

Abstract
We report a catalytic, light-driven protocol for the intramolecular hydroetherification of unactivated alkenols to furnish cyclic ether products. These reactions occur under visible light irradiation in the presence of an Ir(III)-based photoredox catalyst, a Brønsted base catalyst, and a hydrogen atom transfer co-catalyst. Reactive alkoxy radicals are proposed as key intermediates, generated via the direct homolytic activation of alcohol O–H bonds through a proton-coupled electron transfer mechanism. This method exhibits a broad substrate scope and high functional group tolerance, and it accommodates a diverse range of alkene substitution patterns. Results demonstrating the extension of this catalytic system to carboetherification reactions are also presented.
Keywords: alcohols, ethers, hydroetherification, photocatalysis, radicals
Graphical Abstract
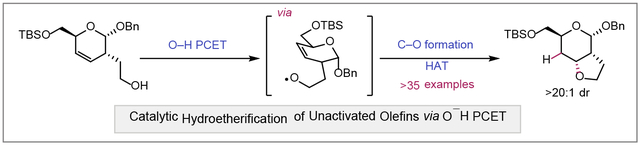
Alkoxy radicals generated directly from the activation of alcohol O–H bonds under catalytic, light-driven conditions are leveraged for the intramolecular hydroetherification of a diverse range of unactivated olefins. This strategy allows for productive C–O bond formation from highly reactive alkoxy radical intermediates and accommodates a broad substrate scope with high functional group tolerance.
The addition of alcohols to alkenes is a powerful approach to C–O bond formation and the construction of oxygen-containing heterocycles, which are common structural motifs in natural products and medicinal agents.[1] Numerous olefin hydroetherification methods have been developed using Brønsted acid- or transition metal-based catalysts (Scheme 1a), which typically operate through alkene activation mechanisms.[2–5] As a result, these reactions are often sensitive to C=C substitution patterns and generally afford Markovnikov-type addition products. More recently, an alternative mode of alkene activation has been developed, wherein single-electron oxidation of an olefin furnishes an electrophilic alkene radical cation that can be intercepted by an alcohol nucleophile.[6] While this method affords complementary anti-Markovnikov regioselectivity, its use is largely limited to oxidizable styrenyl and trisubstituted olefin substrates. Considering these strategies more broadly, the development of a general catalytic protocol for hydroetherification that accommodates electronically unbiased alkenes with diverse substitution remains an outstanding challenge.
Scheme 1.
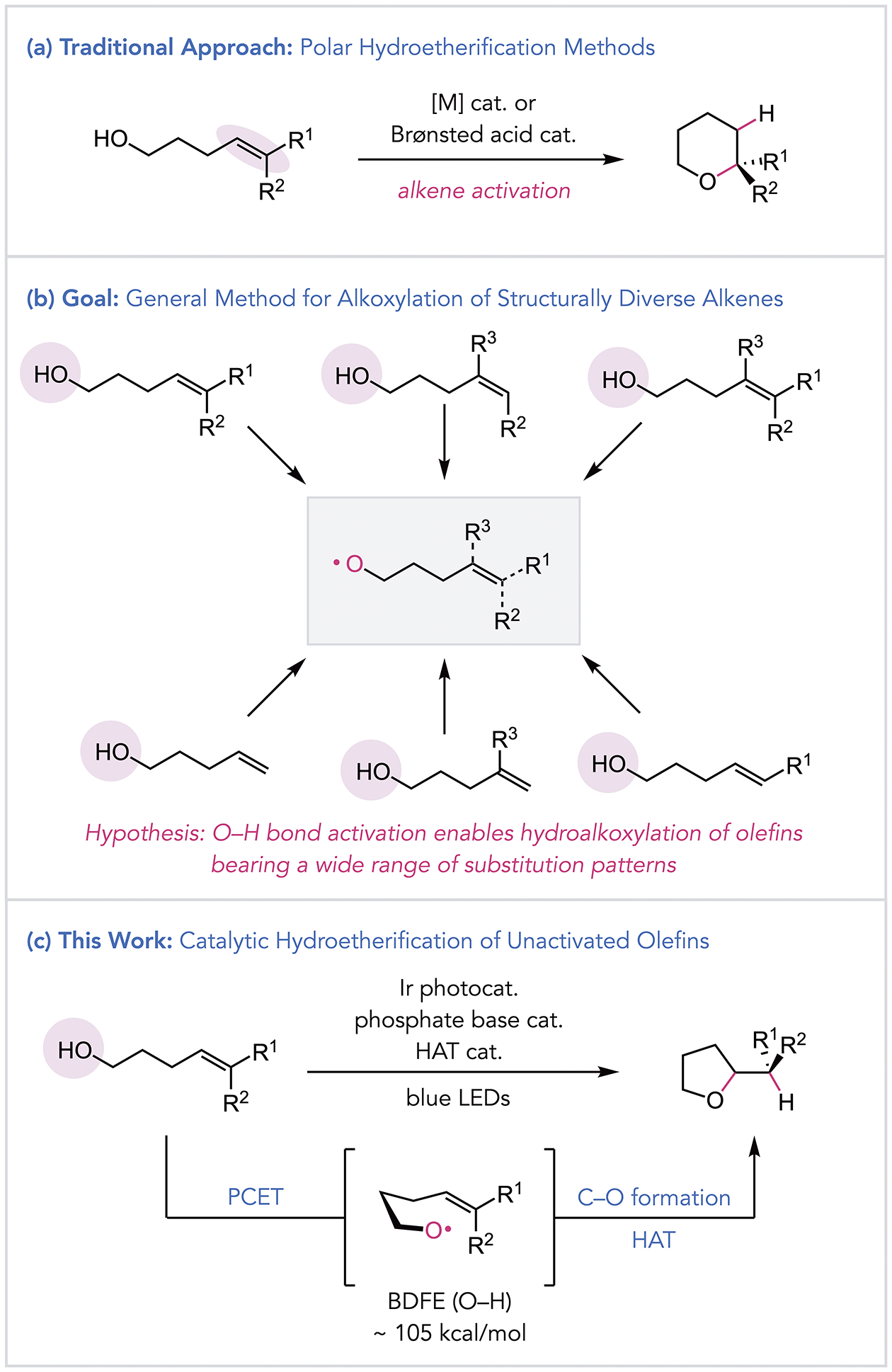
(a) Traditional approaches to hydroetherification typically involve alkene activation mechanisms. (b) Development of a general strategy for hydroalkoxylation via O–H bond activation. (c) PCET-mediated hydroetherification of unactivated alkenes.
In contrast to these alkene activation strategies, we recently became interested in an orthogonal approach to catalytic olefin hydroetherification that employs reactive alkoxy radical intermediates formed through O–H bond activation (Scheme 1b). Alkoxy radicals have been shown to undergo addition to pendent alkenes to furnish cyclic ethers, but their generation typically requires either prefunctionalization of the hydroxyl group or the use of strong stoichiometric oxidants.[7–9] While effective, these conditions can lead to poor atom economy and incompatibility with common functional groups. As such, development of a mild, catalytic protocol for olefin hydroetherification utilizing alkoxy radicals generated directly from alcohol starting materials has the potential to advance the value of radical-based etherification in organic synthesis.
To this end, we envisioned that excited-state proton-coupled electron transfer (PCET) would provide a solution to the existing limitations of radical hydroetherification (Scheme 1c). Our group has developed methods for the homolytic activation of alcohol O–H bonds to access alkoxy radical intermediates, focusing exclusively on C–C bond β-scission reactions.[10,11] As the rates of β-scission and 1,5-hydrogen atom transfer (1,5-HAT) are often comparable to the rate of cyclization,[12,8b,8i] we questioned whether alkoxy radicals could be leveraged instead for productive C–O bond formation, generating cyclic ethers directly from readily accessible alkenols while outcompeting other pathways.[13] Within this context, we endeavored to find catalysts and reaction conditions that could not only effectively bias the reactivity of alkoxy radicals to favor olefin addition but to do so with a broad scope and functional group tolerance.
We began our optimization studies with 1,2-disubstituted alkenol substrate 1 (Table 1) and found that 2 mol% of [Ir(dF(CF3)ppy)2(5,5ʹ-d(CF3)bpy)]PF6 photocatalyst, 20 mol% of diphenyl phosphate, and 20 mol% of 2,4,6-triisopropylthiophenol (TRIP-SH) in trifluorotoluene under blue light irradiation (~450 nm) afforded the desired ether product 2 in 31% yield (entry 1). Notably, this highly oxidizing photocatalyst (EIII*/II = +1.30 V vs. Fc+/Fc in MeCN)[14] is required—in combination with phosphate bases (pKa = ~13 in MeCN)[15]—to achieve effective bond dissociation free energies (BDFEs) approaching that of the alcohol O–H bond (BDFE = 105 kcal/mol).[11b] Less oxidizing Ir(III)-based photocatalysts were ineffective in the reaction, providing 0% yield of 2 (Table S1). The counter-cation of the phosphate base was found to have a marked effect on the reaction efficiency, as the tetrabutylphosphonium ion led to a 40% boost in yield compared to the corresponding ammonium cation (entry 2). This effect is possibly due to the enhanced solubility of tetrabutylphosphonium diphenyl phosphate in trifluorotoluene or to the more dissociated nature of the ion pair.[16] After examining a series of electron-rich and electron-deficient thiophenols (entries 3–8), we found that 2-fluorothiophenol outperformed the other thiophenols surveyed, providing 2 in 75% yield (entry 7)—a notable improvement over the other fluorothiophenol regioisomers (entries 5–6). The corresponding disulfide was equally effective in the reaction (entry 9), and we therefore elected to use 1,2-bis(2-fluorophenyl)disulfide as the HAT co-catalyst due to its ease of handling.[6b] Lowering the substrate concentration increased the yield to 80% (entry 10), and adjusting the HAT co-catalyst loading to 30 mol% gave tetrahydrofuran 2 in 85% yield, providing our optimal reaction conditions (entry 11). Control experiments run in the absence of light and photocatalyst furnished no product, and significantly reduced yields were observed in the absence of either Brønsted base or HAT co-catalyst (entries 12–15).
Table 1.
Reaction Optimization[a]
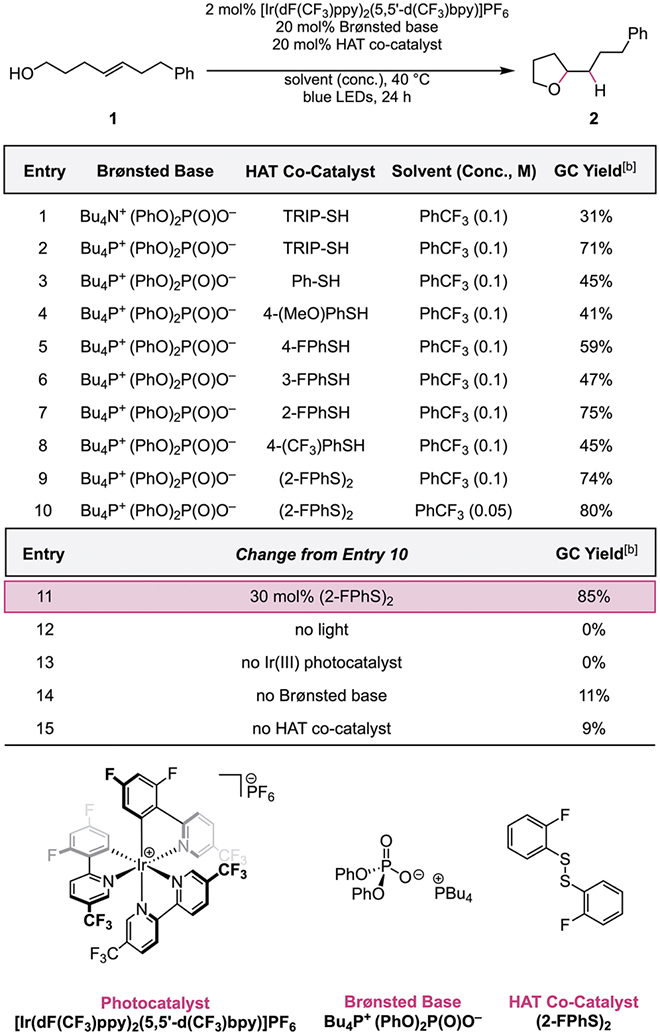
|
Reactions were run on a 0.05 mmol scale.
GC yields determined relative to biphenyl as an internal standard.
Having established the optimized reaction conditions, we next examined the scope of this reaction with respect to various alkene substitution patterns (Table 2). With the success of model substrate 1, we found that a range of other 1,2-disubstituted olefins gave excellent yields of the desired cyclic ether products (3–9). Hydroetherification of disubstituted olefins with secondary alcohols also proved to be viable, albeit with moderate reactivity (10). Notably, this protocol provides direct access to a variety of bicyclic structures from alcohol precursors, forming fused rings (11, 12) and bridged ethers (13) with high diastereoselectivities. Electronically diverse styrenyl alkenes (14–16), as well as pyridine and thiophene derivatives (17, 18), furnished tetrahydrofuran products in good yields.
Table 2.
Substrate Scope[a]
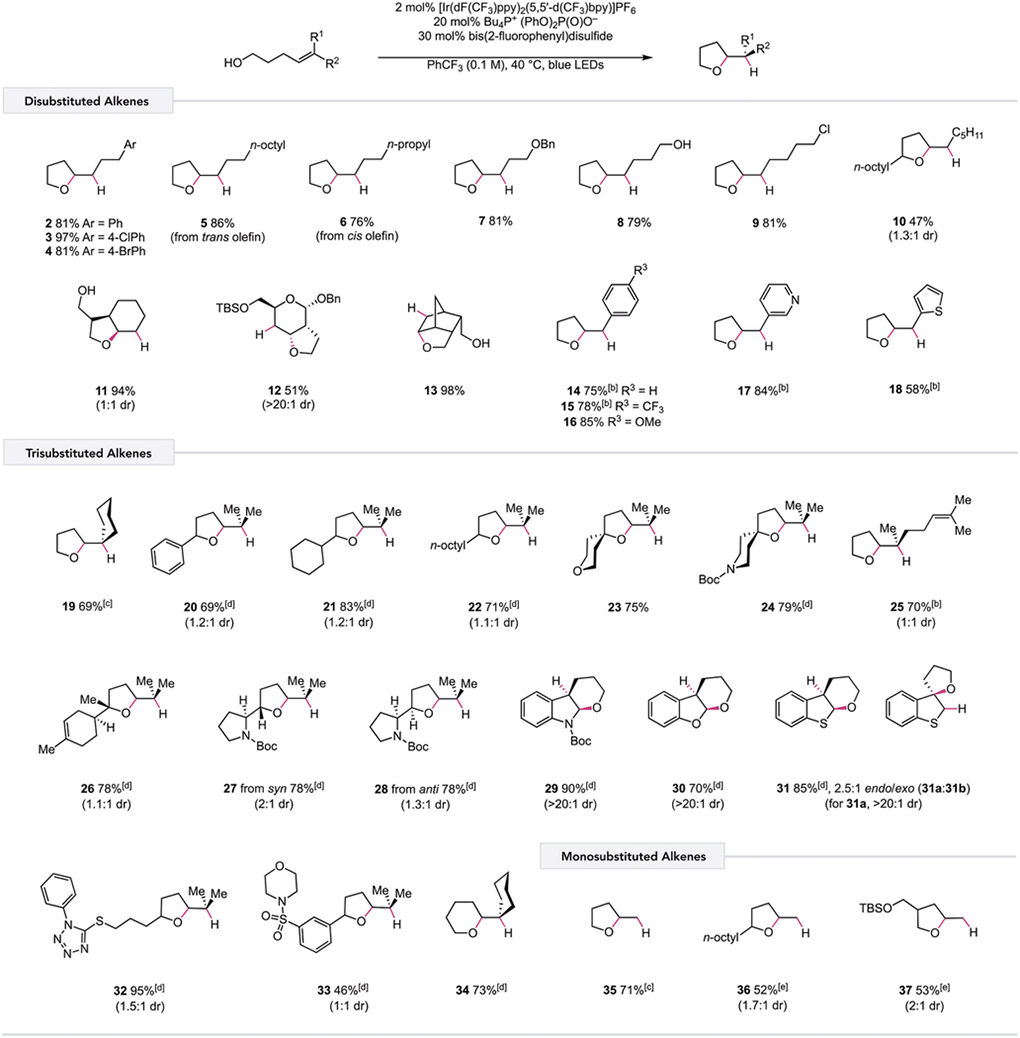
|
Reactions were performed on a 0.5 mmol scale. Reported yields are for isolated and purified material unless otherwise noted and represent the average of two experiments.
Bu4N+ CF3C(O)O– used as the base.
GC yield is reported relative to an internal standard due to volatility.
2-methyl-2-oxazoline used as the base, and 4-trifluoromethylthiophenol used as the HAT co-catalyst.
1,2-Bis(4-methoxyphenyl)disulfide used as the HAT co-catalyst. See Supporting Information for details.
With respect to trisubstituted alkenes, anti-Markovnikov hydroetherification proceeded with primary, secondary, and tertiary alcohols (19–24), providing ether products that are generally not accessible using traditional hydroalkoxylation strategies. Interestingly, products resulting from competing C–C β-scission were not observed during the formation of 23 and 24, enabling facile and efficient synthesis of spirocyclic scaffolds. Derivatives of terpenoids were also obtained in good yields using this protocol (25, 26), leaving the more distal olefins unaffected and demonstrating that 5-exo-trig cyclization is preferred. Both syn and anti diastereomers of an N-Boc-l-prolinol derivative were transformed to their respective tetrahydrofuran products in good yields (27, 28). Additionally, N-Boc-indole and benzofuran derivatives proceeded through 6-endo-trig cyclizations to favor generation of an intermediate tertiary benzylic radical, affording tricyclic ether products in excellent yields and diastereoselectivities (29, 30). In the case of benzothiophene, both 6-endo and 5-exo products (2.5:1) were formed, allowing access to both fused- and spirocyclic structures (31a, 31b). Moreover, this hydroetherification protocol can tolerate the presence of polar functionality, operating in systems containing N-phenyltetrazole thioethers and sulfonamides (32, 33). Hydroetherification proceeding via 6-exo-trig ring closures is also possible, furnishing tetrahydropyran product 34 in 73% yield, outcompeting 1,5-HAT from the allylic C–H bonds. Remarkably, hydroalkoxylation of unactivated monosubstituted alkenes can also be achieved in moderate to good yields, even with generation of a primary C-centered radical after cyclization (35–37). Due to the high oxidation potential of monosubstituted olefins, these substrates cannot be accommodated using current alkene oxidation methods.
Lastly, we evaluated the synthetic versatility of our hydroalkoxylation protocol in the context of olefin carboetherification reactions,[17] wherein electron-deficient olefins were used to intercept the intermediate C-centered radical following C–O bond formation (Table 3). Tetrahydrofuran derivatives were successfully alkylated with α-phenyl methacrylate (38), dimethyl fumarate (39), and dehydroalanine derivative (40), demonstrating that intermolecular C–C bond formation can be achieved following alkoxy radical cyclization. Moreover, acceptors containing heterocyclic scaffolds, such as 2-vinylpyridine (41) and 2-vinylpyrazine (42), provided modest yields of alkylated product. Even in cases where a primary alkyl radical is generated upon cyclization, addition to α-phenyl methacrylate outcompetes other possible reaction pathways to give the desired difunctionalization product (43). Carboetherification of a secondary alkenol with 1,1-bis(phenylsulfonyl)ethylene also proceeded in good yield (44). These results highlight the unique potential for alkoxy radical-mediated C–O bond formation to be adapted for further product derivatization, forming functionalized ethers in one step.
Table 3.
Carboetherification of Unactivated Alkenes[a]
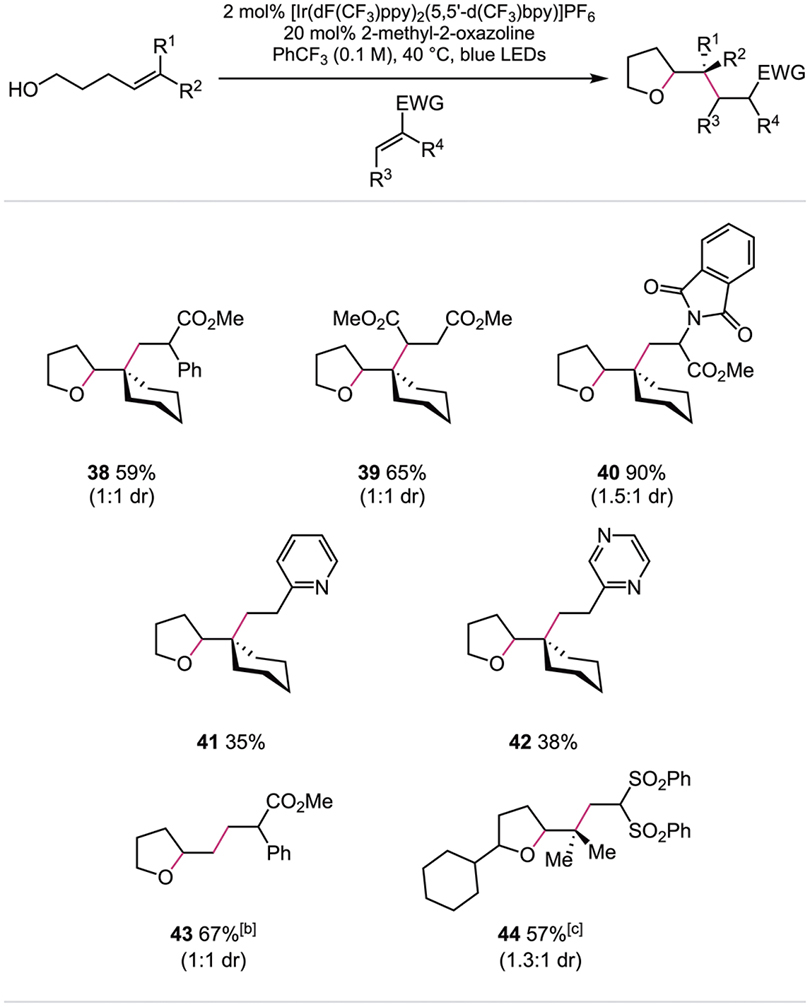
|
Reactions were performed on a 0.5 mmol scale. Reported yields are for isolated and purified material and represent the average of two experiments.
Bu4P+ (PhO)2P(O)O– used as the base.
Reactions were performed on a 0.25 mmol scale in PhCF3 (0.05 M).
A prospective mechanism for the transformation is detailed in Scheme 2. Based on prior work, we envisioned that PCET activation of the alcohol O–H bond of the substrate would form an alkoxy radical intermediate through the concerted action of an Ir(III)-based visible-light photooxidant and a weak Brønsted base catalyst.[10,11] The resulting O-centered radical would undergo addition to a pendent olefin, forming a new C–O bond and an adjacent alkyl radical. Hydrogen-atom transfer from a thiol-derived co-catalyst to the C-centered radical would furnish the desired cyclic ether.[18] Subsequent reduction of the thiyl radical by the Ir(II) state of the photocatalyst and protonation of the resulting thiolate by the conjugate acid of the Brønsted base would close the catalytic cycle. Notably, for more oxidizable trisubstituted and styrenyl olefins, we observed diminished conversion to the desired ether products in the absence of base, suggesting that an alternative but much less efficient alkene oxidation pathway could also be operative.[6]
Scheme 2.
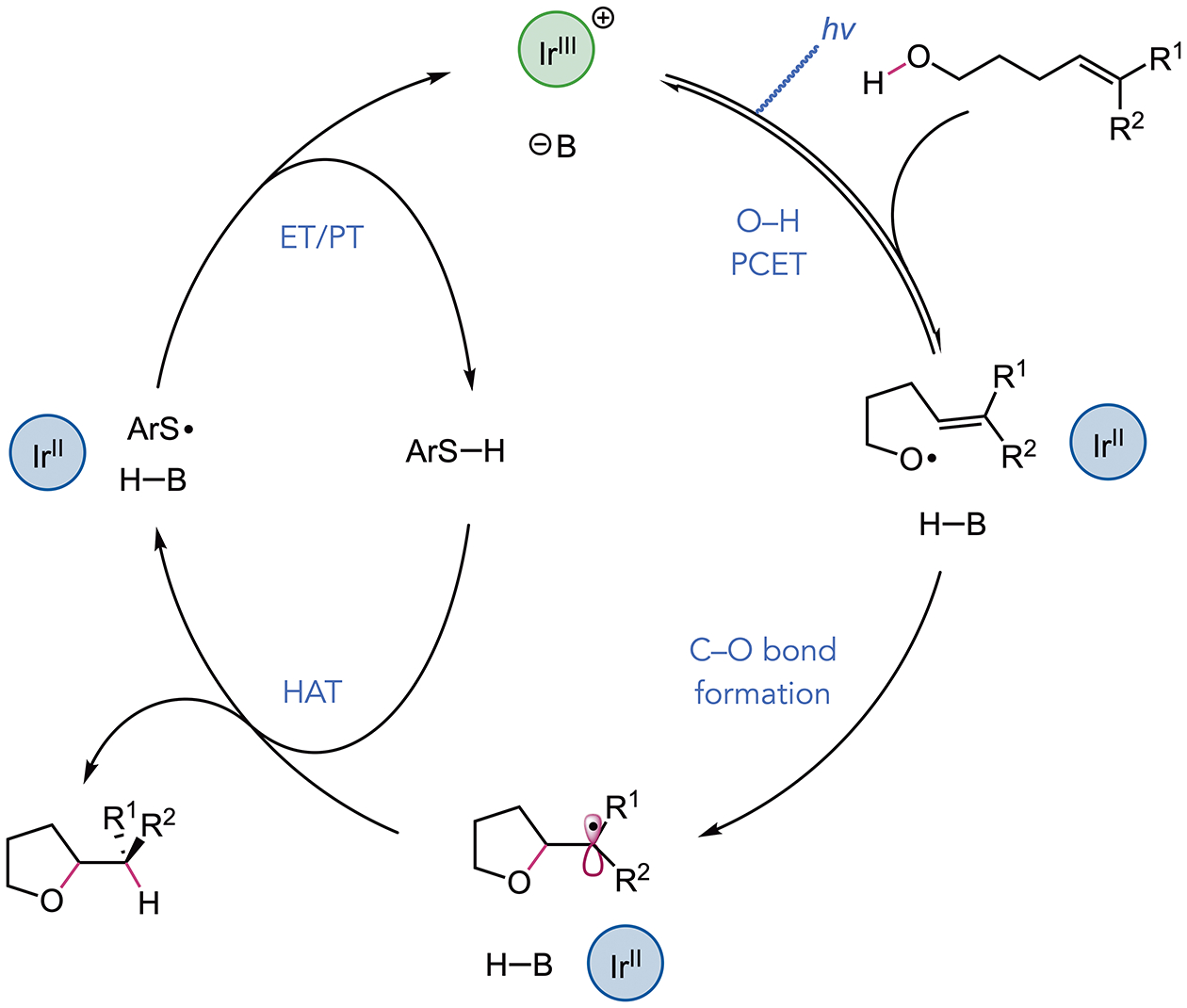
Prospective catalytic cycle.
This observation, however, does not preclude the viability of a dominant alkoxy radical-mediated mechanism under the optimal PCET conditions. Specifically, while investigating the cyclizations to N-Boc-l-prolinol derivatives 27 and 28, we found that the remaining mass balance in these reactions was predominantly comprised of the β-scission product, N-Boc-pyrrolidine. This observation serves as evidence that a discrete alkoxy radical intermediate is formed under these PCET conditions and is consistent with an alkoxy radical-mediated mechanism for C–O bond formation. Moreover, the high yield of cyclized product suggests that these electrophilic O-centered radicals react more rapidly with electron-rich alkenes than they undergo C–C cleavage, even when the C-centered radical resulting from β-scission is stabilized by an adjacent heteroatom.[10c] In contrast, when the monosubstituted and 1,2-disubstituted variants of this substrate were studied under the reaction conditions, only N-Boc-pyrrolidine was formed (Table S7), consistent with a kinetically less favorable C–O bond-forming event.[8i,19]
In summary, we have developed a catalytic protocol for the intramolecular hydroetherification of unactivated alkenes with a wide range of substitution patterns. This work leverages light-driven PCET for the homolysis of strong alcohol O–H bonds, thereby enabling the activation of common hydroxyl groups, for productive C–O bond formation. Taken together, this strategy further illustrates the potential of excited-state PCET to access alkoxy radicals from simple alcohol starting materials under mild, catalytic conditions.
Supplementary Material
Acknowledgements
Financial support was provided by the NIH (R35 GM134893). A.J.M. is grateful to the National Institute of General Medical Sciences of the NIH for a postdoctoral fellowship (F32 GM128245). Y.T. thanks the Institute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University for support. The authors would like to thank Alec Mendelsohn for the synthesis of materials and helpful discussions. We also acknowledge Nick Chiappini and Hunter Ripberger for the preparation of materials.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].(a) Elliott MC, J. Chem. Soc., Perkin Trans 1 1998, 4175–4200; [Google Scholar]; (b) Lorente A, Lamariano-Merketegi J, Albericio F, Álvarez M, Chem. Rev 2013, 113, 4567–4610. [DOI] [PubMed] [Google Scholar]
- [2].(a) Hintermann L, Top. Organomet. Chem 2010, 31, 123–155; [Google Scholar]; (b) Beller M, Seayad J, Tillack A, Jiao H, Angew. Chem. Int. Ed 2004, 43, 3368–3398; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2004, 116, 3448–3479; [Google Scholar]; (c) Zeng X, Chem. Rev 2013, 113, 6864–6900; [DOI] [PubMed] [Google Scholar]; (d) Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF, Org. Lett 2006, 8, 4179–4182. [DOI] [PubMed] [Google Scholar]
- [3].(a) Qian H, Han X, Widenhoefer RA, J. Am. Chem. Soc 2004, 126, 9536–9537; [DOI] [PubMed] [Google Scholar]; (b) Sevov CS, Hartwig JF, J. Am. Chem. Soc 2013, 135, 9303–9306; [DOI] [PubMed] [Google Scholar]; (c) Schlüter J, Blazejak M, Boeck F, Hintermann L, Angew. Chem. Int. Ed 2015, 54, 4014–4017; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 4086–4089; [Google Scholar]; (d) Marcyk PT, Cook SP, Org. Lett 2019, 21, 1547–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Coulombel L, Duñach E, Green Chem. 2004, 6, 499–501; [Google Scholar]; (b) Li Z, Zhang J, Brouwer C, Yang C-G, Reich NW, He C, Org. Lett 2006, 8, 4175–4178; [DOI] [PubMed] [Google Scholar]; (c) Tsuiji N, Kennemur JL, Buyck T, Lee S, Prévost S, Kaib PSJ, Bykov D, Farès C, List B, Science 2018, 359, 1501–1505. [DOI] [PubMed] [Google Scholar]
- [5].(a) Leger PR, Murphy RA, Pushkarskaya E, Sarpong R, Chem. Eur. J 2015, 21, 4377–4383; [DOI] [PubMed] [Google Scholar]; (b) Fujita S, Abe M, Shibuya M, Yamamoto Y, Org. Lett 2015, 17, 3822–3825; [DOI] [PubMed] [Google Scholar]; (c) Shigehisa H, Hayashi M, Ohkawa H, Suzuki T, Okayasu H, Mukai M, Yamazaki A, Kawai R, Kikuchi H, Satoh Y, Fukuyama A, Hiroya K, J. Am. Chem. Soc 2016, 138, 10597–10604; [DOI] [PubMed] [Google Scholar]; (d) Luo C, Bandar JS, J. Am. Chem. Soc 2018, 140, 3547–3550. [DOI] [PubMed] [Google Scholar]
- [6].(a) Hamilton DS, Nicewicz DA, J. Am. Chem. Soc 2012, 134, 18577–18580; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero N, Nicewicz DA, J. Am. Chem. Soc 2014, 136, 17024–17035; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cheng H, Wang X, Chang L, Chen Y, Chu L, Zuo Z, Sci. Bull 2019, 64, 1896–1901. [DOI] [PubMed] [Google Scholar]; (c) Neunteufel RA, Arnold DR, J. Am. Chem. Soc 1973, 95, 4080–4081; [Google Scholar]; (d) Sutterer A, Moeller KD, J. Am. Chem. Soc 2000, 122, 5636–5637; [Google Scholar]; (e) Weiser M, Hermann S, Penner A, Wagenknecht H-A, Beilstein J Org. Chem 2015, 11, 568–575; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yang Z, Li H, Li S, Zhang M-T, Luo S, Org. Chem. Front 2017, 4, 1037–1041; [Google Scholar]; (g) Cheng H, Wang X, Chang L, Chen Y, Chu L, Zuo Z, Sci. Bull 2019, 64, 1896–1901. [DOI] [PubMed] [Google Scholar]
- [7].(a) Shelton JR, Uzelmeier CW, J. Org. Chem 1970, 35, 1576–1581; [Google Scholar]; (b) Elson IH, Mao SW, Kochi JK, J. Am. Chem. Soc 1975, 97, 335–341; [Google Scholar]; (c) Jones MJ, Moad G, Rizzardo E, Solomon DH, J. Org. Chem 1989, 54, 1607–1611. [Google Scholar]
- [8].For reviews, see:; (a) Hartung J, Eur. J. Org. Chem 2001, 619–632; [Google Scholar]; (b) Hartung J, Gottwald T, Špehar K, Synthesis 2002, 11, 1469–1498; [Google Scholar]; for examples, see:; (c) Surzur JM, Bertrand MP, Nouguier R, Tetrahedron Lett. 1969, 48, 4197–4200; [Google Scholar]; (d) Johns A, Murphy JA, Tetrahedron Lett. 1988, 29, 837–840; [Google Scholar]; (e) Hartung J, Gallou F, J. Org. Chem 1995, 60, 6706–6716; [Google Scholar]; (f) Hartung J, Kneuer R, Tetrahedron: Asymmetry 2003, 14, 3019–3031; [Google Scholar]; (g) Hartung J, Kneuer R, Rummey C, Bringmann G, J. Am. Chem. Soc 2004, 126, 12121–12129; [DOI] [PubMed] [Google Scholar]; (h) Zlotorzynska M, Zhai H, Sammis GM, Org. Lett 2008, 10, 5083–5086; [DOI] [PubMed] [Google Scholar]; (i) Rueda-Becerril M, Leung JCT, Dunbar CR, Sammis GM, J. Org. Chem 2011, 76, 7720–7729; [DOI] [PubMed] [Google Scholar]; (j) Schur C, Kelm H, Gottwald T, Ludwig A, Kneuer R, Hartung J, Org. Biomol. Chem 2014, 12, 8288–8307; [DOI] [PubMed] [Google Scholar]; (k) Kim Y, Lee K, Mathi GR, Kim I, Hong S, Green Chem. 2019, 21, 2082–2087; [Google Scholar]; (l) Ren H, Song J-R, Li Z-Y, Pan W-D, Org. Lett 2019, 21, 6774–6778. [DOI] [PubMed] [Google Scholar]
- [9].Barthelemy A-L, Tuccio B, Magnier E, Dagousset G, Angew. Chem. Int. Ed 2018, 57, 13790–13794; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 13986–13990. [Google Scholar]
- [10].(a) Yayla HG, Wang H, Tarantino KT, Orbe HS, Knowles RR, J. Am. Chem. Soc 2016, 138, 10794–10797; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ota E, Wang H, Frye NL, Knowles RR, J. Am. Chem. Soc 2019, 141, 1457–1462; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao K, Yamashita K, Carpenter JE, Sherwood TC, Ewing WR, Cheng PTW, Knowles RR, J. Am. Chem. Soc 2019, 141, 8752–8757; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nguyen ST, Murray PRD, Knowles RR, ACS Catal. 2020, 10, 800–805. [Google Scholar]
- [11].(a) Reece SY, Nocera DG, Annu. Rev. Biochem 2009, 78, 673–699; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Warren JJ, Tronic TA, Mayer JM, Chem. Rev 2010, 110, 6961–7001; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ, Chem. Rev 2012, 112, 4016–4093; [DOI] [PubMed] [Google Scholar]; (d) Gentry EC, Knowles RR, Acc. Chem. Res 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For kinetic studies, see:; (a) Beckwith ALJ, Hay BP, J. Am. Chem. Soc 1988, 110, 4415–4416; [Google Scholar]; (b) Horner JH, Choi S-Y, Newcomb M, Org. Lett 2000, 2, 3369–3372; [DOI] [PubMed] [Google Scholar]; (c) Bietti M, Lanzalunga O, Salamone M, J. Org. Chem 2005, 70, 1417–1422. [DOI] [PubMed] [Google Scholar]
- [13].For advances in β-scission and 1,5-HAT chemistry, see:; (a) Guo J-J, Hu A, Zuo Z, Tetrahedron Lett. 2018, 59, 2103–2111; [Google Scholar]; (b) Jia K, Chen Y, Chem. Commun 2018, 54, 6105–6112; [DOI] [PubMed] [Google Scholar]; (c) Wu X, Zhu C, Chem. Commun 2019, 55, 9747–9756, and references therein. [DOI] [PubMed] [Google Scholar]
- [14].Zhu Q, Gentry EC, Knowles RR, Angew. Chem. Int. Ed 2016, 55, 9969–9973; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 10123–10127. [Google Scholar]
- [15].Rueping M, Nachtsheim BJ, Ieawsuwan W, Atodiresei I, Angew. Chem. Int. Ed 2011, 50, 6707–6720; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 6838–6853. [Google Scholar]
- [16].Carvalho PJ, Ventura SPM, Batista MLS, Schröder B, Gonçalves F, Esperança J, Mutelet F, Coutinho JAP, J. Chem. Phys 2014, 140, 064505. [DOI] [PubMed] [Google Scholar]
- [17].(a) Wolfe JP, Eur. J. Org. Chem 2007, 571–582; [PMC free article] [PubMed] [Google Scholar]; (b) Miller Y, Miao L, Hosseini AS, Chemler SR, J. Am. Chem. Soc 2012, 134, 12149–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qiu G, Knowles RR, J. Am. Chem. Soc 2019, 141, 16574–16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Attempts to form oxetanes and epoxides via this method have thus far proved unsuccessful.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.