

Abstract
Background:
Cancer-testis genes can serve as prognostic biomarkers and valuable targets for immunotherapy in multiple tumors because of their restricted expression in testis and cancer. However, their expression pattern in hepatocellular carcinoma is still not well understood. The purpose is to comprehensively characterize the cancer-testis gene expression in hepatocellular carcinoma as well as identify prognostic markers and potential targets for immunotherapy.
Methods:
Cancer-testis database and publicly available data sets reporting new cancer-testis genes were integrated, and then restricted them in a testis and hepatocellular carcinoma expression pattern. Pathway enrichment analysis and survival analysis were conducted to evaluate the biological function and prognostic effect of cancer-testis genes. Clustering analysis and coexpression analysis were performed to illustrate cancer-testis gene expression patterns in hepatocellular carcinoma. The association of gene expression of each cancer-testis gene to the corresponding methylation status was detected. Finally, we explored the associations between cancer-testis genes and CD8+ T-cell infiltration in hepatocellular carcinoma by TISIDB, and then validated it in an independent hepatocellular carcinoma cohort with 72 patients.
Results:
A total of 59 testis-specific genes were identified highly expressed in hepatocellular carcinoma. Pathway enrichment analysis revealed that cancer-testis genes in hepatocellular carcinoma significantly involves in the process of cell cycle regulation. Most of the cancer-testis genes were coexpressed, and cluster analysis suggested that cancer-testis gene expressed in hepatocellular carcinoma is independent of sex, hepatitis status, and histology type. We also found that demethylation might be a regulatory mechanism of cancer-testis gene expression in hepatocellular carcinoma. Survival analysis indicated that cancer-testis genes could predict the prognosis of patients with hepatocellular carcinoma. Furthermore, BUB1B was identified contributing to the resistance of CD8+ T-cell infiltration in hepatocellular carcinoma and was an independent prognostic factor both for overall survival and disease-free survival.
Conclusions:
Our analysis enables better understanding of cancer-testis genes in hepatocellular carcinoma and provides potential targets for hepatocellular carcinoma treatment. Experimental and clinical studies are needed for further validations.
Keywords: cancer-testis genes, hepatocellular carcinoma, prognostic marker, immunotherapy, bioinformatics
Introduction
Hepatocellular carcinoma (HCC) is a leading cause of mortality of cancer-related death in many parts of the world.1 The incidence and mortality from HCC are increasing globally.1 Molecular alterations in human tumors have been extensively studied in past decades. The identification of driver mutations as therapeutic targets has been applied in clinical practice in some solid tumors such as melanoma, lung cancer, or breast cancer.2,3 Molecular characterizations have identified the most frequently mutated drivers (such as TP53 and CTNNB1), and deregulated signal pathways (such as WNT, mTOR, or IGF2 signaling, among others) associated with HCC.4-7 Unfortunately, HCC has not reached the point of molecular-based stratification of treatment.2,3 In fact, most patients are still diagnosed at an advanced stage.3 Although the multitarget tyrosine kinase inhibitor sorafenib, as the systemic therapy for advanced HCCs, have clinical benefits, the improvements in patient outcomes have been modest and incremental.3 Therefore, there is a need to reveal new predictive biomarkers and explore alternative approaches to improve the clinical management of patients with HCC, including immunotherapeutic strategies such as tumor-associated antigen (TAA)-targeting vaccines and monoclonal antibody-based therapies.8
Cancer-testis (CT) genes have been indicated as a group of potential targets for TAA-specific immunotherapy because they restrictedly expressed in normal tissues except for the testis and aberrantly expressed in tumor tissues and often trigger humoral or cellular antitumor responses in patients with cancer.9-11 The testis are consequently immune privileged because spermatogenesis occurs after a long period of time after immune system development.12 Thus, aberrantly expressed testis-derived antigens have never been encountered by the immune system and therefore could be immunogenic.10 Over the past decades, clinical trials targeting CT genes (such as NY-ESO-1) have shown positive efficacy in multiple cancers such as melanoma and synovial sarcoma.13,14 On the other hand, immune checkpoint blockade (ICB) has contributed to a novel improvement in the treatment options for multiple tumor types.15,16 However, a majority of patients failed to respond to ICB. Successful responses preferentially occur in tumors with a preexisting CD8+ T-cell infiltration.17 Thus, understanding whether CT genes contributes to the tumor microenvironment remains a need.
Approximately 250 genes were initially recorded as CT genes in Cancer Testis database (CTdatabase).18 After that, several other analyses were performed based on genome-wide transcriptomic screen, and new CT genes across different tumor types were identified.19-21 However, definitions of CT genes are inconsistent among the literature, from genes exclusively expressed in testis to dominant testicular expression with possible additional presence in placenta and ovary.22 Also, the expression pattern of CT genes in HCC remained largely unknown. Therefore, in the present study, we combined CT genes from CT database and other published data sets to provide a comprehensive landscape of CT gene with a testis-specific pattern in HCC. The potential regulatory mechanisms of promoter methylation on CT gene expression were analyzed. Associations between CT genes and HCC prognosis were evaluated. Furthermore, potential CT gene regulating the tumor microenvironment was explored. BUB1B was identified correlating with failed CD8+ T-cell infiltration and was an independent prognostic CT gene marker for patients with HCC. Overall, the present study provided a comprehensive insight into the roles of CT genes in HCC and revealed promising targets for HCC treatment.
Materials and Methods
Databases and Study Samples
CTdatabase (http://www.cta.lncc.br/) was established by the Ludwig Institute for Cancer Research in 2009 listing detailed CT gene information from existing literature and several database.18 Three publicly available data sets identifying new CT genes compared to CTdatabase were integrated in our present study.19-21 Testis-restricted genes were retrieved from CTatlas (CTatlas: http://reprod.njmu.edu.cn/ctatlas/),20 which integrated normal tissue transcriptomic information derived from the Genotype-Tissue Expression (GTEx)23 and the Human Body Map (HBM; GSE30611). Differentially expressed genes in HCC were retrieved from The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov/). P value <.01 and log fold change >1 were set as the inclusion criteria for selection of upregulated genes.
The validation cohort in our analysis contains 72 patients with HCC underwent hepatectomy from 2015 to 2018 in the First Affiliated Hospital of Zhejiang University. The diagnosis of primary HCC was confirmed by pathological examinations. All enrolled patients received no preoperative treatment prior to surgery. Patients’ clinicopathological characteristics and follow-up information were obtained from the electronic records in hospital. This study was approved by Clinical Research Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (approval #2019-564). Written informed consent was obtained from all patients.
Function and Pathway Enrichment Analysis
Gene ontology (GO) analysis is a useful method for annotating genes and gene sets for high-throughput genome or transcriptome data.24 The online biological information Database for Annotation, Visualization, and Integrated Discovery (DAVID: https://david.ncifcrf.gov/)25 was used for GO analysis of CT genes. Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was conducted by using Cluego plug-in in an open-source bioinformatics software platform Cytoscape (3.6.1 version).26 We significantly excavated pathways with P < .05 as the cutoff value.
Protein–Protein Interaction Network Construction and Module Analysis
The online STRING (http://string-db.org) database aims to integrate direct or indirect function interactions between proteins and helps to understand the mechanisms of diseases with a combined score >0.4 as the cutoff criterion.27 In this study, protein–protein interaction (PPI) network was constructed on STRING database, and the network node file was exported to Cytoscape for visualization. Then, the most significant module in the PPI network was extracted using the plugin MCODE of Cytoscape with degree cutoff = 2, node score cutoff = 0.2, K score = 2, and maximum depth = 100.
Hub Genes Identification and Survival Analysis
Nodes with higher connectivity were considered to be more important for maintaining the stability of the entire PPI network. First, scores from the topological analysis method Degree were granted to each node in the PPI network by executing “compute Hubba result” function in the cytoHubba options.28 Then, we ranked top 10 CT genes in the PPI network to be the core candidate genes from highest to lowest according to the degree scores.
Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/index.html)29 is an online database resource originated from The TCGA database and was used in the present analysis for the overall survival (OS) and disease-free survival (DFS) outcomes. The Cox proportional hazards regression model was used to calculate the hazard ratio in GEPIA. As for our validation cohort, the Kaplan-Meier method was used to determine the survival curves, and the log-rank test was used to compare the survival differences between different groups. The independent clinicopathological factors associated with overall and DFS were evaluated by the multivariable Cox proportional hazard regression model. A 2-tailed P value less than .05 was considered to indicate a statistically significant difference.
Correlation Between Methylation Levels and CT Genes
The cBioPortal (http://www.cbioportal.org) online platform,30,31 containing the DNA methylation data from TCGA, displays a scatter plot of messenger RNA (mRNA) expression compared with DNA methylation data of a gene across all selected samples. Spearman test was used to detect the relationship between methylation and gene expression. P value of less than .05 was considered statistically significant. The online MethHC (http://MethHC.mbc.nctu.edu.tw),32 a database comprising a systematic integration of a large collection of DNA methylation profiles in human cancer from TCGA, was utilized to compare the methylation levels between tumor and normal liver tissues.
Cell Culture and 5-Azacytidine Treatment
Five HCC cell lines (Huh 7, HCCLM3, HepG2, SMMC-7721, SK-Hep-1) were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences and was cultured in Minimum Essential Media (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in an incubator at 37 °C and 5% CO2 concentration. For methylation regulation analysis, cells were treated with 5-Azacytidine (AZA; Selleckchem) at a concentration of 5 μM in the medium for 96 hours.
Quantitative Reverse Transcription Polymerase Chain Reaction
In brief, RNA was extracted from cell lines using RNA-Quick Purification Kit (YiShan Biotech). The HiScript II Q RT SuperMix for quantitative polymerase Chain Reaction (Vazyme Biotech) was used to generate the complementary DNA. Quantitative reverse transcription polymerase chain reaction was then conducted using the ABI 7500 FAST RT-PCR system (Applied Biosystems), and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was set as the internal reference. Relative quantitation was calculated using the ΔΔCt method. The primers we used are presented in Supplementary Table 1.
CD8+ T-Cell Infiltration Analysis
The web portal TISIDB,33 which integrated multiple types of data resources in oncoimmunology, facilitates comprehensive investigation of tumor–immune interactions. The relation between the immune features (CD8 expression) and the expression of any given CT gene was analyzed using TISIDB. Spearman test was performed to investigate the relationship between CD8+ T-cell infiltration and corresponding CT gene expression in our validation cohort.
Immunohistochemistry
Primary antibodies used for immunohistochemistry (IHC) of paraffin-embedded 72 HCC tissues and their paired noncancerous tissues included rabbit polyclonal anti-BUB1B antibody (1:200, Proteintech, Cat No. 11504-2-AP), rabbit polyclonal anti-ASPM antibody (1:100, Proteintech, Cat No. 26223-1-AP), rabbit polyclonal anti-beta catenin antibody (1:2000, Proteintech, Cat No. 51067-2-AP), and mouse monoclonal anti-CD8 antibody (1:200, Abcam, Cat No. ab17147). In brief, the paraffin-embedded microarray tissues were baked at 60 °C for 30 minutes and then dewaxed. The endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 15 minutes. After antigen retrieval using microwave oven, the tissues were then blocked with fetal bovine serum for 30 minutes. The tissues were next incubated with primary antibodies at specific dilutions overnight at 4 °C. After incubation with horseradish peroxidase–conjugated secondary antibody at 37 °C for 60 minutes, the sections were washed and then visualized by DAB detection kit.
Two pathologists who were blinded to the clinical data independently evaluate the tissues, and differences in viewpoints were evaluated in consensus with a third viewer. The BUB1B expression was evaluated using the multiplicative Quickscore method, which accounts for staining intensity and the proportion of positive cells.34 In brief, the IHC intensity was scored as 0 for no signal; 1 for light signal; 2 for moderate signal; and 3 for strong signal. The extent of the IHC staining area was scored as 1 for <4%; 2 for 5% to 19%; 3 for 20% to 39%; 4 for 40% to 59%; 5 for 60% to 79%, and 6 for 80% to 100%. The final score was calculated by multiplying the IHC intensity and extent score, ranging from 0 to 18. A positive cutoff score was defined as ≥3 based on Quickscore method.34 The cutoff values for high and low expression were the median of final IHC score of all the corresponding samples. The same strategy was used to record the expression of ASPM and β-catenin in HCC tissue. The expression of CD8 was evaluated by density measurement of CD8 for every stained core of tissue microarray.35 The cutoff values for high and low CD8 expression were the median of the final IHC score.
Results
Identification of CT Genes in HCC
The CTdatabase18 currently recording 276 CT genes. Nevertheless, 21 of them were excluded from the present analysis because of either overlapped with other listed CT genes (n = 19) or could not be retrieved (n = 2; Supplementary Data 1A and B). Further publicly available data sets identifying putative new CT genes were checked through literature searching for the comprehensive exploration in our analysis. And 3 were finally included in our study: Wang et al20 with 1019 CT genes identified, da Silva et al21 with 745 CT genes identified, and Bruggeman et al19 with 756 CT genes identified, respectively. By omitting the overlapped CT genes combined from the above 4 databases, 2121 CT genes were yielded as candidate CT genes in the present study (Supplementary Data 2). Testis-restricted genes were obtained from CTatlas20 (Supplementary Data 3), which integrating a variety of normal tissues’ transcriptomic data from GTEx23 and the HBM (GSE30611). Because CT genes are characterized as aberrantly expressed genes in the tumor compared with normal tissue, we therefore retrieved significantly upregulated genes in HCC from TCGA for the further validation of CT genes expression profile in HCC (Supplementary Data 4). Finally, our comparison yielded 59 putative novel CT genes (see in Supplementary Data 5) that are highly expressed in testis and highly expressed in HCC (Figure 1).
Figure 1.
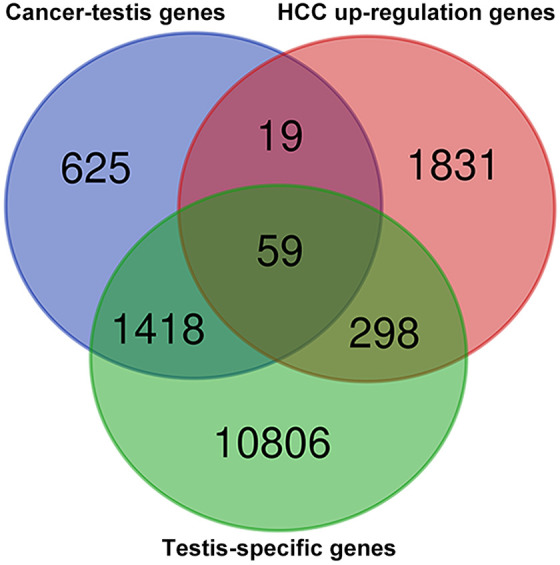
Venn diagram showing the 59 putative cancer-testis (CT) genes in hepatocellular carcinoma (HCC) with testis-specific pattern combined from 3 data sets.
Gene Ontology and KEGG Pathway Enrichment Analysis of CT Genes in HCC
To better understand the biological classification of the 59 putative CT genes in HCC, the online resources DAVID was used to perform GO and KEGG pathway analyses. By subjecting the identified CT genes to enrichment analysis, we observed numerous enriched gene sets. The changes in CT genes in biological processes were significantly enriched in both mitotic and meiotic cell cycle regulation and cell division (Figure 2A). Alterations in cell component (CC) were mainly abundant in chromosome, and its centromeric region, and microtubule cytoskeleton (Figure 2B). The changes in molecular function were significantly enriched in ATP binding, adenyl ribonucleotide binding, and protein serine/threonine kinase activity (Figure 2C). We further investigated the functional implications of these CT genes in the development of HCC by KEGG pathway analysis. A number of CT genes were enriched in 3 KEGG pathways, including cell cycle, oocyte meiosis, and progesterone-mediated oocyte maturation (Figure 2D). In line with previous research,20,36 these processes suggest that CT genes in HCC might be involved in regulating tumor cell proliferation, viability, and metastasis.
Figure 2.

Gene function analysis of the 59 cancer-testis (CT) genes. (A) The biological process (BP) analysis in hepatocellular carcinoma (HCC). (B) The cellular component (CC) analysis in HCC. (C) The molecular function (MF) analysis in HCC. (D) The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis in HCC.
Coexpression Analysis and Cluster Analysis
We analyzed the expression patterns of the 59 CT genes in HCC. As shown in Figure 3, most of the CT genes were coexpressed; 80.4% of the HCC cases expressed the whole of 59 CT genes; 17.8% of the HCC cases expressed 58 CT genes; only 1.8% of the HCC cases expressed less than 58 CT genes. Furthermore, the expression pattern of CT genes was illustrated through hierarchical cluster analysis among the 342 HCC cases derived from TCGA. Intriguingly, CT genes expressed in HCC is independent of sex, hepatitis status, and histology type (Figure 4). The cluster analysis also revealed that many CT genes were prone to express together with one CT gene or several other CT genes simultaneously. As expected, these analyses are consistent with CT gene expression might be the consequence of the activation of a coordinated gene expression program, rather than being independent events.10
Figure 3.
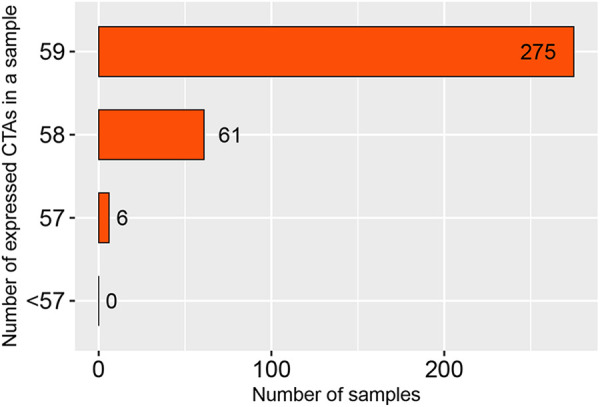
Coexpression analysis of 59 cancer-testis (CT) genes in hepatocellular carcinoma (HCC). Y-axis: the number of CT genes coexpressed in a HCC tumor sample. X-axis: the number of tumor samples.
Figure 4.

Hierarchical cluster analysis of 59 cancer-testis (CT) genes in hepatocellular carcinoma (HCC). The 342 HCC cases were clustered according to the expression of the 59 CT genes identified in our analysis.
Protein–Protein Interaction Network Construction and Hub Gene Identification
To further investigate the interaction among the 59 CT genes, a PPI network was constructed from STRING database and visualized by Cytoscape (Figure 5A). We uploaded 59 CT genes into STRING, a total of 59 nodes and 897 edges were filtered into the PPI network complex, and the most significant module was highlighted by the plugin MCODE (Figure 5B). Following ranked CT genes in the PPI network from the highest to the lowest according to the degree scores, we selected the top 10 as hub CT genes for further analysis (Table 1).
Figure 5.

Protein–protein interaction (PPI) network complex of the 59 cancer-testis (CT) genes in hepatocellular carcinoma (HCC). (A) All 59 CT genes interaction networks. (B) The most significant module selected from the PPI network.
Table 1.
Top 10 Hub Genes With Higher Degree Scores.
| Gene symbol | Functions | Degree |
|---|---|---|
| ASPM | Cell cycle, spermatogenesis, regulation of meiotic cell cycle, protein binding, positive regulation of neuroblast proliferation | 45 |
| DLGAP5 | Protein binding, cell cycle, positive regulation of mitotic metaphase/anaphase transition, cell population proliferation | 45 |
| AURKA | ATP binding, cell cycle, G2/M transition of mitotic cell cycle, histone serine kinase activity | 45 |
| TPX2 | Cell cycle, protein kinase binding, regulation of signal transduction by p53 class mediator, importin-alpha family protein binding | 45 |
| BUB1B | Cell cycle, apoptotic process, ubiquitin-dependent protein catabolic process, anaphase-promoting complex-dependent catabolic process | 45 |
| CCNB2 | Cell cycle, protein kinase activity, cyclin-dependent protein serine/threonine kinase regulator activity, mitotic nuclear envelope disassembly | 45 |
| CDC45 | DNA replication initiation, cell cycle, mitotic DNA replication preinitiation complex assembly, replication fork protection complex | 45 |
| PBK | Mitotic cell cycle, nucleotide binding, negative regulation of protein phosphorylation, negative regulation of stress-activated MAPK cascade | 44 |
| NUF2 | Cell cycle, protein binding, mitotic spindle organization, attachment of mitotic spindle microtubules to kinetochore | 44 |
| BUB1 | Kinase activity, cell cycle, apoptotic process, cell population proliferation | 44 |
The Association Between Methylation Levels and CT Gene Expression in HCC
The activation of CT genes in tumors is often considered to be associated with the methylation level of their promoters.10 We therefore firstly explored the methylation levels of the 59 CT genes in 369 HCC samples and 50 normal liver samples using online MethHC database32 which originated from TCGA. Fifty-eight of the methylation information data of the 59 (98%) CT genes could be obtained. The analysis revealed that 56.9% of the CT genes were hypomethylated in HCC compared to normal liver tissue (Supplementary Data 6). Furthermore, we focused 10 hub CT genes to explored whether hypomethylation was correlated with CT gene expression in HCC using cBioPortal database. The result showed that the expression of 8 CT genes (AURKA, ASPM, BUB1, BUB1B, CCNB2, NUF2, PBK, TPX2) among the 10 hub CT genes in HCC significantly exhibited negative correlations with their methylation levels (Figure 6).
Figure 6.

Correlation between methylation levels and cancer-testis (CT) gene expression. Promoter methylation and gene expression data from The Cancer Genome Atlas (TCGA) were visualized using cBioPortal to demonstrate the correlation among selected hub CT genes, and significant negative relationship was identified in: (A) ARUKA, (B) ASPM, (C) BUB1, (D) BUB1B, (E) CCNB2, (F) NUF2, (G) PBK, (H) TPX2.
BUB1B Is a Potential Prognostic Marker Suppressing CD8+ T-cell Infiltration in HCC
All the above, 10 hub CT genes were analyzed using the prognostic values of OS via the GEPIA website data which originated from the TCGA database. We then identified 9 CT genes of ASPM, ARUKA, BUB1, BUB1B, CDC45, DLGAP5, NUF2, PBK, and TPX2 may lead to poor survival of patients with HCC (all P < .05; Figure 7).
Figure 7.

Prognostic values of 9 cancer-testis (CT) genes identified by overall survival analysis using Kaplan-Meier curve from GEPIA: (A) ASPM, (B) ARUKA, (C) BUB1, (D) BUB1B, (E) CDC45, (F) DLGAP5, (G) NUF2, (H) PBK, (I) TPX2.
Since studies suggest that CT genes might contribute to the presence of CD8+ T-cells within the tumor microenvironment,37-39 we next explore the association between the above 9 potential prognostic markers of CT gene and CD8+ T-cell infiltration in HCC by TISIDB. And the analysis revealed that higher expression of ASPM and BUB1B was associated with poor CD8+ T-cell infiltration (Figure 8A). Then, we tested these 2 genes in our cohort of 72 HCC specimens for potential suppressor of CD8+ T-cell infiltration. However, ASPM was negatively expressed in our HCC specimens based on the Quickscore (score <3), which limits the association analysis. Positive BUB1B was detected expressed in 62 (86.1%) of our HCC specimens, and the analysis showed that BUB1B was associated with a decreased CD8+ T-cell infiltration of our cohort (r = −0.489, P < .001; Figure 8B). Representative immunostaining images of BUB1B and CD8 are shown in Figure 8C. Since the activation of cancer-intrinsic β-catenin pathway frequently results in T-cell exclusion,40,41 we therefore explored the association between BUB1B mRNA expression levels and β-catenin mRNA expression levels via GEPIA, and the result revealed a significant positive relation between them (r = 0.46, P < .001; Figure 8D). Next, we validated a positive association between BUB1B protein expression levels and β-catenin protein expression levels in our cohort (r = 0.588, P < .001; Figure 8C and E).
Figure 8.

BUB1B might be a potential target for immunotherapy in hepatocellular carcinoma (HCC). (A) Association of the BUB1B expression and CD8+ T-cell infiltration in HCC by TISIDB (r = −0.129, P = .013). (B) BUB1B expression level is negatively associated with CD8+ T-cell infiltration level validated in our 72 patients, tested by Spearman test (r = −0.489, P < .001). (C) Representative immunohistochemical staining of BUB1B, β-catenin, and CD8 in HCC. Negative, weak positive, moderate positive, and strong positive expression of BUB1B were shown, respectively. (D) Association of the BUB1B messenger RNA (mRNA) expression and β-catenin mRNA in HCC by GEPIA (r = 0.46, P < .001). (E) BUB1B expression level is positively associated with β-catenin expression level validated in our 72 patients, tested by Spearman test (r = −0.588, P < .001). (F) Overall survival (OS) curves of patients with HCC according to cancer expressed BUB1B levels (P < .001). (G) Disease-free survival (DFS) curves of patients with HCC according to cancer expressed BUB1B levels (P = .008).
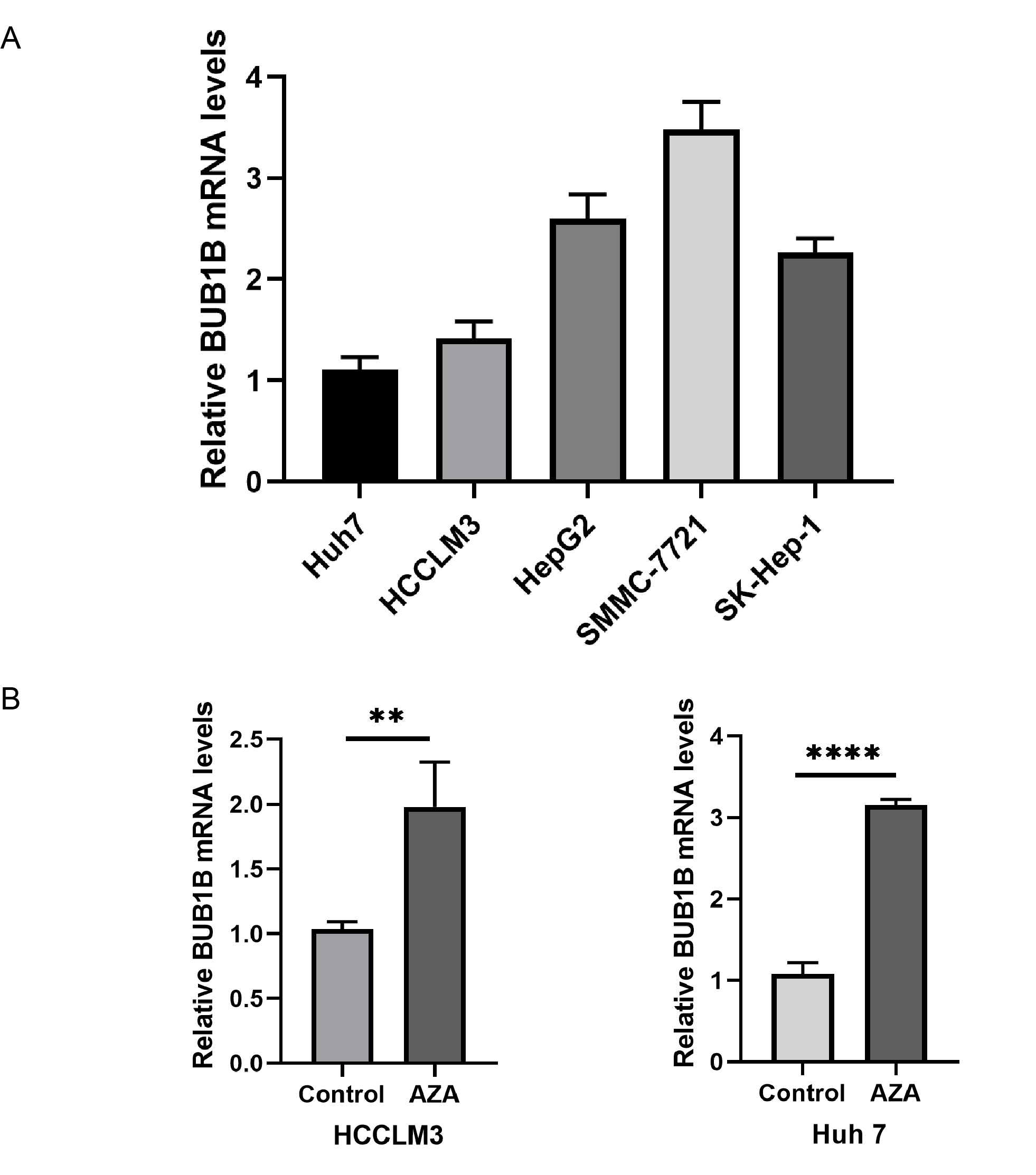
The baseline clinicopathological characteristics of the 72 patients with HCC are shown in Table 2. Multivariate Cox regression analysis showed that the expression of BUB1B is an independent predictive factor both for OS and DFS (all P < .05, Tables 3 and 4; Figure 8F and G). Moreover, we evaluated the effect of 5-AZA, a DNA methyltransferase inhibitor, in 2 BUB1B mRNA relatively low HCC cell lines. As expected, BUB1B mRNA was increased significantly after AZA treatment in both Huh 7 and HCCLM3 cell lines (Supplementary Figure 1). Collectively, these results indicated that BUB1B might be regulated through hypomethylation and is a potential target for immunotherapy in HCC.
Table 2.
Clinical and Pathological Features of the 72 Patients.
| Sex (M/F) | 55 (76%)/17 (24%) |
|---|---|
| Age (median, years) | 46.5 |
| Risk factors | |
| HBV | 55 (76%) |
| HCV | 16 (22%) |
| Nonalcoholic steatohepatitis | 1 (2%) |
| AFP level ≥20 ng/mL | 51 (71%) |
| Mean tumor size (mm, range) | 93.4 (15-250) |
| Tumor numbers | |
| Single | 38 (53%) |
| Multiple | 34 (47%) |
| TNM stage | |
| Ⅰ-Ⅱ | 33 (46%) |
| Ⅲ | 23 (32%) |
| Ⅳ | 16 (22%) |
| Vascular invasion | 19 (26%) |
| Tumor differentiation (Edmondson-Steiner grade III-IV) | 31 (43%) |
| BUB1B | |
| Low | 36 (50%) |
| High | 36 (50%) |
| CD8 | |
| Low | 47 (65%) |
| High | 25 (35%) |
Table 3.
Univariate and Multivariate Analyses of Clinicopathologic Factors in Patients With HCC With Respect to OS.
| Variables | Univariate | Multivariate | ||
|---|---|---|---|---|
| P value | HR (95% CI) | P value | ||
| Sex | Male vs female | .566 | ||
| Age | ≥46.5 vs <46.5 | .852 | ||
| HBV | HBV vs others | .889 | ||
| AFP levels | ≥20 ng/mL vs <20 ng/mL | .013 | 2.37 (0.72-7.78) | .154 |
| Tumor numbers | Single vs multiple | .137 | ||
| Vascular invasion | Present vs absent | .954 | ||
| Differentiation | ES III-IV vs ES Ⅰ-Ⅱ | .534 | ||
| BUB1B | High vs low | <.001 | 6.03 (1.82-19.99) | .003 |
| CD8 | High vs low | .012 | 0.87 (0.26-2.89) | .824 |
Abbreviations: HCC, hepatocellular carcinoma; HR, hazard ratio; OS, overall survival.
Table 4.
Univariate and Multivariate Analyses of Clinicopathologic Factors in Patients With HCC With Respect to DFS.
| Variables | Univariate | Multivariate | ||
|---|---|---|---|---|
| P value | HR (95% CI) | P value | ||
| Sex | Male vs female | .804 | ||
| Age | ≥46.5 vs <46.5 | .14 | ||
| HBV | HBV vs others | .531 | ||
| AFP levels | ≥20 ng/mL vs <20 ng/mL | .01 | 2.47 (0.79-7.74) | .122 |
| Tumor numbers | Single vs multiple | .08 | ||
| Vascular invasion | Present vs absent | .084 | ||
| Differentiation | ES III-IV vs ES Ⅰ-Ⅱ | .69 | ||
| BUB1B | High vs low | .008 | 6.426 (2.22-18.64) | .001 |
| CD8 | High vs low | .098 | ||
Abbreviations: HCC, hepatocellular carcinoma; HR, hazard ratio; DFS, disease-free survival.
Discussion
Cancer-testis genes are considered as promising prognostic markers and targets for cancer treatment because of their specific expression patterns in multiple tumor types.42 However, the classification of CT genes based on its expression pattern in normal tissues is a critical issue.22 Previous study revealed that CT genes can be stratified into testis-restricted or non-testis-restricted group according to their expression profiles.22 Therefore, this imposes a challenge to identify the CT genes that are most suitable for cancer therapies without side effects against other normal tissues.22 Hepatocellular carcinoma, along with other tumors such as melanoma, tend to frequently express CT genes.10 Thus, the systematic evaluation of the CT genes with testis-restricted pattern in HCC may help us better understand the role of them in the development of HCC and identify precision therapeutic targets. To our knowledge, this present study first provides a systematic and comprehensive profiling of the expression of the CT genes in HCC, which contributes to identifying prognostic and therapeutic markers in HCC.
Molecular-based therapies have been used increasingly as an alternative option to chemotherapy in cancer treatment for decades,43 which requires the identified targets play a key role in the progression and survival of cancer cells. Considering the nature of the CT genes being expressed exclusively in the testis and tumors, there is a significant possibility that CT gene expression might contribute to the process of tumorigenesis. Evidence revealed that dysploidy in tumorigenesis shares similar characteristics with meiosis in gametogenesis.10 The breaks of the DNA double strand are essential for proper chromosome segregation at the first step of meiotic division, which makes it a crucial process during meiotic recombination.44 Mitosis inhibits this process and may therefore lead to telomere fusion, leading to diploid development.45 Recently, a study revealed that HORMAD1, a CT gene involved in the process of nonconservative recombination during meiosis, induce chromosome instability in triple-negative breast cancers.46 Furthermore, several studies found that CT genes play fundamental roles in the cell cycle pathway in tumorigenesis.20,47 In line with this, in our present analysis, CT gene in HCC was found mainly involves in the regulation of cell cycle, intriguingly both in mitotic and in meiotic, which is essential for tumorigenesis. This is consistent with the fact that CT gene is highly testicular and cancer-specific, making it a promising candidate molecular-targeted therapies for HCC.
Cancer-testis genes are frequently coexpressed and are thus expected to act concertedly in many cases.10 In a study of CT genes in lung cancer, most Cancer-testis antigens (CTAs) were coexpressed except for 5.5% of cases where no CT gene was detected.48 Similarly, in our analysis, we found that 80.4% of the HCC cases expressed the whole of 59 CT genes and only 1.8% of the HCC cases expressed less than 58 CT genes. It is therefore possible that CT genes may play more effective role when jointly expressed in tumor cells. Moreover, several CT genes were reported to have physical interactions with other CT genes49 or to act as transcriptional regulators for other CT genes,50 thus suggesting functional cooperativity. On the other hand, cooperation between CT genes can also be regulated by comprehensively affecting common signaling pathways. For example, the Melanoma Antigen Gene (MAGE) family is shown to target p53 depend of apoptosis,51 while other CT genes might prevent the process by affecting other regulators in the apoptotic pathway.36
This similar regulatory mechanism and coordinated expression pattern of CT genes might also be supported by the methylation analysis of CT genes. In our study, we revealed that in HCC, over 50% of the CT genes were hypomethylated compared with normal liver samples; moreover, most CT genes were demonstrated to have negative correlations between methylation and their expression levels. Indeed, it is commonly recognized that DNA demethylation might be a primary mechanism of transcriptional activation for many CT genes. There is evidence that demethylation of the CT gene promoter by experimental methods can induce the expression of antigens that are not normally expressed in cells.52 Mechanically, global DNA hypomethylation occurs during tumorigenesis and has been associated with CT gene expression.10 In line with this, we found that AZA treatment significantly increased BUB1B mRNA expression. In HCC, epigenome disruption has emerged as a major hallmark.2 Therefore, it is of clinical importance to better understand the mechanisms of CT gene activation through epigenetic changes.
Prognostic values of CT genes were reported in many tumor types, such as multiple myeloma,53 clear cell renal cell carcinoma,54 testicular germ cell tumor,55 and esophageal squamous cell carcinoma.47 In our study, survival analysis revealed that 9 CT genes of our identified hub CT genes were associated with the poor survival of HCC, which makes them promising candidate prognostic biomarkers for HCC. These findings lead to a possibility that these CT gene products might have a functional role in the pathogenesis of cancer resulting in a bad evolution. However, this oncogenic role might be influenced by complex confounders and thus be weakened at the late stage of HCC process since the survival difference disappeared after 80 months of OS in some cases. Studies validating CT gene oncogenic functions are needed. The prognostic potential of some of these CT genes was reported in recent studies. In support of this, NUF2, one of a potential prognostic marker detected in our analysis, was reported to be a prognostic biomarker for early HCC recurrence in another HCC cohort.56 In addition, ASPM, another potential prognostic marker reported in our analysis, which mRNA levels was overexpressed in HCC tissues and was found as a marker predicting enhanced invasive potential and poor prognosis of HCC by Lin et al.57 The correlation between mRNA and protein expression is a matter of scientific issue, and studies have revealed that the process of mRNA translation to protein is complicated.20 Of note, the expression of some CT proteins might be regulated posttranscriptionally; thus, the expression of CT gene mRNA may not guarantee protein expression, which therefore limits its generalization as candidate therapeutic targets.9 Positive ASPM expression was detected in another HCC cohort,58 and its prognostic value was also highlighted. The negative ASPM findings in our study might due to the heterogeneous expression pattern of CT genes, or the low sensitivity of the IHC antibody used. More studies are needed to investigate the regulation of ASPM expression as well as other CT genes.
Except for the oncogenic role of CT genes in driving tumorigenesis, these genes might also influence the tumor microenvironment. Negative regulation pathways, such as the immune checkpoint of PD-1/PD-L1 axis, can be used by cancers to evade immune-mediated tumor clearance.17 Recently, antibodies targeting PD-1/PD-L1 axis have shown broad applicability across a range of tumor types including HCC.8 However, only a small group of patients benefit from ICB therapies. The presence of a preexisting CD8+ T-cell infiltration within the tumor microenvironment positively correlates with immune checkpoint immunotherapy.59 Thus, understanding the mechanism of tumors developing the non-T cell-inflamed phenotype is of vital importance for fighting against treatment failure for many patients. A recent study found that the expression of SAGE, a CT gene, was associated with a non-T cell-inflamed phenotype.38 In line with this, we revealed that the expression of BUB1B was associated with a decreased CD8+ T-cell infiltration in HCC. This finding suggests patients with higher BUB1B expression in HCC might confront resistance against immune checkpoint therapy. T-cell infiltration is associated with chemokines in the tumor microenvironment, which are likely to mediate their recruitment.60 Importantly, we also found that BUB1B expression was significantly associated with decreased expression of other T cell-inflamed gene signatures, such as CCL2 and CCL3, through TISIDB (data not shown). Mechanical studies using in vivo tumor models revealed that activation of the Peroxisomal Proliferator Receptor gamma (PPARγ) pathway results in reduced CD8+ T-cell infiltration through the reduction of CCL2 and other chemokines.61 This lead to a hypothesis that overexpression of BUB1B activate the PPARγ pathway mediated reduction of CCL2, which resists the CD8+ T-cell infiltration. Further studies should validate this hypothesis. On the other hand, cancer-intrinsic β-catenin pathway frequently results in T-cell exclusion.40,41 We observed a positive correlation between BUB1B expression and β-catenin in HCC, which suggested that BUB1B might influence CD8+ T-cell infiltration through activating the oncogenic β-catenin signaling pathway. Besides, we also find that BUB1B was correlated with poor OS as well as DFS in our HCC cohort through multivariate Cox regression analysis. Overall, BUB1B might be a promising target for immunotherapy given its potential not only for prognosis prediction but also for expanding the proportion of patients responding to current immunotherapies.
Conclusion
Our study provides a systematic and comprehensive profiling of the expression of the CT genes in HCC. The detailed profiling provides us a better understanding of the role of CT genes in HCC, and further experimental or clinic studies should be performed to confirm the potential candidate targets for immunotherapy.
Supplemental Material
Supplemental Material, Supplementary_data for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
{kind=link}
Supplemental Material, Supplementary_Figure_1 for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Supplemental Material, supplementary_materials for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Supplemental Material, Supplementary_table_1_(1) for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Abbreviations
- AZA
azacytidine
- CT gene
cancer-testis gene
- CTdatabase
Cancer Testis database
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- DFS
disease-free survival
- GTEx
Genotype-Tissue Expression
- GO
gene ontology
- HBM
Human Body Map
- HCC
hepatocellular carcinoma
- ICB
immune checkpoint blockade
- IHC
immunohistochemistry
- OS
overall survival
- PPI
protein–protein interaction
- TAA
tumor-associated antigen
- TCGA
The Cancer Genome Atlas
Footnotes
Authors’ Note: Y-.P.Z. and Z-.W.B. contributed equally to this work. Written informed consent was obtained from all patients.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Innovative Research Groups of National Natural Science Foundation of China (No. 81721091) and National S&T Major Project (No. 2017ZX10203205). This study was approved by Clinical Research Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (approval #2019-564).
ORCID iD: Shu-Sen Zheng  https://orcid.org/0000-0003-1459-8261
https://orcid.org/0000-0003-1459-8261
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Villanueva A, Llovet JM. Liver cancer in 2013: mutational landscape of HCC—the end of the beginning. Nat Rev Clin Oncol. 2014;11(2):73–74. [DOI] [PubMed] [Google Scholar]
- 3. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616. [DOI] [PubMed] [Google Scholar]
- 4. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 5. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149(5):1226–1239.e1224. [DOI] [PubMed] [Google Scholar]
- 6. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267–1273. [DOI] [PubMed] [Google Scholar]
- 8. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12(12):681–700. [DOI] [PubMed] [Google Scholar]
- 9. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100(11):2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5(8):615–625. [DOI] [PubMed] [Google Scholar]
- 11. Knuth A, Danowski B, Oettgen HF, Old LJ. T-cell-mediated cytotoxicity against autologous malignant melanoma: analysis with interleukin 2-dependent T-cell cultures. Proc Natl Acad Sci U S A. 1984;81(11):3511–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao S, Zhu W, Xue S, Han D. Testicular defense systems: immune privilege and innate immunity. Cell Mol Immunol. 2014;11(5):428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhodapkar MV, Sznol M, Zhao B, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6(9):990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Almeida LG, Sakabe NJ, deOliveira AR, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37(Database issue):D816–D819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bruggeman JW, Koster J, Lodder P, Repping S, Hamer G. Massive expression of germ cell-specific genes is a hallmark of cancer and a potential target for novel treatment development. Oncogene. 2018;37(42):5694–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang C, Gu Y, Zhang K, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun. 2016;7:10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. da Silva VL, Fonseca AF, Fonseca M, et al. Genome-wide identification of cancer/testis genes and their association with prognosis in a pan-cancer analysis. Oncotarget. 2017;8(54):92966–92977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hofmann O, Caballero OL, Stevenson BJ, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105(51):20422–20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 26. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(suppl 4):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang WY, Hsu SD, Huang HY, et al. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res. 2015;43(Database issue): D856–D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ru B, Wong CN, Tong Y, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35(20):4200–4202. [DOI] [PubMed] [Google Scholar]
- 34. Detre S, Saclani Jotti G, Dowsett M. A “quickscore” method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J Clin Pathol. 1995;48(9):876–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Lee WS, Kong SJ, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. 2019;130:4350–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maxfield KE, Taus PJ, Corcoran K, et al. Comprehensive functional characterization of cancer-testis antigens defines obligate participation in multiple hallmarks of cancer. Nat Commun. 2015;6:8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan H, Wang X, Shi C, et al. Plac1 is a key regulator of the inflammatory response and immune tolerance in mammary tumorigenesis. Sci Rep. 2018;8(1):5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spranger S, Luke JJ, Bao R, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113(48):E7759–E7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eng KH, Tsuji T. Differential antigen expression profile predicts immunoreactive subset of advanced ovarian cancers. PLoS One. 2014;9(11):e111586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. [DOI] [PubMed] [Google Scholar]
- 41. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gordeeva O. Cancer-testis antigens: unique cancer stem cell biomarkers and targets for cancer therapy. Semin Cancer Biol. 2018;53:75–89. [DOI] [PubMed] [Google Scholar]
- 43. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baudat F, Imai Y, de Massy B. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet. 2013;14(11):794–806. [DOI] [PubMed] [Google Scholar]
- 45. Orthwein A, Fradet-Turcotte A, Noordermeer SM, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344(6180):189–193. [DOI] [PubMed] [Google Scholar]
- 46. Watkins J, Weekes D, Shah V, et al. Genomic complexity profiling reveals that HORMAD1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov. 2015;5(5):488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu J, Zhu C, Yu Y, et al. Systematic cancer-testis gene expression analysis identified CDCA5 as a potential therapeutic target in esophageal squamous cell carcinoma. EBioMedicine. 2019;46:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Djureinovic D, Hallstrom BM, Horie M, et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight. 2016;1(10):e86837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cho HJ, Caballero OL, Gnjatic S, et al. Physical interaction of two cancer-testis antigens, MAGE-C1 (CT7) and NY-ESO-1 (CT6). Cancer Immun. 2006;6:12. [PubMed] [Google Scholar]
- 50. Kang Y, Hong JA, Chen GA, Nguyen DM, Schrump DS. Dynamic transcriptional regulatory complexes including BORIS, CTCF and Sp1 modulate NY-ESO-1 expression in lung cancer cells. Oncogene. 2007;26(30):4394–4403. [DOI] [PubMed] [Google Scholar]
- 51. Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010;39(6):963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coral S, Sigalotti L, Altomonte M, et al. 5-aza-2’-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res. 2002;8(8):2690–2695. [PubMed] [Google Scholar]
- 53. Condomines M, Hose D, Raynaud P, et al. Cancer/testis genes in multiple myeloma: expression patterns and prognosis value determined by microarray analysis. J Immunol. 2007;178(5):3307–3315. [DOI] [PubMed] [Google Scholar]
- 54. Yao J, Caballero OL, Yung WK, et al. Tumor subtype-specific cancer-testis antigens as potential biomarkers and immunotherapeutic targets for cancers. Cancer Immunol Res. 2014;2(4):371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chang Y, Wang X, Xu Y, et al. Comprehensive characterization of cancer-testis genes in testicular germ cell tumor. Cancer Med. 2019;8(7):3511–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Y, Tan PY, Handoko YA, et al. NUF2 is a valuable prognostic biomarker to predict early recurrence of hepatocellular carcinoma after surgical resection. Int J Cancer. 2019;145(3):662–670. [DOI] [PubMed] [Google Scholar]
- 57. Lin SY, Pan HW, Liu SH, et al. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res. 2008;14(15):4814–4820. [DOI] [PubMed] [Google Scholar]
- 58. Liao WY, Hsu CC, Chan TS, et al. Dishevelled 1-regulated superpotent cancer stem cells mediate Wnt heterogeneity and tumor progression in hepatocellular carcinoma. Stem Cell Reports. 2020;14(3):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Korpal M, Puyang X, Jeremy Wu Z, et al. Evasion of immunosurveillance by genomic alterations of PPARgamma/RXRalpha in bladder cancer. Nat Commun. 2017;8(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material, Supplementary_data for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Supplemental Material, Supplementary_Figure_1 for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Supplemental Material, supplementary_materials for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment
Supplemental Material, Supplementary_table_1_(1) for Cancer-Testis Gene Expression in Hepatocellular Carcinoma: Identification of Prognostic Markers and Potential Targets for Immunotherapy by Yan-Peng Zhang, Zhi-Wei Bao, Jing-Bang Wu, Yun-Hao Chen, Jun-Ru Chen, Hai-Yang Xie, Lin Zhou, Jian Wu and Shu-Sen Zheng in Technology in Cancer Research & Treatment