

Abstract
Nocardioides sp. PD653 genes hcbA1, hcbA2, and hcbA3 encode enzymes that catalyze the oxidative dehalogenation of hexachlorobenzene (HCB), which is one of the most recalcitrant persistent organic pollutants (POPs). In this study, HcbA1, HcbA2, and HcbA3 were heterologously expressed and characterized. Among the flavin species tested, HcbA3 showed the highest affinity for FMN with a Kd value of 0.75±0.17 µM. Kinetic assays revealed that HcbA3 followed a ping-pong bi–bi mechanism for the reduction of flavins. The Km for NADH and FMN was 51.66±11.58 µM and 4.43±0.69 µM, respectively. For both NADH and FMN, the Vmax and kcat were 2.21±0.86 µM and 66.74±5.91 sec−1, respectively. We also successfully reconstituted the oxidative dehalogenase reaction in vitro, which consisted of HcbA1, HcbA3, FMN, and NADH, suggesting that HcbA3 may be the partner reductase component for HcbA1 in Nocardioides sp. PD653.
Keywords: Nocardioides sp. PD653, hexachlorobenzene, flavin reductase, TC-FDM, HcbA3
Introduction
Hexachlorobenzene (C6Cl6; HCB) was first introduced in the 1940s as an organochlorine fungicide for crop seeds1) and is one of the most persistent environmental pollutants, whose average half-life in soil is approximately 9 years. Due to its persistence in the environment and tendency to bioaccumulate, it was listed as a persistent organic pollutant at the Stockholm Convention in 2001. Nocardioides sp. PD653 (PD653) is a wild-type bacterium capable of mineralizing HCB under aerobic conditions2) and has been isolated and studied for the bioremediation of HCB-contaminated sites. PD653 can dehalogenate and hydroxylate HCB in the first step of the HCB mineralization pathway to form pentachlorophenol (C6HCl5O; PCP), and the genes hcbA1, hcbA2, and hcbA3 have been identified as being involved in catalyzing this reaction.3) However, the links between phenotypes and genotypes in PD653 remain unclear due to the lack of gene deletions and complementation systems in Nocardioides.
From the deduced amino acid sequences of HcbA1, HcbA2, and HcbA3, we hypothesized that the mechanism was closely related to the two-component flavin-diffusible monooxygenase (TC-FDM) systems, which are comprised of flavin-dependent monooxygenase and its partner reductase component.4,5) In these systems, the reductase components generate reduced flavins, which are transferred to oxygenase enzymes for monooxygenation reactions accompanied by the formation of a hydroperoxyflavin or peroxyflavin intermediate. Purified HcbA1 dehalogenates HCB in the presence of an Escherichia coli flavin reductase (Fre),6) FMN, and NADH, resulting in the formation of flavin-N5-oxide.7) These results are consistent with our hypothesis and imply the presence of a partner reductase component of HcbA1. Notably, the co-expression of hcbA1 and hcbA3 leads to HCB dehalogenase activity in E. coli cells; hence, HcbA3 is believed to supply reduced flavin in vivo. However, detailed characterization and identification of HcbA3 functions remain to be achieved. To test our hypothesis noted above, we purified C-terminally His-tagged HcbA1, HcbA2, and HcbA3 and evaluated HcbA3 as an NADH:FMN oxidoreductase. Then the oxidative HCB dechlorination was reconstituted in vitro with HcbA1C-His and HcbA3C-His.
Materials and Methods
1. Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1. A preculture medium for PD653 was prepared as described previously.3) Rhodococcus erythropolis and E. coli strains were routinely cultured in lysogeny broth (LB; 1% Bacto tryptone, 0.5% Bacto yeast extract, and 1% NaCl) with 1.5% agar added for culturing on agar plates. Escherichia coli DH5α and Rhodococcus transformants were selected using an LB medium containing 100 µg mL−1 ampicillin and 20 µg mL−1 chloramphenicol, respectively.
Table 1. Bacterial strain and plasmid used in this study.
| Strain or plasmid | Relevant characteristics | Reference or origin |
|---|---|---|
| Strains | ||
| Nocardioides sp. PD653 | HCB+ | 2 |
| E. coli DH5α | F−, l−, f80dlacZDM15, D(lacZYA-argF)U169, deoR, recA1, endA1, hsdR17(rk−, mk+), phoA, supE44, thi-1, gyrA96, relA1 | TOYOBO |
| Rhodococcus erythropolis L88 | A lysozyme sensitive mutant derived from Rhodococcus erythropolis JCM3201 | Hokkaido system science Co., Ltd. |
| Plasmids | ||
| pNitRC1 | E. coli-R. erythropolis shuttle vector, Apr for DH5α, Cmr for L88, Pnit, rep (pRE8424) | Hokkaido system science Co., Ltd. |
| pCpiRC1 | E. coli-R. erythropolis shuttle vector, Apr for DH5α, Cmr for L88, Pcpi, rep (pRE8424) | Hokkaido system science Co., Ltd. |
| pCpiRC1(hcbA1)C-His | pCpiRC1 with PCR-amplified DNA fragment of hcbA1 | This study |
| pCpiRC1(hcbA2)C-His | pCpiRC1 with PCR-amplified DNA fragment of hcbA2 | This study |
| pNitRC1(hcbA3)C-His | pNitRC1 with PCR-amplified DNA fragment of hcbA3 | This study |
HCB+, ability of degrade HCB; Apr, ampicillin resistant; Cmr, chloramphenicol resistant.
2. Chemicals
HCB was purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany). PCP was purchased from Wako Pure Chemical Industries (Osaka, Japan). NADH, nicotinamide adenine dinucleotide phosphate (NADPH), riboflavin, flavin adenine dinucleotide (FAD), and FMN were purchased from TCI Tokyo Kasei (Tokyo, Japan).
3. DNA isolation
Total DNA was purified from PD653 grown on a preculture medium using a DNeasy Tissue Kit (Qiagen, Valencia, CA, USA). Plasmids from recombinant E. coli cells were isolated using sodium dodecyl sulfate (SDS) and alkaline lysis as described elsewhere.36)
4. Construction of plasmids pCpiRC1(hcbA1)C-His, pCpiRC1(hcbA2)C-His, and pNitRC1(hcbA3)C-His
The primers synthesized to amplify hcbA genes are listed in Table 2. Genomic DNA from PD653 was used as a template for the PCR experiments described below. The region containing the open reading frame (ORF) of HCB oxidative dehalogenase HcbA1 was amplified using primer set pCRA1_F with pCpiRC1(hcbA1) His_R as a template. A PCR-amplified fragment containing hcbA1 was subjected to a seamless ligation cloning extract (SLiCE) reaction8) with NcoI- and XhoI-digested pCpiRC1. The resultant plasmid was designated as pCpiRC1(hcbA1)C-His. Plasmid pCpiRC1(hcbA2)C-His, which included the ORF of the putative flavin reductase component HcbA2, was constructed in a similar manner using primer set pCRA2_F and pCpiRC1(hcbA2)His_R. Plasmid pNitRC1(hcbA3)C-His was generated by PCR amplifying the fragment containing hcbA3 using primer set pTRA3_F and pNitRC1(hcbA3)His_R. The PCR-amplified fragment containing ORF3 was subjected to a SLiCE reaction with NcoI- and XhoI- digested pNitRC1. The recombinant plasmids were sequenced for verification. One-hundred nanograms of plasmids was added to 40 µL of R. erythropolis L88 electrocompetent cells containing 10% glycerol in a 2 mm gap electroporation cuvette. Cells were transformed using a MicroPulser Electroporator (Bio-Rad, Hercules, CA, USA) with a 2.5 kV pulse (time constant 4.8–5.2), rescued with 1 mL of LB, and incubated for 3 hr at 28°C with shaking at 180 rpm. Cells were plated onto LB agar plates containing chloramphenicol and incubated at 28°C for 5–7 day.
Table 2. Specific primers used for amplification of hcbA genes.
| Primer | 5′ to 3′ |
|---|---|
| pCRA1_F | GAGATGGAGTCTTGCCATGGTGCGGGATACCCTTGTA |
| pCpiRC1(hcbA1) His_R | TGGTGATGGTGATGCTCGAAGGAGAAGATGCCCCGAT |
| pCRA2_F | GAGATGGAGTCTTGCCATGAACCTCGTCACCGTCATC |
| pCpiRC1(hcbA2)His_R | TGGTGATGGTGATGCTCGAATGAGCGAGTGCTTTCCA |
| pTRA3_F | AAGAAGGAGATATACCATGACCACCTCCGCACCGATC |
| pNitRC1(hcbA3)His_R | TGGTGATGGTGATGCTCGAAGGCGGTGGTGAGGCGCT |
5. Heterologous expression and purification of C-terminally His-tagged proteins
Cells from a single colony of recombinant R. erythropolis cells were inoculated into 20 mL of LB and incubated with shaking at 28°C, 180 rpm, for 3–5 day until they reached the stationary phase. The culture was diluted to 1 : 10 in 100 mL of LB, and protein expression was induced with shaking at 28°C, 180 rpm, for 48 hr. The expression of HcbA3C-His was induced at 20°C to increase the yield of the soluble form of the enzyme. The lysates were cleared and subjected to immobilized metal affinity chromatography (Bio-Rad) at 4°C, and the target proteins were purified according to the manufacturer’s instructions. The concentration of imidazole for elution was 100 mM for both HcbA1C-His and HcbA3C-His and 200 mM for HcbA2C-His.
6. pH and ionic strength optimization
The optimal pH for HcbA3C-His activity was determined in a 200 mM acetate buffer (pH 4.6–5.6), 200 mM phosphate buffer (pH 5.7–8.0), 200 mM Tris–HCl buffer (pH 7.2–8.6), and 200 mM glycine-NaOH buffer (pH 8.6–10) at 20°C in total volumes of 1 mL. The enzyme activity at ionic strengths ranging from 10 to 160 mM KPi (pH 7.5) was also tested using a similar approach.
7. Fluorescence measurement of dissociation constants
The binding of flavins (riboflavin, FAD, and FMN) to HcbA3C-His was determined by spectrofluorometric titration, monitoring the decreased relative flavin emission intensity due to the fluorescence quenching of flavin upon HcbA3C-His binding. A 50 nM solution of flavin in 20 mM KPi buffer, pH 7.5, at 20°C was titrated with increasing concentrations of HcbA3C-His (0.157–2.814 µM). Spectra were recorded using a BioSpectrometer fluorescence (Eppendorf, Hamburg, Germany) with an excitation wavelength of 470 nm and emission measurements at 520 nm. Emission and excitation bandwidths were set at 25 and 15 nm, respectively. Titrations were performed in triplicate, and Kd values were obtained by determining the amount of bound FMN according to the following equation [Eq. (1)], which was modified from Gao and Ellis9):
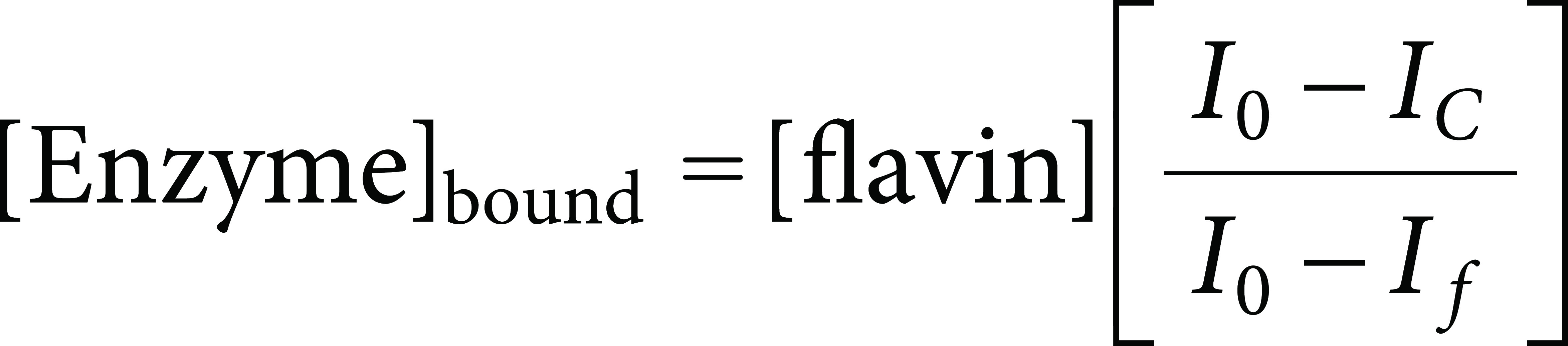 |
(1) |
where [Enzyme]bound represents the concentration of HcbA3C-His bound to flavin, [flavin] represents the initial concentration of flavin, I0 is the initial fluorescence intensity of flavin prior to the addition of a substrate, IC is the fluorescence intensity of the flavin following each substrate addition, and If is the final fluorescence intensity. The concentrations of bound HcbA3C-His were plotted against total HcbA3C-His concentrations and fitted according to equation Eq. (2) as previously described.9)
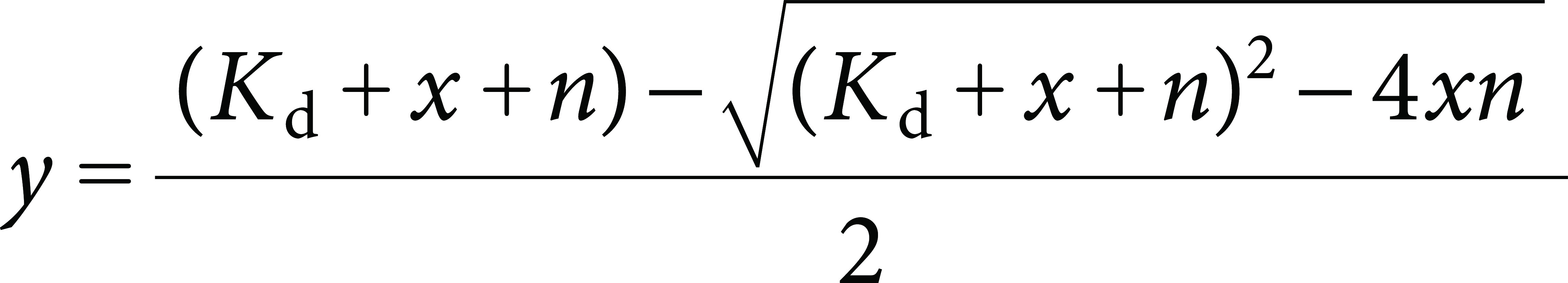 |
(2) |
Briefly, y and x represent the concentration of bound and total HcbA3C-His, respectively, following each substrate addition, and n represents the binding capacity of HcbA3C-His.
8. HcbA1 and HcbA3 enzymatic assays
8.1. Steady-state kinetic measurements of HcbA3 NADH:FMN oxidoreductase activity
Enzymatic assays to measure HcbA3C-His activity were performed by monitoring the oxidation of NADH to NAD+ (ε340=6.22 mM−1 cm−1) using a V-630Bio UV-Visible spectrophotometer (JASCO, Tokyo, Japan). Reactions were initiated by the addition of HcbA3C-His (25 nM) to 1 mL reaction mixtures containing 10–120 µM NADH and 1–8 µM FMN in a 20 mM KPi buffer (pH 7.5) at 20°C. Experiments were performed in triplicate. Enzyme activities were determined during the linear portions of the progress curves, with less than 10% of the reduced pyridine nucleotide being utilized over the time course of the reaction. The data from assays were fitted using the Enzfitter software package (Biosoft, Cambridge, UK) according to the ping-pong mechanism [Eq. (3)]:
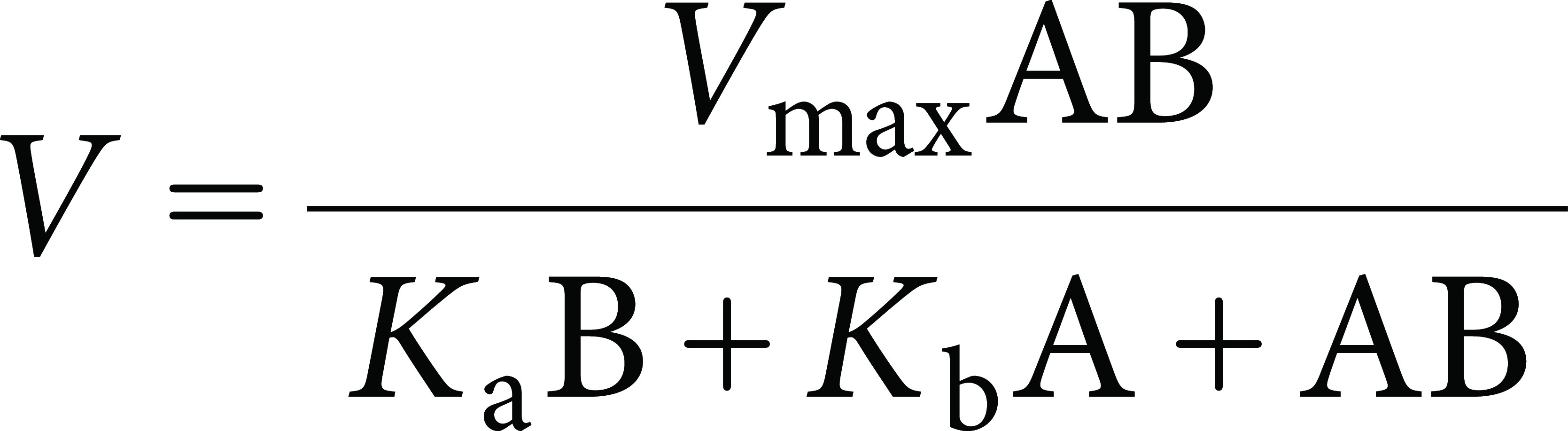 |
(3) |
where A and B are the substrate concentrations and Ka and Kb represent the Michaelis constants for substrates A and B, respectively. According to this equation, the actual value of Vmax and kcat were the same for both substrates (NADH and FMN).
8.2. HcbA1 oxidative dehalogenase activity against HCB
Oxidative dehalogenase assays were performed by measuring the HcbA1 activity at 28°C, 150 rpm, for 60 min. The reaction mixture composition was as follows: 20 mM phosphate buffer (pH 7.5), 240 µM NADH, 50 mM glucose, 0.7 U mL−1 glucose dehydrogenase, 555 U mL−1 catalase, 8 µM FMN, 17.5 µM HCB, 500 nM HcbA1C-His, and 71.4 nM HcbA3C-His (at a molar ratio of 7 : 1). The total volume of the reaction mixture was 500 µL. A stock solution of HCB was prepared at 500 mg L−1 in methanol, and 5 µL of this solution was added to each of a series of the reaction mixtures in 10 mL glass-stoppered test tube. The reactions were initiated by adding NADH to the reaction mixture, then stopped by adding 1 mL of acetonitrile at 0, 10, 20, 40 and 60 min. After centrifugation at 15,500×g for 10 min, the concentrations of HCB and PCP in the supernatants were quantified using high-performance liquid chromatography (HPLC).
9. Analytical methods
Protein concentrations were determined using the Bradford assay with bovine serum albumin as a standard. Enzyme purity and size were estimated via SDS polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad) using 12% polyacrylamide gels under denaturing conditions and a Mini-PROTEAN Tetra cell (Bio-Rad). HCB and PCP amounts were determined via HPLC using a Hewlett-Packard series 1100 system (Hewlett-Packard, Waldbronn, Germany) with UV absorption measured at 220 nm. Analysis was performed using a ZORBAX Eclipse XDB-C18 column (4.4 mm ×150 mm, 5 µm particle size; Agilent Technologies, Tokyo, Japan). The mobile phase was acetonitrile/0.1% aqueous phosphoric acid (v/v=90/10).
Results
1. Expression and purification of C-terminally histidine-tagged HcbA1 and HcbA3
Recombinant HcbA1, HcbA2, and HcbA3 were overexpressed in R. erythropolis L88 and purified (Fig. 1). Each purified protein preparation was colorless and did not exhibit any typical flavin adsorption spectrum of flavin-containing flavoproteins, suggesting that the purified proteins did not contain any bound flavin cofactor.
Fig. 1. Purified C-terminally histidine-tagged HcbA1, HcbA2, and HcbA3 as shown by SDS-PAGE. Lane M, marker proteins; lane 1, oxidative HCB dehalogenase HcbA1C-His; lane 2, putative flavin reductase HcbA2C-His; lane 3, NADH:FMN oxidoreductase HcbA3C-His.

2. Optimal conditions for enzymatic activity of the purified HcbA3C-His
The effects of temperature, pH, and ionic strength on HcbA3C-His activity were determined. The optimal pH for HcbA3C-His flavin reductase activity was between 7.0 and 8.6. At pH 7.0, 7.2, and 8.0, the enzyme activity levels in 100 mM phosphate buffer were approximately 50.2, 88.1, and 81.1%, respectively, relative to that at pH 7.5. HcbA3C-His showed the highest flavin reductase activity in 60 mM KPi buffer.
3. Flavin-binding studies
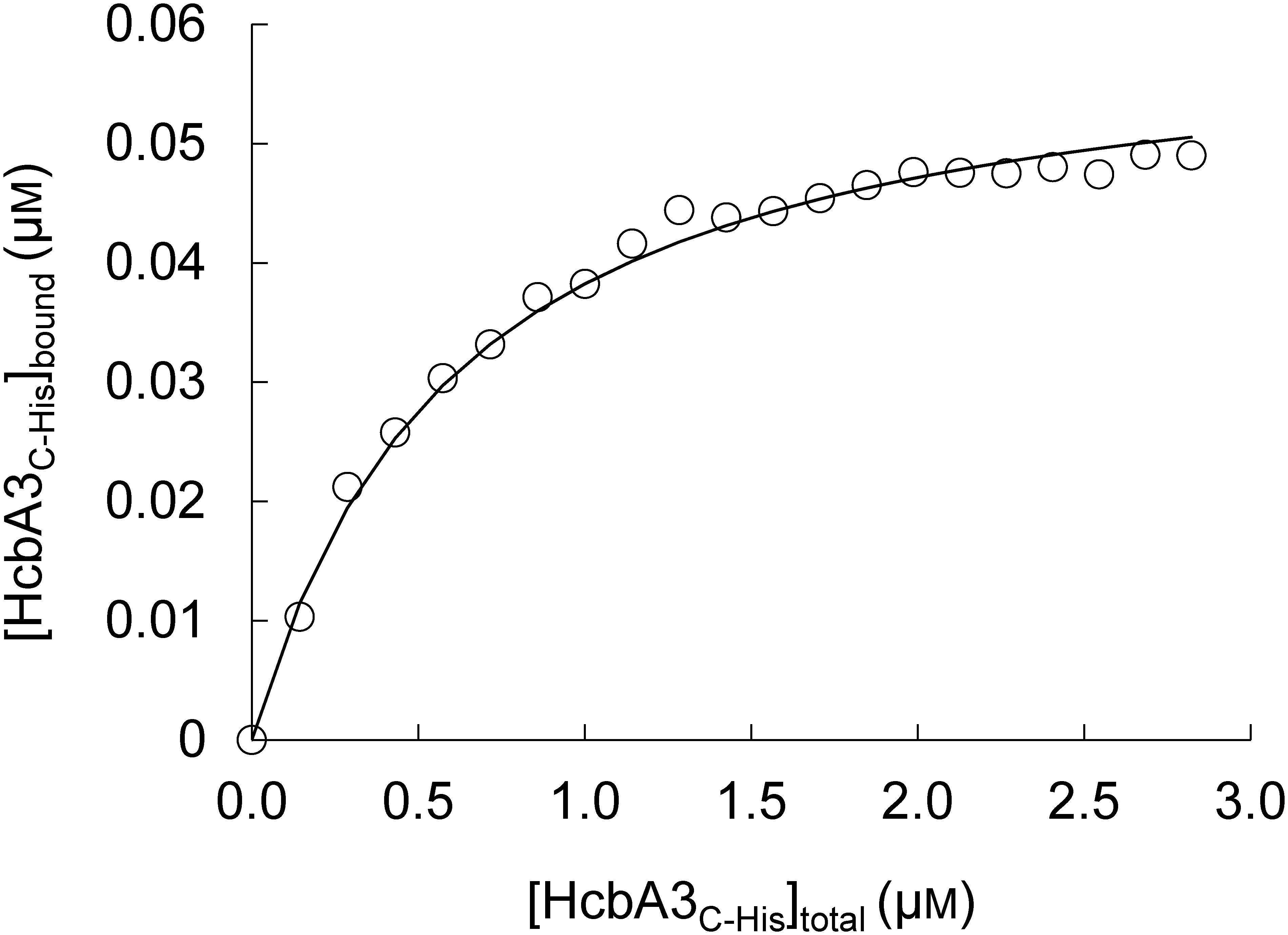
Upon the binding of FMN to HcbA3C-His, the fluorescence-emission intensity of FMN decreased, while this was not seen with riboflavin and FAD. This quenching of FMN fluorescence by HcbA3C-His was used to evaluate the dissociation constant of FMN binding by HcbA3C-His. Fifty-nanomolar FMN was titrated with 0.157–2.814 µM HcbA3C-His, with the quenching of FMN fluorescence being monitored at 520 nm. The concentrations of HcbA3C-His bound to flavin were plotted against the total HcbA3C-His concentrations added to each aliquot (Fig. 2). A Kd value of 0.75±0.17 µM for HcbA3C-His binding to FMN were calculated according to Eq. (2). No fluorescence quenching was observed when flavins were titrated with HcbA1C-His (0.073–0.16 µM) or HcbA2C-His (0.014–0.28 µM).
Fig. 2. Fluorometric titration of FMN with the HcbA3C-His enzyme. Changes in the fluorescence of flavin following the addition of HcbA3C-His were used to estimate the concentration of bound HcbA3C-His.
4. Steady-state kinetic analysis of HcbA3C-His
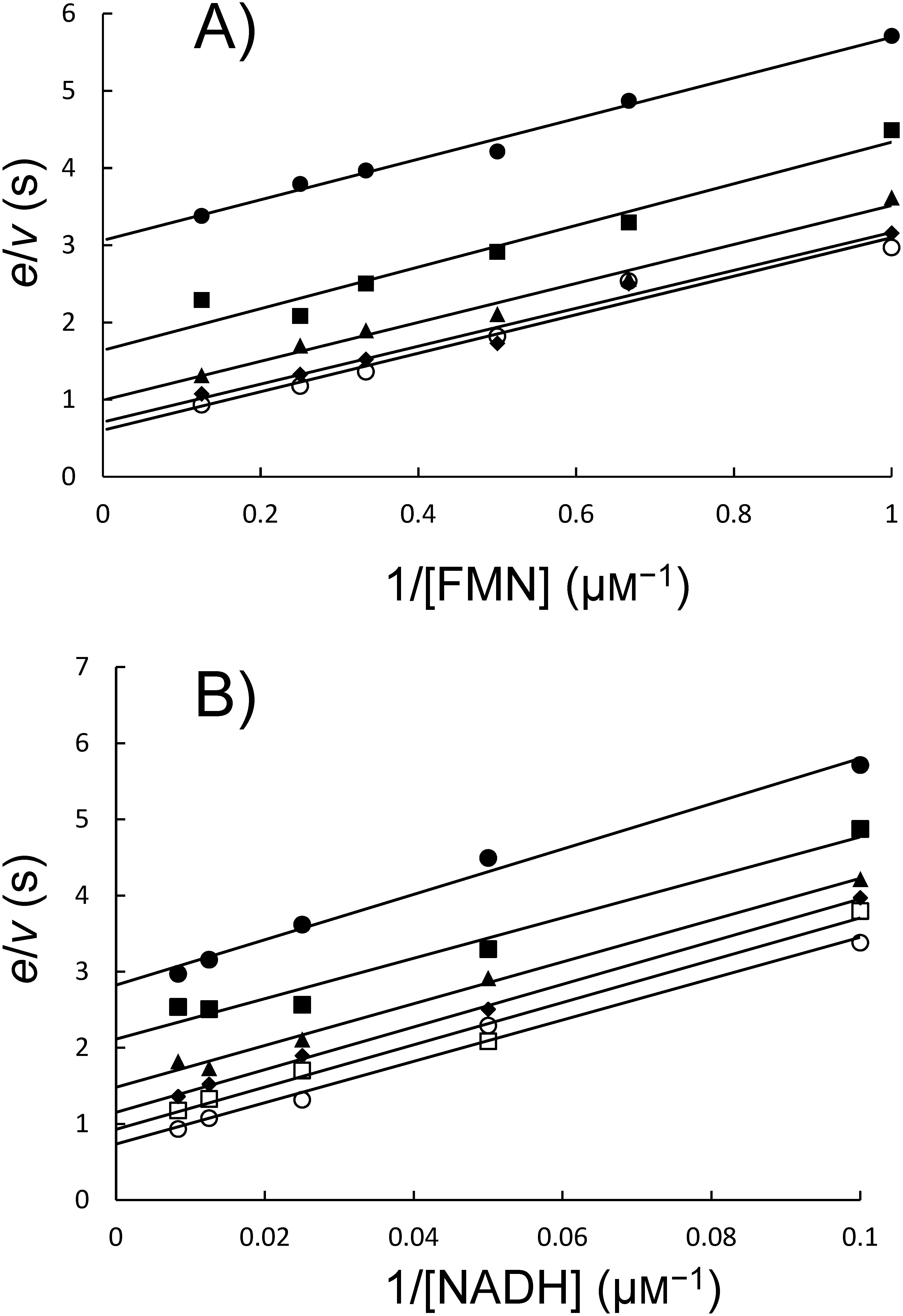
When NADH was used as a pyridinic substrate of HcbA3C-His in combination with FMN, a distinct activity was detected, while it was not with NADPH. FMN was the preferred flavin substrate of HcbA3C-His as compared to FAD and riboflavin. Therefore, the steady-state kinetic parameters of HcbA3C-His were measured in the presence of both FMN and NADH. The initial rates of NADH oxidation followed typical Michaelis–Menten kinetics (Fig. S1), and Lineweaver–Burk plots showed parallel patterns for a ping-pong mechanism (Fig. 3). Therefore, the kinetic parameters could be calculated using Eq. (3) to yield Km values for NADH and FMN, which were 51.66±11.58 µM and 4.43±0.69 µM, respectively. The Vmax and kcat values for both substrates were 2.21±0.86 µM and 66.74±5.91 sec−1, respectively. We concluded that HcbA3 is an NADH:FMN oxidoreductase.
Fig. 3. Double reciprocal plots of HcbA3C-His steady-state kinetics. (a) Assays were performed using 25 nM HcbA3C-His, 1–8 µM FMN, and various fixed concentrations of NADH, including 10 µM (●), 20 µM (■), 40 µM (▲), 80 µM (◆), and 120 µM (○). (b) Assays were performed using 25 nM HcbA3C-His, 10–120 µM NADH, and various fixed concentrations of FMN, including 1 µM (●), 1.5 µM (■), 2 µM (▲), 3 µM (◆), 4 µM (○), and 8 µM (□).
5. Reconstitution of HcbA3C-His oxidative dehalogenase activity against HCB
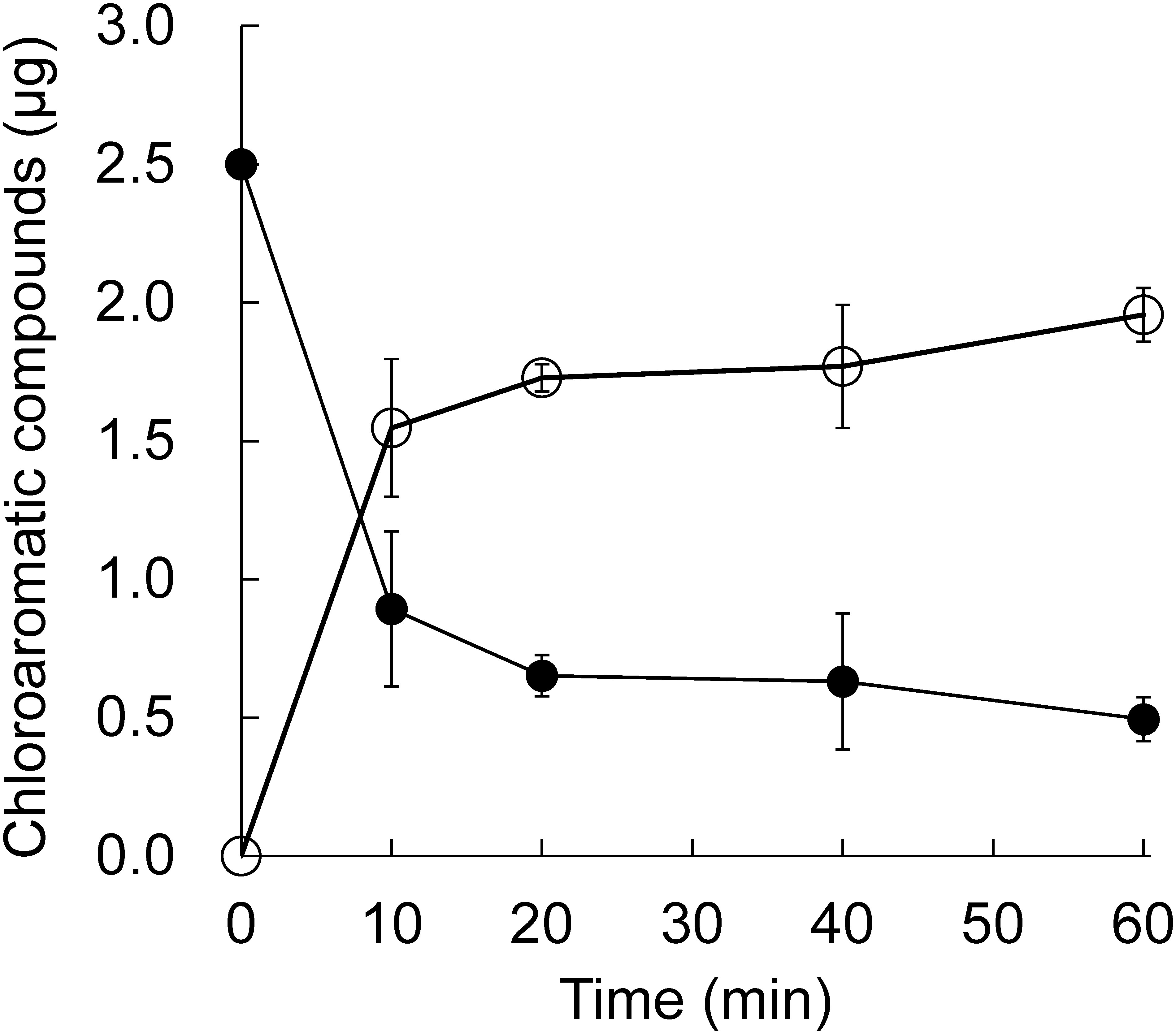
Although we demonstrated that HcbA3C-His generated reduced flavin in vitro, it was important to investigate whether this activity led to the dehalogenation of HCB by HcbA1C-His. First, the optimal molar ratio of HcbA1 and HcbA3 was found to be 7 : 1. The degradation curve of HCB by the enzymes at this ratio is shown in Fig. 4. Briefly, 0.129 µg of HCB was dehalogenated, and 0.124 µg of PCP was generated by 1 µg of HcbA1C-His in 10 min.
Fig. 4. Conversion of HCB (●) to PCP (○) via oxidative dehalogenation of HcbA1C-His–HcbA3C-His. Each error bar indicates the standard error for triplicate samples.
Discussion
Bacteria utilize TC-FDM family enzymes for various reactions, including bioluminescence,10) oxidation of aromatic compounds,11) degradation of chelating agents,12–14) desulfurization of sulfonated compounds,15–17) and biosynthesis of antibiotics.18–20) Notably, with respect to the degradation of POPs, single monooxygenase (Ese)21) has been identified as the enzyme responsible for the metabolism of endosulfan and classified as a TC-FDM. A common feature of all of TC-FDM enzymes is that the flavin reductase supplies reduced flavin for the partner oxygenase component by catalyzing the reduction of oxidized flavin with reducing equivalents provided by a pyridine nucleotide. Furthermore, the genes expressing the reductase and monooxygenase enzymes are often located in the same operon. These features are shared with the oxidative HCB dehalogenase gene hcbA1 and putative flavin reductase gene hcbA3. Purified HcbA1 displays HCB dehalogenase activity with photoreduced FMN and molecular oxygen in vitro.7) However, due to the lack of biochemical analysis of the flavin reductase component, it remained unclear whether the oxidative HCB dehalogenation was catalyzed by a TC-FDM system. Thus, the goal of the current study was to first characterize the flavin reductase component in a single-enzyme assay and then clarify whether HCB dehalogenation was catalyzed in a coupled assay with HcbA1.
Three proteins were expressed by R. erythropolis L88 and purified: HcbA1, HcbA2, and HcbA3. Binding analysis revealed that HcbA3C-His was able to bind to FMN, while HcbA1C-His and HcbA2C-His could not. In addition, as shown in the steady-state kinetic analysis, HcbA3C-His oxidized NADH in the presence of FMN, indicating that this enzyme can be classified as an NADH:FMN oxidoreductase. The fact that HcbA3 demonstrated a significant affinity for FMN was consistent with a major property of the reductase component in TC-FDM systems. In general, the flavin reductase has a greater affinity for oxidized flavin than that for monooxygenase.9) The distinct specificity of this flavin reductase indicates that FMN may be a major cosubstrate for the HCB oxidative halogenation reaction by HcbA1 and HcbA3 in PD653. Since the deduced amino acid sequence of HcbA2 was similar to the sequence in EmoB, an NADH:FMN oxidoreductase,22) we previously predicted that HcbA2 would function as a flavin reductase. Nevertheless, HcbA2C-His did not show any affinity for the flavins tested, nor flavin reductase activity, but only significant inhibition of HCB dehalogenase activity in our coupled assays (data not shown). Hence, the true function of HcbA2 remains unknown.
Based on its secondary structure, flavin reductase HcbA3 is predicted to be similar to TftC,23) HpaC,24) PheA2,25) and CobR.26) Reactions with TftC and PheA2 proceed according to a sequential and ping-pong bi–bi mechanism, respectively.25,27) To determine the kinetic mechanism of HcbA3C-His, steady-state kinetic analyses in association with both FMN and NADH were performed. It was determined that HcbA3 reduced flavins via a ping-pong bi-bi mechanism. In the TC-FDM family of enzymes, in addition to PheA2, the NADPH-preferring flavin reductase FRP also follows this type of mechanism in single enzyme assays.28) Both PheA2 and FRP contain flavins as prosthetic groups, which mediate the proton transfer from pyridinic substrates to exogenously added flavin substrates. On the other hand, HcbA3C-His does not have such flavin cofactors. Thus, it will be hypothesized that a mechanism independent on flavin cofactor is involved in HcbA3 oxidoreductase activity.
We determined that HcbA3 was a possible flavin reductase partner component of HcbA1 in the oxidative HCB dehalogenation system of PD653. Successful dehalogenation of HCB by these two components will allow us to investigate the mechanism involved in reduced flavin transfer. Since reduced flavins generated in HcbA3-catalyzed reactions are unstable due to susceptibility to oxidation by molecular oxygen, they should be rapidly diffused from reductase to monooxygenase. The flavin transfer may occur either through a diffusion mechanism or via direct flavin transfer involving protein–protein interactions (PPIs). There are several cases involving PPIs between monooxygenase and a flavin reductase for the transfer of reduced flavin, including FRP–luciferase in Vibrio harveyi,29) SsuE–SsuD in E. coli,30) EmoA–EmoB in Mesorhizobium sp. BNC1,22) and PrnF–PrnD in Pseudomonas fluorescens Pf-5.31) To evaluate the PPIs between HcbA1 and HcbA3, we performed a pull-down assay using a metal affinity column and C-terminally 6×His-tagged proteins. In this assay, each enzyme was independently bound to the column as the bite protein and cell lysates containing other proteins that lacked a fusion tag as a prey protein were loaded onto the columns. These assays, however, provided no conclusive evidence for the presence of specific interactions of these components. Additionally, fluorometric titration of flavin-binding HcbA3C-His exhibited no fluorescence changes following each addition of HcbA1C-His (unpublished data). From these findings, it may be speculated that there was no transient interaction between these two components.
In summary, the NADH:FMN oxidoreductase HcbA3 was purified and characterized for the first time in this study. We demonstrated that HCB was dehalogenated to PCP by HcbA1C-His in concert with HcbA3C-His, suggesting that oxidative HCB dehalogenation may occur through a TC-FDM system in bacteria. We believe these results provide greater insight into how bacteria dehalogenate POPs and identify a potentially novel enzymatic bioremediation technique. Eibes et al. summarized works on the bioremediation of organic pollutants in soil using enzymes.32) Several papers argue that enzymatic bioremediation is suitable for extracellular enzymes that maintain stable activity under operational conditions and have no dependency on expensive coenzymes or cofactors such as NAD(P)H, reduced flavin, and glutathione.33–35) Given these requirements, the application of HcbA1 to soils is likely to be difficult, but eliminating these bottlenecks should expand the use of bacterial enzymes with diverse metabolic capacities.
Acknowledgments
This work was funded by grant from JSPS KAKENHI (grant number 18H02319).
Electronic supplementary materials
The online version of this article contains supplementary materials (Supplemental Figure S1), which are available at http://www.jstage.jst.go.jp/browse/jpestics/.
Supplementary Data
References
- 1).J. L. Barber, A. J. Sweetman, D. van Wijk and K. C. Jones: Sci. Total Environ. 349, 1–44 (2005). [DOI] [PubMed] [Google Scholar]
- 2).K. Takagi, A. Iwasaki, I. Kamei, K. Satsuma, Y. Yoshioka and N. Harada: Appl. Environ. Microbiol. 75, 4452–4458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).K. Ito, K. Takagi, A. Iwasaki, N. Tanaka, Y. Kanesaki, F. Martin-Laurent and S. Igimi: Appl. Environ. Microbiol. 83, e00824–e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).H. R. Ellis: Arch. Biochem. Biophys. 497, 1–12 (2010). [DOI] [PubMed] [Google Scholar]
- 5).M. M. E. Huijbers, S. Montersino, A. H. Westphal, D. Tischler and W. J. H. van Berkel: Arch. Biochem. Biophys. 544, 2–17 (2014). [DOI] [PubMed] [Google Scholar]
- 6).L. Xun and E. R. Sandvik: Appl. Environ. Microbiol. 66, 481–486 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).S. Adak and T. P. Begley: Biochemistry 58, 1181–1183 (2019). [DOI] [PubMed] [Google Scholar]
- 8).Y. Okegawa and K. Motohashi: Biochem. Biophys. Rep. 4, 148–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).B. Gao and H. R. Ellis: Biochem. Biophys. Res. Commun. 331, 1137–1145 (2005). [DOI] [PubMed] [Google Scholar]
- 10).E. A. Meighen: Microbiol. Rev. 55, 123–142 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).U. Arunachalam, V. Massey and C. S. Vaidyanathan: J. Biol. Chem. 267, 25848–25855 (1992). [PubMed] [Google Scholar]
- 12).T. Uetz, R. Schneider, M. Snozzi and T. Egli: J. Bacteriol. 174, 1179–1188 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).H. R. Knobel, T. Egli and J. R. van der Meer: J. Bacteriol. 178, 6123–6132 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Y. Xu, M. W. Mortimer, T. S. Fisher, M. L. Kahn, F. J. Brockman and L. Xun: J. Bacteriol. 179, 1112–1116 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).K. A. Gray, G. T. Mrachko and C. H. Squires: Curr. Opin. Microbiol. 6, 229–235 (2003). [DOI] [PubMed] [Google Scholar]
- 16).J. R. van der Ploeg, R. Iwanicka-Nowicka, T. Bykowski, M. M. Hryniewicz and T. Leisinger: J. Biol. Chem. 274, 29358–29365 (1999). [DOI] [PubMed] [Google Scholar]
- 17).E. Eichhorn, J. R. van der Ploeg and T. Leisinger: J. Biol. Chem. 274, 26639–26646 (1999). [DOI] [PubMed] [Google Scholar]
- 18).S. G. Kendrew, S. E. Harding, D. A. Hopwood and E. N. Marsh: J. Biol. Chem. 270, 17339–17343 (1995). [DOI] [PubMed] [Google Scholar]
- 19).L. Filisetti, M. Fontecave and V. Niviére: J. Biol. Chem. 278, 296–303 (2003). [DOI] [PubMed] [Google Scholar]
- 20).J. Valton, C. Mathevon, M. Fontecave, V. Niviére and D. P. Ballou: J. Biol. Chem. 283, 10287–10296 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).K. M. Weir, T. D. Sutherland, I. Horne, R. J. Russell and J. G. Oakeshott: Appl. Environ. Microbiol. 72, 3524–3530 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).J. Bohuslavek, J. W. Payne, Y. Liu, H. J. Bolton Jr. and L. Xun: Appl. Environ. Microbiol. 67, 688–695 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).M. R. Gisi and L. Xun: J. Bacteriol. 185, 2786–2792 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).B. Galán, E. Díaz, M. A. Prieto and J. L. García: J. Bacteriol. 182, 627–636 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).U. Kirchner, A. H. Westphal, R. Müller and J. H. van Berkel: J. Biol. Chem. 279, 47545–47553 (2003). [DOI] [PubMed] [Google Scholar]
- 26).A. D. Lawrence, S. L. Taylor, A. Scott, M. L. Rowe, C. M. Johnson, S. E. J. Rigby, M. A. Geeves, R. W. Pickersgill, M. J. Howard and M. J. Warren: Biosci. Rep. 34, e00120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).B. N. Webb, J. W. Ballinger, E. Kim, S. M. Belchik, K. S. Lam, B. Youn, M. S. Nissen, L. Xun and C. Kang: J. Biol. Chem. 285, 2014–2027 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).B. Lei and S. C. Tu: Biochemistry 37, 14623–14629 (1998). [DOI] [PubMed] [Google Scholar]
- 29).J. C. Low and S. C. Tu: Photochem. Photobiol. 77, 446–452 (2003). [DOI] [PubMed] [Google Scholar]
- 30).K. Abdurachim and H. R. Ellis: J. Bacteriol. 188, 8153–8159 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).J. K. Lee and H. Zhao: J. Bacteriol. 189, 8556–8563 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).G. Eibes, A. Arca-Ramos, G. Feijoo, J. M. Lema and M. T. Moreira: Appl. Microbiol. Biotechnol. 99, 8815–8829 (2015). [DOI] [PubMed] [Google Scholar]
- 33).C. Scott, G. Pandey, C. Hartley, C. Jackson, M. Cheesman, M. Taylor, R. Pandey, J. Khurana, M. Teese, C. Coppin, K. Weir, R. Jain, R. Lal, R. Russell and J. Oakeshott: Indian J. Microbiol. 48, 65–79 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).T. D. Sutherland, I. Horne, K. M. Weir, C. W. Coppin, M. R. Williams, M. Selleck, R. J. Russell and J. G. Oakshott: Clin. Exp. Pharmacol. Physiol. 31, 817–821 (2004). [DOI] [PubMed] [Google Scholar]
- 35).R. J. Russell, R. L. Harcourt and J. G. Oakshott: ACIAR Proc. 85, 341-348 (1998).
- 36).H. C. Birnboim and J. Doly: Nucleic Acids Res. 7, 1513–1523 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.