

Abstract
Substance use disorder (SUD) is a major public health crisis worldwide, and effective treatment options are limited. During the past 2 decades, researchers have investigated the impact of a variety of pharmacological approaches to treat SUD, one of which is the use of medical cannabis or cannabinoids. Significant progress was made with the discovery of rimonabant, a selective CB1 receptor (CB1R) antagonist (also an inverse agonist), as a promising therapeutic for SUDs and obesity. However, serious adverse effects such as depression and suicidality led to the withdrawal of rimonabant (and almost all other CB1R antagonists/inverse agonists) from clinical trials worldwide in 2008. Since then, much research interest has shifted to other cannabinoid-based strategies, such as peripheral CB1R antagonists/inverse agonists, neutral CB1R antagonists, allosteric CB1R modulators, CB2R agonists, fatty acid amide hydrolase (FAAH) inhibitors, monoacylglycerol lipase (MAGL) inhibitors, fatty acid binding protein (FABP) inhibitors, or nonaddictive phytocannabinoids with CB1R or CB2R-binding profiles, as new therapeutics for SUDs. In this article, we first review recent progress in research regarding the endocannabinoid systems, cannabis reward versus aversion, and the underlying receptor mechanisms. We then review recent progress in cannabinoid-based medication development for the treatment of SUDs. As evidence continues to accumulate, neutral CB1R antagonists (such as AM4113), CB2R agonists (JWH133, Xie2–64), and nonselective phytocannabinoids (cannabidiol, β-caryophyllene, Δ9-tetrahydrocannabivarin) have shown great therapeutic potential for SUDs, as shown in experimental animals. Several cannabinoid-based medications (e.g., dronabinol, nabilone, PF-04457845) that entered clinical trials have shown promising results in reducing withdrawal symptoms in cannabis and opioid users.
1. Introduction
Substance use disorder (SUD) remains a serious problem worldwide and has an enormous economic burden in the USA and many other countries because of costs related to crimes, loss of productivity, and healthcare. Sadly, the greatest cost of SUDs is paid in human lives caused by either drug overdose or by SUD-related diseases. In recent years, there has been a dramatic increase in opioid overdose incidents, bringing the number of fatal incidents to nearly 48,000 in 2017 in the USA [1, 2]. Although the US FDA has approved several medications (methadone and buprenorphine for the treatment of opioid use disorders [3]; disulfiram, naltrexone, and acamprosate for alcohol use disorders; nicotine-replacement therapy, bupropion, and varenicline for cigarette smoking cessation [4]), the rate of relapse is extremely high. Less than 3% of individuals with SUDs remain abstinent for > 1 year after pharmacotherapy is terminated [3, 4]. No medication for the treatment of psychostimulant use disorders has been approved by the FDA. The lack of effective treatments for the prevention of relapse has encouraged scientific communities to focus on developing more effective pharmacotherapies to combat SUDs. Significant progress has been made in various receptor mechanism-based and clinic-based medication development strategies [5–8]. In this article, we focus on recent progress in cannabinoid or cannabinoid receptor-based medication development for the treatment of SUDs. The information was gathered using PubMed, Web of Science, and Biological Abstract search engines and the following key terms: addiction (or substance use disorder) and endocannabinoid system, CB1, CB2, allosteric modulators, fatty acid amide hydrolase (FAAH) inhibitor, monoacylglycerol lipase (MAGL) inhibitor, fatty acid-binding protein (FABP), and phytocannabinoids.
2. Mesolimbic Dopamine System
To understand the mechanisms through which cannabis produces rewarding, aversive, or anti-addictive effects, we briefly review the mesolimbic dopamine (DA) system, a neural substrate critically involved in drug reward and addiction [9, 10]. Although our understanding of the etiology of drug abuse and addiction remains limited, a well-accepted view is that the rewarding effects of drugs of abuse are mediated primarily by the activation of the DA system that originates in the ventral tegmental area (VTA) of the midbrain and projects to the prefrontal cortex (PFC), nucleus accumbens (NAc), and amygdala, the brain regions critically involved in addiction [11]. Addictive drugs may directly or indirectly activate this system by stimulating or blocking distinct molecular or cellular mechanisms [9]. For example, the psychostimulant cocaine may activate this system mainly by blocking DA transporters (DATs), whereas nicotine activates VTA DA neurons directly by stimulating nicotinic cholinergic receptors or indirectly by stimulating glutamate release in the VTA and NAc [3]. On the other hand, opioids may activate the VTA DA neurons mainly by stimulation of opioid receptors located on VTA γ-aminobutyric acid (GABA)ergic interneurons and/or GABAergic afferents from other brain regions, which causes a decrease in GABA release and consequently DA neuron disinhibition [3, 5]. Therefore, the VTA–NAc DA pathway has been thought to play a central role in drug reward and addiction [9], making it a crucial target in medication development for the treatment of SUDs. Significance progress has been made in DA-, glutamate-, and GABA-based medication development for the treatment of SUDs [6, 7, 9]. In addition, increasing attention has been given to cannabis or cannabinoids that alter the brain DA reward system [12–14]. Therefore, we first review how cannabis and cannabinoids modulate the mesolimbic DA system to produce reward or aversion; we then survey potential cannabinoid receptor-based medications that ameliorate effects produced by different drugs of abuse.
3. Endocannabinoid System
The endocannabinoid (eCB) system consists of endogenous cannabinoids, such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG), G-protein-coupled cannabinoid receptors (CB1Rs, CB2Rs), enzymes for eCB synthesis and degradation, intracellular signaling molecules, and their putative transport systems (Fig. 1) [15]. In 1992, the first eCB, AEA, was discovered [16]. AEA is biosynthesized from membrane N-arachidonoyl phosphatidylethan-olamine (NAPE) via multiple pathways by enzymes such as phospholipase A2, phospholipase C, and NAPE-phospholipase D (NAPE-PLD). It is degraded primarily by the enzyme FAAH, which converts AEA into ethanolamine and arachidonic acid (AA). AEA is an endogenous CB1R agonist (inhibitory constant [Ki] = 87.7 nM for rat CB1R; Ki = 239.2 nM for human CB1R) and a weak CB2R agonist (Ki = 267.8 nM for rat CB2; Ki = 439.5 nM for human CB2) [17]. The effects of AEA are thought to be mediated mainly by stimulation of CB1Rs and CB2Rs in the brain and periphery. Endogenous AEA is present at very low levels and has a very short half-life because of the action of FAAH. As such, inhibition of FAAH leads to elevated AEA levels; therefore, FAAH inhibitors are being pursued for therapeutic use [18].
Fig. 1.
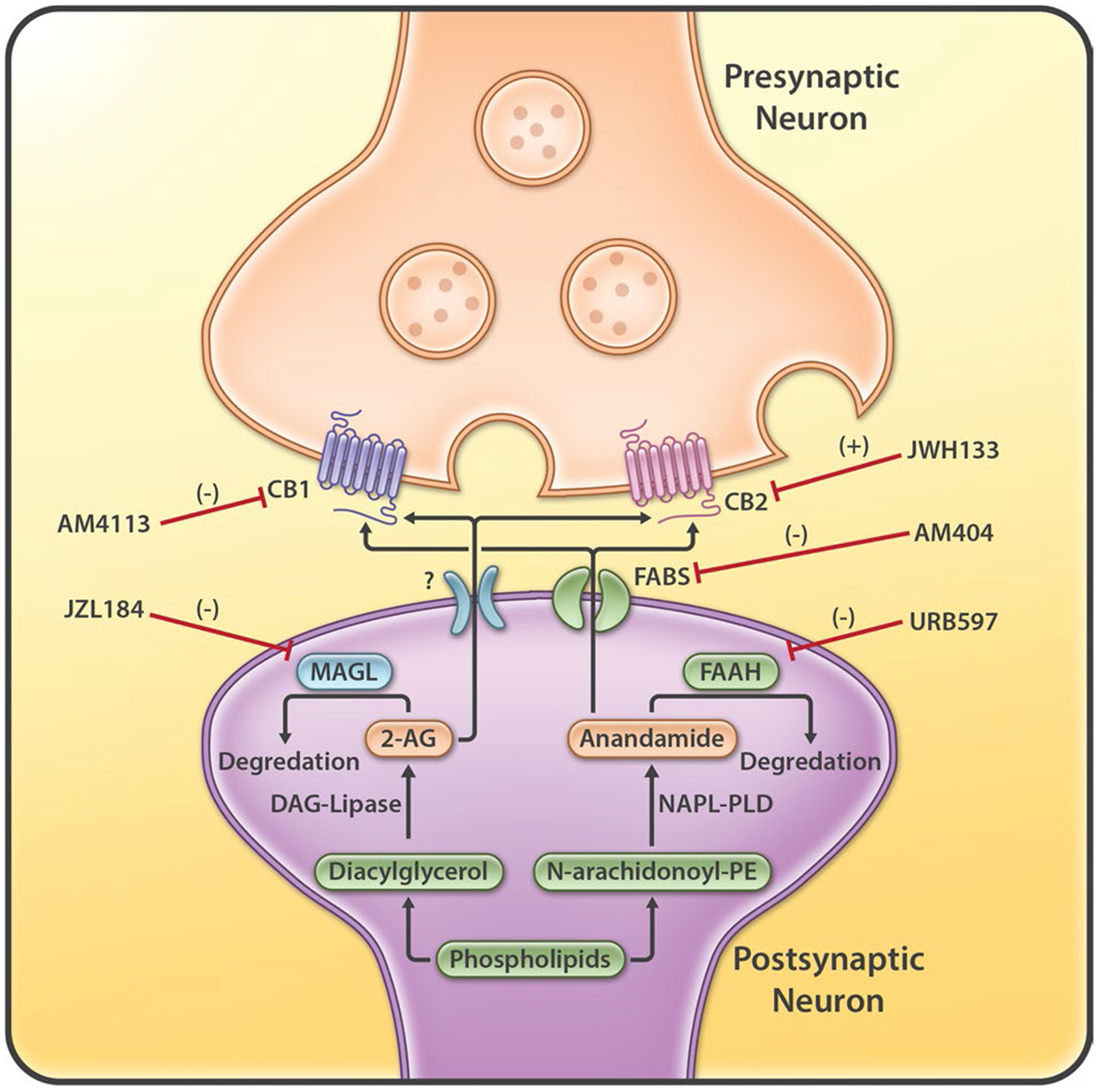
The endocannabinoid system, including the synthesis and degradation enzymes (FAAH, MAGL) of endocannabinoids (AEA and 2-AG), cannabinoid receptors (CB1 and CB2), anandamide transporter (FABPs), and the five major therapeutic targets (CB1, CB2, FAAH, MAGL, FABPS). AEA anandamide, DAG diacylglycerol, FAAH fatty acid amide hydrolase, FABPs fatty acid-binding proteins, MAGL monoacylglycerol lipase, NAPE-PLD N-arachidonoyl phosphatidylethanolamine-phospholipase D, PE phosphatidylethanolamine, 2-AG 2-arachidonoylglycerol
In 1995, 2-AG, the second eCB, was discovered [19]. It is an endogenous agonist of the CB1Rs (Ki = 1180 nM for rat CB1R; Ki = 3423 nM for human CB1R) and CB2Rs (Ki = 1900 nM for rat CB2R; Ki = 1193 nM for human CB2R) [17]. Unlike AEA, 2-AG is present at relatively high levels in the central nervous system (CNS). 2-AG is synthesized from AA-containing diacylglycerol (DAG) by the action of DAG lipase and metabolized by the enzyme MAGL that converts 2-AG to free AA and glycerol. Inactivation of MAGL by JZL184 or others results in immense elevations of brain 2-AG levels that lead to cannabinoid behavioral effects in mice [20]. Therefore, both FAAH and MAGL constitute two additional major targets (in addition to CB1Rs and CB2Rs) in medication development for the treatment of neuropsychiatric disorders, including SUDs (Fig. 1). eCB transporters for AEA include the heat shock proteins (Hsp70s) and FABPs [21, 22]. FABP inhibitors attenuate the breakdown of AEA by the FAAH. The blockade of AEA transport may, at least partly, be the mechanism through which FABP inhibitors exert their pharmacological effects. Unlike AEA transporters, which are more extensively studied, only a few studies have explored the presence and function of the transporters of 2-AG [23].
3.1. Neuronal CB1 Receptor (CB1R) Signaling
Endocannabinoids regulate physiological functions in the brain mainly through activation of CB1Rs because of their high density of expression in the brain. Since CB1Rs are abundant at neuronal terminals [24], most functional studies have focused on how eCBs modulate synaptic signal transmission and plasticity via a retrograde eCB-CB1R mechanism (Fig. 2) [25–27]. Specifically, presynaptic neuronal depolarization increases glutamate release at excitatory synapses by activation of voltage-gated Ca2+ channels (VGCCs), which subsequently activates postsynaptic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) receptors and depolarizes post-synaptic neurons. Meanwhile, glutamate may also activate postsynaptic group I metabotropic glutamate receptors (mGluR1, mGluR5), participate in synaptic signal transmission, and cause an increase in 2-AG synthesis in postsynaptic neurons. In addition, postsynaptic neuronal depolarization may also elevate intracellular Ca2+ via VGCCs and elicit 2-AG production, most likely via phospholipase C. After being released from postsynaptic neurons, 2-AG retrogradely travels across the synapse to activate presynaptic CB1Rs. Presynaptic CB1Rs are Gi/o protein-coupled receptors. Their activation leads to inhibition of VGCCs and activation of inwardly rectifying K+ channels via the stimulated cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) signal pathway and, therefore, inhibits neurotransmitter release by, for example, inhibiting glutamate release at excitatory synapses (Fig. 2) or GABA release at inhibitory synapses [28]. This neuronal CB1-mediated neuronal inhibition leads to several types of short-term or long-term synaptic plasticity, such as depolarization-induced suppression of excitation (DSE) at excitatory synapses, depolarization-induced suppression of inhibition (DSI) at inhibitory synapses, or long-term depression [26, 27, 29], which are associated with eCB involvement in various brain functions [25, 26, 29].
Fig. 2.
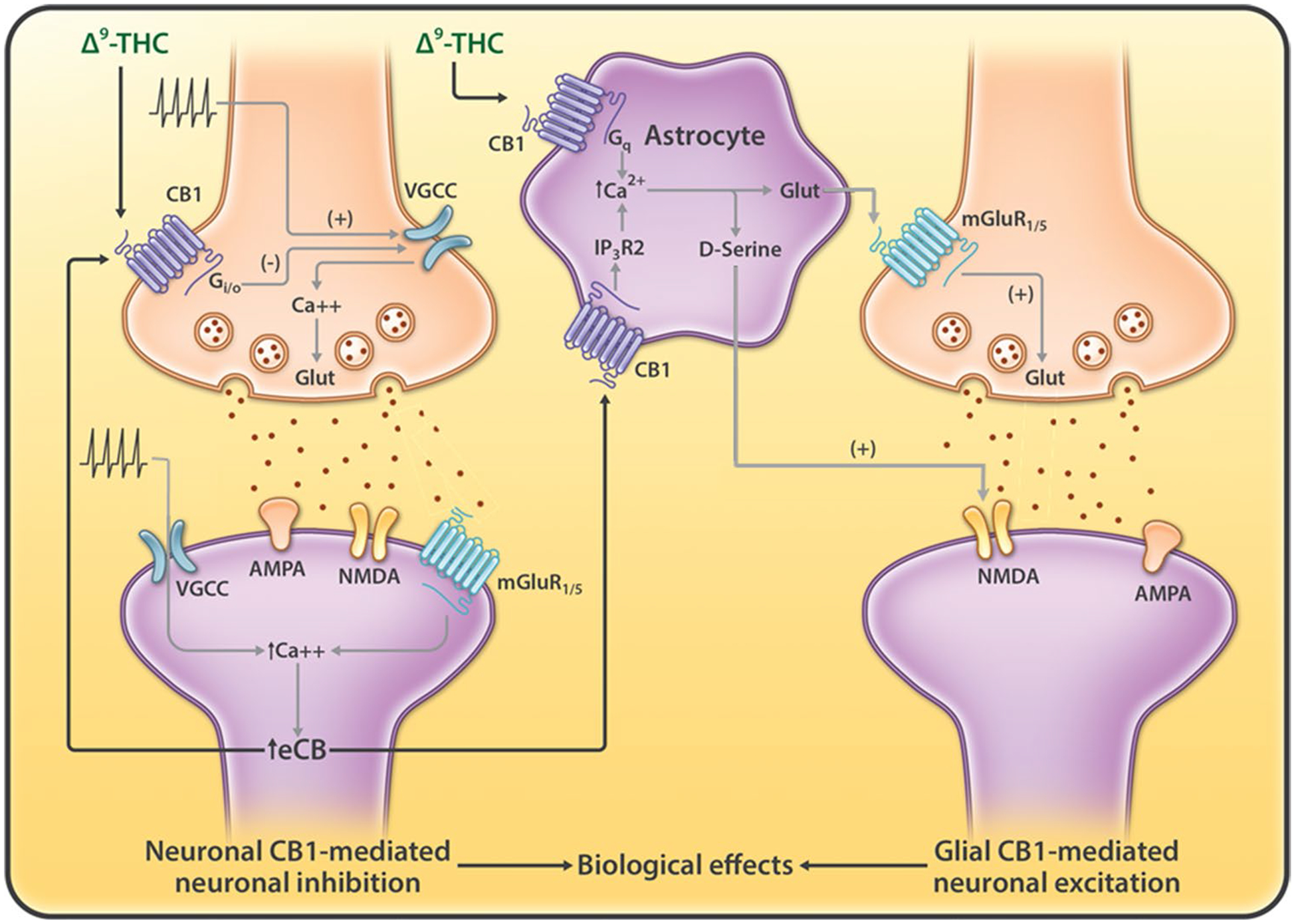
Neuronal vs. glial CB1 receptor signaling that regulates synaptic transmission. Presynaptic or postsynaptic neuronal depolarization may increase eCB (2-AG) release from postsynaptic neurons by glutamate-mGluR1/5 and Ca2+-dependent mechanisms. eCBs released by the postsynaptic neurons directly activate CB1Rs in the presynaptic terminals, which leads to synaptic depression at eCB-releasing synapses (homoneuronal synapses). In addition, eCBs activate CB1Rs in astrocytes, elevate their intracellular Ca2+ level, and stimulate the release of gliotransmitters (such as glutamate, ATP, D-serine) that potentiates synaptic transmission in adjacent neurons (heteroneuronal synapses). The biological effects of cannabinoids (such as Δ9-THC) may be mediated by stimulation of CB1Rs in both neurons and/or astrocytes cells, depending upon the cellular distributions of CB1Rs in different brain regions. AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, ATP adenosine triphosphate, CB1R CB1 receptor, eCB endocannabinoid, Glut glutamate, IP3R inositol trisphosphate receptor, mGluR1 type 1 metabotropic glutamate receptor, NMDA N-methyl-D-aspartate receptor, VGCC voltage-dependence calcium channel, 2-AG 2-arachidonoylglycerol, Δ9-THC Δ9-tetrahydrocannabinol
3.2. Glial CB1R Signaling
In addition to neurons, CB1Rs are also detected in ~ 40% of astrocytes [30, 31]. Astrocytes were traditionally thought to metabolically support neurons and to maintain a stable homeostatic environment for neuronal functions. However, recent studies have indicated that astrocytes can control synaptic plasticity and memory functions [32]. Navarrete and Araque [33] first reported in 2008 that hippocampal astrocytes express functional CB1Rs that increase intracellular Ca2+ levels in astrocytes following electrical stimulation of adjacent neurons. This effect is mediated by a Gαq protein-phospholipase C signal pathway, rather than the Gαi/o protein-cAMP signal pathway observed in neurons. This increase in astrocyte Ca2+ levels causes release of gliotransmitters such as glutamate and D-serine (an NMDA receptor co-activator) [34, 35], causing hetero-synaptic potentiation at excitatory or inhibitory synapses [33, 36] (Fig. 2). This glial CB1R-mediated neuronal excitation is opposite to neuronal CB1R-mediated homosynaptic inhibition. Thus, the functional effect of eCB release or cannabinoids may depend on the balance between these opposing actions (Fig. 2).
It is well-documented that systemic administration of high doses of Δ9-tetrahydrocannabinol (Δ9-THC) or WIN55, 212–2 may impair learning and memory [37]. Strikingly, deletion of CB1Rs from astrocytes (in GFAP-CB1-KO mice) abolished cannabinoid-induced memory impairment, whereas deletion of CB1Rs from specific neurons (i.e., cortical glutamatergic or forebrain GABAergic neurons lacking the CB1R gene) failed to alter it [31, 36]. These findings suggest that glial CB1Rs may play a dominant role in cannabinoid-induced cognitive impairment (Fig. 2). Clearly, more studies are required to confirm these findings and to explore whether glial CB1Rs play a similar role in other effects of cannabinoids such as analgesia, hypothermia, catalepsy, hypolocomotion, and cannabis reward or aversion.
4. Cannabis Reward and the Receptor Mechanisms
4.1. Cannabis Reward
Cannabis is commonly used worldwide, as many people find its effects pleasurable. However, reports regarding the rewarding properties of cannabis are conflicting, and the underlying mechanisms are poorly understood. Accumulative evidence indicates that eCBs may modulate the mesolimbic DA system directly or indirectly via stimulation of CB1Rs and CB2Rs (Fig. 3). CB1Rs are expressed at high density in the cortex, striatum, hippocampus, thalamus, hypothalamus, substantia nigra pars reticulata, and cerebellum (Fig. 4) [24, 38]. These unique distributions account for their functional involvement in cognition, memory, emotions, appetite, and reward. High expressions of CB1Rs have been found on glutamatergic and GABAergic neurons in the VTA and striatum [29, 39, 40], two critical brain regions involved in drug reward and relapse. As stated, CB1Rs participate in retrograde signaling at glutamatergic or GABAergic synapses to control neurotransmitter release [29]. Since CB1Rs are highly expressed in the brain, whereas CB2Rs are expressed predominantly in peripheral tissues in healthy subjects, it is widely accepted that the rewarding effects of cannabinoids are mediated by the stimulation of brain CB1Rs [41, 42]. Several lines of evidence support this hypothesis. For example, stimulation of CB1Rs can excite DA neurons and increase DA release in the NAc [43–46]. Cannabinoid receptor agonists such as Δ9-THC, CP55, 940, WIN55, 212–2, and HU 210 can modulate the DA reward system [47] (possibly through stimulation of CB1Rs), producing conditioned place preference (CPP) [48], enhanced brain-stimulation reward (BSR) [49, 50], self-administration [51–53], and reinstatement of extinguished drug-seeking behavior [54].
Fig. 3.
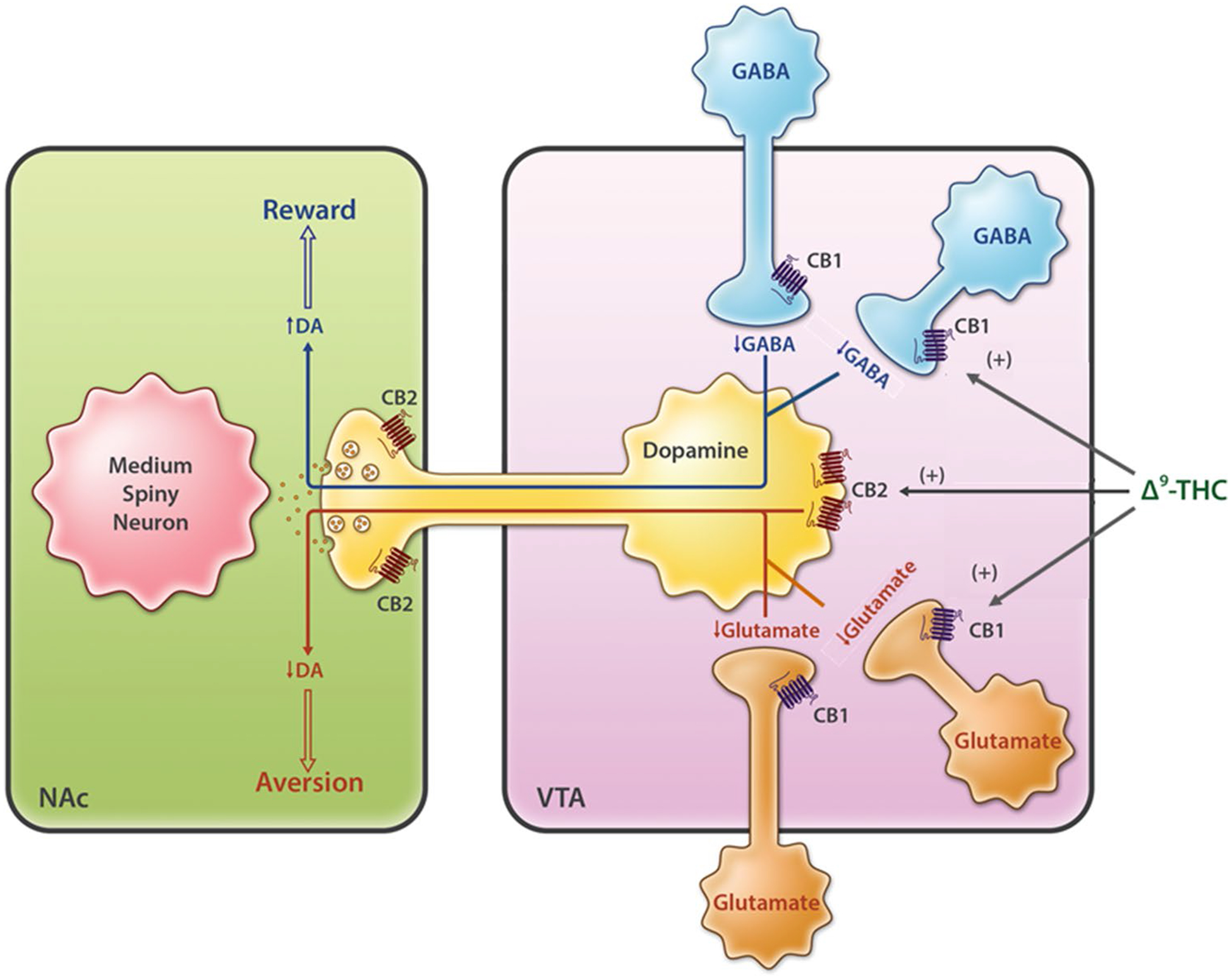
Neural mechanisms underlying cannabis reward vs. aversion. Cannabinoid CB1Rs are expressed in VTA GABAergic interneurons and glutamatergic neurons as well as their afferent projections to the VTA DA neurons, whereas CB2Rs are found in VTA DA neurons. Cannabis or cannabinoids modulate the mesolimbic DA system via activation of brain CB1Rs and CB2Rs. Cannabinoids such as Δ9-THC may produce rewarding effects by binding to CB1R on VTA GABAergic interneurons and/or their afferents, thereby reducing GABA-mediated inhibition of VTA DA neurons and increasing DA release in the NAc. Conversely, cannabinoids may produce aversive effects by activating CB1R on glutamatergic neurons in the VTA, and/or CB2Rs on DA neurons, thereby inhibiting VTA DA release to the NAc. The subjective effects of cannabinoids may thus depend on the balance of opposing CB1R and CB2R effects and individual differences in neural CB1R and CB2R expression. CB1/2R CB1/2 receptor, DA dopamine, GABA γ-aminobutyric acid, NAc nucleus accumbens, VTA ventral tegmental area, Δ9-THC Δ9-tetrahydrocannabinol
Fig. 4.
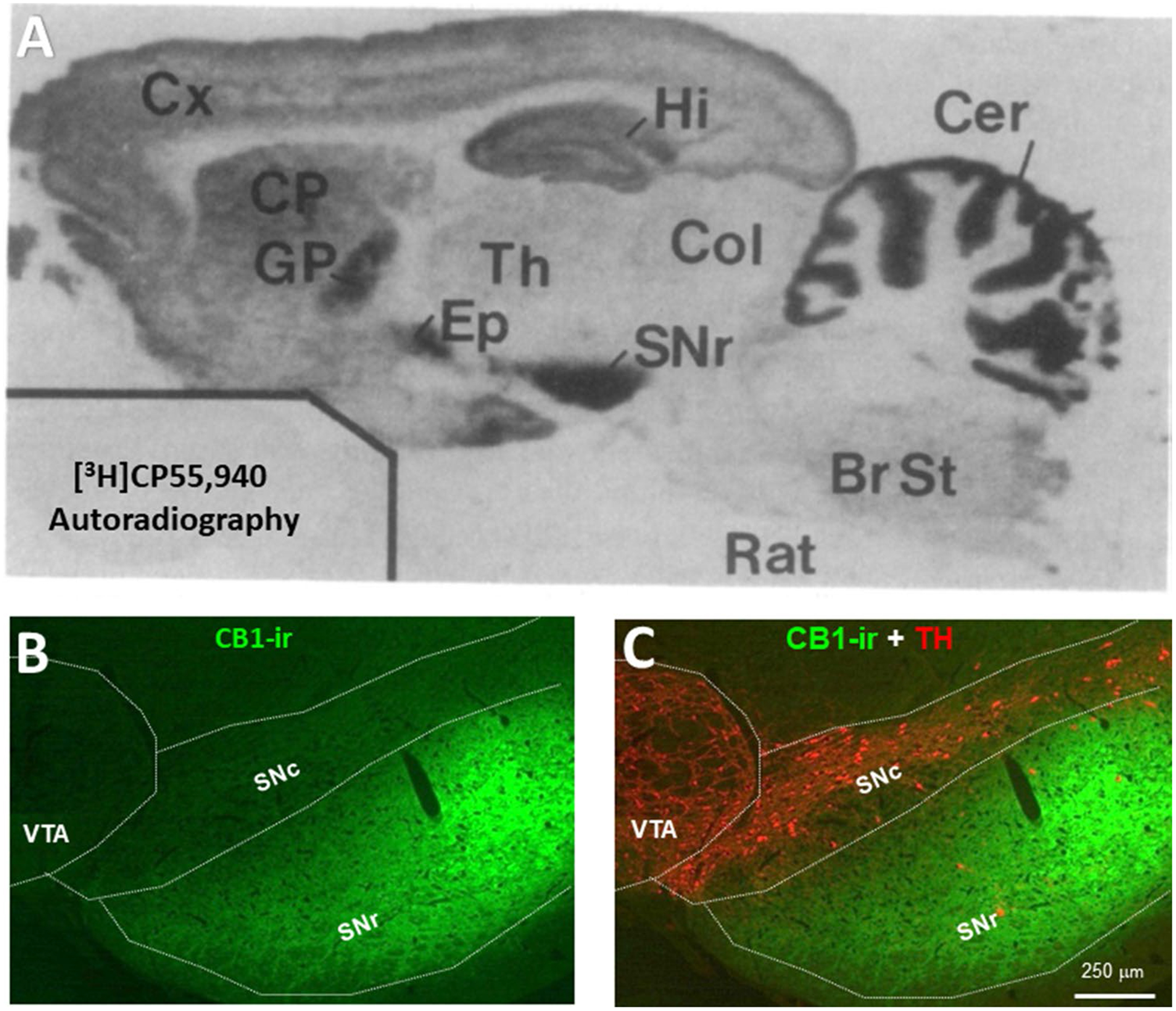
Distributions of CB1Rs in the brain. A [35H]CP55,940 autoradiography image showing a high density of CB1R expression in the SNr, globus pallidus, hippocampus, and cerebellum [24]. B CB1R immunostaining in the midbrain, illustrating that CB1Rs are highly expressed in the SNr, not in the VTA or SNc. BrS Brain stem, CB1R CB1 receptor, Cer cerebellum, Col colliculi, CP caudate-putamen, Cx Cortex, Ep entopeduncular nucleus, GP globus pallidus, Hi hippocampus, SNc substantia nigra pars compacta, SNr substantia nigra pars reticulata, Th thalamus, VTA ventral tegmental area
4.2. GABA-CB1 hypothesis
Electrophysiological studies demonstrate that VTA DA neurons receive both inhibitory GABAergic and excitatory glutamatergic inputs. CB1Rs are expressed in GABAergic neurons in the VTA and NAc [40]. Stimulation of CB1Rs on VTA GABAergic neurons caused VTA DA neuron activation via GABAergic disinhibition [55–58]. These findings suggest that cannabis reward could be mediated by the activation of CB1Rs on VTA GABAergic neurons (Fig. 3). However, there is no direct in vivo behavioral evidence supporting this GABA-CB1R hypothesis of cannabis reward. In addition, morphological evidence indicates that CB1Rs are mainly expressed in the substantia nigra pars reticulata (SNr), cerebellum, hippocampus, and striatum but at much lower density in the VTA (Fig. 4) [24]. Thus, it is more likely that GABAergic afferents projecting to the VTA DA neurons from those high CB1R-expressed regions rather than from VTA GABAergic interneurons may play a more important role in modulating DA neuron activity and cannabis reward. Clearly, more studies are needed to test this GABAergic disinhibition hypothesis of cannabinoid reward.
5. Cannabis Aversion and the Receptor Mechanisms
5.1. Cannabis Aversion
Not all cannabis users enjoy the effects of cannabis, with some experiencing euphoria, pleasure, and relaxation [12, 59, 60] but others experiencing dysphoria, anxiety, and depression [61, 62]. Even in the same person, cannabis may produce positive effects at one time but negative effects at another [63, 64]. Similar paradoxical effects of Δ9-THC have been found in non-human primates. For example, Δ9-THC was reported to be self-administered by squirrel monkeys [65, 66] but not by rhesus monkeys [67, 68]. Unlike many other drugs of abuse, such as opioids, stimulants, nicotine, or alcohol, Δ9-THC or other cannabinoid agonists cannot be reliably self-administered by rats or mice. Δ9-THC might produce rewarding effects, as indicated by enhanced electrical BSR [69–71], but more recent studies have indicated that Δ9-THC or other cannabinoid agonists might also be aversive and inhibit BSR [72–75]. Conflicting findings have also been reported in studies using CPP, in which Δ9-THC or other cannabinoids are either not rewarding or produce overt aversive effects [76, 77]. Similar conflicting findings are also reported in studies using in vivo microdialysis, indicating that Δ9-THC failed to alter the extracellular DA level in the NAc [78]. The neurobiological mechanisms underlying such paradoxical effects are poorly understood. One possible explanation is that distinct cellular distributions of CB1 or CB2 receptors in the brain may underlie the different effects of cannabinoids, as discussed below (Sects. 5.2 and 5.3).
5.2. Glutamate–CB1 Hypothesis
Clearly, the above GABAergic disinhibition hypothesis cannot explain how cannabis produces aversive effects. We recently reported that CB1Rs are expressed not only in VTA GABAergic neurons but also in VTA glutamatergic neurons (Fig. 3) [40]. Optogenetic activation of VTA glutamatergic neurons has been shown to produce potent DA-dependent rewarding effects, as assessed by CPP and intracranial self-stimulation (ICSS) [40, 79]. Strikingly, systemic administration of Δ9-THC dose dependently inhibited such glutamatemediated optical ICSS only in wild-type (WT) mice and not in conditional CB1R-knockout (CB1-KO) mice, in which CB1Rs are deleted selectively from subcortical VgluT2-expressing glutamatergic neurons [40]. These findings suggest that stimulation of CB1Rs on glutamatergic neurons may produce aversive effects by decreasing glutamatergic inputs to VTA DA neurons. It also suggests that stimulation of brain CB1Rs is not always rewarding but could be reward attenuation or aversive. Therefore, we proposed that the hedonic effects of cannabis might depend on a final net effect of at least two opposing actions of cannabinoids on both VTA GABAergic neurons and VTA glutamatergic neurons or their afferent terminals (Fig. 3). If more CB1Rs are expressed in VTA GABAergic neurons or GABAergic afferents, cannabis will be rewarding since GABAergic disinhibition of VTA DA neurons is dominant. In contrast, if more CB1Rs are expressed in VTA glutamatergic neurons or glutamatergic afferents, cannabis will be aversive since CB1R stimulation-induced reduction of glutamatergic inputs to VTA DA neurons is dominant. Congruently, if CB1R levels are equivalent on both neuronal types, cannabis should have no effect on the brain reward function (Fig. 3). This hypothesis appears to explain why Δ9-THC or cannabinoids are rewarding in some human subjects or nonhuman primates (squirrel monkeys), in which more CB1Rs might be expressed in VTA GABAergic neurons or GABAergic afferent terminals, but are ineffective or aversive in other species such as rats and mice in which more CB1Rs might be expressed in VTA glutamatergic neurons or glutamatergic afferent terminals (Fig. 3).
5.3. Dopamine–CB2 Hypothesis
In addition to the abovementioned glutamate–CB1R hypothesis, activation of CB2Rs in VTA DA neurons may also contribute to the aversive effects of cannabis (Fig. 3). A break-through finding in the past decade was the discovery of functional CB2Rs in the brain, particularly in the mesolimbic DA system [80, 81]. Since CB2Rs are Gi-coupled, stimulation of CB2Rs inhibits VTA DA neuron activity and NAc DA release [82–85]. Stimulation of CB2Rs by JWH133 inhibits cocaine self-administration, cocaine-induced CPP, and hyperactivity [82, 86, 87], whereas overexpression of CB2Rs in the brain inhibits cocaine self-administration and cocaine-enhanced locomotion in mice [88]. We recently reported that Δ9-THC and WIN55, 212–2 produce biphasic effects—rewarding at lower doses and aversive at higher doses—as assessed in the electrical BSR paradigm [50]. Pretreatment with a CB1R antagonist (AM251) attenuated the low-dose-enhanced BSR, whereas a CB2R antagonist (AM630) attenuated the high-dose-inhibited BSR. Congruent with these findings are reports that selective CB1R and CB2R agonists produced significant BSR enhancement and inhibition, respectively [50]. Taken together, these new findings suggest that both glutamate–CB1R and DA-CB2R mechanisms may underlie the aversive effects of cannabis or cannabinoids (Fig. 3). Thus, the subjective effects of cannabis may depend on the balance of multiple distinct cell type-specific receptor mechanisms. Cannabis may be either rewarding, ineffective, or aversive in different subjects or species according to differences in brain CB1R and CB2R expression in different types of neurons.
Such different pharmacological effects (reward vs. aversion) of cannabis and the distinct CB1R/CB2R mechanisms of the action produced by different cannabinoids may underlie the bases of cannabinoid receptor-based medication development strategies for the treatment of SUDs, as discussed below (Sects. 6–14). Next, we review multiple cannabinoid receptor-based strategies and emphasize the efficacy of compounds that selectively target CB1Rs, CB2Rs, FAAH, MAGL, and FABP. Lastly, we review recent progress in studying the potential utility of nonaddictive phytocannabinoids such as Δ9-THC, CBD, BCP, and Δ9-tetrahydrocannabivarin (Δ9-THCV) in the treatment of SUDs.
6. CB1R Antagonists/Inverse Agonists as Potential Treatments for Substance Use Disorders (SUDs)
6.1. Rationale
Over the past 3 decades, CB1Rs have been a primary target in medication discovery and medicinal chemistry research as they are highly expressed in the brain. As already stated, Δ9-THC is the major psychoactive component of cannabis, and CB1Rs are the major targets responsible for the physiological and psychological effects of cannabinoids. Further-more, the cannabinoid receptor agonist WIN55, 212–2 has been found to increase motivation to self-administer nicotine and facilitate cue-induced reinstatement in rats [89], whereas Δ9-THC increased heroin self-administration in rats [90]. WIN55, 212–2 and CP55, 940 have also been shown to facilitate alcohol self-administration, alcohol CPP, and binge-like alcohol intake in animals [91, 92]. In addition, cocaine, heroin, and alcohol have been shown to increase 2-AG release in the VTA and NAc [93, 94]. Thus, it was hypothesized that CB1R antagonism may be useful for the treatment of other SUDs [95–98], similar to the use of naloxone for opioid use disorder [3].
6.2. Rimonabant
In 1994, Sanofi Pharmaceutical Inc. (France) developed rimonabant (SR141716A), the first CB1R antagonist [99]. In the absence of cannabinoid agonists, rimonabant itself acts as a CB1R inverse agonist, producing the opposite effect to CB1R agonists such as Δ9-THC on intracellular signaling [100]. Compelling evidence indicates that rimonabant not only blocks the action of Δ9-THC, both in vitro and in vivo, but also has significant anti-addiction efficacy against a broad range of addictive substances [95, 101–103] as well as anti-obesity potential [104]. On these grounds, rimonabant was approved by the European Commission in 2006 for the treatment of obesity and became available for prescribed use in the UK. By 2008, rimonabant was available in 56 countries. Although intended to control over-eating and obesity, rimonabant was soon recognized as able to prolong abstinence from smoking compared with placebo in phase III clinical trials [103, 105]. It was also effective at reducing self-administration of many other drugs of abuse (for a comprehensive review, see Sloan et al. [106]). Unfortunately, rimonabant also produced significant anxiety, depression, and suicidality [107, 108]. In October 2008, the European Medicines Agency recommended suspension of clinical use of rimonabant after concluding that its risks outweighed its benefits, and its approval was withdrawn by the European Commission in January 2009. Consequently, clinical trials with such CB1R antagonists with inverse agonist profiles were terminated worldwide [108, 109]. Therefore, rimonabant-related studies are not covered in detail in this article.
7. Peripheral CB1R Antagonists as Potential Treatments for SUDs
7.1. Rationale
To circumvent the above mentioned adverse effects of rimonabant, peripherally restricted CB1R antagonists or inverse agonists were proposed for controlling binge eating (feeding), obesity, or metabolic diseases because they cannot penetrate the blood–brain barrier [110]. To date, several peripheral CB1R antagonists or inverse agonists have been developed, including JD5037 and AM6545. Like rimonabant, both of these agents significantly inhibit food intake and decrease body weight [110–114], but, unlike rimonabant, neither agent affects behavioral responses mediated by CB1Rs in the brain, such as catalepsy, hypomotility, and hypothermia [115].
7.2. JD5037
The peripheral CB1R inverse agonist, JD5037, also dose dependently reduces alcohol drinking in mice via CB1Rs in ghrelin-producing stomach cells [116]. It is believed that JD5037-induced blockade of gastric vagal afferents abrogated the ability of JD5037 to reduce ethanol drinking. This is a very interesting finding, suggesting that peripheral CB1R blockade may have therapeutic potential in the treatment of alcoholism. Whether these compounds are also effective at attenuating the abuse of drugs such as nicotine, heroin, or cocaine remains unknown, and more studies are required to test this hypothesis.
8. Neutral CB1R Antagonists as Potential Treatments for SUDs
8.1. Rationale
The neural mechanisms underlying the adverse effects of rimonabant remain unknown. Presumably, these effects result from CB1R inverse agonism [95]. This premise appears to be supported by recent findings that rimonabant alone produces anhedonic-like effects, as assessed by electrical BSR [117, 118], in vivo brain microdialysis [119], elevated plus maze, and forced swim test [120]. Therefore, it has been proposed that neutral CB1R antagonists without inverse agonist profiles should retain the therapeutic anti-addictive effects without the unwanted side effects. Accordingly, several neutral CB1R antagonists have been developed, including AM4113, LH-21, and PIMSR. Preclinical studies with those compounds have shown promising results in combating obesity, metabolic diseases, and alcoholic steatosis [121, 122] (Table 1).
Table 1.
Effects of the CB1 receptor neutral antagonist AM4113 on drug-taking and drug-seeking behavior in experimental animals
| Dose (mg/kg) | Species | Results | References |
|---|---|---|---|
| IM 0.32–5.6 | Monkey | ↓ Discriminative stimulus of CB1 agonists | [129] |
| IP 1, 2.5 | Rat | ↓ Naloxone-precipitated CPA | [130] |
| No effect on reinstatement of naloxone CPA | |||
| IP 0.3–10 | Rat | ↓ PR responding for cocaine | [120] |
| IM 0.3–3 | Monkey | No effect on cocaine self-administration | [232] |
| ↓ Cue-induced reinstatement of cocaine seeking | |||
| ↓ Drug-primed reinstatement of cocaine seeking | |||
| ↓ Nicotine self-administration | |||
| ↓ Cue-induced reinstatement of nicotine seeking | |||
| ↓ Drug primed-reinstatement of nicotine seeking | |||
| ↓ Δ9-THC self-administration | |||
| ↓ Cue-induced reinstatement of Δ9-THC seeking | |||
| ↓ Drug-primed reinstatement of Δ9-THC seeking | |||
| IP 0.3–10 | Rat | ↓ Nicotine self-administration | [120] |
| ↓ Cue-induced reinstatement of nicotine seeking | |||
| ↓ Drug induced-reinstatement of nicotine seeking | |||
| IP 3, 10 | Rat | No effect on cocaine self-administration | [118] |
| ↓ Methamphetamine self-administration | |||
| ↓ Heroin self-administration | |||
| No effect on brain-stimulation reward | |||
| IP 1, 3 | Mouse | No effect on alcohol consumption | [127] |
| ↓ Alcohol consumption |
CB1 type 1 cannabinoid receptor, CPA conditioned place aversion, IM intramuscularly, IP intraperitoneally, PR progressive-ratio, Δ9-THC Δ9-tetrahydrocannabinol, ↓ indicates decrease
8.2. AM4113
AM4113 is a novel CB1R neutral antagonist developed by Salamone et al. [123] in 2007. It is a pyrazole-3-carboxamide analog of SR141716A [124]. Unlike the inverse agonists (e.g., rimonabant and AM251), AM4113 does not alter forskolin-stimulated cAMP formation in vitro [125], suggesting a lack of an inverse agonist profile. In experimental animals, AM4113 inhibited food intake to a similar degree as AM251 but, unlike AM251, did not potentiate vomiting in the ferret or promote nausea [124, 125]. It also did not produce malaise or anxiety-like effects [126]. In a BSR paradigm, rimonabant decreased BSR but AM4114 did not, suggesting that AM4113 did not produce aversive or rewarding effects [118]. All these data suggest that AM4113 has an improved safety profile over CB1R inverse agonists.
Recent studies indicated that AM4113 reduced alcohol consumption and preference in mice [127] but not in alcohol-preferring rats [96]. Similarly, AM4113 caused significant reductions in nicotine self-administration under fixed ratio 5 (FR5) and progressive-ratio (PR) schedules of reinforcement [120], suggesting reductions in nicotine reward and motivation to seek the drug. AM4113 also dose dependently reduced reinstatement of nicotine seeking induced by drug priming, stress, and drug cues, suggesting substantial therapeutic value in relapse prevention [120]. In line with these reports, we recently found that AM4113 also dose dependently inhibited heroin self-administration but was less effective at reducing cocaine or methamphetamine self-administration in rats [118]. In squirrel monkeys, AM4113 dose dependently attenuated self-administration of nicotine or Δ9-THC but not cocaine [128]. During the reinstatement test, upon presentation of the drug cue, squirrel monkeys reinstated their extinguished response for nicotine, Δ9-THC, or cocaine, whereas AM4113 pretreatment significantly reduced drug seeking. In addition, AM4113 was reported to reduce discriminative effects of CB1R agonists [129]. During a substitution test, when the drug of abuse (e.g., nicotine, Δ9-THC, or cocaine) was replaced with saline and drug priming was introduced, animals reinstated drug seeking but showed a significant reduction in responding under AM4113 treatment [128]. AM4113 also reduced conditioned place aversion (CPA) induced by naloxone-precipitated morphine withdrawal but failed to interfere with the reinstatement of the CPA [130]. Although not all findings are fully consistent, most of the results are promising, suggesting that this CB1R-neutral antagonist has therapeutic value in the treatment of SUDs, particularly cigarette smoking cessation (Table 1).
8.3. LH-21
LH-21 is a triazole-derived compound with anorectic properties that was initially identified as a neutral CB1R antagonist [131] and later considered a weak CB1R inverse agonist [132]. In vitro and in vivo assays have shown that LH-21 has low brain-penetration ability [133, 134]. For example, intraperitoneal systemic administration of LH-21 did not antagonize the motor inhibition induced by central administration of CP55940 in Wistar rats [133]. Acute administration of LH-21 reduced feeding behavior in food-deprived rats, whereas subchronic administration reduced food intake and body weight gain in genetically obese Zucker rats (i.e., leptin receptor mutant rats) [133]. It also reduced feeding and body weight gain in Wistar rats fed a high-fat diet [135]. In addition, just like rimonabant, LH-21 dose dependently inhibited alcohol self-administration and alcohol intake but to a lesser degree [133]. These data suggest that the effects of LH-21 on feeding and alcohol-taking behavior could be mediated, mainly by acting on peripheral CB1Rs. However, this compound has yet to be determined as a potential treatment for other SUDs.
8.4. PIMSR
PIMSR (or PIMSR1) is another neutral CB1R antagonist [122]. In experimental animals, both rimonabant and PIMSR significantly inhibited cocaine-enhanced BSR in rats [136]. However, rimonabant itself produced dose-dependent dysphoric effects in the BSR model, whereas PIMSR did not, suggesting that PIMSR may antagonize the acute rewarding effects of drugs of abuse but does not itself produce aversive effects [125]. These findings suggest that this neutral CB1R antagonist deserves further study as potential pharmacotherapy for SUDs.
9. Negative Allosteric Modulators of CB1Rs as Potential Treatments for SUDs
9.1. Rationale
A new emerging approach in medication discovery research is to target allosteric sites of CB1Rs. Allosteric modulators are ligands that bind to the allosteric binding site of a receptor and can remotely modify the conformation of the primary orthosteric binding site. Consequently, conformational changes affect the affinity of orthosteric ligands and respective signaling. Positive allosteric modulators (PAMs) can increase the binding of the orthosteric agonists. Conversely, negative allosteric modulators (NAMs) can decrease orthosteric agonist binding. One beneficial characteristic of allosteric modulators over traditional orthosteric ligands is that they do not block, but only modulate, eCB binding to the orthosteric binding site. Therefore, allosteric modulators have no significant effect on basal eCB function and, therefore, possibly have fewer unwanted side effects. In addition, highly selective CB1R allosteric modulators may also circumvent adverse side effects that often derive from the activation of multiple signaling pathways by nonselective orthosteric ligands.
9.2. Org27569
Org27569 is a positive allosteric modulator of CB1R [137]. Studies in vitro suggest that it binds to an allosteric regulatory site on the CB1R, causing a conformational change that increases the binding affinity of CB1R agonists such as CP 55,940. However, once bound, Org27569 decreases their efficacy at stimulating second messenger signaling and so behaves as a functional antagonist of CB1R [137]. Jing et al. [138] reported that rats treated with Org27569 showed a reduction in cue-induced or drug-induced reinstatement of cocaine and methamphetamine seeking. Similar results were observed in rats treated with rimonabant, indicating that both ligands produce similar effects [138]. However, unlike rimonabant, Org27569 did not produce CB1R-related adverse effects [139]. Org27569 was also reported to decrease food intake and body weight in rodents [139, 140], suggesting its therapeutic potential for weight loss and obesity. Surprisingly, Org27569 also affected food consumption in CB1R-KO mice, raising a possibility that its pharmacological effects might be mediated by other non-CB1Rs [139]. There are also conflicting reports regarding its effect on CB1R-dependent tetrad effects. Ding et al. reported that Org27469 can attenuate hypothermia induced by CP55, 940 [140], whereas others found that Org27569 was ineffective at reducing analgesia, hypothermia, and catalepsy induced by Δ9-THC, AEA, or CP55, 940 [139]. Org27469 did not substitute for either AEA or Δ9-THC, nor did it modify the discriminative stimulus effects of these ligands in the drug discrimination paradigm [139].
9.3. Pepcan-12
Pepcan-12 (also known as RVD-Hpα and RVD-hemopressin) was recently localized in discrete cells of the CNS and adrenal glands [141] and identified as an NAMs of CB1Rs based on its ability to cause a loss in agonist binding, negatively modulate agonist-induced accumulation of cAMP, and abolish agonist-induced guanosine 5′-O-[γ-thio]triphosphate (GTPγS) binding [142]. There is also a report that pepcan-12 acts as a positive CB2R allosteric modulator (CB2R agonist) (Ki value ~ 50 nM) and potentiates (five- to tenfold) the effects of CB2R agonists in both [35S]GTPγS binding and intracellular cAMP assays [143]. Given the unique characteristics of pepcan-12 as an allosteric modulator of CB1Rs and CB2Rs, several groups evaluated its therapeutic potential. Pepcan-12 has been shown to reduce food consumption in rats, increase locomotor activity [144, 145], and induce anxiolytic and antidepressive effects, as assessed in locomotor activity/open field, light–dark exploration, and forced swim tests [146]. To date, no study has evaluated its therapeutic potential for the treatment of SUDs.
In summary, in the last 2 decades, significant progress has been made in CB1R-based medication development for the treatment of obesity and SUDs. Although the initial studies showed promising results with rimonabant, a selective CB1R antagonist or inverse agonist, serious adverse side effects associated with this compound led the cannabinoid research community to shift their interest to peripheral CB1R antagonists, neutral CB1R antagonists, or NAMs of CB1Rs. Peripheral CB1R antagonists such as JD5037 and LH-21 have shown promise as therapeutics for obesity and alcohol abuse as they can inhibit food intake and alcohol consumption. Neutral CB1 antagonists such as AM4113 can reduce nicotine, alcohol, and heroin self-administration and impede relapse without producing negative side effects, making AM4113 a great therapeutic candidate for the treatment of SUDs.
10. CB2R Agonists as Potential Treatments for SUDs
10.1. Rationale
An important finding in cannabinoid research in the last decade is the identification of functional CB2Rs in the brain, particularly in the mesolimbic DA system (Fig. 2) [80, 81]. Therefore, interest is growing in CB2Rs as potential therapeutic targets for the treatment of SUDs and other disorders. CB2Rs were previously regarded as peripheral cannabinoid receptors [147]. However, recent advances in genetics and molecular techniques have allowed us to detect low levels of CB2R expression in the brain [80, 81]. CB2Rs have been found on neurons of multiple brain regions, including the cerebellum, hippocampus, globus pallidus, and midbrain [148, 149]. We and others have identified the presence of CB2Rs on cell bodies of DA neurons in the VTA [83, 84] as well as on the terminals of these neurons in the NAc [85, 88]. Although our understanding of the physiological relevance of brain CB2Rs remains incomplete, evidence suggests they play an important role in reward and addiction. For example, genetic deletion of CB2Rs has been reported to reduce nicotine reward and intake [82, 150–152] but also to increase preference for and vulnerability to alcohol consumption [153, 154]. Correspondingly, overexpression of CB2Rs in the brain has been associated with reduced cocaine sensitization, cocaine self-administration, and cocaine CPP [88]. Similarly, CB2R expression in rats with a history of cocaine self-administration decreased in the NAc and medial globus pallidus but increased in the PFC, VTA, and lateral septal nuclei after cocaine priming [155, 156]. These findings suggest that brain CB2Rs are important in many addiction-related behaviors through interaction with the DA reward circuit. As a result, several research avenues are currently being explored to identify efficacious CB2R agonists in the treatment of SUDs (Table 2).
Table 2.
Effects of CB2 agonists on drug-taking and drug-seeking behavior in experimental animals
| Drug and dosage | Species | Results | References |
|---|---|---|---|
| JWH133 | |||
| IP 10, 20 mg/kg | Mouse | ↓ Cocaine self-administered in WT, CB1-KO, not in CB2-KO | [82] |
| ↓ PR responding for cocaine in mice | |||
| ↓ DA release in the NAc | |||
| IP 10, 20 mg/kg | Rat | No effect on FR cocaine self-administration | [87] |
| ↑ PR responding for cocaine | |||
| IP 10, 20 mg/kg | Mouse | ↓ Cocaine self-administration | [87] |
| ↓ PR responding for cocaine | |||
| IP 3, 10 mg/kg | Rat | ↓ Acquisition of cocaine CPP | [86] |
| ↓ Expression of cocaine CPP | |||
| ↓ Cocaine-induced hyperlocomotion | |||
| IP 3 mg/kg | Mouse | ↓ Expression of cocaine CPP | [159] |
| ↓ Nicotine CPP | |||
| IP 10, 20 mg/kg | Mouse | ↓ Sucrose self-administration in WT and CB1-KO, not in CB2-KO | [257] |
| 1–100 μM, local | Mouse | ↓ VTA DA neuronal firing | [82, 158] |
| GW405833 | |||
| IP 3, 10 mg/kg | Mouse | ↓ Cocaine self-administration in mice | [82] |
| O-1966 | |||
| IP 1–20 mg/kg | Mouse | ↑ Nicotine CPP at low doses | [150] |
| IP 20 mg/kg | ↓ Cocaine CPP | ||
| AM1241 | |||
| IP 1, 3, 10 mg/kg | Rat | No effect on nicotine self-administration | [89] |
| No effect on drug-induced reinstatement of nicotine seeking | |||
| No effect on cue-induced reinstatement of nicotine seeking | |||
| IP 1, 3 mg/kg | Mouse | ↓ Morphine tolerance and withdrawal | [164] |
| Xie2–64 | |||
| IP 10, 20 mg/kg | Mouse | ↓ Cocaine self-administration in WT, not in CB2-KO | [169] |
| ↓ Cocaine-enhanced optical ICSS; ↓ Optical ICSS maintained by stimulation of VTA DA neurons | |||
| IP 10, 20 mg/kg | Rat | ↓ Cocaine self-administration; ↓ PR responding for cocaine;↓ DA release in the NAc | |
| AM1710 | |||
| IP 1, 3, 10 mg/kg | Mouse | ↓ Morphine physical dependence | [165] |
| JHW015 | |||
| IP 1–100 mg/kg | Mouse | ↑ Morphine analgesia | [166] |
| ↓ Morphine CPP | |||
| ↓ Morphine-induced DA release |
CB1 type 1 cannabinoid receptor, CB2 type 2 cannabinoid receptor, CPP conditioned place preference, DA dopamine, DAT dopamine transporter, DAT-cre cre-recombinase under the control of the dopamine transporter promoter, ICSS intracranial self-stimulation, IP intraperitoneally, KO knock-out, NAc nucleus accumbens, FR fixed-ratio, PR progressive-ratio, VTA ventral tegmental area, WT wild-type, ↓ and ↑ represent decrease and increase, respectively
10.2. JWH133
JWH133 is a highly selective CB2R agonist with about 200-fold selectivity for CB2Rs versus CB1Rs [157]. Stimulation of CB2Rs by JWH133 has been shown to inhibit VTA DA neuron firing and DA release in the NAc [82, 84]. Specifically, JWH133 reduces DA neuron firing by prolonging action potential initiation, enhancing after hyperpolarization amplitude, and hyperpolarizing membrane potential through CB2R-mediated reduction of intracellular cAMP and enhancement of M-type K+ currents [158]. We previously reported that systemic and local administration of JWH133 into the VTA or NAc reduced cocaine self-administration in WT and CB1-KO mice but not in CB2-KO mice [82–84, 87]. Although JWH133 was less effective at attenuating cocaine self-administration in rats [87], it blocked the acquisition and expression of cocaine-induced CPP and reduced cocaine-elicited conditioned hyperactivity in rats and mice [86, 159]. In addition, JWH133 also reduced nicotine CPP [159], and GW405833—another highly potent and selective CB2R agonist [160]—inhibited cocaine self-administration in mice [82]. These data suggest that this CB2R agonist can reduce drug reward (Table 2). In contrast, it was also reported that SR144,528 (a CB2R antagonist/inverse agonist) significantly reduced drug-primed reinstatement of cocaine seeking [161].
10.3. O-1966
O-1966 is another highly selective synthetic CB2R agonist with over 200-fold selectivity for CB2Rs versus CB1Rs (Ki = 23 vs. 5055 nM, respectively) [162]. Like the attenuating effects of JWH133 on cocaine-related behaviors, O-1966 also blocked cocaine-induced CPP [150]. In addition, it reduced or delayed the development of opioid tolerance and morphine physical dependence [163–165]. Likewise, O-1966 and JHW015 (another CB2R agonists) also potentiated the antinociceptive effects of morphine and blocked morphine CPP [163, 166], suggesting their therapeutic potential in the management of cocaine or opiate abuse. However, when given in combination with a subthreshold dose of nicotine, O-1966 produced nicotine CPP [150], suggesting a differential role of CB2Rs in cocaine versus nicotine reward. This is supported by findings that genetic deletion or pharmacological blockade of CB2Rs by AM630 or SR144528 attenuated nicotine-induced CPP [150, 151], nicotine self-administration [151], and nicotine withdrawal symptoms [151] (but see Ignatowska-Jankowska et al. [150]) (Table 2).
10.4. AM1241
AM1241 is another selective CB2R agonist with ~ 70-fold selectivity for CB2Rs over CB1Rs [167]. Both agonists AM1241 and AM1710 reduced or delayed the development of opioid tolerance and morphine physical dependence [163–165]. In contrast, it was also reported that AM1241 at lower doses (1–10 mg/kg intraperitoneally), failed to alter nicotine self-administration or reinstatement of nicotine seeking [89], suggesting a differential role of CB2Rs in nicotine reward, as stated.
10.5. Xie2–64
Xie2–64 is a novel CB2R inverse agonist with high affinity for CB2R (Ki = 0.5 nM) and high selectivity for CB2Rs versus CB1s (> 2500-fold) [168]. Given the findings with various CB2R agonists already described, we also evaluated the potential utility of this novel CB2R inverse agonist for the treatment of cocaine addiction using animal models of drug abuse (Table 2). We found that systemic administration of Xie2–64 dose dependently inhibited cocaine self-administration and shifted cocaine dose-response curves downward in rats and WT mice but not in CB2-KO mice [169]. Xie2–64 also dose dependently attenuated cocaine-enhanced BSR maintained by optical stimulation of VTA DA neurons in transgenic mice expressing Cre recombinase under the promoter of DAT-cre mice. In vivo microdialysis studies in rats revealed that Xie2–64 dose-dependently reduced the extracellular DA level in the NAc [169]. Together, these results suggest that Xie2–64 can reduce cocaine self-administration and cocaine intake, most likely via a DA-dependent mechanism.
In summary, several studies have provided evidence supporting the premise that CB2R agonists have therapeutic potential for the treatment of obesity and SUDs. Compounds such as JWH 133, Xie2–64, and BCP (see Sect. 14) share the ability to reduce drug self-administration and drug seeking in rodents, making them frontier candidates in medication development for nicotine, heroin, and cocaine use disorders (Table 2).
11. Fatty Acid Amide Hydrolase Inhibitors as Potential Therapeutics for SUDs
11.1. Rationale
Fatty acid amide hydrolase (FAAH) is an enzyme responsible for degradation of AEA, an eCB produced on demand in response to elevated intracellular calcium levels or depolarization of DA neurons [57, 170]. Enhancement of endogenous AEA levels activates CB1Rs and CB2R, producing effects similar to those of systemically administered cannabinoid receptor agonists (Fig. 1). Therefore, FAAH inhibitors could be promising therapeutics for the treatment of SUDs (Table 3) [171, 172].
Table 3.
Effects of fatty acid amide hydrolase inhibitors on drug-taking and drug-seeking behavior in experimental animals
| Drug and dosage | Species | Results | References |
|---|---|---|---|
| URB597 | |||
| IP 0.01–0.3 mg/kg | Rat | No effect on PR responding for heroin | [177] |
| IP 0.3–3 mg/kg | Rat | ↓ Brain-stimulation reward | [230] |
| IP 0.5 mg/kg | Mouse | ↓ Preference for alcohol | [189] |
| IV 0.1 mg/kg | Rat | ↓ VTA DA neuron response to nicotine | [187] |
| IP 0.1, 0.3 mg/kg | Rat | ↓ Acquisition of nicotine self-administration | [182] |
| ↓ Acquisition of nicotine CPP | |||
| ↓ Drug-primed reinstatement of nicotine seeking | |||
| IP 0.1–1 mg/kg | Rat | No effect on voluntary alcohol drinking | [191] |
| No effect on PR responding for alcohol | |||
| No effect on drug-, cue-, or stress-induced reinstatement of alcohol seeking | |||
| ↓ Anxiety during alcohol withdrawal | |||
| IV 0.3 mg/kg | Monkey | No effect on reinstatement of alcohol seeking | [174] |
| ↓ Sensitivity to alcohol sedation | |||
| ↑ Recovery from alcohol-induced motor incoordination | |||
| IP 1–3 mg/kg | Rat | No effect on cocaine self-administration | [175] |
| ↓ Drug-primed reinstatement of cocaine seeking | |||
| ↓ Cue-induced reinstatement of cocaine seeking | |||
| IP 0.3, 1 mg/kg | Rat | ↓ Cue-induced reinstatement of nicotine seeking | [183] |
| ↓ Drug-induced reinstatement of nicotine seeking | |||
| ↓ Drug-induced reinstatement of nicotine seeking | |||
| IP 0.03–0.3 mg/kg | Rat | ↑ Extinction of CPA in morphine-treated rats | [179] |
| IV 0.1 mg/kg | Rat | ↓ NAc GABA neuron response to nicotine or cocaine | [188] |
| No effect on VTA DA neuron response to cocaine or morphine | |||
| IP 0.3 mg/kg | Rat | No effect on morphine CPP | [178] |
| No effect on CPA induced by the morphine withdrawal | |||
| IP 0.1–1 mg/kg | Rat | ↓ Naloxone-induced opioid withdrawal symptoms | [181] |
| IP 0.1, 0.3 mg/kg | Rat | ↓ Anxiety during nicotine withdrawal | [186] |
| No effect on somatic symptoms of nicotine withdrawal | |||
| IP 0.3, 1, 3 mg/kg | Rat | ↓ Cue-induced reinstatement of cocaine seeking | [176] |
| ↓ Stress-induced reinstatement of cocaine seeking | |||
| IV 0.1–1 mg/kg | Monkey | No effect on cocaine self-administration | [185] |
| ↓ Nicotine self-administration | |||
| ↓ Cue-induced reinstatement of nicotine seeking | |||
| IP 0.3 mg/kg | Rat | ↓ Drug-primed reinstatement of nicotine seeking | [183] |
| IP 0.5 mg/kg | Mouse | ↓ Alcohol consumption | [192] |
| ↓ Alcohol tolerance | |||
| IP 10 mg/kg | Mouse | ↓ Withdrawal symptoms from Δ9-THC | [218] |
| PF-3845 | |||
| IP 10 mg/kg | Mouse | ↓ Naloxone-precipitated withdrawal from morphine | [180] |
| IP 1–10 mg/kg | Mouse | No effect on morphine withdrawal symptoms | [218] |
| No effect on morphine withdrawal-induced CPA |
CPA conditioned place aversion, CPP conditioned place preference, DA dopamine, GABA γ-aminobutyric acid, IP intraperitoneally, IV intravenously, NAc nucleus accumbens, PR progressive-ratio, VTA ventral tegmental area, Δ9-THC Δ9-tetrahydrocannabinol, ↓ and ↑ represent decrease and increase, respectively
11.2. URB597
URB597 is a relatively selective FAAH inhibitor [173] that almost completely suppresses FAAH activity, causing significant increases in AEA levels in brain regions involved in motivational, emotional, and cognitive functions [174]. It has been reported that acute treatment with URB597 did not alter cocaine self-administration [174, 175]. It also failed to alter cocaine-induced reinstatement of drug seeking in monkeys [174] but effectively reduced cocaine seeking in rats [175, 176]. These findings suggest limited applications for this FAAH inhibitor in the treatment of cocaine use disorder.
Similarly, the acute administration of URB597 neither altered heroin self-administration [177] nor interfered with CPA induced by morphine withdrawal [130, 178]. However, URB597 facilitated the extinction of CPA in rats with a history of morphine exposure [179] and attenuated naloxone-precipitated opioid withdrawal symptoms [180, 181], suggesting that inactivation of FAAH might help alleviate opiate withdrawal symptoms.
However, chronic administration of URB597 blocked the acquisition of nicotine self-administration and nicotine-induced CPP [182]. In addition, the acute administration of URB597 significantly inhibited reinstatement of nicotine seeking [182–185] and abolished anxiogenic responses during withdrawal from nicotine [186]. URB597 did not alter the neuronal activity of the VTA DA neurons in response to nicotine or cocaine, while it inhibited the neuroal activity of NAc shell GABAergic neurons in response to nicotine or cocaine [187, 188], suggesting that a DA-dependent mechanism may be involved in the therapeutic effects of URB597 [189]. Together, these findings support the potential utility of URB597 in the treatment of nicotine use disorder.
FAAH inhibitors have also been extensively studied in relation to alcohol consumption and abuse. In a two-bottle choice paradigm, FAAH-KO mice, especially females, showed higher preference for alcohol and increased alcohol consumption than did WT mice [189, 190]. FAAH-KO mice can develop alcohol CPP and show alcohol-induced withdrawal symptoms [189]. Similar behavioral phenotypes (increased alcohol consumption) were observed in WT mice treated with URB597, suggesting that FAAH plays a critical role in regulation of alcohol intake [189]. However, acute administration of URB597 failed to alter voluntary home-cage alcohol drinking or self-administration of alcohol in rats under a PR schedule of reinforcement [191]. In the reinstatement tests, URB597 also failed to alter reinstatement of alcohol seeking induced by cue, foot-shock stress, and yohimbine [191]. In contrast, URB597 was reported to reduce alcohol consumption in mice after 1 day or 1 week of withdrawal from chronic alcohol drinking [192]. Repeated administration of URB597 also prevented the development of tolerance and reduced escalation of alcohol consumption [192]. In addition, URB597 abolished anxiogenic responses during withdrawal from alcohol [191]. These findings suggest that chronic administration of URB597 may have therapeutic potential for the treatment of alcohol use disorder (Table 3).
It is worth noting that FAAH inhibitors themselves have low abuse liability as they do not increase extracellular DA levels in the NAc [193], do not produce CPP [194], and are not readily self-administered [174, 195]. Several studies have also reported that URB597 does not affect the consumption of or response to saccharin or sucrose [179, 189, 190]. When administered at high doses that profoundly inhibit brain FAAH activity, URB597 itself does not cause catalepsy or hypothermia or increase appetite, effects that are commonly observed with cannabinoid administration [196, 197]. Other beneficial characteristics of URB597 are its good oral bioavailability and low toxicity profile, even with a chronic regimen of up to 1500 mg/kg per treatment in rodents [197].
11.3. BIA10–2474
Based on these findings, some FAAH inhibitors entered clinical trials [198–200]. Despite encouraging results at the preclinical and clinical levels, a recent clinical trial in France with BIA10–2474, another FAAH inhibitor, tempered initial enthusiasm in the research community after healthy volunteers developed severe neurological impairments and one become brain dead. This tragic outcome led to immediate discontinuation of FAAH inhibitor testing [201, 202] and termination of related research projects by major pharmaceutical companies for some time [203, 204]. However, after a comprehensive review of safety information relevant to BIA10–2474, the FDA found that other FAAH inhibitors did not pose similar safety risks [205, 206]. Consequently, clinical trials have now restarted with new generations of FAAH inhibitors (e.g., PF-0447845, V158866, ASP3652) that do not produce toxicity and are well-tolerated by healthy individuals [198–200, 207, 208]. The clinical study assessing the therapeutic potential of PF-0447845 for cannabis use disorder found that treatment with PF-04457845 was associated with reduced cannabis withdrawal symptoms, self-reported cannabis use, and drug-positive urine samples [209] (Table 6).
Table 6.
Effects of Δ9-tetrahydrocannabinoid, cannabidiol, and PF-04457845 (a fatty acid amide hydrolase inhibitor) on cannabis or opioid use disorders in humans
| Drug and dosage | Participants | Results | References |
|---|---|---|---|
| Dronabinol (Δ9-THC) | |||
| 50 mg, 6 days | Healthy cannabis users (n = 7). Placebo-controlled within-subject study | ↓ Withdrawal severity from cannabis | [240] |
| ↓ Withdrawal cravings for cannabis | |||
| 30, 90 mg, 5 days | Healthy cannabis users (n = 8). Placebo-controlled within-subject study | ↓ Withdrawal severity from cannabis | [239] |
| 60 mg, 7 days | Healthy cannabis users (n = 8). Placebo-controlled within-subject study | ↓ Withdrawal severity from cannabis | [238] |
| No effect on abstinence rate from cannabis | |||
| 40 mg, 8 weeks | Individuals with CUD (n = 156). Randomized double-blind, placebo-controlled study | ↓ Withdrawal severity from cannabis | [233] |
| No effect on abstinence rate from cannabis | |||
| 30, 60, 120 mg, 5 days | Healthy cannabis users (n = 13). Placebo-controlled within-subject study | ↓ Withdrawal severity from cannabis | [235] |
| No effect on subjective effect of cannabis | |||
| 60 mg, 8 weeks | Individuals with CUD (n = 156). Randomized double-blind, placebo-controlled study | No effect on abstinence rate from cannabis | [237] |
| 10, 20, 30 mg, 5 weeks (outpatient) in combination with naltrexone | Individuals with OUD (n = 60). Randomized double-blind, placebo-controlled, between-subject study | ↓ Withdrawal symptoms from opioids | [242] |
| 5, 10, 20, 30, 40 mg, 5 days | Individuals with CUD (n = 12). Randomized double-blind, placebo-controlled, within-subject study | ↓ Withdrawal symptoms from opioids | [241] |
| 5, 10, 20, 30, 40 mg, 5 weeks | Individuals with CUD (n = 12). Randomized double-blind, placebo-controlled, within-subject study | ↓ Withdrawal symptoms from opioids | [243] |
| Nabilone (Δ9-THC analog) | |||
| 6, 8 mg, 7 days | Healthy cannabis users (n = 11). Placebo-controlled within-subject study | ↓ Withdrawal severity from cannabis | [236] |
| ↓ Abstinence rate from cannabis | |||
| 6 mg nabilone + 12.5 mg zolpidem, 7 days | Healthy cannabis users (n = 11). Placebo-controlled within-subject study | ↓ Cannabis use (intake) | [234] |
| ↓ Withdrawal severity from cannabis | |||
| Nabiximols (Δ9-THC + CBD) | |||
| 86.4 + 80 mg, 6 days | Individuals with CUD (n = 78). Randomized double-blind, placebo-controlled, between-subject study | No effect on cannabis use (intake) | [274] |
| ↓ Withdrawal severity from cannabis | |||
| 113.4 + 105 mg, 12 weeks | Individuals with CUD (n = 40). Randomized double-blind, placebo-controlled, between-subject study | ↓ Cannabis use | [275] |
| No effect on withdrawal symptoms | |||
| ↓ Craving for cannabis | |||
| CBD | |||
| 750, 1500, 4500 mg, single dose | Healthy polydrug users (n = 43). Randomized, double-blind, double-dummy, placebo- and active-controlled crossover study | Well-tolerated | [267] |
| Very low abuse potential | |||
| Very low subjective effects | |||
| 400, 800 mg, 1 week | Healthy opioid users (n = 6). Randomized double-blind, placebo-controlled, between-subject study | Well-tolerated | [268] |
| No effect on subjective effect of fentanyl | |||
| No effect on fentanyl-induced adverse effect | |||
| 150 mg/kg, single dose | Healthy individuals (n = 15). Double-blind, within-subject study | No effect on subjective effects of Δ9-THC | [269] |
| 15, 30, 60 mg, single dose | Healthy individuals (n = 40). Double-blind, placebo-controlled, between-subject study | No effect on subjective effects of Δ9-THC | [270] |
| ↓ Δ9-THC-induced anxiety | |||
| 1 mg/kg, single dose | Healthy individuals (n = 8). Double-blind, placebo- and diazepam-controlled between-subject study | No effect on subjective effects of Δ9-THC | [271] |
| ↓ Δ9-THC-induced anxiety | |||
| 200, 400, 800 mg, single dose | Healthy cannabis users (n = 31). Randomized double-blind, placebo-controlled, within-subject study | No effect on the subjective, rewarding, or cardiovascular effects of cannabis | [272] |
| 4, 400 mg, single dose | Healthy cannabis users (n = 36). Randomized, placebo-controlled study | Low dose enhanced and high dose reduced the acute intoxicating effects of Δ9-THC | [273] |
| PF-04457845 | |||
| 4 mg × 5–8 days (inpatient) + 3 weeks (outpatient) | Individuals with CUD (n = 70). Randomized, double-blind, placebo-controlled, between-subject study | ↓ Cannabis withdrawal symptoms | [209] |
| ↓ Cannabis use |
CBD cannabidiol, CUD cannabis use disorders, OUD opioid use disorders, wk week(s), Δ9-THC Δ9-tetrahydrocannabinol, ↓ represents decrease
12. Monoacylglycerol Lipase Inhibitors as Potential Treatment for SUDs
12.1. Rationale
Monoacylglycerol lipase (MAGL) is an enzyme responsible for the metabolism of 2-AG in the brain and is also involved in lipid signaling [210] (Fig. 1). Blockade of MAGL, much like that of FAAH, elevates brain 2-AG levels and produces Δ9-THC–like effects such as hypolocomotion, hypothermia, and analgesia via activation of brain CB1Rs and CB2Rs [20, 75, 211, 212]. A growing number of reports indicates that MAGL inhibitors produce antinociceptive, anti-inflammatory, anxiolytic, and antiemetic effects by enhancing eCB signaling via the CB1Rs [213, 214]. There is also evidence that MAGL inhibitors may have neuroprotective functions through CB2R stimulation [215]. Drugs of abuse such as cocaine and heroin stimulate 2-AG release and modulate the MAGL expression in the hippocampus, PFC, and cerebellum [216, 217], suggesting that MAGL inhibitors may serve as indirect cannabinoid receptor agonists for the treatment of SUDs (Table 4).
Table 4.
Effects of monoacylglycerol lipase inhibitors on drug-taking and drug-seeking behavior in experimental animals
| Drug and dosage | Species | Results | References |
|---|---|---|---|
| JZL184 | |||
| IP 16 mg/kg | Mouse | ↓ Withdrawal symptoms from Δ9-THC | [218] |
| IP 40 mg/kg | Mouse | ↓ Somatic signs of morphine withdrawal | [180] |
| IP 16 mg/kg | Mouse | ↓ Methamphetamine-induced DA neurotoxicity | [215] |
| IP 4, 40 mg/kg | Mouse | ↓ Brain-stimulation reward | [75] |
| IP 4–40 mg/kg | Mouse | ↓ Nicotine withdrawal symptoms | [219] |
| IP 4, 40 mg/kg | Mouse | No effect on naloxone-precipitated CPA | [220] |
| ↓ Somatic signs of morphine withdrawal | |||
| IP 8, 16 mg/kg | Mouse | No effect on food taking | [13] |
| No effect on nicotine taking | |||
| No effect on PR responding for nicotine | |||
| ↑ Cue-induced reinstatement of nicotine seeking | |||
| IP 2–18 mg/kg | Mouse | ↑ PR responding for alcohol | [226] |
| SA-57 | |||
| IP 1–13 mg/kg | Mouse | ↓ Somatic signs of morphine withdrawal | [220] |
| No effect on naloxone-induced CPA | |||
| IP 3–18 mg/kg | Mouse | ↓ Brain-stimulation reward | [75] |
| IP 1–13 mg/kg | Mouse | ↓ Heroin self-administration | [221] |
| ↓ PR responding for heroin | |||
| ↓ Cue-induced reinstatement of heroin seeking |
CPA conditioned place aversion, DA dopamine, IP intraperitoneally, PR progressive-ratio, Δ9-THC Δ9-tetrahydrocannabinol, ↓ and ↑ represent decrease and increase, respectively
12.2. JZL184
JZL184 is an irreversible MAGL inhibitor [20]. Acute administration of JZL184 did not affect food or nicotine taking or reduce motivation for nicotine, as assessed on fixed ratio 1 (FR1, i.e., one lever press leads to one drug infusion) and PR schedules of reinforcement, but it increased cue-induced nicotine seeking [13], suggesting that JZL184 or other MAGL inhibitors may not be the optimal therapeutic choice for the management of SUDs. However, pharmacological inactivation of MAGL by JZL184 or genetic deletion of MAGL has been associated with a reduction in somatic or aversive withdrawal symptoms from nicotine and morphine [180, 218–220]. JZL184 (and URB597) also dampened methamphetamine-induced neurotoxicity in the striatum by a CB2R-dependent mechanism [215], suggesting its possible utility in the management of drug withdrawal and neurotoxicity (Table 4).
12.3. SA-57
Interestingly, concurrent FAAH–MAGL inhibitors such as SA-57 can reduce heroin self-administration and heroin seeking in mice [221] and morphine withdrawal CPA in rats [220]. SA-57 also produces enhanced morphine analgesia, making it a great therapeutic candidate for the treatment of chronic pain and opioid abuse. In an electrical BSR paradigm, concurrent inhibition of FAAH and MAGL by SA-57 or coadministration of JZL184 and PF-3845 (an FAAH inhibitor) significantly inhibited electrical BSR [75] (Table 4). The CB1R inverse agonist rimonabant, but not CB2 antagonist SR144528, blocked the attenuating effects of JZL184 or SA-57, suggesting that FAAH and MAGL may function as regulators that prevent CB1R overstimulation by eCBs [75].
In summary, FAAH- or MAGL-based strategies are currently being explored as potential treatments for SUDs. At the preclinical level, FAAH inhibitors (e.g., URB597) have showed promise as potential therapeutics for nicotine and alcohol but not cocaine and opioid use disorders. Clinical studies have shown that a new generation of FAAH inhibitors might be useful for reducing withdrawal symptoms and cannabis use in cannabis use disorders. There is little evidence suggesting that MAGL inhibitors are effective in reducing food or drug-taking behavior.
13. Fatty Acid Binding Proteins Inhibitors as Potential Treatment for SUDs
13.1. Rationale
In addition to the breakdown of AEA by FAAH, it is also terminated through reuptake into neurons [222–224]. FABPs are proteins that transport AEA, 2-AG, and AA derivatives between extra- and intracellular membranes and to the FAAH for breakdown (Fig. 1). FABP inhibitors can extend the availability of AEA, thus prolonging stimulation of cannabinoid receptors, which ultimately reduces the rewarding and relapse-inducing effects of illicit drugs.
13.2. AM404
AM404 is an FABP or AEA transporter inhibitor [225] and reduces alcohol self-administration and intake [186, 226], but its effect on relapse-like behaviors remains ambiguous. AM404 either reduced or failed to alter alcohol seeking [186, 226], suggesting that inactivation of AEA transporters can be an effective therapeutic approach to ameliorate the acute effects of alcohol but not relapse. In regard to the receptor mechanisms of action by which AM404 reduces alcohol consumption, it appears that AM404 action does not require the stimulation of CB1, CB2, or transient receptor potential vanilloid 1 (TRPV1) receptors, as antagonists to these receptors did not block the effects of AM404 [227].
AM404 has also been evaluated as a potential therapeutic agent for nicotine use disorder. AM404 dose dependently reduced cue- or drug-primed reinstatement of nicotine seeking but had no effects on nicotine self-administration under FR or PR schedules of reinforcement [228]. Paradoxically, AM404 was reported to reduce nicotine-induced DA elevation in the NAc [229], suggesting that it has the capacity to reduce the rewarding effects of nicotine. When administered during conditioning, AM404 prevented the development of nicotine CPP, presumably by reducing the rewarding effects of nicotine. It also reduced nicotine-induced reinstatement of CPP [229], suggesting that AM404 might have therapeutic value.
Although evidence supporting the efficacy of AM404 for SUDs is accumulating, the therapeutic utility of FABP inhibitors for cocaine or opioid use disorders remains to be elucidated. In a BSR paradigm, FABP inhibitors such as AM404 or OMDM-2 blocked the rewarding effects of BSR and cocaine-enhanced BSR [230, 231]. However, in a self-administration paradigm, AM404 failed to reduce motivation for heroin [177] or cocaine [232] (Table 5).
Table 5.
Effects of the fatty acid-binding protein inhibitor AM404 on drug-taking and drug-seeking behavior in experimental animals
| Drug and dosage | Species | Results | References |
|---|---|---|---|
| IP 1–10 mg/kg | Rat | No effect on PR responding for heroin | [177] |
| IP 0.4–10 mg/kg | Rat | ↓ Ethanol self-administration | [227] |
| No effect on alcohol seeking | |||
| ↑ Δ9-THC seeking | |||
| IP 1–30 mg/kg | Rat | ↓ Cocaine-enhanced brain-stimulation reward | [231] |
| ↓ Cocaine-induced hyperlocomotion | |||
| IP 1–10 mg/kg | Rat | ↓ Acquisition of nicotine CPP | [229] |
| ↓ Reinstatement of nicotine CPP | |||
| No effect on nicotine-induced hypolocomotion | |||
| ↓ NAc DA response to nicotine | |||
| IP 1–10 mg/kg | Rat | No effect on nicotine self-administration | [228] |
| No effect on PR responding for nicotine | |||
| ↓ Cue-induced reinstatement of nicotine seeking | |||
| ↓ Drug-primed reinstatement of nicotine seeking | |||
| IV 0.03–0.3 mg/kg | Monkey | No effect on cocaine self-administration | [128] |
| ↑ Cocaine-seeking | |||
| ↑ AEA self-administration | |||
| IP 10 mg/kg | Mouse | ↓ Ethanol approach | [226] |
| ↓ Ethanol self-administration | |||
| ↓ Alcohol seeking |
AEA anandamide, CPP conditioned place preference, DA dopamine, IP intraperitoneally, IV intravenously, NAc nucleus accumbens, PR progressive-ratio, Δ9-THC Δ9-tetrahydrocannabinol, ↓ and ↑ represent decrease and increase, respectively
In regard to its abuse potential, AM404 was readily self-administered by rhesus monkeys [232], but it neither produced CPP nor enhanced BSR [228, 231]. Clearly, more research is needed to determine the potential abuse liability of AM404.
In summary, preclinical studies have produced mixed results for FABP inhibitors as potential candidates for the treatment of SUDs. AM404 was reported to ameliorate the acute effects of alcohol but not relapse. However, AM404 shows efficacy for nicotine seeking but not nicotine consumption. Therefore, the potential utility of FABP inhibitors in the clinical setting would require more research evaluating the therapeutic potential of these compounds.
14. Phytocannabinoids as Potential Treatments for SUDs
14.1. Rationale
Phytocannabinoids refer to chemicals derived from cannabis sativa and non-cannabis plants. They can modulate the eCB system, producing therapeutic effects for several medical and neuropsychological disorders, including nausea, anorexia, obesity, epilepsy, Alzheimer disease, multiple sclerosis, cancer, anxiety, and depression. There are 113 known phytocannabinoids in the cannabis plant. Among the most prevalent and extensively studied are Δ9-THC, cannabidiol (CBD), BCP, and Δ9-THCV. They act at multiple pharmacological targets, not solely via CB1Rs and CB2Rs. We briefly review recent progress in treatment-oriented animal research on Δ9-THC, CBD, BCP, and Δ8-THCV (a Δ9-THCV analog) (Table 6).
14.2. Δ9-Tetrahydrocannabinol
As stated, cannabis is rewarding in some subjects under certain circumstances. Accordingly, cannabinoid agonist substitution has been proposed as a potential treatment for cannabis use disorder as it may reduce withdrawal symptoms, in a way similar to methadone or buprenorphine for opioid use disorder or the nicotine patch for nicotine use disorder [3, 233]. Δ9-THC is a partial CB1R agonist that has been proposed as an agonist therapy for SUDs, similar to methadone and buprenorphine for opioid use disorders and nicotine-replacement therapy for cigarette smoking cessation [3, 4]. Dronabinol (a stereoisomer of Δ9-THC) and nabilone (a synthetic analog of Δ9-THC), which are approved by the FDA for the management of anorexia with body weight loss in patients with AIDS and treatment-resistant nausea and vomiting associated with cancer chemotherapy, have both shown promising results in reducing withdrawal symptoms from cannabis [234–240] and opioid use in human clinical trials [241–243] (Table 6). However, dronabinol and nabilone may not prevent cannabis use or relapse [233]. These findings suggest that Δ9-THC-based agonist therapies may be useful in reducing cannabis or opioid withdrawal symptoms but not in preventing relapse to drug-seeking behavior [106].
14.3. Cannabidiol
Cannabidiol (CBD) is the second major component in cannabis plants, with a wide range of medical applications; it was recently approved by the FDA for the treatment of epilepsy [244, 245]. CBD has low affinity for the orthosteric binding sites of CB1Rs or CB2Rs [246–248]. More recent studies have provided evidence that CBD behaves as a potent NAM at CB1Rs or CB2Rs with nM range affinity [247–251]. CBD also has binding affinity to many other targets, such as GPR55, TRPV1, 5-HT1A, and opioid receptors.
Preclinical studies have shown that CBD can reduce drug reward, as demonstrated in various paradigms of addiction (Table 6). High-dose CBD can reduce self-administration of cocaine [252, 253], methamphetamine [254], and ethanol [255] but not heroin [256]. We recently reported that CBD reduced self-administration of cocaine maintained by low doses of cocaine, and this effect was mediated by multiple receptor mechanisms, including CB2R, 5-HT1A, and TRPV1 [253]. In addition, CBD attenuated oral sucrose self-administration in rats and WT mice but not in CB2-KO mice [257]. CBD also attenuated the rewarding effects of cocaine in a BSR paradigm via multiple receptor mechanisms, including CB2Rs [253]. An earlier study indicated that CBD had no effect on cocaine-enhanced BSR but attenuated morphine-enhanced BSR via stimulation of 5-HT1A receptors [258]. In a CPP paradigm, CBD disrupted the development of cocaine CPP [252] and blocked the expression of CPP induced by cocaine [252] or morphine [259] but not by amphetamine [260] (Table 7).
Table 7.
Effects of phytocannabinoids on drug-taking and drug-seeking behavior in experimental animals
| Drug and dosage | Species | Results | References |
|---|---|---|---|
| CBD | |||
| IP 5–20 mg/kg | Mouse | ↓ Naloxone-precipitated withdrawal from morphine | [264] |
| IP 5 mg/kg | Rat | No effect on acquisition of cocaine CPP | [260] |
| ↑ Extinction of cocaine CPP | |||
| No effect on reinstatement of cocaine CPP | |||
| No effect on acquisition of amphetamine CPP | |||
| ↑ Extinction of amphetamine CPP | |||
| IP 5, 20 mg/kg | Rat | No effect on heroin self-administration | [256] |
| No effect on heroin induced-reinstatement | |||
| ↓ Cue-induced reinstatement of heroin seeking | |||
| IP 5 mg/kg | Rat | No effect on cocaine-enhanced BSR | [258] |
| ↓ Expression of cocaine CPP | |||
| ↓ Morphine-enhanced BSR | |||
| No effect on BSR by itself | |||
| IP 5, 10 mg/kg | Rat | ↓ Reinstatement of morphine CPP | [262] |
| ↓ Reconsolidation of cocaine or morphine CPP | |||
| ↓ CPP following naltrexone-precipitated morphine withdrawal | |||
| Transdermal gel (2.5 g CBD/100g gel) for 7 days | Rat | ↓ Context-induced reinstatement of cocaine seeking | [261] |
| ↓ Stress-induced reinstatement of cocaine seeking | |||
| ↓ Stress-induced reinstatement of alcohol seeking | |||
| IP 80 mg/kg | ↓ Reinstatement of methamphetamine seeking | [254] | |
| ICV 10 μg | Rat | ↓ Reinstatement of methamphetamine CPP | [263] |
| IP 30, 60, 120 mg/kg | Rat | ↓ Alcohol self-administration | [255] |
| ↓ Alcohol consumption and preference | |||
| ↓ Context-induced reinstatement of alcohol seeking | |||
| IP 2–20 mg/kg | Mouse | ↓ Expression of morphine CPP | [259] |
| ↓ Cue-induced reinstatement of heroin seeking | |||
| IP 20, 40 mg/kg | Rat | ↓ Sucrose self-administration | [257] |
| IP 10, 20 mg/kg | Mouse | ↓ Sucrose self-administration in WT, not in CB2-KO | |
| IP 20–40 mg/kg | Rat | ↓ Cocaine self-administration | [253] |
| ↓ PR responding for cocaine | |||
| ↓ Cocaine-enhanced BSR | |||
| ↓ Cocaine-induced DA release | |||
| No effect on BSR by itself | |||
| BCP | |||
| IP 25–100 mg/kg | Mouse | ↓ Alcohol consumption | [284] |
| ↓ Alcohol CPP | |||
| ↑ Alcohol-induced loss of righting reflex | |||
| IP 25, 50 mg/kg | Rat | ↓ Nicotine self-administration | [286] |
| ↓ PR responding for nicotine | |||
| ↓ Nicotine-enhanced BSR | |||
| IP 25, 50 mg/kg | Mouse | ↓ Optical ICSS of VTA DA neurons | |
| Δ9-thcv | |||
| IP 10, 20 mg/kg | Rat | ↓ Nicotine self-administration | [301] |
| IP 3, 10 mg/kg | Rat | ↓ Cue-induced reinstatement of nicotine seeking | |
| SC 0.03–30 mg/kg | Mouse | ↓ Acquisition of nicotine CPP | |
| ↓ Anxiety during nicotine withdrawal | |||
| ↓ Withdrawal signs from nicotine |
BSR brain stimulation reward, CBD cannabidiol, CB2 type 2 cannabinoid receptor, CPP conditioned place preference, DA dopamine, ICV intracerebral ventricular, ICSS intracranial self-stimulation, IP intraperitoneally, KO knock-out, PR progressive-ratio, SC subcutaneously, VTA ventral tegmental area, WT wild-type, Δ9-THC Δ9-tetrahydrocannabinol, ↓ and ↑ represent decrease and increase, respectively
CBD has a promising potential to reduce relapse, as preclinical studies have reported that CBD reduced stress- or context cue-induced reinstatement of alcohol and cocaine seeking [255, 261]. Cue-induced heroin seeking also reduced after administration of CBD, the effects of which lasted up to 2 weeks [256]. In addition, CBD effectively reduced drug-primed reinstatement of methamphetamine [254], reinstatement of psychostimulants and opiate CPP [261–263], and reconsolidation of cocaine CPP mice [262]. CBD also reduced naloxone-precipitated withdrawal from morphine [262, 264] (Table 7).
CBD appears to have low reinforcing properties with limited abuse potential as it does not induce CPP on its own [260], nor does it affect BSR in rodents [258]. An additional CBD effect of therapeutic interest might be protective against acute cocaine toxicity, particularly liver damage and seizures [265]. Given its favorable effects on drug reward and seeking, CBD appears to be a promising therapeutic for cocaine use disorder.
In clinical studies, CBD was well-tolerated by healthy individuals [266] and had low abuse potential, as assessed in poly-drug users [267]. Although CBD did not reduce fentanyl-induced drug cravings and physiological responses [268], it attenuated cannabis consumption in smokers and the euphoric and intoxicating effects of Δ9-THC [269–271]. However, a more recent clinical study showed that CBD had no significant effects on the reinforcing, subjective, cognitive, or physiological effects of smoked cannabis [272]. These observations contrast with the clinical findings that low doses of CBD in combination with Δ9-THC enhanced, and high doses of CBD reduced, the intoxicating effects of Δ9-THC [273]. In cannabis users, CBD improved impaired performance on the emotional facial affect recognition task, including fearful, angry, happy, sad, surprise, and disgust faces. When combined with Δ9-THC, it also exhibited low abuse potential in humans [267]. In a double-blind randomized clinical inpatient trial with nabiximols that contained Δ9-THC and CBD, nabiximols attenuated withdrawal-related irritability, depression, and cannabis cravings and improved patient retention in the cognitive behavioral treatment program [274] (Table 6). However, no significant change in abstinence from cannabis use was observed in patients treated with nabiximols [275].
14.4. β-Caryophyllene
BCP is a volatile phytocannabinoid present in high proportions in cannabis [276] and is also found in the essential oils of many spice and food plants, including black pepper, thyme, and cloves [277]. BCP was first synthesized in 1964 [278] and later identified as a natural selective agonist of CB2Rs (Ki = 155 nM) with favorable CNS penetration [279]. Given that BCP is an FDA-approved dietary food additive with good oral bioavailability [280], favorable pharmacokinetics [281], and low toxicity [282, 283], it offers great promise as a therapeutic compound (Table 7). BCP decreased alcohol consumption and ethanol-induced CPP in rodents [284]. In a clinical study, nicotine smokers inhaling vapor from an essential oil of black pepper (Piper nigrum), which contains rich BCP, reported reduced nicotine cravings [285]. Although this effect was attributed to irritation of the bronchial tree and no molecular mechanisms of action were proposed, it is conceivable that stimulation of CB2Rs might be responsible for the reduced nicotine cravings. In addition, we recently showed that BCP reduced the rewarding effects of electrical optical brain stimulation or nicotine in rodents [286]. BCP, at lower doses, was also effective at attenuating nicotine self-administration in mice and rats but not in CB2-KO mice. Given that the inhibitory effects of BCP on nicotine reward can be blocked by AM630 but not by AM251 (a CB1R antagonist), CB2Rs might be the targets through which BCP produces its effects. In addition, high doses of intraperitoneal BCP (50, 100 mg/kg) inhibited food self-administration in both WT and CB2-KO mice [286], suggesting an effect mediated by other non-CB2R mechanisms. In regard to a BCP safety profile, BCP had no effects on spontaneous locomotor activity or rotarod performance and has been reported to be safe even at high doses of ≤ 700 mg/kg/day for 90 days in rats or 2000 mg/kg/day for 28 days in mice when administered orally [282, 283], placing it at the frontier of medication development for nicotine use disorder and obesity (Table 7).
14.5. Δ9-Tetrahydrocannabivarin
Δ9-THCV is another cannabis-derived phytocannabinoid [287]; it has CB1R antagonist action combined with CB2R agonist action [248, 288, 289]. Since Δ9-THCV does not inhibit or stimulate [35S]GTPγS binding to mouse or rat brain membranes at concentrations ≤ 10 μM, it is considered a neutral CB1R antagonist rather than an antagonist/inverse agonist [290–293]. However, Δ9-THCV can also act at the 5-HT1A receptor, and by doing so it can reduce phencyclidine (PCP)-induced psychosis-like behaviors in rats [294]. In addition, Δ9-THCV can stimulate and desensitize human TRPV1 and TRPV2 channels [295] and inhibit FAAH, MAGL, and AEA uptake [295]. Other targets of Δ9-THCV include TRPV5 and TRPV6 channels [296] and GPR55 receptors [297].
It has been reported that Δ9-THCV, just like AM251, can reduce food consumption and body weight in obese mice when administered acutely or chronically [298]. It was also found that Δ9-THCV produced antiobesity effects by reducing obesity-related glucose intolerance and increasing energy expenditure in obese mice [299]. Δ8-THCV is a synthetic, more stable, and easier-to-synthesize analog of Δ9-THCV with a similar pharmacological profile of combined CB1R antagonist and CB2R agonist action [300]. Using multiple models of drug abuse, we screened the therapeutic potential of Δ8-THCV on nicotine-related behaviors and found that Δ8-THCV reduced nicotine self-administration and nicotine-induced CPP in rats and mice [301]. In addition, Δ8-THCV attenuated context cue-triggered reinstatement of nicotine seeking following forced abstinence and nicotine-triggered reinstatement of nicotine seeking. After 14 days of continuous subcutaneous nicotine administration via osmotic minipumps, removal of the minipumps induced spontaneous nicotine withdrawal, during which, the mice spent more time in the closed arms than in the open arms of the elevated plus maze, showing anxiety-like behaviors. This anxiety-like behavior was significantly ameliorated by Δ8-THCV treatment. In addition, Δ8-THCV robustly ameliorated withdrawal signs in nicotine-treated mice, but it did not alter behavior in saline treated-mice. Δ8-THCV also reversed nicotine withdrawal-induced hyperalgesia, as measured by a hotplate test [301]. Based on these findings (Table 7), we suggest that the THCVs might constitute a safe and non-psychoactive class of potential anti-addiction, anticraving, and antirelapse pharmacotherapies [301].
THCVs are not currently scheduled as controlled or addictive substances by the US federal government or the United Nations Convention on Psychotropic Substances. They appear to be exceptionally safe in human use [302, 303]. Clinical studies have provided supporting evidence for this premise and reported that THCV administered orally was well-tolerated and did not produce negative psychotropic effects. Interestingly, cannabis users reported weaker subjective effects of Δ9-THC and showed less intense Δ9-THC-induced impairment of delayed verbal recall with Δ9-THCV treatment, suggesting Δ9-THCV may have therapeutic and protective effects [302]. In addition, functional magnetic resonance imaging showed that Δ9-THCV increased neural responses for food reward in the midbrain, striatum, and anterior cingulate cortex and for food-related aversive stimuli in the amygdala, insula, mid orbitofrontal cortex, and striatum, suggesting that it may have therapeutic utility for the treatment of obesity and food addiction [304].
In summary, clinical evidence indicates that Δ9-THC or its analog as a substitution therapy is effective in reducing cannabis use and withdrawal symptoms. Given that CBD, BCP, and Δ9-THCV can reduce drug self-administration and relapse without significant adverse effects, these three phytocannabinoids show great promise as medical cannabinoids in translation studies for SUDs.
15. Conclusions
The complexities of the behavioral and neurobiological factors underlying SUDs create enormous challenges in efforts to develop effective pharmacological treatments. In recent years, studies have recognized a critical role of the eCB system (particularly CB1Rs and CB2Rs) in many aspects of addiction and proposed eCB receptors as new targets in the development of therapies for the treatment of SUDs. Although results from early preclinical studies with rimonabant (a CB1R antagonist and CB1R inverse agonist) were promising, the adverse effects tempered initial enthusiasm and encouraged consideration of other strategies to modulate the eCB system. Recent discoveries of functional CB2Rs in the brain and their importance in addictive behaviors opened new avenues for therapeutic exploration. As evidence continues to accumulate, neutral CB1R antagonists (AM4113), selective CB2 agonists (such as JWH133, Xie2–64), and phytocannabinoids with CB1R antagonist and/or CB2R agonist profiles (CBD, BCP, Δ8-THCV, or Δ9-THCV) show great therapeutic potential as they can attenuate drug reward and relapse to drug-seeking behavior without adverse effects. In addition, strategies targeting the allosteric binding sites of CB1Rs and CB2Rs or targeting the metabolizing enzymes (FAAH, MAGL) are currently being pursued with limited success in reducing reward and relapse to drug-seeking behavior. Clinical studies have demonstrated that Δ9-THC-based agonist therapies (dronabinol, nabilone) and a novel FAAH inhibitor (PF-04457845) were effective in reducing cannabis withdrawal symptoms but not in reducing cannabis use or in preventing relapse to drug-seeking behavior. Given the current opioid epidemic and the urgent need for effective treatment, it is important that pursuit of innovative strategies, such as those targeting the eCB system, continues and that repurposing medications already approved by the FDA for other indications is considered in the treatment of SUDs.
Key Points.
Rimonabant or other CB1 receptor (CB1R) antagonists with inverse agonist profiles have failed in clinical trials for the treatment of substance use disorders (SUDs).
Recent preclinical studies with peripheral CB1R antagonists or inverse agonists, neutral CB1R antagonists, CB2R agonists, and allosteric CB1R or CB2R modulators have shown encouraging results for SUDs.
Among the phytocannabinoids, cannabidiol (CBD), β-caryophyllene (BCP), and Δ9-tetrahydrocannabivarin (Δ9-THCV) have shown significant CB1R antagonist and/or CB2R agonist profiles and great therapeutic potential for SUDs.
Clinical studies have shown that dronabinol, nabilone, or CBD fail to prevent relapse but can attenuate the symptoms of withdrawal from cannabis and opioids, making them potential candidates for the treatment of some SUDs.
Funding
This research was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, USA.
Footnotes
Conflict of interest Ewa Galaj and Zheng-Xiong Xi have no conflicts of interest that are directly relevant to the content of this article.
References
- 1.Rudd RA, Aleshire N, Zibbell JE, Gladden RM. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2016;64(50–51):1378–82. [DOI] [PubMed] [Google Scholar]
- 2.Drug Overdose Deaths | Drug Overdose | CDC Injury Center [Internet]; 2018. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Cited 17 May 2019.
- 3.Jordan CJ, Cao J, Newman AH, Xi Z-X. Progress in agonist therapy for substance use disorders: lessons learned from methadone and buprenorphine. Neuropharmacology. 2019. 10.1016/j.neuropharm.2019.04.015. [DOI] [PMC free article] [PubMed]
- 4.Jordan CJ, Xi Z-X. Discovery and development of varenicline for smoking cessation. Expert Opin Drug Discov. 2018;13(7):671–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xi Z-X, Gardner EL. Hypothesis-driven medication discovery for the treatment of psychostimulant addiction. Curr Drug Abuse Rev. 2008;1(3):303–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16(10):974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filip M, Frankowska M, Sadakierska-Chudy A, Suder A, Szumiec L, Mierzejewski P, et al. GABAB receptors as a therapeutic strategy in substance use disorders: focus on positive allosteric modulators. Neuropharmacology. 2015;88:36–47. [DOI] [PubMed] [Google Scholar]
- 8.Volkow ND, Jones EB, Einstein EB, Wargo EM. Prevention and treatment of opioid misuse and addiction: a review. JAMA Psychiatry. 2019;76(2):208–16. [DOI] [PubMed] [Google Scholar]
- 9.Volkow ND, Wise RA, Baler R. The dopamine motive system: implications for drug and food addiction. Nat Rev Neurosci. 2017;18(12):741–52. [DOI] [PubMed] [Google Scholar]
- 10.Ostroumov A, Dani JA. Inhibitory plasticity of mesocorticolimbic circuits in addiction and mental illness. Trends Neurosci. 2018;41(12):898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci. 2015;16(10):579–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trigo JM, Le Foll B. Inhibition of monoacylglycerol lipase (MAGL) enhances cue-induced reinstatement of nicotine-seeking behavior in mice. Psychopharmacology (Berl). 2016;233(10):1815–22. [DOI] [PubMed] [Google Scholar]
- 14.Serrano A, Parsons LH. Endocannabinoid influence in drug reinforcement, dependence and addiction-related behaviors. Pharmacol Ther. 2011;132(3):215–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ligresti A, Cascio MG, Di Marzo V. Endocannabinoid metabolic pathways and enzymes. Curr Drug Targets CNS Neurol Disord. 2005;4(6):615–23. [DOI] [PubMed] [Google Scholar]
- 16.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–9. [DOI] [PubMed] [Google Scholar]
- 17.McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152(5):583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugiura T, Waku K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem Phys Lipids. 2000;108(1–2):89–106. [DOI] [PubMed] [Google Scholar]
- 20.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5(1):37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci USA. 2009;106(15):6375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oddi S, Fezza F, Pasquariello N, D’Agostino A, Catanzaro G, De Simone C, et al. Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol. 2009;16(6):624–32. [DOI] [PubMed] [Google Scholar]
- 23.Hermann A, Kaczocha M, Deutsch DG. 2-Arachidonoylglycerol (2-AG) membrane transport: history and outlook. AAPS J. 2006;8(2):E409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87(5):1932–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piomelli D The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4(11):873–84. [DOI] [PubMed] [Google Scholar]
- 26.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89(1):309–80. [DOI] [PubMed] [Google Scholar]
- 27.Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76(1):70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17(14):1382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83(3):1017–66. [DOI] [PubMed] [Google Scholar]
- 30.Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia. 2010;58(9):1017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;148(5):1039–50. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira da Cruz JF, Robin LM, Drago F, Marsicano G, Metna-Laurent M. Astroglial type-1 cannabinoid receptor (CB1): a new player in the tripartite synapse. Neuroscience. 2016;26(323):35–42. [DOI] [PubMed] [Google Scholar]
- 33.Navarrete M, Araque A. Endocannabinoids mediate neuronastrocyte communication. Neuron. 2008;57(6):883–93. [DOI] [PubMed] [Google Scholar]
- 34.Mothet JP, Parent AT, Wolosker H, Brady RO, Linden DJ, Ferris CD, et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA. 2000;97(9):4926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metna-Laurent M, Marsicano G. Rising stars: modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia. 2015;63(3):353–64. [DOI] [PubMed] [Google Scholar]
- 36.Robin LM, Oliveira da Cruz JF, Langlais VC, Martin-Fernandez M, Metna-Laurent M, Busquets-Garcia A, et al. Astroglial CB1 receptors determine synaptic D-serine availability to enable recognition memory. Neuron. 2018;98(5):935–944.e5. [DOI] [PubMed] [Google Scholar]
- 37.Calabrese EJ, Rubio-Casillas A. Biphasic effects of THC in memory and cognition. Eur J Clin Investig. 2018;48(5):e12920. [DOI] [PubMed] [Google Scholar]
- 38.Mailleux P, Vanderhaeghen JJ. Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience. 1992;48(3):655–68. [DOI] [PubMed] [Google Scholar]
- 39.Mátyás F, Yanovsky Y, Mackie K, Kelsch W, Misgeld U, Freund TF. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience. 2006;137(1):337–61. [DOI] [PubMed] [Google Scholar]
- 40.Han X, He Y, Bi G-H, Zhang H-Y, Song R, Liu Q-R, et al. CB1 receptor activation on VgluT2-expressing glutamatergic neurons underlies Δ9-tetrahydrocannabinol (Δ9-THC)-induced aversive effects in mice. Sci Rep. 2017;7(1):12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackie K Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol. 2005;168:299–325. [DOI] [PubMed] [Google Scholar]
- 42.Fratta W, Fattore L. Molecular mechanisms of cannabinoid addiction. Curr Opin Neurobiol. 2013;23(4):487–92. [DOI] [PubMed] [Google Scholar]
- 43.Gueudet C, Santucci V, Rinaldi-Carmona M, Soubrié P, Le Fur G. The CB1 cannabinoid receptor antagonist SR 141716A affects A9 dopamine neuronal activity in the rat. Neuroreport. 1995;6(10):1421–5. [DOI] [PubMed] [Google Scholar]
- 44.De Luca MA, Bimpisidis Z, Melis M, Marti M, Caboni P, Valentini V, et al. Stimulation of in vivo dopamine transmission and intravenous self-administration in rats and mice by JWH-018, a Spice cannabinoid. Neuropharmacology. 2015;99:705–14. [DOI] [PubMed] [Google Scholar]
- 45.De Luca MA, Castelli MP, Loi B, Porcu A, Martorelli M, Miliano C, et al. Native CB1 receptor affinity, intrinsic activity and accumbens shell dopamine stimulant properties of third generation SPICE/K2 cannabinoids: BB-22, 5F-PB-22, 5F-AKB-48 and STS-135. Neuropharmacology. 2016;105:630–8. [DOI] [PubMed] [Google Scholar]
- 46.Mateo Y, Johnson KA, Covey DP, Atwood BK, Wang H-L, Zhang S, et al. Endocannabinoid actions on cortical terminals orchestrate local modulation of dopamine release in the nucleus accumbens. Neuron. 2017;96(5):1112–1126.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport. 1997;8(3):649–52. [DOI] [PubMed] [Google Scholar]
- 48.Braida D, Iosuè S, Pegorini S, Sala M. Delta9-tetrahydrocannabinol-induced conditioned place preference and intracer-ebroventricular self-administration in rats. Eur J Pharmacol. 2004;506(1):63–9. [DOI] [PubMed] [Google Scholar]
- 49.Gardner EL. Endocannabinoid signaling system and brain reward: emphasis on dopamine. Pharmacol Biochem Behav. 2005;81(2):263–84. [DOI] [PubMed] [Google Scholar]
- 50.Spiller KJ, Bi G-H, He Y, Galaj E, Gardner EL, Xi Z-X. Cannabinoid CB1 and CB2 receptor mechanisms underlie cannabis reward and aversion in rats. Br J Pharmacol. 2019;176(9):1268–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi RN, Singer G. Self-administration of delta 9-tetrahydrocannabinol by rats. Pharmacol Biochem Behav. 1979;11(6):737–40. [DOI] [PubMed] [Google Scholar]
- 52.Fattore L, Cossu G, Martellotta CM, Fratta W. Intravenous self-administration of the cannabinoid CB1 receptor agonist WIN 55,212–2 in rats. Psychopharmacology (Berl). 2001;156(4):410–6. [DOI] [PubMed] [Google Scholar]
- 53.Lefever TW, Marusich JA, Antonazzo KR, Wiley JL. Evaluation of WIN 55,212–2 self-administration in rats as a potential cannabinoid abuse liability model. Pharmacol Biochem Behav. 2014;118:30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spencer S, Neuhofer D, Chioma VC, Garcia-Keller C, Schwartz DJ, Allen N, et al. A model of Δ9-tetrahydrocannabinol self-administration and reinstatement that alters synaptic plasticity in nucleus accumbens. Biol Psychiatry. 2018;84(8):601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci. 2002;15(12):2057–61. [DOI] [PubMed] [Google Scholar]
- 56.Lupica CR, Riegel AC, Hoffman AF. Marijuana and cannabinoid regulation of brain reward circuits. Br J Pharmacol. 2004;143(2):227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48(8):1105–16. [DOI] [PubMed] [Google Scholar]
- 58.Lupica CR, Hoffman AF. Cannabinoid disruption of learning mechanisms involved in reward processing. Learn Mem. 2018;25(9):435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006;29(4):225–32. [DOI] [PubMed] [Google Scholar]
- 60.Fattore L, Fadda P, Spano MS, Pistis M, Fratta W. Neurobiological mechanisms of cannabinoid addiction. Mol Cell Endocrinol. 2008;286(1–2 Suppl 1):S97–107. [DOI] [PubMed] [Google Scholar]
- 61.Raft D, Gregg J, Ghia J, Harris L. Effects of intravenous tetrahydrocannabinol on experimental and surgical pain. Psychological correlates of the analgesic response. Clin Pharmacol Ther. 1977;21(1):26–33. [DOI] [PubMed] [Google Scholar]
- 62.D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Wu Y-T, et al. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology. 2004;29(8):1558–72. [DOI] [PubMed] [Google Scholar]
- 63.Gregg JM, Small EW, Moore R, Raft D, Toomey TC. Emotional response to intravenous delta9tetrahydrocannabinol during oral surgery. J Oral Surg. 1976;34(4):301–13. [PubMed] [Google Scholar]
- 64.Farris SG, Zvolensky MJ, Boden MT, Bonn-Miller MO. Cannabis use expectancies mediate the relation between depressive symptoms and cannabis use among cannabis-dependent veterans. J Addict Med. 2014;8(2):130–6. [DOI] [PubMed] [Google Scholar]
- 65.Tanda G, Munzar P, Goldberg SR. Self-administration behavior is maintained by the psychoactive ingredient of marijuana in squirrel monkeys. Nat Neurosci. 2000;3(11):1073–4. [DOI] [PubMed] [Google Scholar]
- 66.Justinova Z, Tanda G, Redhi GH, Goldberg SR. Self-administration of delta9-tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology (Berl). 2003;169(2):135–40. [DOI] [PubMed] [Google Scholar]
- 67.Mansbach RS, Nicholson KL, Martin BR, Balster RL. Failure of Delta(9)-tetrahydrocannabinol and CP 55,940 to maintain intravenous self-administration under a fixed-interval schedule in rhesus monkeys. Behav Pharmacol. 1994;5(2):219–25. [DOI] [PubMed] [Google Scholar]
- 68.John WS, Martin TJ, Nader MA. Behavioral determinants of cannabinoid self-administration in old world monkeys. Neuropsychopharmacology. 2017;42(7):1522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardner EL, Paredes W, Smith D, Donner A, Milling C, Cohen D, et al. Facilitation of brain stimulation reward by delta 9-tetrahydrocannabinol. Psychopharmacology. 1988;96(1):142–4. [DOI] [PubMed] [Google Scholar]
- 70.Lepore M, Liu X, Savage V, Matalon D, Gardner EL. Genetic differences in delta 9-tetrahydrocannabinol-induced facilitation of brain stimulation reward as measured by a rate-frequency curve-shift electrical brain stimulation paradigm in three different rat strains. Life Sci. 1996;58(25):PL365–72. [DOI] [PubMed] [Google Scholar]
- 71.Katsidoni V, Kastellakis A, Panagis G. Biphasic effects of Δ9-tetrahydrocannabinol on brain stimulation reward and motor activity. Int J Neuropsychopharmacol. 2013;16(10):2273–84. [DOI] [PubMed] [Google Scholar]
- 72.Vlachou S, Nomikos GG, Stephens DN, Panagis G. Lack of evidence for appetitive effects of Delta 9-tetrahydrocannabinol in the intracranial self-stimulation and conditioned place preference procedures in rodents. Behav Pharmacol. 2007;18(4):311–9. [DOI] [PubMed] [Google Scholar]
- 73.Kwilasz AJ, Negus SS. Dissociable effects of the cannabinoid receptor agonists Δ9-tetrahydrocannabinol and CP55940 on pain-stimulated versus pain-depressed behavior in rats. J Pharmacol Exp Ther. 2012;343(2):389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Negus SS, Miller LL. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol Rev. 2014;66(3):869–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wiebelhaus JM, Grim TW, Owens RA, Lazenka MF, Sim-Selley LJ, Abdullah RA, et al. Δ9-tetrahydrocannabinol and endocannabinoid degradative enzyme inhibitors attenuate intracranial self-stimulation in mice. J Pharmacol Exp Ther. 2015;352(2):195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panagis G, Vlachou S, Nomikos GG. Behavioral pharmacology of cannabinoids with a focus on preclinical models for studying reinforcing and dependence-producing properties. Curr Drug Abuse Rev. 2008;1(3):350–74. [DOI] [PubMed] [Google Scholar]
- 77.Vlachou S, Panagis G. Regulation of brain reward by the endocannabinoid system: a critical review of behavioral studies in animals. Curr Pharm Des. 2014;20(13):2072–88. [DOI] [PubMed] [Google Scholar]
- 78.Castañeda E, Moss DE, Oddie SD, Whishaw IQ. THC does not affect striatal dopamine release: microdialysis in freely moving rats. Pharmacol Biochem Behav. 1991;40(3):587–91. [DOI] [PubMed] [Google Scholar]
- 79.Wang H-L, Qi J, Zhang S, Wang H, Morales M. Rewarding effects of optical stimulation of ventral tegmental area glutamatergic neurons. J Neurosci. 2015;35(48):15948–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manzanares J, Cabañero D, Puente N, García-Gutiérrez MS, Grandes P, Maldonado R. Role of the endocannabinoid system in drug addiction. Biochem Pharmacol. 2018;157:108–21. [DOI] [PubMed] [Google Scholar]
- 81.Jordan CJ, Xi Z-X. Progress in brain cannabinoid CB2 receptor research: from genes to behavior. Neurosci Biobehav Rev. 2019;98:208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xi Z-X, Peng X-Q, Li X, Song R, Zhang H-Y, Liu Q-R, et al. Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat Neurosci. 2011;14(9):1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang H-Y, Gao M, Liu Q-R, Bi G-H, Li X, Yang H-J, et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci USA. 2014;111(46):E5007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang H-Y, Gao M, Shen H, Bi G-H, Yang H-J, Liu Q-R, et al. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict Biol. 2017;22(3):752–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Foster DJ, Wilson JM, Remke DH, Mahmood MS, Uddin MJ, Wess J, et al. Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron. 2016;91(6):1244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delis F, Polissidis A, Poulia N, Justinova Z, Nomikos GG, Goldberg SR, et al. Attenuation of cocaine-induced conditioned place preference and motor activity via cannabinoid CB2 receptor agonism and CB1 receptor antagonism in rats. Int J Neuropsychopharmacol. 2017;20(3):269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang H-Y, Bi G-H, Li X, Li J, Qu H, Zhang S-J, et al. Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology. 2015;40(4):1037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aracil-Fernández A, Trigo JM, García-Gutiérrez MS, Ortega-Álvaro A, Ternianov A, Navarro D, et al. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB2 receptors. Neuropsychopharmacology. 2012;37(7):1749–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gamaleddin I, Zvonok A, Makriyannis A, Goldberg SR, Le Foll B. Effects of a selective cannabinoid CB2 agonist and antagonist on intravenous nicotine self administration and reinstatement of nicotine seeking. PLoS One. 2012;7(1):e29900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Solinas M, Panlilio LV, Goldberg SR. Exposure to delta-9-tetrahydrocannabinol (THC) increases subsequent heroin taking but not heroin’s reinforcing efficacy: a self-administration study in rats. Neuropsychopharmacology. 2004;29(7):1301–11. [DOI] [PubMed] [Google Scholar]
- 91.Colombo G, Serra S, Brunetti G, Gomez R, Melis S, Vacca G, et al. Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology (Berl). 2002;159(2):181–7. [DOI] [PubMed] [Google Scholar]
- 92.Linsenbardt DN, Boehm SL. Agonism of the endocannabinoid system modulates binge-like alcohol intake in male C57BL/6J mice: involvement of the posterior ventral tegmental area. Neuroscience. 2009;164(2):424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caillé S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci. 2007;27(14):3695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang H, Treadway T, Covey DP, Cheer JF, Lupica CR. Cocaine-induced endocannabinoid mobilization in the ventral tegmental area. Cell Rep. 2015;12(12):1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Le Foll B, Gorelick DA, Goldberg SR. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology. 2009;205(1):171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maccioni P, Colombo G, Carai MAM. Blockade of the cannabinoid CB1 receptor and alcohol dependence: preclinical evidence and preliminary clinical data. CNS Neurol Disord Drug Targets. 2010;9(1):55–9. [DOI] [PubMed] [Google Scholar]
- 97.Gamaleddin IH, Trigo JM, Gueye AB, Zvonok A, Makriyannis A, Goldberg SR, et al. Role of the endogenous cannabinoid system in nicotine addiction: novel insights. Front Psychiatry. 2015;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stern CAJ, de Carvalho CR, Bertoglio LJ, Takahashi RN. Effects of cannabinoid drugs on aversive or rewarding drug-associated memory extinction and reconsolidation. Neuroscience. 2018;01(370):62–80. [DOI] [PubMed] [Google Scholar]
- 99.Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350(2–3):240–4. [DOI] [PubMed] [Google Scholar]
- 100.Mato S, Pazos A, Valdizán EM. Cannabinoid receptor antagonism and inverse agonism in response to SR141716A on cAMP production in human and rat brain. Eur J Pharmacol. 2002;443(1–3):43–6. [DOI] [PubMed] [Google Scholar]
- 101.Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther. 2005;312(3):875–83. [DOI] [PubMed] [Google Scholar]
- 102.Wiskerke J, Pattij T, Schoffelmeer ANM, De Vries TJ. The role of CB1 receptors in psychostimulant addiction. Addict Biol. 2008;13(2):225–38. [DOI] [PubMed] [Google Scholar]
- 103.Cahill K, Ussher MH. Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst Rev. 2011;3:CD005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gaal LFV, Rissanen AM, Scheen AJ, Ziegler O, Rössner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365(9468):1389–97. [DOI] [PubMed] [Google Scholar]
- 105.Elrashidi MY, Ebbert JO. Emerging drugs for the treatment of tobacco dependence: 2014 update. Expert Opin Emerg Drugs. 2014;19(2):243–60. [DOI] [PubMed] [Google Scholar]
- 106.Sloan ME, Gowin JL, Ramchandani VA, Hurd YL, Le Foll B. The endocannabinoid system as a target for addiction treatment: trials and tribulations. Neuropharmacology. 2017;15(124):73–83. [DOI] [PubMed] [Google Scholar]
- 107.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup AV. A meta-analysis of the efficacy and safety of the anti-obesity agent Rimonabant. Ugeskr Laeg. 2007;169(50):4360–3. [PubMed] [Google Scholar]
- 108.Sam AH, Salem V, Ghatei MA. Rimonabant: from RIO to ban. J Obes. 2011;2011:432607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morgan CJA, Das RK, Joye A, Curran HV, Kamboj SK. Cannabidiol reduces cigarette consumption in tobacco smokers: preliminary findings. Addict Behav. 2013;38(9):2433–6. [DOI] [PubMed] [Google Scholar]
- 110.Chorvat RJ. Peripherally restricted CB1 receptor blockers. Bioorg Med Chem Lett. 2013;23(17):4751–60. [DOI] [PubMed] [Google Scholar]
- 111.Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161(3):629–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kunos G, Tam J. The case for peripheral CB1 receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br J Pharmacol. 2011;163(7):1423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chorvat RJ, Berbaum J, Seriacki K, McElroy JF. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg Med Chem Lett. 2012;22(19):6173–80. [DOI] [PubMed] [Google Scholar]
- 114.Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16(2):167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tai S, Nikas SP, Shukla VG, Vemuri K, Makriyannis A, Järbe TUC. Cannabinoid withdrawal in mice: inverse agonist vs neutral antagonist. Psychopharmacology (Berl). 2015;232(15):2751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Godlewski G, Cinar R, Coffey NJ, Liu J, Jourdan T, Mukhopadhyay B, et al. Targeting peripheral CB1 receptors reduces ethanol intake via a gut–brain axis. Cell Metab. 2019;29(6):1320–1333. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xi Z-X, Spiller K, Pak AC, Gilbert J, Dillon C, Li X, et al. Cannabinoid CB1 receptor antagonists attenuate cocaine’s rewarding effects: experiments with self-administration and brain-stimulation reward in rats. Neuropsychopharmacology. 2008;33(7):1735–45. [DOI] [PubMed] [Google Scholar]
- 118.He X-H, Jordan CJ, Vemuri K, Bi G-H, Zhan J, Gardner EL, et al. Cannabinoid CB1 receptor neutral antagonist AM4113 inhibits heroin self-administration without depressive side effects in rats. Acta Pharmacol Sin. 2019;40:365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gardner EL, Gamaleddin I, Manzanares RJ, Rodrigues FF. The endocannabinoid system: useful targets for anti-addiction treatments? Subst Abuse. 2013;34:324–5. [Google Scholar]
- 120.Gueye AB, Pryslawsky Y, Trigo JM, Poulia N, Delis F, Antoniou K, et al. The CB1 neutral antagonist AM4113 retains the therapeutic efficacy of the inverse agonist rimonabant for nicotine dependence and weight loss with better psychiatric tolerability. Int J Neuropsychopharmacol. 2016. 10.1093/ijnp/pyw068. [DOI] [PMC free article] [PubMed]
- 121.Alvarado M, Decara J, Luque MJ, Hernandez-Folgado L, Gómez-Cañas M, Gómez-Ruiz M, et al. Novel antiobesity agents: synthesis and pharmacological evaluation of analogues of Rimonabant and of LH21. Bioorg Med Chem. 2013;21(7):1708–16. [DOI] [PubMed] [Google Scholar]
- 122.Seltzman HH, Maitra R, Bortoff K, Henson J, Reggio PH, Wesley D, et al. Metabolic profiling of CB1 neutral antagonists. Methods Enzymol. 2017;593:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salamone JD, McLaughlin PJ, Sink K, Makriyannis A, Parker LA. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: effects on food intake, food-reinforced behavior and food aversions. Physiol Behav. 2007;91(4):383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sink KS, McLaughlin PJ, Wood JAT, Brown C, Fan P, Vemuri VK, et al. The novel cannabinoid CB1 receptor neutral antagonist AM4113 suppresses food intake and food-reinforced behavior but does not induce signs of nausea in rats. Neuropsychopharmacology. 2008;33(4):946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chambers AP, Vemuri VK, Peng Y, Wood JT, Olszewska T, Pittman QJ, et al. A neutral CB1 receptor antagonist reduces weight gain in rat. Am J Physiol Regul Integr Comp Physiol. 2007;293(6):R2185–93. [DOI] [PubMed] [Google Scholar]
- 126.Järbe TUC, LeMay BJ, Olszewska T, Vemuri VK, Wood JT, Makriyannis A. Intrinsic effects of AM4113, a putative neutral CB1 receptor selective antagonist, on open-field behaviors in rats. Pharmacol Biochem Behav. 2008;91(1):84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Balla A, Dong B, Shilpa BM, Vemuri K, Makriyannis A, Pandey SC, et al. Cannabinoid-1 receptor neutral antagonist reduces binge-like alcohol consumption and alcohol-induced accumbal dopaminergic signaling. Neuropharmacology. 2018;15(131):200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schindler CW, Redhi GH, Vemuri K, Makriyannis A, Le Foll B, Bergman J, et al. Blockade of nicotine and cannabinoid reinforcement and relapse by a cannabinoid CB1-receptor neutral antagonist AM4113 and inverse agonist rimonabant in squirrel monkeys. Neuropsychopharmacology. 2016;41(9):2283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kangas BD, Delatte MS, Vemuri VK, Thakur GA, Nikas SP, Subramanian KV, et al. Cannabinoid discrimination and antagonism by CB(1) neutral and inverse agonist antagonists. J Pharmacol Exp Ther. 2013;344(3):561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wills KL, Vemuri K, Kalmar A, Lee A, Limebeer CL, Makriyannis A, et al. CB1 antagonism: interference with affective properties of acute naloxone-precipitated morphine withdrawal in rats. Psychopharmacology (Berl). 2014;231(22):4291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jagerovic N, Hernandez-Folgado L, Alkorta I, Goya P, Navarro M, Serrano A, et al. Discovery of 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1h-1,2,4-triazole, a novel in vivo cannabinoid antagonist containing a 1,2,4-triazole motif. J Med Chem. 2004;47(11):2939–42. [DOI] [PubMed] [Google Scholar]
- 132.Chen RZ, Frassetto A, Lao JZ, Huang R-RC, Xiao JC, Clements MJ, et al. Pharmacological evaluation of LH-21, a newly discovered molecule that binds to cannabinoid CB1 receptor. Eur J Pharmacol. 2008;584(2–3):338–42. [DOI] [PubMed] [Google Scholar]
- 133.Pavon FJ, Bilbao A, Hernández-Folgado L, Cippitelli A, Jagerovic N, Abellán G, et al. Antiobesity effects of the novel in vivo neutral cannabinoid receptor antagonist 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1H-1,2,4-triazole–LH 21. Neuropharmacology. 2006;51(2):358–66. [DOI] [PubMed] [Google Scholar]
- 134.Pavón FJ, Serrano A, Pérez-Valero V, Jagerovic N, Hernández-Folgado L, Bermúdez-Silva FJ, et al. Central versus peripheral antagonism of cannabinoid CB1 receptor in obesity: effects of LH-21, a peripherally acting neutral cannabinoid receptor antagonist, in Zucker rats. J Neuroendocrinol. 2008;20(Suppl 1):116–23. [DOI] [PubMed] [Google Scholar]
- 135.Alonso M, Serrano A, Vida M, Crespillo A, Hernandez-Folgado L, Jagerovic N, et al. Anti-obesity efficacy of LH-21, a cannabinoid CB1 receptor antagonist with poor brain penetration, in diet-induced obese rats. Br J Pharmacol. 2012;165(7):2274–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gardner EL, Bi G-H, Thakur GA, Makriyannis A, Seltzman HH, He X-Y, et al. Preclinical evaluation of neutral cannabinoid CB1 receptor antagonists and cannabinoid CB1 receptor negative allosteric modulators for treating drug addiction. Int J Neuropsychopharmacol. 2016. Report No.: Meeting Abstract-PM288. [Google Scholar]
- 137.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68(5):1484–95. [DOI] [PubMed] [Google Scholar]
- 138.Jing L, Qiu Y, Zhang Y, Li J-X. Effects of the cannabinoid CB1 receptor allosteric modulator ORG 27569 on reinstatement of cocaine-and methamphetamine-seeking behavior in rats. Drug Alcohol Depend. 2014;1(143):251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gamage TF, Ignatowska-Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behav Pharmacol. 2014;25(2):182–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y, Li J-X. Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacol Res Perspect. 2014;2(6):e00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hofer SC, Ralvenius WT, Gachet MS, Fritschy J-M, Zeilhofer HU, Gertsch J. Localization and production of peptide endocannabinoids in the rodent CNS and adrenal medulla. Neuropharmacology. 2015;98:78–89. [DOI] [PubMed] [Google Scholar]
- 142.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, et al. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem. 2012;287(44):36944–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Petrucci V, Chicca A, Glasmacher S, Paloczi J, Cao Z, Pacher P, et al. Pepcan-12 (RVD-hemopressin) is a CB2 receptor positive allosteric modulator constitutively secreted by adrenals and in liver upon tissue damage. Sci Rep. 2017;7(1):9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gomes I, Grushko JS, Golebiewska U, Hoogendoorn S, Gupta A, Heimann AS, et al. Novel endogenous peptide agonists of cannabinoid receptors. FASEB J. 2009;23(9):3020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ferrante C, Recinella L, Leone S, Chiavaroli A, Di Nisio C, Martinotti S, et al. Anorexigenic effects induced by RVD-hemopressin(α) administration. Pharmacol Rep. 2017;69(6):1402–7. [DOI] [PubMed] [Google Scholar]
- 146.Leone S, Recinella L, Chiavaroli A, Martinotti S, Ferrante C, Mollica A, et al. Emotional disorders induced by Hemopressin and RVD-hemopressin(α) administration in rats. Pharmacol Rep. 2017;69(6):1247–53. [DOI] [PubMed] [Google Scholar]
- 147.Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160(3):467–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lanciego JL, Barroso-Chinea P, Rico AJ, Conte-Perales L, Callén L, Roda E, et al. Expression of the mRNA coding the cannabinoid receptor 2 in the pallidal complex of Macaca facicularis. J Psychopharmacol (Oxford). 2011;25(1):97–104. [DOI] [PubMed] [Google Scholar]
- 149.Sierra S, Luquin N, Rico AJ, Gómez-Bautista V, Roda E, Dopeso-Reyes IG, et al. Detection of cannabinoid receptors CB1 and CB2 within basal ganglia output neurons in macaques: changes following experimental parkinsonism. Brain Struct Funct. 2015;220(5):2721–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl). 2013;229(4):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Navarrete F, Rodríguez-Arias M, Martín-García E, Navarro D, García-Gutiérrez MS, Aguilar MA, et al. Role of CB2 cannabinoid receptors in the rewarding, reinforcing, and physical effects of nicotine. Neuropsychopharmacology. 2013;38(12):2515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu Q-R, Canseco-Alba A, Zhang H-Y, Tagliaferro P, Chung M, Dennis E, et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci Rep. 2017;7(1):17410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ortega-Álvaro A, Ternianov A, Aracil-Fernández A, Navarrete F, García-Gutiérrez MS, Manzanares J. Role of cannabinoid CB2 receptor in the reinforcing actions of ethanol. Addict Biol. 2015;20(1):43–55. [DOI] [PubMed] [Google Scholar]
- 154.Powers MS, Breit KR, Chester JA. Genetic versus pharmacological assessment of the role of cannabinoid type 2 receptors in alcohol reward-related behaviors. Alcohol Clin Exp Res. 2015;39(12):2438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bystrowska B, Frankowska M, Smaga I, Pomierny-Chamioło L, Filip M. effects of cocaine self-administration and its extinction on the rat brain cannabinoid CB1 and CB2 receptors. Neurotox Res. 2018;34:547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bystrowska B, Frankowska M, Smaga I, Niedzielska-Andres E, Pomierny-Chamioło L, Filip M. cocaine-induced reinstatement of cocaine seeking provokes changes in the endocannabinoid and N-acylethanolamine levels in rat brain structures. Molecules. 2019;24(6):E1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huffman JW. CB2 receptor ligands. Mini Rev Med Chem. 2005;5(7):641–9. [DOI] [PubMed] [Google Scholar]
- 158.Ma Z, Gao F, Larsen B, Gao M, Luo Z, Chen D, et al. Mechanisms of cannabinoid CB2 receptor-mediated reduction of dopamine neuronal excitability in mouse ventral tegmental area. EBioMedicine. 2019;42:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Canseco-Alba A, Schanz N, Sanabria B, Zhao J, Lin Z, Liu Q-R, et al. Behavioral effects of psychostimulants in mutant mice with cell-type specific deletion of CB2 cannabinoid receptors in dopamine neurons. Behav Brain Res. 2018;30(360):286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48(5):658–72. [DOI] [PubMed] [Google Scholar]
- 161.Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegaliński E. The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res. 2012;20(1444):45–54. [DOI] [PubMed] [Google Scholar]
- 162.Wiley JL, Beletskaya ID, Ng EW, Dai Z, Crocker PJ, Mahadevan A, et al. Resorcinol derivatives: a novel template for the development of cannabinoid CB(1)/CB(2) and CB(2)-selective agonists. J Pharmacol Exp Ther. 2002;301(2):679–89. [DOI] [PubMed] [Google Scholar]
- 163.Alavi MS, Hosseinzadeh H, Shamsizadeh A, Roohbakhsh A. The effect of O-1602, an atypical cannabinoid, on morphine-induced conditioned place preference and physical dependence. Pharmacol Rep. 2016;68(3):592–7. [DOI] [PubMed] [Google Scholar]
- 164.Zhang M, Dong L, Zou H, Li J, Li Q, Wang G, et al. Effects of cannabinoid type 2 receptor agonist AM1241 on morphine-induced antinociception, acute and chronic tolerance, and dependence in mice. J Pain. 2018;19(10):1113–29. [DOI] [PubMed] [Google Scholar]
- 165.Li A-L, Lin X, Dhopeshwarkar AS, Thomaz AC, Carey LM, Liu Y, et al. Cannabinoid CB2 agonist AM1710 differentially suppresses distinct pathological pain states and attenuates morphine tolerance and withdrawal. Mol Pharmacol. 2019;95(2):155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Grenald SA, Young MA, Wang Y, Ossipov MH, Ibrahim MM, Largent-Milnes TM, et al. Synergistic attenuation of chronic pain using mu opioid and cannabinoid receptor 2 agonists. Neuropharmacology. 2017;116:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100(18):10529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yang P, Wang L, Feng R, Almehizia AA, Tong Q, Myint KZ, et al. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: discovery, optimization, and biological evaluation. J Med Chem. 2013;56:2045–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jordan C, Feng XW, Bi G-H, Liang Y, Han X, Xie X-Q, et al. Xie2–64 is a promising cannabinoid CB2 receptor ligand that reduces cocaine abuse-related behaviors in rodents. Addict Biol. 2019. (in press). [Google Scholar]
- 170.Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24(1):53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Panlilio LV, Justinova Z, Goldberg SR. Inhibition of FAAH and activation of PPAR: New approaches to the treatment of cognitive dysfunction and drug addiction. Pharmacol Ther. 2013;138(1):84–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Deutsch DG. A personal retrospective: elevating anandamide (AEA) by targeting fatty acid amide hydrolase (FAAH) and the fatty acid binding proteins (FABPs). Front Pharmacol [Internet]; 2016. Cited 6 Mar 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12(11):1179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, et al. Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry. 2008;64(11):930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Adamczyk P, McCreary AC, Przegalinski E, Mierzejewski P, Bienkowski P, Filip M. The effects of fatty acid amide hydrolase inhibitors on maintenance of cocaine and food self-administration and on reinstatement of cocaine-seeking and food-taking behavior in rats. J Physiol Pharmacol. 2009;60(3):119–25. [PubMed] [Google Scholar]
- 176.Chauvet C, Nicolas C, Thiriet N, Lardeux MV, Duranti A, Solinas M. Chronic stimulation of the tone of endogenous anandamide reduces cue- and stress-induced relapse in rats. Int J Neuropsychopharmacol. 2014. 10.1093/ijnp/pyu025. [DOI] [PMC free article] [PubMed]
- 177.Solinas M, Panlilio LV, Tanda G, Makriyannis A, Matthews SA, Goldberg SR. Cannabinoid agonists but not inhibitors of endogenous cannabinoid transport or metabolism enhance the reinforcing efficacy of heroin in rats. Neuropsychopharmacology. 2005;30(11):2046–57. [DOI] [PubMed] [Google Scholar]
- 178.McCallum AL, Limebeer CL, Parker LA. Reducing endocannabinoid metabolism with the fatty acid amide hydrolase inhibitor, URB597, fails to modify reinstatement of morphine-induced conditioned floor preference and naloxone-precipitated morphine withdrawal-induced conditioned floor avoidance. Pharmacol Biochem Behav. 2010;96(4):496–500. [DOI] [PubMed] [Google Scholar]
- 179.Manwell LA, Satvat E, Lang ST, Allen CP, Leri F, Parker LA. FAAH inhibitor, URB-597, promotes extinction and CB(1) antagonist, SR141716, inhibits extinction of conditioned aversion produced by naloxone-precipitated morphine withdrawal, but not extinction of conditioned preference produced by morphine in rats. Pharmacol Biochem Behav. 2009;94(1):154–62. [DOI] [PubMed] [Google Scholar]
- 180.Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, et al. Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther. 2011;339(1):173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shahidi S, Hasanein P. Behavioral effects of fatty acid amide hydrolase inhibition on morphine withdrawal symptoms. Brain Res Bull. 2011;86(1–2):118–22. [DOI] [PubMed] [Google Scholar]
- 182.Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, et al. Inhibition of anandamide hydrolysis by cyclohexyl carbamic acid 3′-carbamoyl-3-yl ester (URB597) reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther. 2008;327(2):482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Forget B, Guranda M, Gamaleddin I, Goldberg SR, Le Foll B. Attenuation of cue-induced reinstatement of nicotine seeking by URB597 through cannabinoid CB1 receptor in rats. Psychopharmacology (Berl). 2016;233(10):1823–8. [DOI] [PubMed] [Google Scholar]
- 184.Forget B, Coen KM, Le Foll B. Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration—comparison with CB(1) receptor blockade. Psychopharmacology (Berl). 2009;205(4):613–24. [DOI] [PubMed] [Google Scholar]
- 185.Justinova Z, Panlilio LV, Moreno-Sanz G, Redhi GH, Auber A, Secci ME, et al. Effects of fatty acid amide hydrolase (FAAH) inhibitors in non-human primate models of nicotine reward and relapse. Neuropsychopharmacology. 2015;40(9):2185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stop-poni S, et al. Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One. 2011;6(11):e28142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, et al. Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci. 2008;28(51):13985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, et al. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol. 2010;15(3):277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Blednov YA, Cravatt BF, Boehm SL, Walker D, Harris RA. Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology. 2007;32(7):1570–82. [DOI] [PubMed] [Google Scholar]
- 190.Basavarajappa BS, Yalamanchili R, Cravatt BF, Cooper TB, Hungund BL. Increased ethanol consumption and preference and decreased ethanol sensitivity in female FAAH knockout mice. Neuropharmacology. 2006;50(7):834–44. [DOI] [PubMed] [Google Scholar]
- 191.Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, et al. Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology (Berl). 2008;198(4):449–60. [DOI] [PubMed] [Google Scholar]
- 192.Zhou Y, Schwartz BI, Giza J, Gross SS, Lee FS, Kreek MJ. Blockade of alcohol escalation and “relapse” drinking by pharmacological FAAH inhibition in male and female C57BL/6J mice. Psychopharmacology (Berl). 2017;234(19):2955–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Solinas M, Justinova Z, Goldberg SR, Tanda G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem. 2006;98(2):408–19. [DOI] [PubMed] [Google Scholar]
- 194.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102(51):18620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wiley JL, Walentiny DM, Wright MJ, Beardsley PM, Burston JJ, Poklis JL, et al. Endocannabinoid contribution to Δ9-tetrahydrocannabinol discrimination in rodents. Eur J Pharmacol. 2014;15(737):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9(1):76–81. [DOI] [PubMed] [Google Scholar]
- 197.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, et al. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006;12(1):21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153(9):1837–46. [DOI] [PubMed] [Google Scholar]
- 199.Li GL, Winter H, Arends R, Jay GW, Le V, Young T, et al. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol. 2012;73(5):706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pawsey S, Wood M, Browne H, Donaldson K, Christie M, Warrington S. Safety, tolerability and pharmacokinetics of FAAH inhibitor V158866: a double-blind, randomised, placebo-controlled phase i study in healthy volunteers. Drugs R D. 2016;16(2):181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kaur R, Sidhu P, Singh S. What failed BIA 10–2474 phase I clinical trial? Global speculations and recommendations for future Phase I trials. J Pharmacol Pharmacother. 2016;7(3):120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kerbrat A, Ferré J-C, Fillatre P, Ronzière T, Vannier S, Carsin-Nicol B, et al. Acute neurologic disorder from an inhibitor of fatty acid amide hydrolase. N Engl J Med. 2016;375(18):1717–25. [DOI] [PubMed] [Google Scholar]
- 203.Janssen Research & Development, LLC Voluntarily Suspends Dosing in Phase 2 Clinical Trials of Experimental Treatment for Mood Disorders | Janssen [Internet]; 2016. https://web.archive.org/web/20160125052230/http://www.janssen.com/janssen-research-development-llc-voluntarily-suspends-dosing-phase-2-clinical-trials-experimental. Cited 12 May 2019.
- 204.FAAH inhibitor safety under microscope after Bial drug trial death [Internet]. in-pharmatechnologist.com. https://www.in-pharmatechnologist.com/Article/2016/01/19/FAAH-inhibitor-safety-under-microscope-after-Bial-drug-trial-death. Cited 12 May 2019.
- 205.van Esbroeck ACM, Janssen APA, Cognetta AB, Ogasawara D, Shpak G, van der Kroeg M, et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10–2474. Science. 2017;356(6342):1084–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.FDA finds drugs under investigation in the U.S. related to French BIA 10–2474 drug do not pose similar safety risks. FDA [Internet]; 2018. http://www.fda.gov/drugs/drug-safety-and-availability/fda-finds-drugs-under-investigation-us-related-french-bia-10-2474-drug-do-not-pose-similar-safety. Cited 6 Aug 2019.
- 207.Postnov A, Schmidt ME, Pemberton DJ, de Hoon J, van Hecken A, van den Boer M, et al. Fatty acid amide hydrolase inhibition by JNJ-42165279: a multiple-ascending dose and a positron emission tomography study in healthy volunteers. Clin Transl Sci. 2018;11(4):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wagenlehner FME, van Till JWO, Houbiers JGA, Martina RV, Cerneus DP, Melis JHJM, et al. Fatty acid amide hydrolase inhibitor treatment in men with chronic prostatitis/chronic pelvic pain syndrome: an adaptive double-blind, randomized controlled trial. Urology. 2017;103:191–7. [DOI] [PubMed] [Google Scholar]
- 209.D’Souza DC, Cortes-Briones J, Creatura G, Bluez G, Thurnauer H, Deaso E, et al. Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: a double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry. 2019;6(1):35–45. [DOI] [PubMed] [Google Scholar]
- 210.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66(5):1260–4. [DOI] [PubMed] [Google Scholar]
- 211.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16(7):744–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci USA. 2009;106(48):20270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334(6057):809–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013;92(8–9):492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nader J, Rapino C, Gennequin B, Chavant F, Francheteau M, Makriyannis A, et al. Prior stimulation of the endocannabinoid system prevents methamphetamine-induced dopaminergic neurotoxicity in the striatum through activation of CB2 receptors. Neuropharmacology. 2014;87:214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Blanco E, Pavón FJ, Palomino A, Luque-Rojas MJ, Serrano A, Rivera P, et al. Cocaine-induced behavioral sensitization is associated with changes in the expression of endocannabinoid and glutamatergic signaling systems in the mouse prefrontal cortex. Int J Neuropsychopharmacol. 2014. 10.1093/ijnp/pyu024. [DOI] [PMC free article] [PubMed]
- 217.Li W, Zhang C-L, Qiu Z-G. Differential expression of endocannabinoid system-related genes in the dorsal hippocampus following expression and reinstatement of morphine conditioned place preference in mice. Neurosci Lett. 2017;16(643):38–44. [DOI] [PubMed] [Google Scholar]
- 218.Schlosburg JE, Carlson BLA, Ramesh D, Abdullah RA, Long JZ, Cravatt BF, et al. Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J. 2009;11(2):342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Muldoon PP, Chen J, Harenza JL, Abdullah RA, Sim-Selley LJ, Cravatt BF, et al. Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. Br J Pharmacol. 2015;172(3):869–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gamage TF, Ignatowska-Jankowska BM, Muldoon PP, Cravatt BF, Damaj MI, Lichtman AH. Differential effects of endocannabinoid catabolic inhibitors on morphine withdrawal in mice. Drug Alcohol Depend. 2015;1(146):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wilkerson JL, Ghosh S, Mustafa M, Abdullah RA, Niphakis MJ, Cabrera R, et al. The endocannabinoid hydrolysis inhibitor SA-57: intrinsic antinociceptive effects, augmented morphine-induced antinociception, and attenuated heroin seeking behavior in mice. Neuropharmacology. 2017;01(114):156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372(6507):686–91. [DOI] [PubMed] [Google Scholar]
- 223.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277(5329):1094–7. [DOI] [PubMed] [Google Scholar]
- 224.Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem. 1997;69(2):631–8. [DOI] [PubMed] [Google Scholar]
- 225.Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, et al. Anandamide transport is independent of fattyacid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci USA. 2004;101(23):8756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gianessi CA, Groman SM, Thompson SL, Jiang M, van der Stelt M, Taylor JR. Endocannabinoid contributions to alcohol habits and motivation: relevance to treatment. Addict Biol. 2019;6:e12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cippitelli A, Bilbao A, Gorriti MA, Navarro M, Massi M, Piomelli D, et al. The anandamide transport inhibitor AM404 reduces ethanol self-administration. Eur J Neurosci. 2007;26(2):476–86. [DOI] [PubMed] [Google Scholar]
- 228.Gamaleddin I, Guranda M, Scherma M, Fratta W, Makriyannis A, Vadivel SK, et al. AM404 attenuates reinstatement of nicotine seeking induced by nicotine-associated cues and nicotine priming but does not affect nicotine- and food-taking. J Psychopharmacol. 2013;27(6):564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Scherma M, Justinová Z, Zanettini C, Panlilio LV, Mascia P, Fadda P, et al. The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br J Pharmacol. 2012;165(8):2539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Vlachou S, Nomikos GG, Panagis G. Effects of endocannabinoid neurotransmission modulators on brain stimulation reward. Psychopharmacology (Berl). 2006;188(3):293–305. [DOI] [PubMed] [Google Scholar]
- 231.Vlachou S, Stamatopoulou F, Nomikos GG, Panagis G. Enhancement of endocannabinoid neurotransmission through CB1 cannabinoid receptors counteracts the reinforcing and psychostimulant effects of cocaine. Int J Neuropsychopharmacol. 2008;11(7):905–23. [DOI] [PubMed] [Google Scholar]
- 232.Schindler CW, Scherma M, Redhi GH, Vadivel SK, Makriyannis A, Goldberg SR, et al. Self-administration of the anandamide transport inhibitor AM404 by squirrel monkeys. Psychopharmacology (Berl). 2016;233(10):1867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Levin FR, Mariani JJ, Brooks DJ, Pavlicova M, Cheng W, Nunes EV. Dronabinol for the treatment of cannabis dependence: a randomized, double-blind, placebo-controlled trial. Drug Alcohol Depend. 2011;116(1–3):142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Herrmann ES, Cooper ZD, Bedi G, Ramesh D, Reed SC, Comer SD, et al. Effects of zolpidem alone and in combination with nabilone on cannabis withdrawal and a laboratory model of relapse in cannabis users. Psychopharmacology (Berl). 2016;233(13):2469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Vandrey R, Stitzer ML, Mintzer MZ, Huestis MA, Murray JA, Lee D. The dose effects of short-term dronabinol (oral THC) maintenance in daily cannabis users. Drug Alcohol Depend. 2013;128(1–2):64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Haney M, Cooper ZD, Bedi G, Vosburg SK, Comer SD, Foltin RW. Nabilone decreases marijuana withdrawal and a laboratory measure of marijuana relapse. Neuropsychopharmacology. 2013;38(8):1557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Levin FR, Mariani JJ, Pavlicova M, Brooks D, Glass A, Mahony A, et al. Dronabinol and lofexidine for cannabis use disorder: a randomized, double-blind, placebo-controlled trial. Drug Alcohol Depend. 2016;1(159):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Haney M, Hart CL, Vosburg SK, Comer SD, Reed SC, Foltin RW. Effects of THC and lofexidine in a human laboratory model of marijuana withdrawal and relapse. Psychopharmacology (Berl). 2008;197(1):157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Budney AJ, Vandrey RG, Hughes JR, Moore BA, Bahrenburg B. Oral delta-9-tetrahydrocannabinol suppresses cannabis withdrawal symptoms. Drug Alcohol Depend. 2007;86(1):22–9. [DOI] [PubMed] [Google Scholar]
- 240.Haney M, Hart CL, Vosburg SK, Nasser J, Bennett A, Zubaran C, et al. Marijuana withdrawal in humans: effects of oral THC or divalproex. Neuropsychopharmacology. 2004;29(1):158–70. [DOI] [PubMed] [Google Scholar]
- 241.Jicha CJ, Lofwall MR, Nuzzo PA, Babalonis S, Elayi SC, Walsh SL. Safety of oral dronabinol during opioid withdrawal in humans. Drug Alcohol Depend. 2015;1(157):179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bisaga A, Sullivan MA, Glass A, Mishlen K, Pavlicova M, Haney M, et al. The effects of dronabinol during detoxification and the initiation of treatment with extended release naltrexone. Drug Alcohol Depend. 2015;1(154):38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lofwall MR, Babalonis S, Nuzzo PA, Elayi SC, Walsh SL. Opioid withdrawal suppression efficacy of oral dronabinol in opioid dependent humans. Drug Alcohol Depend. 2016;1(164):143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Samanta D Cannabidiol: a review of clinical efficacy and safety in epilepsy. Pediatr Neurol. 2019;96:24–9. [DOI] [PubMed] [Google Scholar]
- 245.Maroon J, Bost J. Review of the neurological benefits of phytocannabinoids. Surg Neurol Int. 2018;9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Bisogno T, Hanuš L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134(4):845–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Martínez-Pinilla E, Varani K, Reyes-Resina I, Angelats E, Vincenzi F, Ferreiro-Vera C, et al. Binding and signaling studies disclose a potential allosteric site for cannabidiol in cannabinoid CB2 receptors. Front Pharmacol. 2017;8:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Navarro G, Reyes-Resina I, Rivas-Santisteban R, Sánchez de Medina V, Morales P, Casano S, et al. Cannabidiol skews biased agonism at cannabinoid CB1 and CB2 receptors with smaller effect in CB1–CB2 heteroreceptor complexes. Biochem Pharmacol. 2018;157:148–58. [DOI] [PubMed] [Google Scholar]
- 251.Tham M, Yilmaz O, Alaverdashvili M, Kelly MEM, Denovan-Wright EM, Laprairie RB. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br J Pharmacol. 2019;176(10):1455–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Luján MÁ, Castro-Zavala A, Alegre-Zurano L, Valverde O. Repeated Cannabidiol treatment reduces cocaine intake and modulates neural proliferation and CB1R expression in the mouse hippocampus. Neuropharmacology. 2018;143:163–75. [DOI] [PubMed] [Google Scholar]
- 253.Galaj E, Bi G-H, Yang H-J, Xi Z-X. Cannabidiol attenuates the rewarding effects of cocaine by CB2, 5-TH1A and TRPV1 receptor mechanisms. Neuropharmacology. 2019. 10.1016/j.neuropharm.2019.107740. [DOI] [PMC free article] [PubMed]
- 254.Hay GL, Baracz SJ, Everett NA, Roberts J, Costa PA, Arnold JC, et al. Cannabidiol treatment reduces the motivation to self-administer methamphetamine and methamphetamine-primed relapse in rats. J Psychopharmacol (Oxford). 2018;32(12):1369–78. [DOI] [PubMed] [Google Scholar]
- 255.Viudez-Martínez A, García-Gutiérrez MS, Navarrón CM, Morales-Calero MI, Navarrete F, Torres-Suárez AI, et al. Cannabidiol reduces ethanol consumption, motivation and relapse in mice. Addict Biol. 2018;23(1):154–64. [DOI] [PubMed] [Google Scholar]
- 256.Ren Y, Whittard J, Higuera-Matas A, Morris CV, Hurd YL. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci. 2009;29(47):14764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Bi G-H, Galaj E, He Y, Xi Z-X. Cannabidiol inhibits sucrose self-administration by CB1 and CB2 receptor mechanisms in rodents. Addict Biol. 2019;19:e12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Katsidoni V, Anagnostou I, Panagis G. Cannabidiol inhibits the reward-facilitating effect of morphine: involvement of 5-HT1A receptors in the dorsal raphe nucleus. Addict Biol. 2013;18(2):286–96. [DOI] [PubMed] [Google Scholar]
- 259.Markos JR, Harris HM, Gul W, ElSohly MA, Sufka KJ. Effects of cannabidiol on morphine conditioned place preference in mice. Planta Med. 2018;84(4):221–4. [DOI] [PubMed] [Google Scholar]
- 260.Parker LA, Burton P, Sorge RE, Yakiwchuk C, Mechoulam R. Effect of low doses of delta9-tetrahydrocannabinol and cannabidiol on the extinction of cocaine-induced and amphetamine-induced conditioned place preference learning in rats. Psychopharmacology (Berl). 2004;175(3):360–6. [DOI] [PubMed] [Google Scholar]
- 261.Gonzalez-Cuevas G, Martin-Fardon R, Kerr TM, Stouffer DG, Parsons LH, Hammell DC, et al. Unique treatment potential of cannabidiol for the prevention of relapse to drug use: preclinical proof of principle. Neuropsychopharmacology. 2018;43(10):2036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.de Carvalho CR, Takahashi RN. Cannabidiol disrupts the reconsolidation of contextual drug-associated memories in Wistar rats. Addict Biol. 2017;22(3):742–51. [DOI] [PubMed] [Google Scholar]
- 263.Karimi-Haghighi S, Haghparast A. Cannabidiol inhibits priming-induced reinstatement of methamphetamine in REM sleep deprived rats. Prog Neuropsychopharmacol Biol Psychiatry. 2018;02(82):307–13. [DOI] [PubMed] [Google Scholar]
- 264.Bhargava HN. Effect of some cannabinoids on naloxone-precipitated abstinence in morphine-dependent mice. Psychopharmacology (Berl). 1976;49(3):267–70. [DOI] [PubMed] [Google Scholar]
- 265.Vilela LR, Gomides LF, David BA, Antunes MM, Diniz AB, Moreira F de A, et al. Cannabidiol rescues acute hepatic toxicity and seizure induced by cocaine. Mediators Inflamm [Internet]; 2015. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4427116/. Cited 2 Mar 2019. [DOI] [PMC free article] [PubMed]
- 266.Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Schoedel KA, Szeto I, Setnik B, Sellers EM, Levy-Cooperman N, Mills C, et al. Abuse potential assessment of cannabidiol (CBD) in recreational polydrug users: a randomized, double-blind, controlled trial. Epilepsy Behav. 2018;1(88):162–71. [DOI] [PubMed] [Google Scholar]
- 268.Manini AF, Yiannoulos G, Bergamaschi MM, Hernandez S, Olmedo R, Barnes AJ, et al. Safety and pharmacokinetics of oral cannabidiol when administered concomitantly with intravenous fentanyl in humans. J Addict Med. 2015;9:204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dalton WS, Martz R, Lemberger L, Rodda BE, Forney RB. Influence of cannabidiol on delta-9-tetrahydrocannabinol effects. Clin Pharmacol Ther. 1976;19(3):300–9. [DOI] [PubMed] [Google Scholar]
- 270.Karniol IG, Shirakawa I, Kasinski N, Pfeferman A, Carlini EA. Cannabidiol interferes with the effects of delta 9—tetrahydrocannabinol in man. Eur J Pharmacol. 1974;28(1):172–7. [DOI] [PubMed] [Google Scholar]
- 271.Zuardi AW, Shirakawa I, Finkelfarb E, Karniol IG. Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology (Berl). 1982;76(3):245–50. [DOI] [PubMed] [Google Scholar]
- 272.Haney M, Malcolm RJ, Babalonis S, Nuzzo PA, Cooper ZD, Bedi G, et al. Oral cannabidiol does not alter the subjective, reinforcing or cardiovascular effects of smoked cannabis. Neuropsychopharmacology. 2016;41(8):1974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Solowij N, Broyd S, Greenwood L-M, van Hell H, Martelozzo D, Rueb K, et al. A randomised controlled trial of vaporised Δ9-tetrahydrocannabinol and cannabidiol alone and in combination in frequent and infrequent cannabis users: acute intoxication effects. Eur Arch Psychiatry Clin Neurosci. 2019;269(1):17–35. [DOI] [PubMed] [Google Scholar]
- 274.Allsop DJ, Copeland J, Lintzeris N, Dunlop AJ, Montebello M, Sadler C, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry. 2014;71(3):281–91. [DOI] [PubMed] [Google Scholar]
- 275.Trigo JM, Soliman A, Quilty LC, Fischer B, Rehm J, Selby P, et al. Nabiximols combined with motivational enhancement/cognitive behavioral therapy for the treatment of cannabis dependence: a pilot randomized clinical trial. PLoS One. 2018;13(1):e0190768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mediavilla V, Steinemann S. Essential oil of Cannabis sativa L strains. J Int Hemp Assoc. 1997;4:82–4. [Google Scholar]
- 277.Sharma C, Al Kaabi JM, Nurulain SM, Goyal SN, Kamal MA, Ojha S. Polypharmacological properties and therapeutic potential of β-caryophyllene: a dietary phytocannabinoid of pharmaceutical promise. Curr Pharm Des. 2016;22(21):3237–64. [DOI] [PubMed] [Google Scholar]
- 278.Corey EJ, Mitra RB, Hisashi U. Total synthesis of d, l-caryophyllene and d, l-isocaryophyllene. J Am Chem Soc. 1964;86(3):485–92. [Google Scholar]
- 279.Gertsch J, Leonti M, Raduner S, Racz I, Chen J-Z, Xie X-Q, et al. Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci USA. 2008;105(26):9099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Varga ZV, Matyas C, Erdelyi K, Cinar R, Nieri D, Chicca A, et al. β-Caryophyllene protects against alcoholic steatohepatitis by attenuating inflammation and metabolic dysregulation in mice. Br J Pharmacol. 2018;175(2):320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Liu H, Yang G, Tang Y, Cao D, Qi T, Qi Y, et al. Physicochemical characterization and pharmacokinetics evaluation of β-caryophyllene/β-cyclodextrin inclusion complex. Int J Pharm. 2013;450(1–2):304–10. [DOI] [PubMed] [Google Scholar]
- 282.Schmitt D, Levy R, Carroll B. Toxicological evaluation of β-caryophyllene oil: subchronic toxicity in rats. Int J Toxicol. 2016;35(5):558–67. [DOI] [PubMed] [Google Scholar]
- 283.Oliveira GL da S, Machado KC, Machado KC, da Silva APD-SCL, Feitosa CM, de Castro Almeida FR. Non-clinical toxicity of β-caryophyllene, a dietary cannabinoid: absence of adverse effects in female Swiss mice. Regul Toxicol Pharmacol. 2018;92:338–46. [DOI] [PubMed] [Google Scholar]
- 284.Al Mansouri S, Ojha S, Al Maamari E, Al Ameri M, Nurulain SM, Bahi A. The cannabinoid receptor 2 agonist, β-caryophyllene, reduced voluntary alcohol intake and attenuated ethanol-induced place preference and sensitivity in mice. Pharmacol Biochem Behav. 2014;124:260–8. [DOI] [PubMed] [Google Scholar]
- 285.Rose JE, Behm FM. Inhalation of vapor from black pepper extract reduces smoking withdrawal symptoms. Drug Alcohol Depend. 1994;34(3):225–9. [DOI] [PubMed] [Google Scholar]
- 286.He Y, Galaj E, Bi GH, Wang XF, Gardner EL, Xi Z-X. Beta-caryophyllene: a dietary cannabis terpene, inhibits nicotine-taking and nicotine-seeking behavior in rodents. Br J Pharmacol. 2019. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gill EW, Paton WDM, Pertwee RG. Preliminary experiments on the chemistry and pharmacology of cannabis. Nature. 1970;228:134–6. [DOI] [PubMed] [Google Scholar]
- 288.Bolognini D, Costa B, Maione S, Comelli F, Marini P, Di Marzo V, et al. The plant cannabinoid Delta9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br J Pharmacol. 2010;160(3):677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ(9)-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. 2015;172(3):737–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, et al. Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146(7):917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Pertwee RG, Thomas A, Stevenson LA, Ross RA, Varvel SA, Lichtman AH, et al. The psychoactive plant cannabinoid, Delta9-tetrahydrocannabinol, is antagonized by Delta8- and Delta9-tetrahydrocannabivarin in mice in vivo. Br J Pharmacol. 2007;150(5):586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Dennis I, Whalley BJ, Stephens GJ. Effects of Δ9-tetrahydrocannabivarin on [35S]GTPγS binding in mouse brain cerebellum and piriform cortex membranes. Br J Pharmacol. 2008;154(6):1349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hill AJ, Weston SE, Jones NA, Smith I, Bevan SA, Williamson EM, et al. Δ9-Tetrahydrocannabivarin suppresses in vitro epileptiform and in vivo seizure activity in adult rats. Epilepsia. 2010;51(8):1522–32. [DOI] [PubMed] [Google Scholar]
- 294.Cascio MG, Zamberletti E, Marini P, Parolaro D, Pertwee RG. The phytocannabinoid, Δ9-tetrahydrocannabivarin, can act through 5-HT1A receptors to produce antipsychotic effects. Br J Pharmacol. 2015;172(5):1305–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163(7):1479–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Janssens A, Silvestri C, Martella A, Vanoevelen JM, Di Marzo V, Voets T. Δ9-tetrahydrocannabivarin impairs epithelial calcium transport through inhibition of TRPV5 and TRPV6. Pharmacol Res. 2018;136:83–9. [DOI] [PubMed] [Google Scholar]
- 297.Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J Biol Chem. 2012;287(1):91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Riedel G, Fadda P, McKillop-Smith S, Pertwee RG, Platt B, Robinson L. Synthetic and plant-derived cannabinoid receptor antagonists show hypophagic properties in fasted and non-fasted mice. Br J Pharmacol. 2009;156(7):1154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wargent ET, Zaibi MS, Silvestri C, Hislop DC, Stocker CJ, Stott CG, et al. The cannabinoid Δ(9)-tetrahydrocannabivarin (THCV) ameliorates insulin sensitivity in two mouse models of obesity. Nutr Diabetes. 2013;27(3):e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Bátkai S, Mukhopadhyay P, Horváth B, Rajesh M, Gao RY, Mahadevan A, et al. Δ8-Tetrahydrocannabivarin prevents hepatic ischaemia/reperfusion injury by decreasing oxidative stress and inflammatory responses through cannabinoid CB2 receptors. Br J Pharmacol. 2012;165(8):2450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Xi Z-X, Muldoon P, Wang XF, Bi G-H, Damaj IM, Lichtman AH, et al. Δ8-Tetrahydrocannabivarin has potent anti-nicotine effects in multiple rodent models of nicotine dependence. Br J Pharmacol. 2019. 10.1111/bph.14844. [DOI] [PMC free article] [PubMed]
- 302.Englund A, Atakan Z, Kralj A, Tunstall N, Murray R, Morrison P. The effect of five day dosing with THCV on THC-induced cognitive, psychological and physiological effects in healthy male human volunteers: a placebo-controlled, double-blind, crossover pilot trial. J Psychopharmacol (Oxford). 2016;30(2):140–51. [DOI] [PubMed] [Google Scholar]
- 303.Jadoon KA, Ratcliffe SH, Barrett DA, Thomas EL, Stott C, Bell JD, et al. Efficacy and safety of cannabidiol and tetrahydrocannabivarin on glycemic and lipid parameters in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, parallel group pilot study. Diabetes Care. 2016;39(10):1777–86. [DOI] [PubMed] [Google Scholar]
- 304.Tudge L, Williams C, Cowen PJ, McCabe C. Neural effects of cannabinoid CB1 neutral antagonist tetrahydrocannabivarin on food reward and aversion in healthy volunteers. Int J Neuropsychopharmacol. 2014. 10.1093/ijnp/pyu094. [DOI] [PMC free article] [PubMed]