

Abstract
Purpose
Maintaining levels of nicotinamide adenine dinucleotide (NAD+), a coenzyme critical for cellular energetics and biosynthetic pathways, may be therapeutic in retinal disease because retinal NAD+ levels decline during retinal damage and degeneration. The purpose of this study was to investigate whether systemic treatment with nicotinamide riboside (NR), a NAD+ precursor that is orally deliverable and well-tolerated by humans, is protective in a mouse model of light-induced retinal degeneration.
Methods
Mice were injected intraperitoneally with vehicle or NR the day before and the morning of exposure to degeneration-inducing levels of light. Retinal function was assessed by electroretinography and in vivo retinal morphology and inflammation was assessed by optical coherence tomography. Post mortem retina sections were assessed for morphology, TUNEL, and inflammatory markers Iba1 and GFAP. Retinal NAD+ levels were enzymatically assayed.
Results
Exposure to degeneration-inducing levels of light suppressed retinal NAD+ levels. Mice undergoing light-induced retinal degeneration exhibited significantly suppressed retinal function, severely disrupted photoreceptor cell layers, and increased apoptosis and inflammation in the outer retina. Treatment with NR increased levels of NAD+ in retina and prevented these deleterious outcomes.
Conclusions
This study is the first to report the protective effects of NR treatment in a mouse model of retinal degeneration. The positive outcomes, coupled with human tolerance to NR dosing, suggest that maintaining retinal NAD+ via systemic NR treatment should be further explored for clinical relevance.
Keywords: nicotinamide adenine dinucleotide, retinal degeneration, nicotinamide riboside
Metabolic dysregulation in photoreceptor or RPE cells is associated with blinding diseases such as Leber congenital amaurosis type 9,1–3 Leber hereditary optic neuropathy,4 and Age-Related Macular Degeneration (AMD).5 Nicotinamide adenine dinucleotide (NAD+) is the central hydride-transferring cofactor in metabolism and a substrate for NAD+-consuming enzymes.6–8 Maintaining NAD+ levels is critical to retinal health. Retinal NAD+ levels decrease with age in mice,9 are diminished in several models of retinal degeneration,9–12 and are low in post mortem RPE from AMD patients.5 Diminished activities of nicotinamide mononucleotide adenylyltransferase-1 or nicotinamide phosphoribosyltransferase, enzymes required for NAD+ biosynthesis, cause retinal dystrophy in humans1–3 and retinal degeneration in mice.7,9,13 Conversely, treatment with NAD+ biosynthetic precursors nicotinamide or nicotinamide mononucleotide, protects against retinal degeneration in mice and rats.9,10
Brenner and colleagues discovered an alternative NAD+ salvage biosynthesis pathway in which nicotinamide riboside (NR), a form of vitamin B3 that was found in milk and other foods,14,15 can be taken up from oral dosing in humans.16 NR enters cells through nucleoside transporters and is then converted to NAD+ by NR kinases (NMRK1 and NMRK2) and nicotinamide mononucleotide adenylyltransferase isozymes.17,18 The NMRK enzymes are upregulated after neuronal injury or during extreme energetic stress.6 Treatment with NR maintains NAD+ levels in target tissues and cells and is protective in several models of neurodegeneration, including models of Alzheimer's disease, amyotrophic lateral sclerosis, and Parkinson's disease.19–27
We hypothesized that NR treatment will be protective in retinal degeneration. In this study, we tested whether systemic delivery of NR is protective in a mouse model of light-induced retinal degeneration (LIRD).28–30 NR treatment increased retinal NAD+, prevented LIRD-induced retinal NAD+ suppression, and protected retinal morphology and function.
Methods
Animal Models
All mouse procedures were approved by the Emory Institutional Animal Care and Use Committee and followed the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Adult (3 months old) male BALB/cAnNCrl (BALB/c) mice were obtained from Charles River Laboratory (Wilmington, MA, USA) and were housed under a 12:12-hour light–dark cycle (7 AM on and 7 PM off). During the light cycle, light levels measured at the bottom of mouse cages ranged from 5 to 45 lux. Induction of LIRD was previously described.28 Briefly, mice were exposed to 3000 lux light for 4 hours, then returned to home cages under normal lighting conditions for the remainder of the experiment. At the time of light induction they weighed 22 to 28 g. NR treatment did not significantly alter weight (data not shown). Mice were euthanized by asphyxiation with CO2 gas for all experiments.
Drug Administration
NR chloride was from ChromaDex (Item #ASB-00014332-101, Lot# 40C910-18209-21). The drug vehicle was PBS (VWRVK813, Cat#97063-660), which has a 1× solution composition of 137 mM NaCl, 2.7 mM KCl, and 9.5 mM phosphate buffer. Solutions were made fresh each day. BALB/c mice received intraperitoneal injections of either vehicle or NR (1000 mg/kg dose in PBS unless otherwise specified by experiment as reported in the Results) using an injection volume of 10 µL of solution per gram of mouse body weight in accordance with in vivo rodent experiments of Brown et al.20 Unless otherwise specified, two injections were administered before toxic light exposure. One injection was performed the day before (at 4 PM), and the other injection was performed the morning of (at 9 AM) light insult. Toxic light exposure was from 10 AM to 2 PM.
Electroretinograms (ERG)
The complete ERG protocol was previously detailed.31 Briefly, mice dark-adapted overnight were anesthetized with intraperitoneal injections of ketamine (10 mg/mL; AmTech Group Inc., Tempe, AZ) and xylazine (100 mg/mL; AKORN, Lake Forest, IL),31 placed on a heating pad inside a Faraday cage in front of a UBA-4200 Series desktop Ganzfeld stimulator (MDIT-100, LKC Technologies, Gaithersburg, MD), a DTL fiber active electrode placed on top of each cornea, reference electrodes inserted into the cheeks, and a ground electrode placed in the tail. ERGs were recorded for the scotopic condition (0.00039-25.3 cd s/m2 with increasing flash stimulus intervals from 2.0 to 62.6 seconds). In the LIRD model, ERGs were performed 1 week after toxic light exposure.
In Vivo Ocular Imaging
Spectral domain optical coherence tomography (SD-OCT) was conducted immediately after ERG measurement, when mice were still anesthetized and their pupils were still dilated. A Micron IV SD-OCT system with fundus camera (Phoenix Research Labs, Pleasanton, CA) and a Heidelberg Spectralis HRA+OCT instrument with +25D lens (Heidelberg Engineering, Heidelberg, Germany) were used in tandem sequentially to assess ocular posterior segment morphology in section and en face. Using the Micron IV system, image-guided OCT images were obtained for the left and right eyes after a sharp and clear image of the fundus (with the optic nerve centered) was obtained. SD-OCT imaging was a circular scan about 100 µm from the optic nerve head. Fifty scans were averaged. The retinal layers were identified according to published nomenclature.32 Total photoreceptor and retinal thickness were analyzed using Adobe Photoshop CS6 (Adobe Systems Inc., San Jose, CA). The number of pixels was converted into micrometers by multiplying by the micrometers/pixel conversion factor (1.3 microns = 1 pixel). Immediately after imaging on the Micron IV system, a rigid contact lens was placed on the eye (back optic zone radius: 1.7 mm, diameter: 3.2 mm, power: PLANO) and autofluorescence imaging at the layer of the photoreceptor-RPE interface was conducted using the Heidelberg Spectralis HRA+OCT instrument. During imaging and afterwards through anesthesia recovery, mice were kept on a water circulating heat pad set to 39 °C to maintain body temperature.
Histology, Immunofluorescence, and Morphometrics
BALB/c mice were euthanized 1 week after light exposure. We followed the recommendations of Howland and Howland33 for nomenclature of axes and planes in the mouse eye, as indicated in Figure 1. Histologic and morphometric procedures followed standard techniques as previously described.34,35 Briefly, eyes were dehydrated, embedded in paraffin, and sectioned through the sagittal plane on a microtome at 5-µm increments through the optic nerve head and the center of the cornea to ensure that consistent regions were examined between animals. The slides were deparaffinized across five Coplin jars with 100 mL of xylene for 2 minutes each, consecutively. Then the slides were rehydrated in a series of 100 mL ethanol solutions for 2 minutes each: 100%, 90%, 80%, 70%, 60%, and 50% ethanol. After that, the slides were immersed in PBS for 5 minutes.
Figure 1.
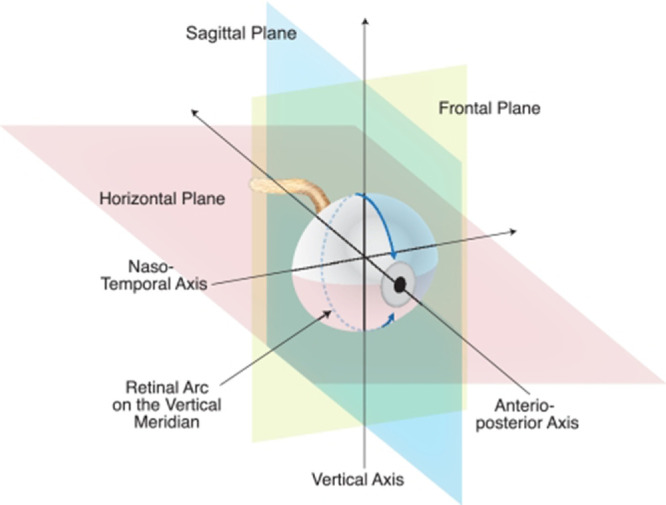
Naming conventions recommended by Howland and Howland for planes and axes of the vertebrate eye regardless of species. A histologic section cut on the vertical plane through the great meridian is illustrated.79 The horizontal, vertical, and frontal planes are marked, as are the anterior–posterior (A–P) axis (also known as optic axis), the nasal–temporal (N–T) axis, and the superior–inferior (S–I; vertical) axis. Reprinted with permission from Wisard J, Faulkner A, Chrenek MA, et al. Exaggerated eye growth in IRBP-deficient mice in early development. Invest Ophthalmol Vis Sci 2011;52:5804–5811. Copyright 2011 CC BY-NC-ND 4.0 license.
After rehydration, a TUNEL assay was performed on some sections according to the protocol for the DeadEnd Fluorometric TUNEL Kit (Promega, Fitchburg, WI). Stained sections were imaged using fluorescent microscopy and TUNEL-positive cells in the outer nuclear layer (ONL) were manually counted for each whole retina using Adobe Photoshop CS6. Some sections were used for hematoxylin and eosin (H&E) staining. ONL nuclei were counted within 100 µm-wide segments spaced at 250, 750, 1250, and 1750 µm from the optic nerve head in both the inferior and superior directions. Mean counts of n = 3 to 6 mice per group were plotted as a spidergram.
For detecting Ionized calcium binding adaptor molecule 1 (Iba1), sections were blocked for 30 minutes in 0.1 M Tris-buffered saline (TBS; Corning 46012CM pH 7.4, Fisher Scientific, Suwanee, GA) containing 2.5% donkey serum, then incubated in the primary antibody (Rabbit anti-Iba1; 1:500; ab178847; Abcam; Cambridge, UK) diluted in the blocking serum overnight at 4 °C. Sections were rinsed twice with 0.1 M TBST (0.1% Tween-20) for 2 minutes each following primary antibody incubation, then incubated for 30 minutes at room temperature with the secondary antibody solution (donkey Anti-Rabbit IgG; 1:1000; Alexa Fluor 488; A21206; Life Technologies, Waltham, MA). Sections were then washed twice with 0.1 M TBST for 2 minutes each and then incubated in diluted PI solution (1:500 in 0.1 M TBS) for 2 minutes at room temperature. The sections were washed in 0.1 M TBS for 5 minutes and mounted with mounting medium, and coverslipped. Bitmaps from the raw images were used for quantification. Using ImageJ, the neural retina, including retinal ganglion cell layer, inner plexiform layer, inner nuclear layer, outer plexiform layer, ONL, and outer segment were each selected with the lasso tool. The total pixels in this selection were recorded. The selection was isolated and split into RGB channels. The green channel was thresholded into a black and white image, with the threshold set at 38 bits. Total black pixels were recorded. Data are reported as the thresholded green pixels divided by the total pixels in the neural retina.
For detecting glial fibrillary acidic protein (GFAP) in ocular sections, sections were blocked for 30 minutes in 0.1 M TBS containing 5% donkey serum, then incubated in the primary antibody (rabbit anti-GFAP; 1:500; Z0334; Agilent Dako, Santa Clara, CA) diluted in the blocking serum overnight at 4 °C. Sections were rinsed three times with 0.1 M TBS for 15 minutes each following primary antibody incubation, then incubated for 2 hours at room temperature with the secondary antibody solution (donkey anti-rabbit IgG; 1:1000; Alexa Fluor 568; A10042; Life Technologies). Sections were then washed three times with 0.1 M TBS for 15 minutes each, mounted with a 4',6-diamidino-2-phenylindole (DAPI) mounting medium, and coverslipped.
The slides immunostained for Iba1 or GFAP were stored in the dark at 4 °C until imaging using a Nikon Ti inverted microscope with C1 confocal scanner (Nikon Instruments Inc., Melville, NY). Using an automated XY stage control within the EZ-C1 software, the flatmount was imaged with a 20× objective lens. Then, confocal images from the entire section were photomerged using Adobe Photoshop CS6. Bitmaps from the raw images were used for quantification. Using ImageJ, all pixels outside the neural retina were deleted. The remaining image was split into RGB channels. The red channel was thresholded into a black and white image, with the threshold set at 38 bits. Total black pixels were recorded.
NAD+ Measurements
Levels of NAD+ in homogenates were measured using a commercially available kit by following manufacturer's instructions (Abcam; ab. 65348; #Lot: GR3226737-3; San Francisco, CA). In brief, retina samples were homogenized in extraction buffer. Extracted samples were separated into two aliquots. One was used to measure total NAD (NADt). For NADH-specific measurements, samples were heated to 60 °C for 30 minutes to decompose NAD+. Extracted samples were placed in a 96-well plate and the NADH developer was added into each well. The plate was placed into a hybrid reader and read every 30 minutes at OD 450 nm while the color was developing. Data from the 2-hour time point are presented. NADt and NADH concentrations were quantified against an NADH standard curve. In the end, NAD+ was calculated with the equation NAD+ = NADt – NADH.
Statistical Analyses
Statistical analyses were conducted using Prism 8.4.2 Software (GraphPad Software Inc. La Jolla, CA). One-way or two-way ANOVA with Newman-Keuls’ post hoc test,36 were performed for ERG, biochemical, and morphometric data. Unless otherwise noted, n is the number of animals per experimental group. For all analyses, results were considered statistically significant if P < 0.05. All graphs display data as mean ± SEM.
Results
NR Treatment Prevents Light-Induced Diminution of Retinal NAD+ Levels
NR is a NAD+ precursor that when given systemically increases NAD+ levels in many tissues, including central nervous system structures.19,25,26 The protective effects of NR treatment are ascribed in part to local NAD+ increases in target tissues.20–22,24,26 To test whether this is the case in retina, BALB/c mice were intraperitoneal injected with NR (1000 mg/kg) or vehicle (PBS). Each of these groups was split in half, one remaining in normal maintenance light and the other exposed to a LIRD-inducing light level as described in the Methods. Forty-eight hours after the end of light exposure, retinas were harvested and assayed for NAD+ content. NAD+ concentration significantly decreased in retinas exposed to LIRD-inducing light compared with those in control (“dim”) lighting (Fig. 2). NR treatment prevented this decrease in NAD+, and indeed increased retinal NAD+ in both lighting conditions (Fig. 2).
Figure 2.
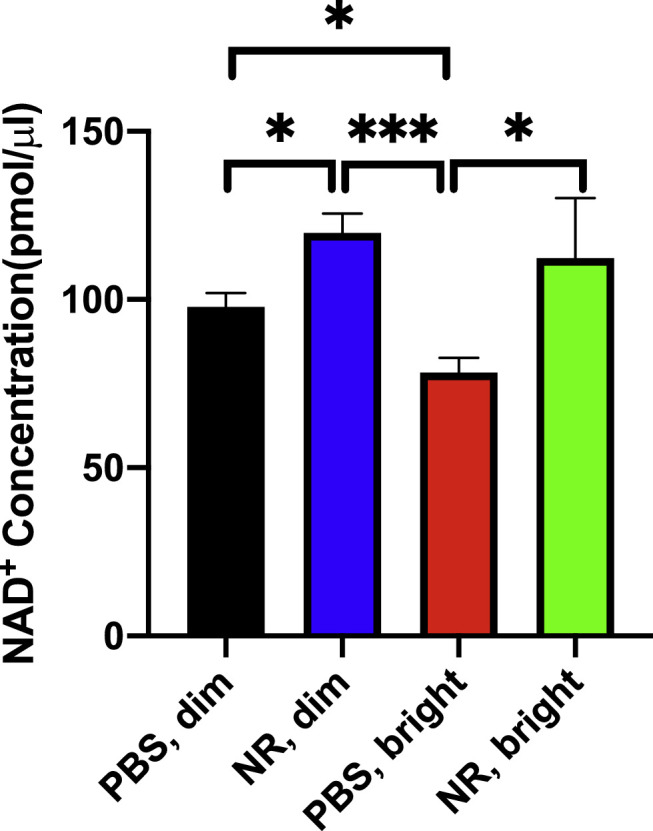
NR treatment increases retinal NAD+ and maintains retinal NAD+ after toxic light exposure. Mice were treated with either NR (1000 mg/kg) or vehicle (PBS). The retinas were harvested and assayed 48 hours after the end of light exposure. NAD+ concentration significantly decreased in retinas exposed to LIRD-inducing light (red bar) compared with those in control lighting (black and blue bars). NR treatment prevented this decrease in NAD+ (green bar). *P < 0.05, ***P < 0.001 by one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 4–9 retinas per group. Error bars represent SEM.
NR Treatment Preserves Retinal Function and Morphology in LIRD Mice
ERG a- and b-wave mean amplitudes of PBS-treated BALB/c mice were significantly decreased 1 week after exposure to 3000 lux light for 4 hours. Representative ERG waveforms of one eye of a single mouse from each treatment group are shown in Figure 3A. This functional loss was entirely prevented in mice treated with NR (Figs. 3B, 3C).
Figure 3.

NR (1000 mg/kg) treatment preserves retinal function in LIRD mice. Representative ERG waveforms (A) of one eye of a single mouse from each treatment group. Scotopic ERG a-wave (B) and b-wave (C) mean amplitudes from LIRD mice at 1 week after LIRD induction. Mice treated with PBS and exposed to 3000-lux light for 4 hours to induce degeneration exhibited a- and b-wave mean amplitudes (red) that were statistically significantly diminished compared with those of dim groups (blue and black). However, the mean ERG amplitudes of mice undergoing retinal degeneration and treated with NR (green) were statistically indistinguishable from those treated with PBS and exposed to bright light. **P < 0.01, ***P < 0.001, ****P < 0.0001 versus other groups by two-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 4–6 mice per group. Error bars represent SEM.
As imaged in vivo by fundus photography and SD-OCT 1 week after degeneration induction, the retinas of LIRD mice treated with PBS exhibited significant damage (Fig. 4). The representative fundus image of Figure 4A shows the region being measured. Corresponding SD-OCT images show considerable thinning of the photoreceptor layer that is prevented by NR treatment (Fig. 4B). Quantification of total retina and photoreceptor layer thicknesses shows that mice undergoing LIRD had significantly thinner retinas compared with noninduced mice, largely owing to nearly complete loss of the photoreceptor layer. This was entirely prevented with NR treatment (Figs. 4C, 4D). Preservation of retinal thickness in mice undergoing LIRD was dose dependent, with significant protection observed with a 500 mg/kg dose (Fig. 4E), the lowest concentration tested, and complete protection obtained at 1000 mg/kg (Fig. 4E) relative to non-LIRD control retinas (see Figs. 4C, 4D).
Figure 4.

NR treatment preserves photoreceptor layer thickness and total retinal thickness as assessed in vivo following LIRD. (A) Representative fundus image. (B) Representative OCT images from each group. The OCT image is a circular scan about 100 µm from the optic nerve head. Photoreceptor thickness (C) and retinal thickness (D) from BALB/c mice 1 week after retinal degeneration induction. Mice treated with PBS and exposed to 3000-lux light for 4 hours (red bar) exhibited great losses in thickness of the photoreceptor and retina layers, whereas induced mice treated with NR (green bar) exhibited statistically significant preservation of layer thickness. (E) Treating with increasing concentrations of NR up to 1000 mg/kg results in significantly increasing retina thicknesses in mice exposed to 3000-lux light for 4 hours. *P < 0.05 between two adjacent groups; ****P < 0.0001 versus all other group means; one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 3–9 mice per group. Error bars represent SEM. Size marker = 200 µm or 100 µm.
Photomicroscopy of H&E–stained sections of eyes harvested 1 week after LIRD induction showed marked degradation of morphology in the outer retina in LIRD mice treated with PBS compared with noninduced mice (Figs. 5A, 5B and Supplementary Fig. S1). In induced mice treated with PBS, photoreceptor cell inner and outer segments and most of the nuclei of the ONL were eliminated, with loss predominantly centrally (Figs. 5A–D and Supplementary Fig. S1). Nearly all of this degeneration was prevented in mice treated with NR (Fig. 5C and Supplementary Fig. S1). Quantification of ONL nuclei counts across retinal sections confirmed significant losses owing to degeneration in PBS-treated mice and confirmed nearly-complete preservation in NR-treated mice (Fig. 5D and Supplementary Fig. S1).
Figure 5.

NR treatment preserves nuclei of the outer nuclear layer in the LIRD mice. (A–C) Representative H&E images of retina sections from each group at the region of 250 to 750 µm from the optic nerve head. Complete sections are shown in Supplementary Figure S1. (D). One week after LIRD induction, nuclei were counted in eight discrete regions of retinal sections starting at 250 µm from the optic nerve head and extending every 500 µm outward along both the dorsal/superior (positive values on abscissa) and ventral periphery/inferior (negative numbers on abscissa). PBS, bright–treated mice (red) showed significant loss of nuclei at six distances from the optic nerve head compared with the control group (black). However, NR treated mice (green), exhibited mean nuclei counts statistically indistinguishable from that of the control group (black) throughout the length of the retina. ***P < 0.001, ****P < 0.0001 versus other groups by two-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 3–6 mice per group. Error bars represent SEM. Size marker = 50 µm.
NR Treatment Prevents Accumulation of TUNEL Signal in Photoreceptor Cells of LIRD Mice
Paraffin-embedded ocular sections from mice euthanized 1 week after toxic light induction were stained for TUNEL and DAPI to label nuclei that contained double-stranded DNA breaks (a marker of programmed cell death [apoptosis] or other forms of cell death).37 ONL TUNEL signal was high in retinas from LIRD mice treated with PBS compared with retinas from uninduced mice (Figs. 6A, 6B and Supplementary Fig. S2). Induced mice treated with NR exhibited significantly less TUNEL signal (Fig. 6C and Supplementary Fig. S2). These data suggest that NR treatment diminished or delayed apoptosis in photoreceptor cells.
Figure 6.

NR treatment prevents increases of TUNEL-positive cells following induction of LIRD. Both PBS- and NR-treated BALB/c mice received toxic light exposure and were euthanized at 1 week after exposure. Retinas from mice treated as described in text were fixed, sectioned, and used in a TUNEL assay. (A–C) Representative morphologic images of each group; the green signal is TUNEL and the blue signal is DAPI staining. Complete sections are shown in Supplementary Figure S2. (D) TUNEL-positive nuclei in ONL were counted from the entire retina. NR treated mice (green bar) exhibited significantly fewer TUNEL-positive cells compared with the PBS treated group (red bar). *P < 0.05, **P < 0.01 by one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 4 mice per group. Error bars represent SEM. Size marker = 50 µm.
NR Treatment Prevents Inflammatory Responses Following Induction of LIRD
Subretinal autofluorescent spots observed in vivo by fundus examination in patients and in animal models are considered diagnostic markers for inflammatory responses in retinal damage and disease.38–42 To test whether NR treatment alters inflammatory responses to LIRD, in vivo fundus examination at the level of the subretinal space was conducted 1 week after exposure of mice to toxic light. Eyes from mice not exposed to toxic light and not undergoing LIRD showed few autofluorescent spots at the level of the subretinal space in vivo (Fig. 7A). In eyes of mice undergoing LIRD and treated with PBS, numerous and widespread autofluorescent spots were observed (Fig. 7B). This outcome was prevented in mice treated with NR, which exhibited fewer autofluorescent spots (Fig. 7C), similar in number and pattern to the uninduced group, confirmed by statistical testing on counts of these spots across several autofluorescent fundus images (Fig. 7D). These in vivo data suggest that LIRD leads to an inflammatory response that is largely prevented by NR treatment.
Figure 7.

NR treatment prevented subretinal autofluorescence observed in vivo in LIRD mice. (A–C) Representative morphology images from each group at the level of the photoreceptor-RPE interface. In vivo Spectralis HRA+OCT images (with autofluorescence detection) were taken 1 week after induction of degeneration. (D) Autofluorescent spots were counted across the fundus image field. Few were detected in uninduced mice (black). Toxic light-exposed mice treated with NR exhibited significantly fewer autofluorescent spots compared with the PBS treated group. ****P < 0.0001 one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 3–6 mice per group. Error bars represent SEM. Size marker = 200 µm.
To further assess the effects of NR treatment on LIRD inflammatory responses, ocular sections from mice euthanized 1 week after light exposure were stained immunohistologically for Iba1, a marker for microglia and monocytes.43,44 Eyes from mice not exposed to toxic light and not undergoing LIRD showed few Iba1-positive cells in the outer retina (Fig. 8A). In the eyes of mice that had undergone LIRD and treated with PBS, numerous and widespread Iba1-positive cells were observed (Fig. 8B). This was prevented in mice treated with NR, which exhibited fewer Iba1-positive cells in outer retina (Fig. 8C), similar in number and pattern to the uninduced group, confirmed by quantification of total Iba1 immunosignal in outer retina across several sections (Fig. 8D). These post mortem data suggest that LIRD leads to the appearance of Iba1-postive cells in outer retina, a response that is largely prevented by NR treatment.
Figure 8.

NR treatment prevents activation of Iba1-positive cells after induction of LIRD. Both PBS- and NR-treated BALB/c mice received toxic light exposure and were euthanized at 1 week after exposure. (A–C) Representative morphologic images of each group; green is Iba1 immunofluorescence signal and red is PI staining. (D) Iba1-positive signals were quantified from the entire retina. NR treated mice (green bar) exhibited significantly less Iba1 signals compared with the PBS treated group (red bar). *P < 0.05 by one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 4–5 mice per group. Error bars represent SEM. Size marker = 50 µm.
Müller glia cell reactive gliosis was assessed by immunofluorescence staining for GFAP, a marker for reactive Müller cells and astrocytes,45 on post mortem ocular sections prepared from mice euthanized 1 week after toxic light exposure. In ocular sections from mice not exposed to toxic light and not undergoing LIRD, GFAP staining largely localized near the inner limiting membrane and less so in the outer plexiform layer (Fig. 9A). In eyes of mice undergoing LIRD and treated with PBS, heavy GFAP staining was observed in processes spanning the inner plexiform layer (Fig. 9B). This additional signal was not in mice undergoing LIRD but treated with NR (Fig. 9C). Quantification of GFAP immunosignal confirmed that GFAP expression was statistically significantly increased in LIRD tissue, but that this increase was prevented with NR treatment (Fig. 9D).
Figure 9.

NR treatment prevents GFAP accumulating after induction of LIRD. Both PBS and NR treated BALB/c mice received toxic light exposure and were euthanized at 1 week after exposure. (A–C) Representative morphologic images of each group; red is GFAP immunofluorescence signal and blue is DAPI staining. (D) GFAP signals were quantified from the entire retina. NR-treated mice (green bar) exhibited significantly less GFAP signals compared with the PBS-treated group (red bar). *P < 0.05, **P < 0.01 by one-way ANOVA with Newman-Keuls multiple comparisons post hoc test. N = 4–5 mice per group. Error bars represent SEM. Size marker = 50 µm.
Discussion
Treatment with NR has been shown to be protective in many models of neurodegenerative diseases, such as Parkinson's disease25 and Alzheimer's disease,19 and is well-tolerated via oral dosing in humans.46,47 In this study, we found that systemic treatment with NR increased and maintained retinal levels of NAD+ and protected photoreceptors in a model of retinal degeneration. Our study is the first to demonstrate protective effects of NR in a retinal degeneration mouse model.
Light-induced retinal damage is an acute model of retinal degeneration with a well-described oxidative stress response.48 Similar to a report from Lin et al.,9 we found that retinal NAD+ levels were suppressed as early as 2 days after bright light exposure (Fig. 2). NR treatment increased retinal NAD+ levels in maintenance light conditions and prevented NAD+ diminution following toxic light exposure (Fig. 2).
To determine whether this maintenance of NAD+ could protect the retina, we assessed (by ERG) the efficacy of NR treatment in preserving photoreceptor function in mice undergoing LIRD (Fig. 3). Remarkably, just two NR intraperitoneal injections completely preserved scotopic a- and b- wave amplitudes. In vivo OCT imaging and fundus imaging showed that systemic NR treatment protected photoreceptors and whole retina from degeneration in a dose-dependent manner, with a maximal effect (no discernable loss) at 1000 mg/kg dosage and a significant effect down to 500 mg/kg (Fig. 4). Of consideration, a 500 mg/kg/d dose in mice allometrically scales to a 2.4 g/d human equivalent dose (normalization of dose to body surface area).49,50 Oral dosing of humans at 2000 mg/d is tolerated well, and even lower doses increase circulating NAD+ levels and suppress circulating levels of inflammatory cytokines.46,47,51 Postmortem H&E staining provided more detailed information on retinal morphology, indicating that NR treatment protected retinal structure, both inferior and superior (Fig. 5). Retinas of mice undergoing LIRD exhibited marked TUNEL staining, especially in the ONL, suggesting massive apoptosis of photoreceptor cells (Fig. 6, compare Fig. 6A with Fig. 6B). NR treatment largely prevented this outcome (Fig. 6C), suggesting that apoptosis was suppressed. This effect, although not complete, apparently prevented enough subsequent cell death that bright light exposure had no discernable impact in other measurements taken 1 week after induction of LIRD, such as ERG amplitudes (Fig. 3) or morphometrics derived from OCT (Fig. 4) or post mortem ONL nuclei counts (Fig. 5). Possibly these measurements are not sensitive enough to detect the impact of a small population of cells that were not protected from apoptosis and, presumbably, subsequent death. Possibly taking functional and morphologic measurements at times later than 1 week after light exposure (e.g., 2 weeks, 1 month, etc.) would show effects on these measurements.
Photoreceptors and RPE have high metabolic demands52 and metabolic dysregulation, including suppressed NAD+ levels in either cell type, which are associated with retinal degeneration or disease.10,53,54 It may be that the observed increase in NAD+ after NR treatment, even after exposure to levels of light that would induce LIRD (Fig. 2), allowed increased or maintained retinal metabolic capacity that contributed to protection.55 Additionally, maintaining NAD+ levels may have permitted continued activity of enzymes for which NAD+ is a substrate and that have been shown to be critical to retinal health or function (e.g., sirtuins,5,9,56–58 poly[ADP-ribose] polymerases,9,59 isocitrate dehydrogenases60). For instance, maintaining NAD+ levels by treating with NR may allow continued enzyme-mediated DNA repair, which may underlie the observed prevention of accumulation of DNA damage (Fig. 6), similar to effects of NR treatment in a mouse Alzheimer's disease model.26
Similar to what we and others reported previously,29,61–63 retinas undergoing LIRD exhibited inflammatory responses (Figs. 7, 8, and 9). In vivo fundus imaging revealed autofluorescent spots in retinas undergoing LIRD. These spots are postulated to be components of inflammatory responses, including activated microglial cells, lipofuscin in RPE cells, and bisretinoids in the photoreceptors,64–67 and RPE cells undergoing epithelial-mesenchymal transition that are migrating into the neural retina.68,69 Confocal immunofluorescence microscopy on post mortem retina sections revealed that retinas undergoing LIRD had increased expression of Iba1 (Figs. 8B, 8C). This finding may indicate the presence of migrating, activated microglia.44 Retina sections from LIRD mice also exhibited increased GFAP signal in processes spanning the inner plexiform layer and into the inner nuclear layer, suggesting that Müller glia cells were activated.45 In retina sections from mice undergoing LIRD but that had received intraperitoneal injections of NR, the appearance of these inflammatory markers in vivo and post-mortem was significantly diminished (Figs. 7C, 7D, and 8C, 8D, and 9C, 9D). It is known that photoreceptors undergoing apoptosis induced by LIRD communicate with microglia and Müller cells, causing inflammatory responses (e.g., Ccl2 upregulation and release from photoreceptor cells).70 Suppression of photoreceptor cell apoptosis by NR treatment may preclude such signaling. Thus, it may be that NR treatment prevented inflammatory stress responses indirectly by preventing early rod photoreceptor injury that would have led to apoptosis and subsequent stress signaling. NR treatment may also more directly affect microglia and Müller cells by some as-yet undefined actions. For instance, NR treatment could increase intracellular NAD+ in microglia or Müller cells, altering the transcriptional regulatory activities of C-terminal–binding protein,71,72 or silent information regulator 1,56,73 leading to decreased expression of nuclear factor-κB and other proinflammatory genes. Such direct suppression of microglia activation could partially prevent retinal degeneration by preventing phagocytosis of nonapoptotic photoreceptor cells, a mechanism known to play a role in retinal degeneration.74
In summary, this study is the first to demonstrate that systemic treatment with NR is protective in an in vivo retinal degeneration model. The protection is significant, and may support the proposition of prospective human subject studies. Future studies ought to test the effects of NR treatment in aged mice, since retinal health and NAD+ levels are known to decline with age,9,75 NR effects in RPE-selective damage models,76,77 and putative protective effects of NR oral dosing in other mouse models of retinal degeneration.
Supplementary Material
Acknowledgments
Funded by the Abraham J. and Phyllis Katz Foundation (JHB); The joint training program between Emory University School of Medicine and Xiangya School of Medicine, Central South University. China Scholarship Council (201806370277 XZ); NIH R01EY028859 (JHB); NIH R01EY021592 (JMN); NIH R01EY028450 (JMN); VA I01RX002806 (JHB); VA I21RX001924 (JHB); VARR&D C9246C (Atlanta VAMC); NIH P30EY06360 (Emory); an unrestricted departmental award from Research to Prevent Blindness. Inc. to the Ophthalmology Department at Emory University, and the Roy J. Carver Trust (CB). Some data presented here were presented in abstract form at the annual meeting of the Association for Research in Vision and Ophthalmology.78
Disclosure: X. Zhang, None; N.F. Henneman, None; P.E. Girardot, None; J.T. Sellers, None; M.A. Chrenek, None; Y. Li, None; J. Wang, None; C. Brenner, (I, C, P); J.M. Nickerson, None; J.H. Boatright, None
References
- 1. Chiang PW, Wang J, Chen Y, et al.. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet. 2012; 44: 972–974. [DOI] [PubMed] [Google Scholar]
- 2. Falk MJ, Zhang Q, Nakamaru-Ogiso E, et al.. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012; 44: 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koenekoop RK, Wang H, Majewski J, et al.. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012; 44: 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liao C, Ashley N, Diot A, et al.. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology. 2017; 88: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang M, Jiang N, Chu Y, et al.. Dysregulated metabolic pathways in age-related macular degeneration. Sci Rep. 2020; 10: 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fletcher RS, Lavery GG.. The emergence of the nicotinamide riboside kinases in the regulation of NAD+ metabolism. J Mol Endocrinol. 2018; 61: R107–R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin JB, Apte RS. NAD(+) and sirtuins in retinal degenerative diseases: a look at future therapies. Prog Retin Eye Res. 2018; 67: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007; 32: 12–19. [DOI] [PubMed] [Google Scholar]
- 9. Lin JB, Kubota S, Ban N, et al.. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016; 17: 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai S, Sheline CT. NAD(+) maintenance attenuates light induced photoreceptor degeneration. Exp Eye Res. 2013; 108: 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tam D, Tam M, Maynard KI. Nicotinamide modulates energy utilization and improves functional recovery from ischemia in the in vitro rabbit retina. Ann N Y Acad Sci. 2005; 1053: 258–268. [DOI] [PubMed] [Google Scholar]
- 12. Sheline CT, Zhou Y, Bai S. Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light. Mol Vis. 2010; 16: 2639–2652. [PMC free article] [PubMed] [Google Scholar]
- 13. Zabka TS, Singh J, Dhawan P, et al.. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol Sci. 2015; 144: 163–172. [DOI] [PubMed] [Google Scholar]
- 14. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008; 28: 115–130. [DOI] [PubMed] [Google Scholar]
- 15. Trammell SA, Yu L, Redpath P, Migaud ME, Brenner C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. J Nutr. 2016; 146: 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trammell SA, Schmidt MS, Weidemann BJ, et al.. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016; 7: 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004; 117: 495–502. [DOI] [PubMed] [Google Scholar]
- 18. Ratajczak J, Joffraud M, Trammell SA, et al.. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016; 7: 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gong B, Pan Y, Vempati P, et al.. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging. 2013; 34: 1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown KD, Maqsood S, Huang JY, et al.. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014; 20: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trammell SA, Weidemann BJ, Chadda A, et al.. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016; 6: 26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats. Pain. 2017; 158: 962–972. [DOI] [PubMed] [Google Scholar]
- 23. Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006; 26: 8484–8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vaur P, Brugg B, Mericskay M, et al.. Nicotinamide riboside, a form of vitamin B3, protects against excitotoxicity-induced axonal degeneration. FASEB J. 2017; 31: 5440–5452. [DOI] [PubMed] [Google Scholar]
- 25. Schondorf DC, Ivanyuk D, Baden P, et al.. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson's disease. Cell Rep. 2018; 23: 2976–2988. [DOI] [PubMed] [Google Scholar]
- 26. Hou Y, Lautrup S, Cordonnier S, et al.. NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018; 115: E1876–E1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Q, Zhu L, Qiu W, et al.. Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of ALS SOD1(G93A) mice. Int J Biol Sci. 2020; 16: 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawson EC, Han MK, Sellers JT, et al.. Aerobic exercise protects retinal function and structure from light-induced retinal degeneration. J Neurosci. 2014; 34: 2406–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mees LM, Coulter MM, Chrenek MA, et al.. Low-intensity exercise in mice is sufficient to protect retinal function during light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2019; 60: 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henneman NF, Foster SL, Chrenek MA, et al.. Xanthohumol protects morphology and function in a mouse model of retinal degeneration. Invest Ophthalmol Vis Sci. 2018; 59: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boatright JH, Moring AG, McElroy C, et al.. Tool from ancient pharmacopoeia prevents vision loss. Mol Vis. 2006; 12: 1706–1714. [PubMed] [Google Scholar]
- 32. Huber G, Beck SC, Grimm C, et al.. Spectral domain optical coherence tomography in mouse models of retinal degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 5888–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Howland HC, Howland M. A standard nomenclature for the axes and planes of vertebrate eyes. Vision Res. 2008; 48: 1926–1927. [DOI] [PubMed] [Google Scholar]
- 34. Phillips MJ, Walker TA, Choi HY, et al.. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Invest Ophthalmol Vis Sci. 2008; 49: 2148–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun N, Shibata B, Hess JF, FitzGerald PG. An alternative means of retaining ocular structure and improving immunoreactivity for light microscopy studies. Mol Vis. 2015; 21: 428–442. [PMC free article] [PubMed] [Google Scholar]
- 36. Zivin JA, Bartko JJ. Statistics for disinterested scientists. Life Sci. 1976; 18: 15–26. [DOI] [PubMed] [Google Scholar]
- 37. Loo DT. In situ detection of apoptosis by the TUNEL assay: an overview of techniques. Methods Mol Biol. 2011; 682: 3–13. [DOI] [PubMed] [Google Scholar]
- 38. Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. Int J Retina Vitreous. 2016; 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X, Girardot PE, Sellers JT, et al.. Wheel running exercise protects against retinal degeneration in the I307N rhodopsin mouse model of inducible autosomal dominant retinitis pigmentosa. Mol Vis. 2019; 25: 462–476. [PMC free article] [PubMed] [Google Scholar]
- 40. Massengill MT, Ahmed CM, Lewin AS, Ildefonso CJ. Neuroinflammation in retinitis pigmentosa, diabetic retinopathy, and age-related macular degeneration: a minireview. Adv Exp Med Biol. 2018; 1074: 185–191. [DOI] [PubMed] [Google Scholar]
- 41. Silverman SM, Wong WT. Microglia in the retina: roles in development, maturity, and disease. Annu Rev Vis Sci. 2018; 4: 45–77. [DOI] [PubMed] [Google Scholar]
- 42. Gargini C, Novelli E, Piano I, Biagioni M, Strettoi E. Pattern of retinal morphological and functional decay in a light-inducible, rhodopsin mutant mouse. Sci Rep. 2017; 7: 5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Couturier A, Bousquet E, Zhao M, et al.. Anti-vascular endothelial growth factor acts on retinal microglia/macrophage activation in a rat model of ocular inflammation. Mol Vis. 2014; 20: 908–920. [PMC free article] [PubMed] [Google Scholar]
- 44. Song D, Sulewski ME Jr., Wang C, et al.. Complement C5a receptor knockout has diminished light-induced microglia/macrophage retinal migration. Mol Vis. 2017; 23: 210–218. [PMC free article] [PubMed] [Google Scholar]
- 45. Heynen SR, Tanimoto N, Joly S, Seeliger MW, Samardzija M, Grimm C. Retinal degeneration modulates intracellular localization of CDC42 in photoreceptors. Mol Vis. 2011; 17: 2934–2946. [PMC free article] [PubMed] [Google Scholar]
- 46. Conze D, Brenner C, Kruger CL. Safety and Metabolism of long-term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep. 2019; 9: 9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elhassan YS, Kluckova K, Fletcher RS, et al.. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019; 28: 1717–1728 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Demontis GC, Longoni B, Marchiafava PL. Molecular steps involved in light-induced oxidative damage to retinal rods. Invest Ophthalmol Vis Sci. 2002; 43: 2421–2427. [PubMed] [Google Scholar]
- 49. Nair A, Morsy MA, Jacob S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev Res. 2018; 79: 373–382. [DOI] [PubMed] [Google Scholar]
- 50. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016; 7: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Airhart SE, Shireman LM, Risler LJ, et al.. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017; 12: e0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanow MA, Giarmarco MM, Jankowski CS, et al.. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife. 2017; 6: e28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jadeja RN, Powell FL, Jones MA, et al.. Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging (Albany NY). 2018; 10: 1306–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jadeja RN, Thounaojam MC, Bartoli M, Martin PM. Implications of NAD(+) metabolism in the aging retina and retinal degeneration. Oxid Med Cell Longev. 2020; 2020: 2692794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Okawa H, Sampath AP, Laughlin SB, Fain GL. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol. 2008; 18: 1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duarte DA, Rosales MA, Papadimitriou A, et al.. Polyphenol-enriched cocoa protects the diabetic retina from glial reaction through the sirtuin pathway. J Nutr Biochem. 2015; 26: 64–74. [DOI] [PubMed] [Google Scholar]
- 57. Silberman DM, Ross K, Sande PH, et al.. SIRT6 is required for normal retinal function. PLoS One. 2014; 9: e98831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin JB, Kubota S, Mostoslavsky R, Apte RS. Role of Sirtuins in retinal function under basal conditions. Adv Exp Med Biol. 2018; 1074: 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nakagawa T, Guarente L.. Sirtuins at a glance. J Cell Sci. 2011; 124: 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Du J, Yanagida A, Knight K, et al.. Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2016; 113: 14710–14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mandal MN, Patlolla JM, Zheng L, et al.. Curcumin protects retinal cells from light-and oxidant stress-induced cell death. Free Radic Biol Med. 2009; 46: 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Terao R, Honjo M, Ueta T, et al.. Light stress-induced increase of sphingosine 1-phosphate in photoreceptors and its relevance to retinal degeneration. Int J Mol Sci. 2019; 20:3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bian M, Zhang Y, Du X, et al.. Apigenin-7-diglucuronide protects retinas against bright light-induced photoreceptor degeneration through the inhibition of retinal oxidative stress and inflammation. Brain Res. 2017; 1663: 141–150. [DOI] [PubMed] [Google Scholar]
- 64. Kim SY, Yang HJ, Chang YS, et al.. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Invest Ophthalmol Vis Sci. 2014; 55: 6031–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bubis E, Sher I, Skaat A, et al.. Blue Autofluorescence fundus imaging for monitoring retinal degeneration in Royal College of Surgeons Rats. Transl Vis Sci Technol. 2019; 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sparrow JR, Duncker T. Fundus autofluorescence and RPE lipofuscin in age-related macular degeneration. J Clin Med. 2014; 3: 1302–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sparrow JR, Wu Y, Nagasaki T, Yoon KD, Yamamoto K, Zhou J. Fundus autofluorescence and the bisretinoids of retina. Photochem Photobiol Sci. 2010; 9: 1480–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zanzottera EC, Ach T, Huisingh C, Messinger JD, Freund KB, Curcio CA. Visualizing retinal pigment epithelium phenotypes in the transition to atrophy in neovascular age-related macular degeneration. Retina. 2016; 36(Suppl 1): S26–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zanzottera EC, Ach T, Huisingh C, Messinger JD, Spaide RF, Curcio CA. Visualizing retinal pigment epithelium phenotypes in the transition to geographic atrophy in age-related macular degeneration. Retina. 2016; 36(Suppl 1): S12–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feng C, Wang X, Liu T, Zhang M, Xu G, Ni Y. Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol Vis. 2017; 23: 765–777. [PMC free article] [PubMed] [Google Scholar]
- 71. Shen Y, Kapfhamer D, Minnella AM, et al.. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun. 2017; 8: 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morris G, Puri BK, Maes M, Olive L, Berk M, Carvalho AF. The role of microglia in neuroprogressive disorders: mechanisms and possible neurotherapeutic effects of induced ketosis. Prog Neuropsychopharmacol Biol Psychiatry. 2020; 99: 109858. [DOI] [PubMed] [Google Scholar]
- 73. McGettrick AF, O'Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013; 288: 22893–22898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhao L, Zabel MK, Wang X, et al.. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. 2015; 7: 1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kolesnikov AV, Fan J, Crouch RK, Kefalov VJ. Age-related deterioration of rod vision in mice. J Neurosci. 2010; 30: 11222–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Girardot P, Zhang X, Gefke I, et al.. Systemic pentosan polysulfate administration causes retinal function loss in the C57Bl/6J mouse. Invest Ophthalmol Vis Sci. 2019; 60: 2352–2352. [Google Scholar]
- 77. Zhang N, Zhang X, Girardot P, et al.. Optimization of a retinal pigment epithelium damage model. Invest Ophthalmol Vis Sci. 2020; 61: 4441–4441. [Google Scholar]
- 78. Zhang X, Henneman N, Girardot PE, et al.. Systemic treatment with nicotinamide riboside is protective in four mouse models of retinal degeneration. Invest Ophthalmol Vis Sci. 2020; 61: 2753–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wisard J, Faulkner A, Chrenek MA, et al.. Exaggerated eye growth in IRBP-deficient mice in early development. Invest Ophthalmol Vis Sci. 2011; 52: 5804–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.