

Abstract
Imaging of gases is a major challenge for any modality including MRI. NMR and MRI signals are directly proportional to the nuclear spin density and the degree of alignment of nuclear spins with applied static magnetic field, which is called nuclear spin polarization. The level of nuclear spin polarization is typically very low, i.e., one hundred thousandth of the potential maximum at 1.5 T and a physiologically relevant temperature. As a result, MRI typically focusses on imaging highly concentrated tissue water. Hyperpolarization methods transiently increases nuclear spin polarizations up to unity, yielding corresponding gains in MRI signal level of several orders of magnitude that enable the 3D imaging of dilute biomolecules including gases. Parahydrogen-induced polarization is a fast, highly scalable, and low-cost hyperpolarization technique. The focus of this Minireview is to highlight selected advances in the field of parahydrogen-induced polarization for the production of hyperpolarized compounds, which can be potentially employed as inhalable contrast agents.
Keywords: parahydrogen, NMR, MRI, hyperpolarization, spectroscopy
Graphical Abstract
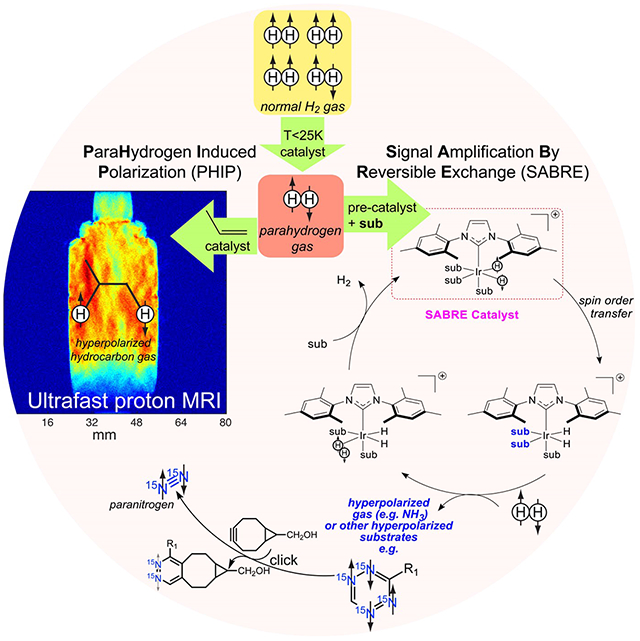
1. Introduction
MRI is an imaging technique based on NMR detection of nuclear spin magnetization. Magnetization is the product of spin density and the degree of alignment (i.e., polarization) of nuclear spin dipoles along the quantization axis determined by the static magnetic field of the MRI scanner. The NMR signal in conventional MRI arises from the very small thermal (Boltzmann) nuclear spin polarization, typically on the order of 10−5 at clinical scanner field strengths of 1.5–3T. This low spin polarization limits the ability of MRI to detect molecules at low concentrations. Hyperpolarization techniques can be used to increase nuclear spin polarization by several orders of magnitude – up to the order of unity – with corresponding gains in detection sensitivity.[1] MRI of hyperpolarized (HP) contrast agents is a revolutionary imaging technique that leverages the 4-6 orders of magnitude of gain in sensitivity for the MRI detection of metabolic and functional exogenous contrast agents at millimolar concentrations.[2, 3] Several HP contrast agents have emerged (most notably 13C-pyruvate [4]) for the visualization of metabolism and organ function in vivo.
HP noble gases have been successfully employed for regional imaging of lung function: ventilation, perfusion and gas exchange, as well as for a wide range of sensing applications.[5, 6] Although 3He was initially the leading HP inhalable contrast agent, its limited supply has acted to significantly decrease the access of 3He HP MRI to the biomedical community.[7] As a result, the bulk of research activities with HP noble gases employ 129Xe, which is hyperpolarized via spin exchange optical pumping (SEOP).[8, 9] Despite the major successes of HP 129Xe in clinical research and basic science studies,[7] the clinical translation of HP 129Xe is hindered by two primary obstacles. First and foremost, the instrumentation employed for 129Xe hyperpolarization is highly complex, very costly, and typically requires highly skilled professional operators to enable the production of only 1-2 clinical doses per hour. Second, MRI of HP 129Xe requires specialized MRI scanners, because 129Xe nucleus resonates at a frequency vastly different from that of protons which are used routinely.[7]
Parahydrogen-induced polarization (PHIP) is a chemical-based hyperpolarization technique relying on parahydrogen (p-H2) as the source of hyperpolarization.[10, 11] PHIP is a highly scalable and relatively low cost method.[12] Recent advances in this field have demonstrated the feasibility of producing HP gases, which may be ultimately suitable for clinical applications, and may mitigate the limitations of HP 129Xe gas. In this Minireview, we focus on selected p-H2-based hyperpolarization approaches that show the potential to produce HP gases, covering the fundamental principles and applications as well as key advances reported in the literature up to September 2019.
2. Production of parahydrogen
Molecular hydrogen exists as two nuclear spin isomers, which are referred to as ortho- and parahydrogen.[13] Orthohydrogen (o-H2) is the triplet, while parahydrogen is the singlet. These two spin isomers, which respectively account for 75% and 25% of H2 at standard temperature and pressure, exhibit different magnetic properties. They also differ in energy as a result of the Pauli exclusion principle, which restricts them to antisymmetric and symmetric rotational states, respectively. Because interconversion between isomers requires a change of two physical properties, the process is forbidden. However, their 120 cm−1 separation in energy means that one can produce nearly 100% parahydrogen at 20 K according to Boltzmann statistics. Fortunately, the rule that prevents interconversion between the two forms at higher temperatures can be broken by passing normal H2 gas over a suitable spin conversion catalyst. The catalyst can be charcoal or ironIII oxide (or another ferromagnetic material) and this process can be rapid but once the gas has moved away from the catalyst its initial stability to spin isomer interchange returns. Cooling hydrogen gas to −196 °C (77 K), using liquid nitrogen is easily achieved and creates a mixture wherein the proportion of parahydrogen is around 50%. Further cooling to −250 °C (20 K) results in >99% parahydrogen. However, 20 K is the boiling point of H2 and cooling H2 gas below this point produces liquid p-H2. Several methods and equipment designs have been suggested for the production of p-H2 for various applications [14–16] including hyperpolarization.[17, 18] A p-H2 converter, suitable for clinical use has been suggested.[19] This apparatus produces a continuous flow at 98% purity (cooling to 25 K) with a maximum output pressure of 50 bar. The half-life of pH2 is 13.7 ± 0.5 hours in a glass vial and 129 ± 36 days in an aluminum storage cylinder.[20] Commercially available parahydrogen generators such as those marketed by Bruker are available, which provide for a large scale use such as that needed for flow studies.[20]
3. Parahydrogen Induced Polarization (PHIP)
3.1. Breaking the symmetry of parahydrogen via pairwise parahydrogen addition
It is well known that p-H2 does not give an observable NMR signal since it has a total zero spin moment. Thus, the observable NMR signal of hydrogen is obtained from o-H2 only. Despite this, it is possible to use the p-H2 singlet spin state to significantly enhance NMR signals by transferring the H atoms of a p-H2 molecule to magnetically nonequivalent environments as, for example, through a hydrogenation reaction involving pairwise H2 addition to an unsymmetrical substrate.[21]
The effects of parahydrogen-induced polarization (PHIP) were first discovered by studying the interaction of transition metal complexes with hydrogen by NMR.[22, 23] These effects were represented by the antiphase (absorption-emission) shape of some NMR signals in the 1H NMR spectra and were initially described via chemically induced dynamic nuclear polarization (CIDNP) theory. In 1985, Weitekamp suggested the possibility of using the singlet spin state of p-H2 to significantly enhance the NMR signals of hydrogenation reaction products.[24] This effect – PASADENA (Parahydrogen And Synthesis Allows Dramatically Enhanced Nuclear Alignment[25]) is observed for the product of the hydrogenation with p-H2 in a strong magnetic field and accounted for the earlier results attributed to CIDNP. It is assumed that only the energy spin levels in the reaction product that have a singlet spin symmetry corresponding to the symmetry of parahydrogen molecule will be populated.[10] In 1987, two research groups, working independently, confirmed experimentally the occurrence of the PASADENA effects.[11, 25]
When the hydrogenation reaction with p-H2 is carried out in a low magnetic field (e.g., the Earth’s field of about 50 μT) and followed by an adiabatic transfer of the sample to the NMR spectrometer, the NMR signals of the reaction products are enhanced; however, the shape of the polarized peaks differs significantly from the PASADENA experiment. This effect was called ALTADENA (Adiabatic Longitudinal Transport After Dissociation Engenders Nuclear Alignment).[26]
It is important to note that in order to observe PHIP in an NMR spectrum, three main conditions should be realized: the rate of the hydrogenation reaction and the time of NMR spectrum acquisition should not significantly exceed the nuclear spin relaxation time of the hyperpolarized (HP) products; the addition of p-H2 to the unsaturated substrate should proceed in a pairwise manner, i.e., with the two H atoms of a p-H2 molecule ending up in the same reaction product, so that the nuclear spin correlation between them is preserved; the H atoms of p-H2 molecule should become magnetically nonequivalent in the product molecule.[27] Therefore, for PHIP effects to be observed, the pairwise hydrogen addition route should be realized during hydrogenation and it turns out despite these constraints, this is quite a common phenomenon.
3.2. Homogeneous catalysis and migration of HP fluids from the liquid to gaseous phase
Homogeneous catalysts can be used successfully for producing HP gases in biphasic catalytic processes. These gas-liquid systems were shown to be useful for the HP gases formation, where transition metal complex (homogeneous catalyst) was present in the liquid phase, while the reactants and products were in the gas phase.[28] The novelty of this approach is that the products of the hydrogenation reaction with p-H2, which are formed in the liquid phase, can return to the gas phase with preservation of spin polarization. This process allows for the rapid separation of reaction products from the homogeneous catalyst, which makes possible to hyperpolarize a broad class of molecules in the gas phase, including vapors of various liquids.
Homogeneous hydrogenation of gaseous C3-hydrocarbons containing double and triple bonds, with the acquisition of 1H NMR spectra of gaseous products, has provided the first evidence of the usefulness of this biphasic approach to prepare HP gaseous molecules. To this end, hydrogenation of propyne or propene was carried out over the following homogeneous catalysts: [Rh(PPh3)3Cl], [Ir(COD)(PCy)3(Py)]PF6, [Rh(NBD)(PPh3)2]BF4,[29] [Rh(P(C6H4SO3Na)3)3Cl]*xH2O, and [Rh(PPh2(CH2)4PPh2)(COD)]BF4. The mixture of the gaseous substrate and p-H2 was bubbled through the catalyst solution and then passed through a Teflon capillary to the NMR tube for recording 1H NMR spectra. It was shown that PHIP effects can be successfully detected. At the same time, due to the relatively short relaxation times of 1H nuclei in the formed gaseous products[30] the NMR signal enhancements did not exceed a factor of 20–50, which is an order of magnitude lower than the typical signal enhancement values observed during homogeneous hydrogenation with p-H2.[31]
3.3. Heterogeneous PHIP of gases over solid catalysts
By combining the use of p-H2 with heterogeneous hydrogenation processes, it is possible to develop new fundamental and practical applications for the production of HP gases.[12, 32] In heterogeneous processes, supported metals are often used as catalysts. It was successfully shown that when the p-H2 based hydrogenation of unsaturated gaseous hydrocarbons is catalyzed by highly dispersed supported metal catalysts, nuclear spin polarized reaction products can be created thus confirming that the pairwise addition of molecular hydrogen can be achieved by a supported metal catalysts.[33] The nature of the catalyst proved to be important with the smallest particle sizes maximizing the signal enhancing PHIP effect.[34]
Once the observation of PHIP effects was successfully demonstrated in heterogeneous hydrogenation reactions, the production of HP gases became an important area of research.[7] Continuous production of HP gases using heterogeneous hydrogenation opens up the way to potential practical applications for void space MR imaging. The first experiments addressing MR imaging with HP propane gas helped visualize various phantoms,[35] and in later studies propane hyperpolarized via PHIP allowed the collection of MR images with ultra-high resolution despite its relatively short relaxation time.[30] One potential solution to increase the magnetic lifetime of HP propane is to create long-lived spin states by using PHIP and a low magnetic field. The spin-locked induced crossing (SLIC) pulse sequence, developed by Rosen et al.[36] has been used to convert this long-lived polarization, created in the Earth’s magnetic field, into observable magnetization at 47.5 mT and hence to use HP gas for MR imaging in the low magnetic fields.[37, 38] In addition to obtaining low field MR images from protonated propane, when the fully deuterated precursor propene was used, there was no need to apply special symmetry breaking pulse sequences at 47.5 mT. Consequently, the deuterated variant can be used to obtain low field MR images.[39]
Recently it was shown that the nature of the substrate is also critical, and heterogeneous hydrogenation of cyclopropane was realized as an alternative route to HP propane. Interestingly, one could expect that ring opening cyclopropane would place the protons obtained from p-H2 into magnetically equivalent positions, i.e., one hydrogen per methyl group. Furthermore, due to the chemical and magnetic equivalence of these two methyl groups, the resulting hyperpolarization should not be detectable directly by NMR; this is similar to the correlated ethylene spin isomers obtained by pairwise addition of p-H2 to acetylene.[40] However, hydrogenation of cyclopropane with p-H2 under the conditions of PASADENA experiment led to the clear-cut observation of PASADENA signals of HP propane, similar to the NMR signals of propane itself observed during hydrogenation of propene.[41] Moreover, it was found that the use of cyclopropane as a precursor to HP propane is promising as it yields surprisingly high polarization levels (ca. >2%).[41]
Because the potential applications will require relatively large amounts of HP gas, the conditions necessary to hydrogenate propene over a Rh/TiO2 catalyst were optimized and the clinical-scale production of HP propane gas using the PHIP technique demonstrated. Hence, more than 0.3 liters of HP propane gas was produced in 2 s which, along with significant NMR signal enhancement, allowed the MRI visualization of a 56 mL phantom in less than 2 seconds at 4.7 T (Figure 2).[42]
Figure 2.
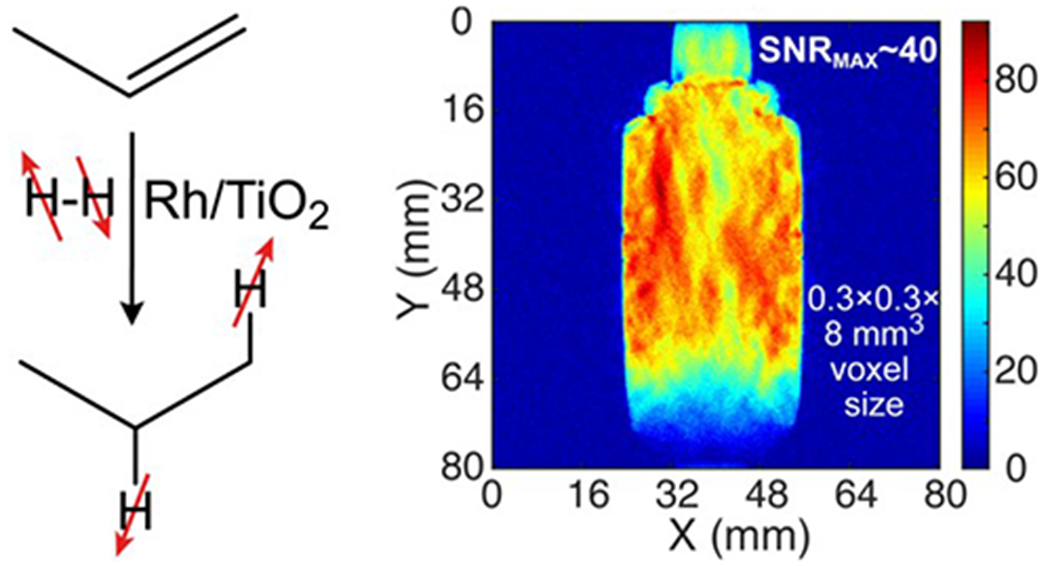
Scheme of heterogeneous hydrogenation of propene with parahydrogen over Rh/TiO2 catalyst, and 2D MRI of 0.2 standard liters of HP propane gas in an ~56 mL collection container after production of an 0.3-standard-liter batch of HP propane in ~2 s using an ~1:2 mixture of propene with parahydrogen. Adapted with permission from Ref. # [42]. Copyright (2019) American Chemical Society.
4. Signal Amplification by Reversible Exchange (SABRE)
4.1. The SABRE phenomenon
In 2009, an extension to the PHIP technique termed Signal Amplification by Reversible Exchange (SABRE) was reported.[43] This approach, which is multinuclear in nature, harnesses p-H2 through its reversible binding to a metal catalyst in association with further reversible binding of a substrate and is therefore not associated with changing the chemical identity of the substrate. SABRE then achieves spin order transfer within the resulting scalar coupling network[43, 44] of the catalyst and harnesses substrate dissociation to catalytically build-up hyperpolarized molecules of it in solution according to Figure 3. One consequence of the interconnected chemical and physical aspects of SABRE is that its efficiency can be rigorously controlled. In one version, low magnetic fields are used to create a propagating spin-topology that enables spontaneous spin order transfer,[43–47] though other approaches employing more traditional radio frequency excitation are also possible. Density matrix theory[43] and level anti-crossing analysis have been used to rationalize this behavior.[45, 48, 49]
Figure 3.
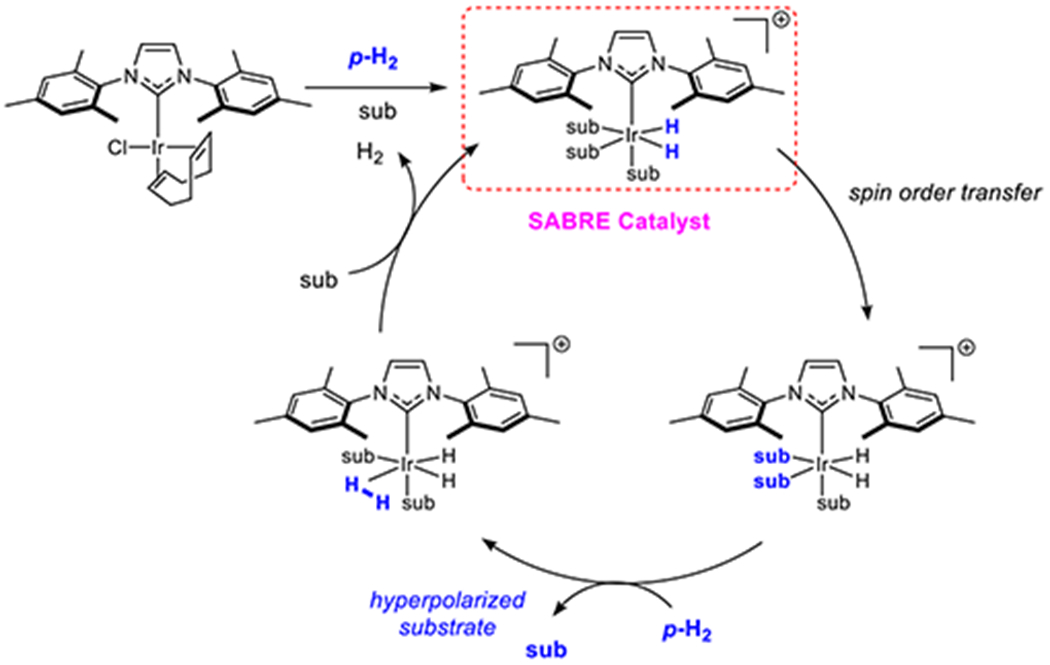
Mechanism of the SABRE process which uses a metal catalyst to harvest the latent magnetism of p-H2 to hyperpolarize materials (sub) without changing their chemical identity.
Chemically, both of these approaches require the SABRE catalyst to remain intact for a time period which is set by the size of the propagating spin-spin couplings between the p-H2 derived hydride ligands of the catalyst and the NMR active nuclei of the recipient substrate. Tessari et al. have quantified some of those couplings involved in 1H-transfer at ~1.2 Hz[50] which are small yet sufficient to enable highly efficient transfer if the complex lifetime is around 0.2 s[46] when the sample experiences an ~5-8 mT field.[51, 52] For transfer to heteronuclei such as 15N that occur through the corresponding couplings that can exceed 20 Hz and involve shorter complex lifetimes, the optimum magnetic fields lie in the 0.2-0.4 μT range and thus necessitate the use of μ-metal magnetic shields.[53] This SABRE variation has been termed “SABRE-SHEATH” (Signal Amplification by Reversible Exchange in SHield Enables Alignment Transfer to Heteronuclei).
Successful magnetization transfer at high field through radio frequency excitation was also exemplified in 2009 for an iridium-phosphine catalyst with pyridine-15N hyperpolarization as the outcome.[54] This approach has then been improved through low-power methods such as LIGHT-SABRE[55, 56] and more recently by matching the reaction dynamics.[57–60] One additional consequence of SABRE happening during the finite lifetime of what is commonly a bulky catalyst is that controlling nuclear spin order relaxation within the catalyst framework reflect a route to improve SABRE efficiency. This can be achieved by 2H labeling, and once the catalyst lifetime, its relaxation properties and polarization transfer field are optimized, 1H hyperpolarization levels have reached 20%.[61] Studies on 15N transfer are also well developed with the resulting high polarization levels allowing the ready detection of MR signals without the need for isotope enrichment.[62] Other studies have demonstrated that 13C, 31P and 19F[43] NMR signals can be substantially amplified in what has become a rapidly expanding range of molecules that are functionalized by nitrogen, oxygen, phosphorus[63, 64] and sulfur[65] based catalyst ligation sites. These studies confirm the multinuclear capabilities of SABRE.
One relaxation-related topic deals with the creation of long lived singlet states, as detailed by Levitt.[66, 67] The SABRE technique readily yields them in a HP form, as exemplified by the strongly coupled 1H-pair in 2-aminothiazole[68] whose singlet state has been accessed with 90% efficiency by rf-methods.[66] This approach has been applied to derivatives of nicotinamide and pyrazinamide that have isolated spin-pairs through selective deuteration.[69] For a pyridazine based 1H-spin pair the SABRE HP signal remained visible for 15 minutes after polarization transfer.[70] When this method has been used in conjunction with 15N2-diazirines[71, 72] similar singlet states can be populated with decay constants that exceed 23 min in an optimum storage field.
4.2. RELAYED-SABRE and hyperpolarization of ammonia and beyond
Ammonia represents a common inorganic ligand that binds to SABRE catalysts. This results in the hyperpolarization of its nitrogen and proton sites in a process that can be readily observed in aprotic solvents. Ammonia is a base that undergoes facile proton exchange with acidic protons like those of alcohols, carboxylic acids and water itself. Consequently, the SABRE hyperpolarization of an amine offers a simple route to place a HP proton into a different molecule through chemical exchange.[73] When this process takes place at low field, spin order transfers through low-field thermal mixing result in the hyperpolarization of coupled spins. When this SABRE-RELAY method (Figure 4) was applied to the study of the straight chain alcohols methanol through octanol, signal gains of up to 650-fold and 600-fold per proton and carbon respectively were achieved through the use of [IrCl(COD)(IMes)] (5 mM), the alcohol (1 μL), NH3 (6-8 eq.) in d2-dichlormethane (0.6 mL). Examples have established that 1H, 13C, 19F, 31P and 15N signals of compounds can be readily enhanced through this approach, meaning that p-H2 can hyperpolarize a much broader range of biologically relevant substrates without the need for their direct interaction with either p-H2 or a SABRE catalyst. It is not surprising that the rate of relaxation of the NH protons responsible for SABRE-RELAY is an important parameter that can be controlled through 2H labeling and the extension to amines such as d7-benzylamine.[74]
Figure 4.
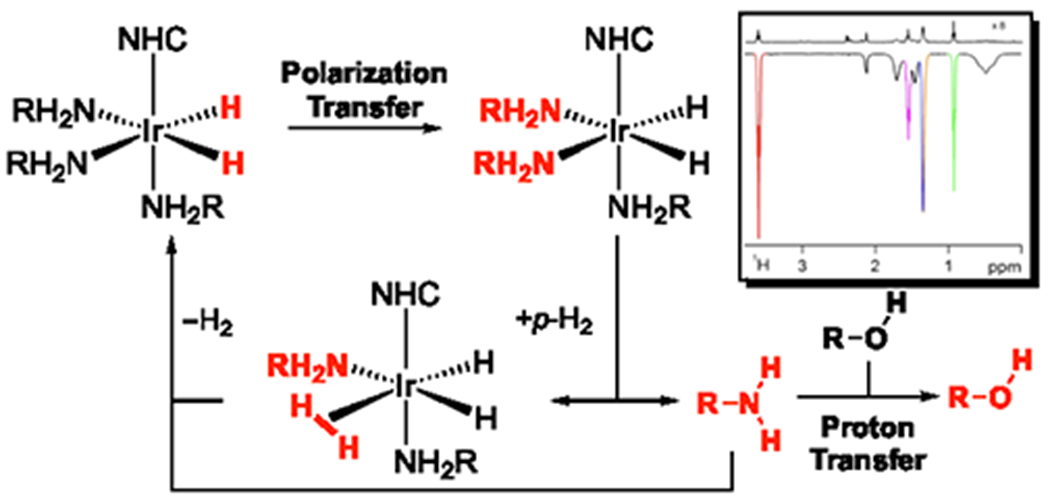
SABRE-RELAY enables substrate hyperpolarization via p-H2 and an amine through proton transfer.
While SABRE-RELAY works with sodium hydrogen carbonate and therefore offers a route to HP carbon dioxide, its use with pyruvic acid is limited by competing Schiff base condensation.[75] The use of dimethylsulfoxide as a weakly binding co-ligand in conjunction with the original SABRE method overcomes this problem, leading to a simple route to HP pyruvate.[76] It should therefore be clear that SABRE offers a truly versatile route to hyperpolarize many classes of chemical compounds in a simple and low-cost manner including HP gases in the liquid phase.
4.3. Production of paranitrogen via SABRE hyperpolarization of 15N-containing heterocycles
Two compelling SABRE approaches have been described for the production of HP 15N2 gas, in particular the para spin isomer which we refer to as 15N2-paranitrogen (p-15N2). The two published approaches are summarized in Figure 6.
Both methods can also be used to hyperpolarize 15N2-orthonitrogen (o-15N2), the ortho spin isomer of 15N2, however, p-15N2 is more intriguing because of its potential to be extremely long lived, in analogy with p-H2 which can be stored for weeks.[19] The first method, depicted in Figure 5a, involves the hyperpolarization of a tetrazine, which is reacted with a strained alkyne in a click reaction to release HP 15N2. Whether p-15N2 or o-15N2 is produced can be controlled by the magnetic field used for SABRE. On one hand, to produce HP o-15N2, magnetization (i.e., spin alignment) has to be created on the 15N4-tetrazine first. This is best accomplished at fields of 5 to 10 mG under SABRE-SHEATH conditions. The subsequent click reaction releases HP o-15N2. On the other hand, to produce p-15N2, SABRE should be conducted at fields above the SABRE-SHEATH[53] condition, yet sufficiently low, as not to induce significant chemical shifts between the two 15N nuclei in the tetrazine (bound or free). Specifically, static magnetic fields between 0.1 to 50 G work well. At these fields singlet states are hyperpolarized on the tetrazine and the subsequent click reaction releases p-15N2.[77]
Figure 5.
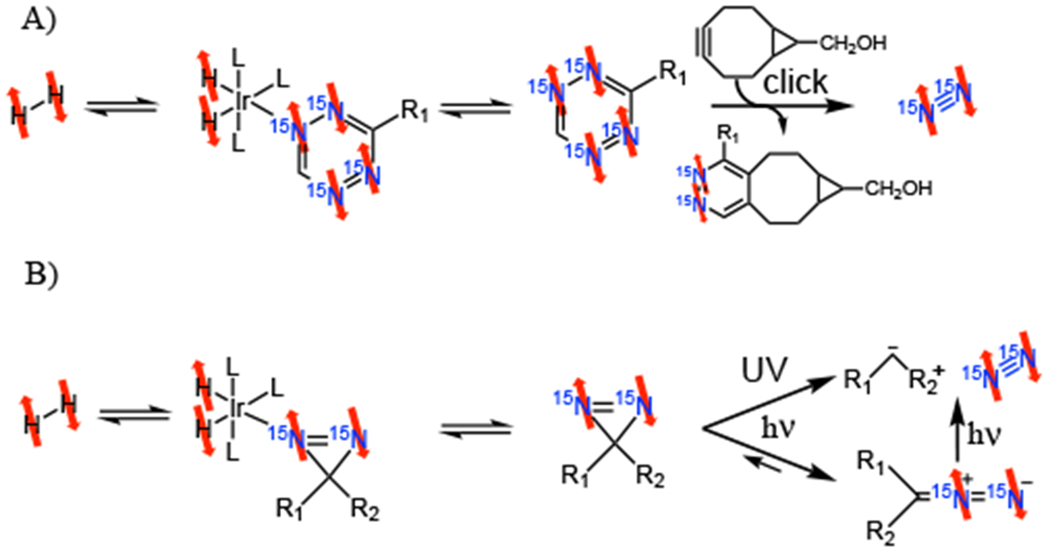
Two routes for the production of 15N2-paranitrogen. A) A 15N4 labeled tetrazine is hyperpolarized by SABRE, which releases HP 15N2, when reacting with a strained alkyne. B) A 15N2-diazirine is hyperpolarized by SABRE, which releases HP 15N2 when irradiated by UV light.
The second method, depicted in Figure 5b, involves the hyperpolarization of a diazirine and subsequent irradiation with UV light to release HP 15N2 gas. Just as in the tetrazine case, para- vs. ortho- is controlled by the magnetic field during SABRE. HP o-15N2 is produced when using μT fields, i.e., SABRE-SHEATH conditions, whereas enriched p-15N2 is produced with slightly higher fields for SABRE. Singlet states in 15N2-diazirines are also enriched well at fields between 0.5 and 50 G. Subsequent UV irradiation releases the p-15N2. Note that, in Figure 5b, UV irradiation of the diazirine can lead to the carbene and nitrogen directly, or indirectly through formation of an intermediate diazo compound.[78]
5. Nuclear spin relaxation considerations in preparation of HP gases
The very long spin-lattice (T1) relaxation times of HP heteronuclei, e.g. 13C, 15N, etc. can be on the order of up to tens of minutes[72, 79, 80] in solution which makes them highly suitable for HP state storage in the context of biomedical applications: i.e. injectable contrast agents.[2] However, the gas-phase T1 of heteronuclei is typically very short, and although 13C T1 increases with pressure, it is only ~0.2 s at 10 atm of CO2.[81] Moreover, 19F fluorocarbons have T1 values on the time scale of less than 0.1 second at clinically relevant conditions in the context of pulmonary imaging applications.[82, 83] On the other hand, proton-hyperpolarized propane has T1 of ~1 s[84] and Ts of ~3 s[38] at 1 atm (and up to more than 13 s at higher pressures[85]) and other clinically relevant conditions in the gas phase. These decay constants of HP state are significantly longer than those of many heteronuclei, and sufficiently long for potential administration as inhalable contrast agents.
5. Summary and Outlook
To summarize, a wide range of HP gases can be produced via PHIP and SABRE hyperpolarization techniques. These HP gases can be potentially employed for many applications including most importantly bioimaging applications. Clinical-scale production of HP propane has been demonstrated, and it can be potentially extended to many others—ongoing active work in our laboratories, which we hope to report in the near future. In vivo bioimaging applications are envisioned for HP gases produced using p-H2. Efforts are now in progress to study the feasibility and effectiveness of lung ventilation imaging in sheep model of acute lung injury using HP propane gas employing 0.35 T clinical MRI scanner. HP propane gas is expected to have Ts of ~3 s at physiologically relevant conditions,[38] and therefore, hyperpolarization process is integrated in the inhalation apparatus to minimize otherwise significant relaxation-associate polarization losses. Future studies will also need to address the potential issues of hydrocarbon flammability, which can be possibly mitigated via administration of HP gas below lower explosive limit (LEL). We envision that paranitrogen may potentially be employed as an inhalable contrast agent—future efforts will be required to perform extraction of paranitrogen from the liquid phase for this application. The key advantages of proton-hyperpolarized HP contrast agents produced using p-H2 include: (i) possibility of detection using clinical MRI scanner, (ii) low-cost and high production speed. The low cost of PHIP and SABRE instrumentation is highly synergistic with the low cost of reagents required for the hyperpolarization process and bode well for future biomedical translation.
Figure 1.
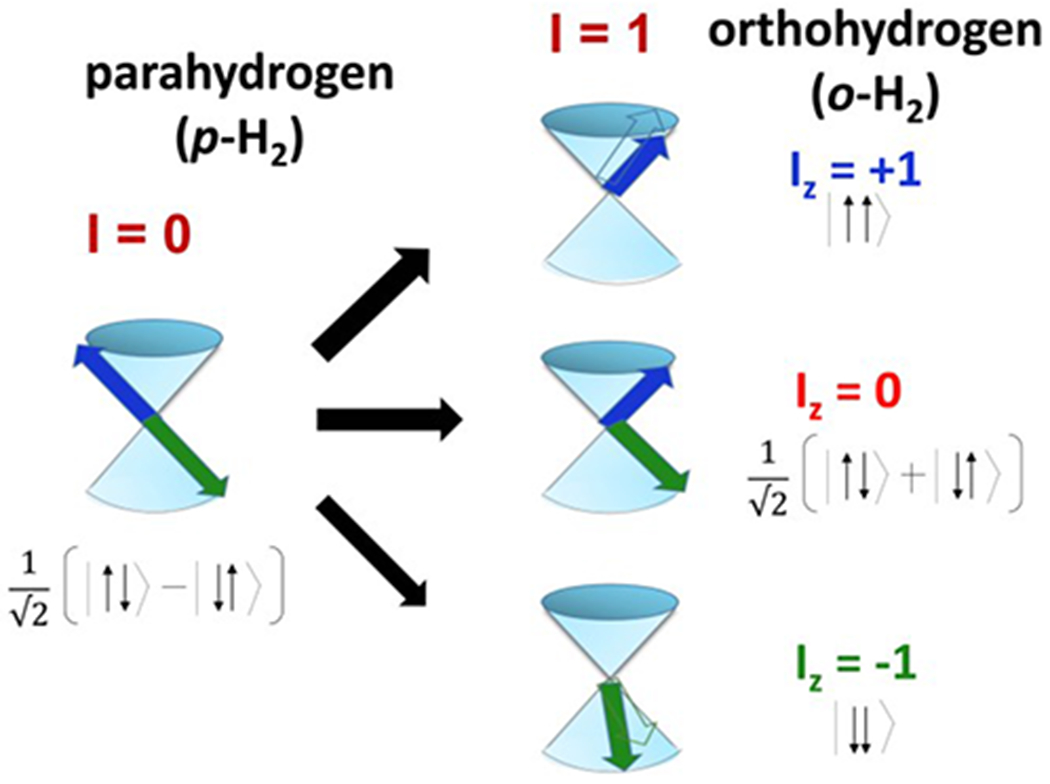
Singlet and triplet spin states of molecular hydrogen.
Acknowledgements
We thank the following award for funding support: NSF CHE-1904780 (EYC), NIH 1R21CA220137 (EYC), R21EB025313 (TT), and 1U01CA202229 (EYC), DOD CDMRP PRMRP W81XWH-15-1-0271 (EYC). KVK and IVK thank RSF (grant # 19-13-00047) for the support of MRI studies and RFBR (grant # 19-29-10003 and 17-54-33037) for the support of catalysts synthesis, and Russian Ministry of Science and Higher Education (AAAA-A16-116121510087-5) for the access to NMR/MRI equipment. SBD thanks financial support from the Wellcome Trust (Grants 092506 and 098335), the MRC (MR/M008991/1). TT also acknowledges support from the Oak Ridge Associated Universities, Ralph E. Powe Junior Faculty Enhancement Award, the North Carolina Biotechnology Center Translational Research Grant as well as support from the Mallinckrodt foundation.
Biographies

Dr. Kirill V. Kovtunov studied chemistry at the Novosibirsk State University, Russia. He completed a PhD in physical-chemistry in 2008 at the International Tomography Center under the supervision of Prof. Igor Koptyug. His research interests include heterogeneous catalysis and utilization of PHIP techniques for NMR/MRI and mechanistic studies of heterogeneous reactions involving hydrogen. Currently he is the leading researcher in the group of Prof. Igor Koptyug.

Prof. Igor V. Koptyug received his Ph.D. degree in 1991; in 1992–1995 he was a postdoctoral researcher in the photochemistry group of Professor N. J. Turro (Columbia University, New York). He earned his Dr. Sci. (Habilitation) degree in catalysis in 2003 and a title of Professor in 2006; currently, he is the deputy director of International Tomography Center, Siberian Branch of the Russian Academy of Sciences, Novosibirsk. His research interests include signal enhancement in NMR and applications of NMR and MRI in catalysis.

Dr. Marianna Fekete graduated from Faculty of Physical Chemistry, University of Belgrade, Serbia in 2003. She obtained her PhD in chemistry in 2008 on the use of reaction kinetics to study catalysis by transitional metal N-heterocyclic carbene complexes at the University of Debrecen, Hungary. Afterwards she undertook postdoctoral work with Prof. M. Botta at the Università del Piemonte Orientale, Italy, studying relaxometric properties of paramagnetic complexes in low field. In 2011, she joined the group of Prof. S. Duckett where she is currently undertaking research on the optimisation, kinetics and catalysis of polarisation transfer reaction.

Prof. Simon Duckett is Director of the Centre for Hyperpolarization in Magnetic Resonance in York. His research involves the development of hyperpolarization techniques and the study of inorganic reactions. He gained a D.Phil. from the University of York in 1990 and then undertook postdoctoral work with Prof. W. D. Jones and Prof. R. Eisenberg at the University of Rochester. He has authored over 130 publications on the parahydrogen effect.

Prof. Thomas Theis is Assistant Professor at North Carolina State University. His research is focused on hyperpolarization technology and unconventional NMR and MRI detection schemes. Dr. Theis completed his masters at the Georg-August University of Goettingen and gained his PhD from UC Berkeley in 2012 working with Prof. A. Pines on “zero-field NMR”. He conducted postdoctoral research at Duke University (USA) with Prof. Warren Warren focused on “singlet states for hyperpolarization storage”, where he was promoted to Research Professor in 2015. Since 2018, Dr. Theis leads the North Carolina State Hyperpolarization Laboratory.

Dr. Baptiste Joalland is a molecular physicist holding a Master degree from the University of Paris XI (2007) and a Ph.D. degree from the University of Toulouse (2011). His primary research interest in fundamental research is geared towards providing a better understanding of the driving forces at play in chemical reactions. Through numerous adventures between France and the USA, he has developed an original set of skills at the boundaries of experimental physical chemistry (with B. R. Rowe, A. G. Suits, and I. R. Sims), quantum chemistry (with C. J. Marsden and S. J. Klippenstein), molecular dynamics simulations (with A. Simon, F. Spiegelman, and T. J. Martínez), and laboratory astrophysics (with C. Joblin and L. Biennier). In 2019, he joined the group of E. Y. Chekmenev as a Senior Postdoctoral Fellow with one overarching goal: turn parahydrogen-induced polarization methods into a clinical reality.

Prof. Eduard Y. Chekmenev, PhD in Physical Chemistry (supervisor Prof. Richard J. Wittebort) 2003, University of Louisville, KY, USA. Postdoctoral Fellow at NHMFL in Tallahassee, FL (Prof. Timothy Cross), Caltech (Prof. Daniel P. Weitekamp) and HMRI (Dr. Brian D. Ross). In 2009, Dr. Chekmenev started his hyperpolarization program at Vanderbilt University, and he was tenured in 2015. He serves as an Associate Professor of Chemistry at Wayne State University since 2018. Research interests include development of methods of hyperpolarization and their Biomedical and industrial use.
References
- [1].Nikolaou P, Goodson BM, Chekmenev EY, Chem. Eur. J 2015, 21, 3156–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY, Comment A, Cunningham CH, DeBerardinis RJ, Green GG, Leach MO, Rajan SS, Rizi RR, Ross BD, Warren WS, Malloy CR, Neoplasia 2011, 13, 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Golman K, in’t Zandt R, Thaning M, Proc. Natl. Acad. Sci. U. S. A 2006, 103, 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PEZ, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, Reed G, Carvajal L, Small EJ, Munster P, Weinberg VK, Ardenkjaer-Larsen JH, Chen AP, Hurd RE, Odegardstuen LI, Robb FJ, Tropp J, Murray JA, Sci. Transl. Med 2013, 5, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schroder L, Phys. Medica 2013, 29, 3–16. [DOI] [PubMed] [Google Scholar]
- [6].Mugler JP, Altes TA, J. Magn. Reson. Imaging 2013, 37, 313–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Barskiy DA, Coffey AM, Nikolaou P, Mikhaylov DM, Goodson BM, Branca RT, Lu GJ, Shapiro MG, Telkki V-V, Zhivonitko VV, Koptyug IV, Salnikov OG, Kovtunov KV, Bukhtiyarov VI, Rosen MS, Barlow MJ, Safavi S, Hall IP, Schröder L, Chekmenev EY, Chem. Eur. J 2017, 23, 725–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goodson BM, J. Magn. Reson 2002, 155, 157–216. [DOI] [PubMed] [Google Scholar]
- [9].Walker TG, Happer W, Rev. Mod. Phys 1997, 69, 629–642. [Google Scholar]
- [10].Bowers CR, Weitekamp DP, Phys. Rev. Lett 1986, 57, 2645–2648. [DOI] [PubMed] [Google Scholar]
- [11].Eisenschmid TC, Kirss RU, Deutsch PP, Hommeltoft SI, Eisenberg R, Bargon J, Lawler RG, Balch AL, J. Am. Chem. Soc 1987, 109, 8089–8091. [Google Scholar]
- [12].Hövener J-B, Pravdivtsev AN, Kidd B, Bowers CR, Glöggler S, Kovtunov KV, Plaumann M, Katz-Brull R, Buckenmaier K, Jerschow A, Reineri F, Theis T, Shchepin RV, Wagner S, Bhattacharya P, Zacharias NM, Chekmenev EY, Angew. Chem. Int. Ed 2018, 57, 11140–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Farkas A, Orthohydrogen, Parahydrogen, and Heavy Hydrogen, Cambridge University Press, Cambridge, 1935. [Google Scholar]
- [14].Tam S, Fajardo ME, Rev. Sci. Instrum 1999, 70, 1926–1932. [Google Scholar]
- [15].Hövener J-B, Chekmenev EY, Harris KC, Perman W, Robertson L, Ross BD, Bhattacharya P, Magn. Reson. Mater. Phy 2009, 22, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tom BA, Bhasker S, Miyamoto Y, Momose T, McCall BJ, Rev. Sci. Instrum 2009, 80, 3. [DOI] [PubMed] [Google Scholar]
- [17].Feng B, Coffey AM, Colon RD, Chekmenev EY, Waddell KW, J. Magn. Reson 2012, 214, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gamliel A, Allouche-Arnon H, Nalbandian R, Barzilay CM, Gomori JM, Katz-Brull R, Appl. Magn. Reson 2010, 39, 329–345. [Google Scholar]
- [19].Hövener J-B, Baer S, Leupold J, Jenne K, Leibfritz D, Hennig J, Duckett SB, von Elverfeldt D, NMR Biomed. 2013, 26, 124–131. [DOI] [PubMed] [Google Scholar]
- [20].Fekete M, Rayner PJ, Green GGR, Duckett SB, Magn. Reson. Chem 2017, 55, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Duckett SB, Sleigh C, J. Prog. Nucl. Magn. Reson. Spectrosc 1999, 34, 71. [Google Scholar]
- [22].Seidler PF, Bryndza HE, Frommer JE, Stuhl LS, Bergman RG, Organometallics 1983, 2, 1701–1705. [Google Scholar]
- [23].Hommeltoft SI, Berry DH, Eisenberg R, J. Am. Chem. Soc 1986, 108, 5345–5347. [Google Scholar]
- [24].Weitekamp DP, in eMagRes (Eds.: Harris RK, Wasylishen RL), 2007, p. 10.1002/9780470034590.emrhp9780470030195. [DOI] [Google Scholar]
- [25].Bowers CR, Weitekamp DP, J. Am. Chem. Soc 1987, 109, 5541–5542. [Google Scholar]
- [26].Pravica MG, Weitekamp DP, Chem. Phys. Lett 1988, 145, 255–258. [Google Scholar]
- [27].Natterer J, Bargon J, Prog. Nucl. Mag. Res. Spectrosc 1997, 31, 293–315. [Google Scholar]
- [28].Kovtunov KV, Zhivonitko VV, Skovpin IV, Barskiy DA, Salnikov OG, Koptyug IV, J. Phys. Chem. C 2013, 117, 22887–22893. [Google Scholar]
- [29].Salnikov OG, Barskiy DA, Coffey AM, Kovtunov KV, Koptyug IV, Chekmenev EY, ChemPhysChem 2016, 17, 3395–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kovtunov KV, Barskiy DA, Coffey AM, Truong ML, Salnikov OG, Khudorozhkov AK, Inozemtseva EA, Prosvirin IP, Bukhtiyarov VI, Waddell KW, Chekmenev EY, Koptyug IV, Chem. Eur. J 2014, 20, 11636–11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Duckett SB, Mewis RE, Acc. Chem. Res 2012, 45, 1247–1257. [DOI] [PubMed] [Google Scholar]
- [32].Kovtunov KV, Pokochueva EV, Salnikov OG, Cousin S, Kurzbach D, Vuichoud B, Jannin S, Chekmenev EY, Goodson BM, Barskiy DA, Koptyug IV, Chem. Asian J. 2018, 13, 1857–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burueva DB, Kovtunova LM, Bukhtiyarov VI, Kovtunov KV, Koptyug IV, Chem. Eur. J 2019, 25, 1420–1431. [DOI] [PubMed] [Google Scholar]
- [34].Burueva DB, Kovtunov KV, Bukhtiyarov AV, Barskiy DA, Prosvirin IP, Mashkovsky IS, Baeva GN, Bukhtiyarov VI, Stakheev AY, Koptyug IV, Chem. Eur. J 2018, 24, 2547–2553. [DOI] [PubMed] [Google Scholar]
- [35].Bouchard LS, Kovtunov KV, Burt SR, Anwar MS, Koptyug IV, Sagdeev RZ, Pines A, Angew. Chem. Int. Ed 2007, 46, 4064–4068. [DOI] [PubMed] [Google Scholar]
- [36].DeVience SJ, Walsworth RL, Rosen MS, Phys. Rev. Lett 2013, 111, 173002-173001-173005. [DOI] [PubMed] [Google Scholar]
- [37].Kovtunov KV, Truong ML, Barskiy DA, Koptyug IV, Coffey AM, Waddell KW, Chekmenev EY, Chem. Eur. J 2014, 20, 14629–14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ariyasingha NM, Salnikov OG, Kovtunov KV, Kovtunova LM, Bukhtiyarov VI, Goodson BM, Rosen MS, Koptyug IV, Gelovani JG, Chekmenev EY, J. Phys. Chem. C 2019, 18, 11734–11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kovtunov KV, Truong ML, Barskiy DA, Salnikov OG, Bukhtiyarov VI, Coffey AM, Waddell KW, Koptyug IV, Chekmenev EY, J. Phys. Chem. C 2014, 118, 28234–28243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhivonitko V, Kovtunov K, Chapovsky P, Koptyug I, Angew. Chem. Int. Ed 2013, 52, 13251–13255. [DOI] [PubMed] [Google Scholar]
- [41].Salnikov OG, Kovtunov KV, Nikolaou P, Kovtunova LM, Bukhtiyarov VI, Koptyug IV, Chekmenev EY, ChemPhysChem 2018, 19, 2621–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Salnikov OG, Nikolaou P, Ariyasingha NM, Kovtunov KV, Koptyug IV, Chekmenev Anal EY. Chem. 2019, 91, 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Adams RW, Aguilar JA, Atkinson KD, Cowley MJ, Elliott PIP, Duckett SB, Green GGR, Khazal IG, Lopez-Serrano J, Williamson DC, Science 2009, 323, 1708–1711. [DOI] [PubMed] [Google Scholar]
- [44].Green RA, Adams RW, Duckett SB, Mewis RE, Williamson DC, Green GGR, Prog. Nucl. Mag. Res. Spectrosc 2012, 67, 1–48. [DOI] [PubMed] [Google Scholar]
- [45].Pravdivtsev AN, Yurkovskaya AV, Vieth H-M, Ivanov KL, Kaptein R, ChemPhysChem 2013, 14, 3327–3331. [DOI] [PubMed] [Google Scholar]
- [46].Barskiy DA, Pravdivtsev AN, Ivanov KL, Kovtunov KV, Koptyug IV, Phys. Chem. Chem. Phys 2016, 18, 89–93. [DOI] [PubMed] [Google Scholar]
- [47].Ivanov KL, Pravdivtsev AN, Yurkovskaya AV, Vieth H-M, Kaptein R, Prog. Nucl. Mag. Res. Spectrosc 2014, 81, 1–36. [DOI] [PubMed] [Google Scholar]
- [48].Knecht S, Ivanov KL, J. Chem. Phys 2019, 150, 124106. [DOI] [PubMed] [Google Scholar]
- [49].Kiryutin AS, Yurkovskaya AV, Zimmermann H, Vieth H-M, Ivanov KL, Magn. Reson. Chem 2018, 56, 651–662. [DOI] [PubMed] [Google Scholar]
- [50].Eshuis N, Aspers RLEG, van Weerdenburg BJA, Feiters MC, Rutjes FPJT, Wijmenga SS, Tessari M, J. Magn. Reson 2016, 265, 59–66. [DOI] [PubMed] [Google Scholar]
- [51].Ducker EB, Kuhn LT, Munnemann K, Griesinger C, J. Magn. Reson 2012, 214, 159–165. [DOI] [PubMed] [Google Scholar]
- [52].Cowley MJ, Adams RW, Atkinson KD, Cockett MCR, Duckett SB, Green GGR, Lohman JAB, Kerssebaum R, Kilgour D, Mewis RE, J. Am. Chem. Soc 2011, 133, 6134–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Theis T, Truong ML, Coffey AM, Shchepin RV, Waddell KW, Shi F, Goodson BM, Warren WS, Chekmenev EY, J. Am. Chem. Soc 2015, 137, 1404–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Atkinson KD, Cowley MJ, Duckett SB, Elliott PIP, Green GGR, López-Serrano J, Khazal IG, Whitwood AC, Inorg. Chem 2009, 48, 663–670. [DOI] [PubMed] [Google Scholar]
- [55].Theis T, Truong M, Coffey AM, Chekmenev EY, Warren WS, J. Magn. Reson 2014, 248, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Svyatova A, Skovpin IV, Chukanov NV, Kovtunov KV, Chekmenev EY, Pravdivtsev AN, Hoevener J-B, Koptyug IV, Chem. Eur. J 2019, 25, 8465–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lindale JR, Eriksson SL, Tanner CPN, Zhou Z, Colell JFP, Zhang G, Bae J, Chekmenev EY, Theis T, Warren WS, Nat. Commun 2019, 10, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Theis T, Ariyasingha NM, Shchepin RV, Lindale JR, Warren WS, Chekmenev EY, J. Phys. Chem. Lett 2018, 9, 6136–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tanner CPN, Lindale JR, Eriksson SL, Zhou Z, Colell JFP, Theis T, Warren WS, J. Chem. Phys 2019, 151, 044201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pravdivtsev AN, Skovpin IV, Svyatova AI, Chukanov NV, Kovtunova LM, Bukhtiyarov VI, Chekmenev EY, Kovtunov KV, Koptyug IV, Hövener J-B, J. Phys. Chem. A 2018, 122, 9107–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rayner PJ, Norcott P, Appleby KM, Iali W, John RO, Hart SJ, Whitwood AC, Duckett SB, Nat. Commun 2018, 9, 4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shchepin RV, Birchall JR, Chukanov NV, Kovtunov KV, Koptyug IV, Theis T, Warren WS, Gelovani JG, Goodson BM, Shokouhi S, Rosen MS, Yen Y-F, Pham W, Chekmenev EY, Chem. Eur. J 2019, 25, 8829–8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhivonitko VV, Skovpin IV, Koptyug IV, Chem. Comm 2015, 51, 2506–2509. [DOI] [PubMed] [Google Scholar]
- [64].Burns MJ, Rayner PJ, Green GGR, Highton LAR, Mewis RE, Duckett SB, J. Phys. Chem. B 2015, 119, 5020–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shchepin RV, Barskiy DA, Coffey AM, Goodson BM, Chekmenev EY, ChemistrySelect 2016, 1, 2552–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Carravetta M, Levitt MH, J. Am. Chem. Soc 2004, 126, 6228–6229. [DOI] [PubMed] [Google Scholar]
- [67].Dumez J-N, Mol. Phys 2019, 1–11. [Google Scholar]
- [68].Olaru AM, Roy SS, Lloyd LS, Coombes S, Green GGR, Duckett SB, Chem. Comm 2016, 52, 7842–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Roy SS, Rayner P, Norcott P, Green GGR, Duckett SB, Phys. Chem. Chem. Phys 2016, 18, 24905–24911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Roy SS, Norcott P, Rayner PJ, Green GGR, Duckett SB, Angew. Chem. Int. Ed 2016, 55, 15642–15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shen K, Logan AWJ, Colell JFP, Bae J, Ortiz GX Jr., Theis T, Warren WS, Malcolmson SJ, Wang Q, Angew. Chem. Int. Ed 2017, 56, 12112–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Theis T, Ortiz GX , Logan AWJ, Claytor KE, Feng Y, Huhn WP, Blum V, Malcolmson SJ, Chekmenev EY, Wang Q, Warren WS, Sci. Adv 2016, 2, e1501438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Iali W, Rayner PJ, Duckett SB, Sci. Adv 2018, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Iali W, Rayner PJ, Alshehri A, Holmes AJ, Ruddlesden AJ, Duckett SB, Chem. Sci 2018, 9, 3677–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tickner BJ, Iali W, Roy SS, Whitwood AC, Duckett SB, ChemPhysChem 2019, 20, 241–245. [DOI] [PubMed] [Google Scholar]
- [76].Iali W, Roy SS, Tickner BJ, Ahwal F, Kennerley AJ, Duckett SB, Angew. Chem. Int. Ed 2019, 58, 10271–10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bae J, Zhou Z, Theis T, Warren WS, Wang Q, Sci. Adv 2018, 4, eaar2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Procacci B, Roy SS, Norcott P, Turner N, Duckett SB, J. Am. Chem. Soc 2018, 140, 16855–16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Nonaka H, Hirano M, Imakura Y, Takakusagi Y, Ichikawa K, Sando S, Sci. Rep 2017, 7, 40104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kurhanewicz J, Vigneron DB, Ardenkjaer-Larsen JH, Bankson JA, Brindle K, Cunningham CH, Gallagher FA, Keshari KR, Kjaer A, Laustsen C, Mankoff DA, Merritt ME, Nelson SJ, Pauly JM, Lee P, Ronen S, Tyler DJ, Rajan SS, Spielman DM, Wald L, Zhang X, Malloy CR, Rizi R, Neoplasia 2019, 21, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Surface JA, Washington University in St. Louis; 2013. [Google Scholar]
- [82].Kuethe DO, Caprihan A, Gach HM, Lowe IJ, Fukushima E, J. Appl. Physiol 2000, 88, 2279–2286. [DOI] [PubMed] [Google Scholar]
- [83].Kuethe DO, Caprihan A, Fukushima E, Waggoner RA, Magn. Reson. Med 1998, 39, 85–88. [DOI] [PubMed] [Google Scholar]
- [84].Barskiy DA, Kovtunov KV, Gerasimov EY, Phipps MA, Salnikov OG, Coffey AM, Kovtunova LM, Prosvirin IP, Bukhtiyarov VI, Koptyug IV, Chekmenev EY, J. Phys. Chem. C 2017, 121, 10038–10046. [Google Scholar]
- [85].Barskiy DA, Salnikov OG, Romanov AS, Feldman MA, Coffey AM, Kovtunov KV, Koptyug IV, Chekmenev EY, J. Magn. Reson 2017, 276, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]