makhnevych T., Vizeacoumar F. J., et al. (2010) The genetic landscape of a cell. Science 327, 425–431 10.1126/science.1180823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kuzmin E., VanderSluis B., Wang W., Tan G., Deshpande R., Chen Y., Usaj M., Balint A., Mattiazzi Usaj M., van Leeuwen J., Koch E. N., Pons C., Dagilis A. J., Pryszlak M., Wang J. Z. Y., et al. (2018) Systematic analysis of complex genetic interactions. Science 360, eaao1729 10.1126/science.aao1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Miller J. E., Zhang L., Jiang H., Li Y., Pugh B. F., and Reese J. C. (2018) Genome-wide mapping of decay factor-mRNA interactions in yeast identifies nutrient-responsive transcripts as targets of the deadenylase Ccr4. G3 (Bethesda) 8, 315–330 10.1534/g3.117.300415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Srivas R., Shen J. P., Yang C. C., Sun S. M., Li J., Gross A. M., Jensen J., Licon K., Bojorquez-Gomez A., Klepper K., Huang J., Pekin D., Xu J. L., Huwate Y., Sivaganesh V., et al. (2016) A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol. Cell 63, 514–525 10.1016/j.molcel.2016.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilmes G. M., Bergkessel M., Bandyopadhyay S., Shales M., Braberg H., Cagney G., Collins S. R., Whitworth G. B., Kress T. L., Weissman J. S., Ideker T., Guthrie C., and Krogan N. J. (2008) A genetic interaction map of RNA processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol. Cell 32, 735–746 10.1016/j.molcel.2008.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Runner V. M., Podolny V., and Buratowski S. (2008) The Rpb4 subunit of RNA polymerase II contributes to cotranscriptional recruitment of 3' processing factors. Mol. Cell Biol. 28, 1883–1891 10.1128/MCB.01714-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Porrua O., Boudvillain M., and Libri D. (2016) Transcription termination: variations on common themes. Trends Genet. 32, 508–522 10.1016/j.tig.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 76. Tirosh I., Berman J., and Barkai N. (2007) The pattern and evolution of yeast promoter bendability. Trends Genet. 23, 318–321 10.1016/j.tig.2007.03.015 [DOI] [PubMed] [Google Scholar]
- 77. Kubik S., Bruzzone M. J., Jacquet P., Falcone J. L., Rougemont J., and Shore D. (2015) Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol. Cell 60, 422–434 10.1016/j.molcel.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 78. de Jonge W. J., O'Duibhir E., Lijnzaad P., van Leenen D., Groot Koerkamp M. J., Kemmeren P., and Holstege F. C. (2017) Molecular mechanisms that distinguish TFIID housekeeping from regulatable SAGA promoters. EMBO J. 36, 274–290 10.15252/embj.201695621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park D., Morris A. R., Battenhouse A., and Iyer V. R. (2014) Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 42, 3736–3749 10.1093/nar/gkt1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Martin M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 81. Schmieder R., and Edwards R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Trapnell C., Pachter L., and Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Eden E., Lipson D., Yogev S., and Yakhini Z. (2007) Discovering motifs in ranked lists of DNA sequences. PLoS Comp. Biol. 3, e39 10.1371/journal.pcbi.0030039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Eden E., Navon R., Steinfeld I., Lipson D., and Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 10, 48 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cleveland W. S. (1981) LOWESS: A program for smoothing scatterplots by robust locally weighted regression. Am. Stat. 35, 54 10.2307/2683591 [DOI] [Google Scholar]
- 86. Fischer J., and Choder M. (2020) NET-seq of S. cerevisiae cells after knocking-out genes encoding various mRNA decay factors, ArrayExpress, E-MTAB-8749
- 87. Churchman L.S., and Weissman J.S. (2011) Native elongationg transcript sequencing (NET-seq) of wild type Saccharomyces cerevisiae and of DST1, RCO1, SET1, SET2, EAF3 deletion strains Gene Expression Omnibus, GSE25107
- 88. Tuck A.C., and Tollervey D. (2013) A transcriptome-wide atlas of RNP compositions reveals diverse classes of mRNAs and lncRNAs Gene Expression Omnibus, GSE46742 [DOI] [PMC free article] [PubMed]
- 89. He F., Celik A., Wu C., and Jacobson A. (2018) General decapping activators target different subsets of inefficiently translated mRNAs. eLife 7, e34409 10.7554/eLife.34409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. He F., Celik A. (2017) Genome-wide identification of decapping substrates in the yeast Saccharomyces cerevisiae Gene Expression Omnibus, GSE107841
- 91. Pérez-Ortín J.E., Medina D.A., Choder M. (2013) Genomic Run-On (GRO): determination of the nascent transcriptional rates and mRNA amount in Xrn1 mutants Gene Expression Omnibus, GSE29619
- 92. Pérez-Ortín J.E., Medina D.A., Choder M., Chávez S. (2015) Genomic Run On (GRO): determination of the nascent transcriptional rates and mRNA levels in several yeast mutants Gene Expression Omnibus, GSE57467
- 93. Sun M. (2014) Global analysis of mRNA degradation reveals Xrn1-dependent buffering of transcript levels ArrayExpress, E-MTAB-1525 [DOI] [PubMed]
- 94. Jordán-Pla A., Pérez-Ortín J.E., de-Miguel L., Chávez S., Pelechano V. (2014) Chromatin-dependent regulation of the RNA polymerases II and II activity throughout the transcription cycle Gene Expression Omnibus, GSE58859 [DOI] [PMC free article] [PubMed]
- 95. Haimovich G., Garber M., Choder M. (2013) Gene expression is a circular process Gene Expression Omnibus, GSE44312
- 96. Iyer V.R. (2014) SMORE-seq maps both ends of transcripts and identifies widespread promoter-associated non-coding RNA governed by TATA elements Gene Expression Omnibus, GSE49026 [DOI] [PMC free article] [PubMed]
- 97. Begley V., Jordán-Pla A., Peñate X., Garrido-Godino A. I., Challal D., Cuevas-Bermúdez A., Mitjavila A., Barucco M., Gutiérrez G., Singh A., Alepuz P., Navarro F., Libri D., Pérez-Ortín J. E., and Chávez S. (2020) Xrn1 influences RNA pol II-dependent transcription elongation rates across the yeast genome and this control is particularly relevant for late elongation of regulatory genes. bioRxiv 10.1101/2020.06.02.129171 [DOI]
- 98. Supek F., Bošnjak M., Škunca N., and Šmuc T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mi H., Muruganujan A., Ebert D., Huang X., and Thomas P. D. (2019) PANTHER version 14: More Genomes, a new PANTHER. GO-SLIM and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426 10.1093/nar/gky1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Perl K. (2015) mHG: Minimum-Hypergeometric Test, RRID:SCR_018776
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Fischer J., and Choder M. (2020) NET-seq of S. cerevisiae cells after knocking-out genes encoding various mRNA decay factors, ArrayExpress, E-MTAB-8749
- Churchman L.S., and Weissman J.S. (2011) Native elongationg transcript sequencing (NET-seq) of wild type Saccharomyces cerevisiae and of DST1, RCO1, SET1, SET2, EAF3 deletion strains Gene Expression Omnibus, GSE25107
- Tuck A.C., and Tollervey D. (2013) A transcriptome-wide atlas of RNP compositions reveals diverse classes of mRNAs and lncRNAs Gene Expression Omnibus, GSE46742 [DOI] [PMC free article] [PubMed]
- He F., Celik A. (2017) Genome-wide identification of decapping substrates in the yeast Saccharomyces cerevisiae Gene Expression Omnibus, GSE107841
- Pérez-Ortín J.E., Medina D.A., Choder M. (2013) Genomic Run-On (GRO): determination of the nascent transcriptional rates and mRNA amount in Xrn1 mutants Gene Expression Omnibus, GSE29619
- Pérez-Ortín J.E., Medina D.A., Choder M., Chávez S. (2015) Genomic Run On (GRO): determination of the nascent transcriptional rates and mRNA levels in several yeast mutants Gene Expression Omnibus, GSE57467
- Sun M. (2014) Global analysis of mRNA degradation reveals Xrn1-dependent buffering of transcript levels ArrayExpress, E-MTAB-1525 [DOI] [PubMed]
- Jordán-Pla A., Pérez-Ortín J.E., de-Miguel L., Chávez S., Pelechano V. (2014) Chromatin-dependent regulation of the RNA polymerases II and II activity throughout the transcription cycle Gene Expression Omnibus, GSE58859 [DOI] [PMC free article] [PubMed]
- Haimovich G., Garber M., Choder M. (2013) Gene expression is a circular process Gene Expression Omnibus, GSE44312
- Iyer V.R. (2014) SMORE-seq maps both ends of transcripts and identifies widespread promoter-associated non-coding RNA governed by TATA elements Gene Expression Omnibus, GSE49026 [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
NET-seq data for WT, xrn1Δ, dhh1Δ, lsm1Δ, ccr4Δ, and rpb4Δ cells were generated as part of this project and are available at ArrayExpression (accession number E-MTAB-8749) (86). Custom analysis scripts are available at: https://github.com/jrfischer/NET-seq_DF_KOs.