

Abstract
De novo lipogenesis (DNL) plays a role in the development of hepatic steatosis. In humans with lipodystrophy, reduced adipose tissue causes lower plasma leptin, insulin resistance, dyslipidemia, and ectopic triglyceride (TG) accumulation. We hypothesized that recombinant leptin (metreleptin) for 6 months in 11 patients with lipodystrophy would reduce DNL by decreasing insulin resistance and glycemia, thus reducing circulating TG and hepatic TG. The percentage of TG in TG-rich lipoprotein particle (TRLP-TG) derived from DNL (%DNL) was measured by deuterium incorporation from body water into palmitate. At baseline, DNL was elevated, similar to levels previously shown in obesity-associated nonalcoholic fatty liver disease (NAFLD). After metreleptin, DNL decreased into the normal range. Similarly, absolute DNL (TRLP-TG × %DNL) decreased by 88% to near-normal levels. Metreleptin improved peripheral insulin sensitivity (hyperinsulinemic-euglycemic clamp) and lowered hemoglobin A1c and hepatic TG. Both before and after metreleptin, DNL positively correlated with insulin resistance, insulin doses, and hepatic TG, supporting the hypothesis that hyperinsulinemia stimulates DNL and that elevated DNL is integral to the pathogenesis of lipodystrophy-associated NAFLD. These data suggest that leptin-mediated improvement in insulin sensitivity increases clearance of blood glucose by peripheral tissues, reduces hepatic carbohydrate flux, and lowers insulinemia, resulting in DNL reductions and improvements in hepatic steatosis and dyslipidemia.
Keywords: Metabolism, Therapeutics
Keywords: Diabetes, Insulin, Leptin
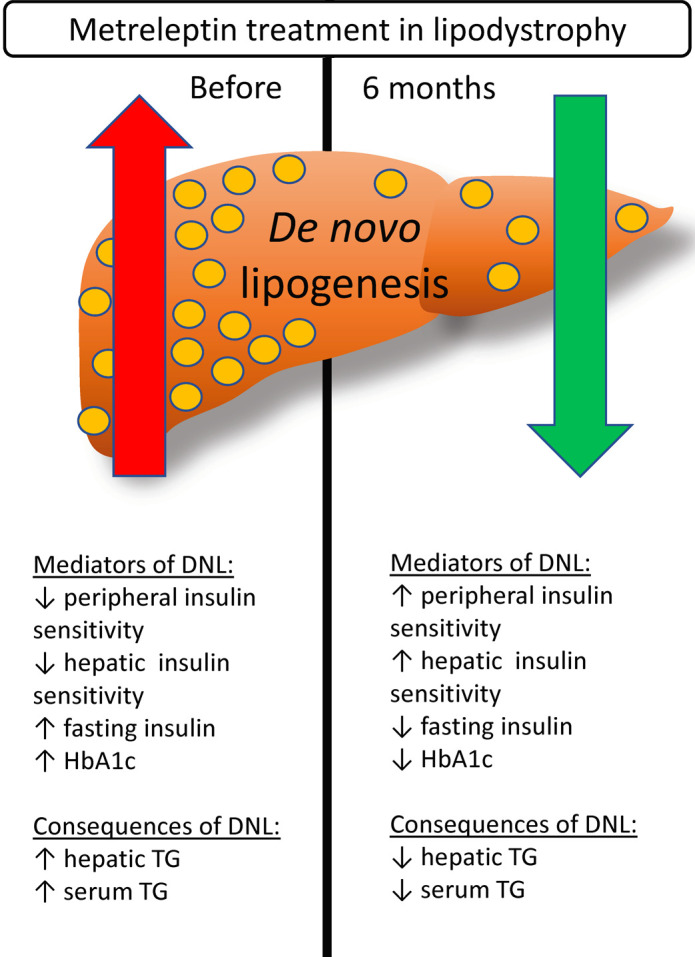
De novo lipogenesis is elevated in lipodystrophy-associated NAFLD and is decreased in association with hepatic steatosis after 6 months of metreleptin in patients with lipodystrophy.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the United States (1), and NAFLD progressing to nonalcoholic steatohepatitis is estimated to overtake hepatitis C as the primary cause of liver transplantation in the United States (2). One source of hepatic triglyceride (TG) is de novo lipogenesis (DNL), the synthesis of fatty acids from nonlipid, primarily carbohydrate, precursors (3). Leptin replacement in leptin-deficient rodents reduces hepatic steatosis by decreasing DNL (4); by contrast, rodent studies suggest that high leptin levels in obesity might contribute to hepatic fibrosis (5, 6). In humans, NAFLD is observed in both the hyperleptinemic state of obesity and the leptin-deficient state of lipodystrophy (7, 8). Therefore, the role of leptin in mediating DNL and NAFLD is unclear.
Lipodystrophy syndromes are characterized by adipose tissue deficiency with metabolic manifestations similar to obesity-associated metabolic syndrome (8). Lipodystrophy syndromes are associated with low circulating leptin levels due to low adipose mass, and thus these syndromes serve as a model to understand effects of leptin deficiency and replacement on metabolic disease. Leptin treatment with recombinant human methionyl leptin (metreleptin) in lipodystrophic patients improves hepatic steatosis and hypertriglyceridemia (9), though the exact mechanisms by which metreleptin mediates these responses have yet to be elucidated. In an earlier report of 3 patients with partial lipodystrophy, DNL was elevated, suggesting that increased DNL may play a role in lipodystrophy-associated NAFLD (10). We hypothesized that metreleptin treatment in patients with lipodystrophy would decrease DNL by lowering hepatic insulin exposure and carbohydrate flux, and that reductions in DNL would be associated with reductions in circulating TG and hepatic TG. Using mass isotopomer analysis, we assessed the effects of metreleptin on DNL in 11 patients with lipodystrophy, by labeling of body water with deuterium and measuring its incorporation into TG-rich lipoproteins (TRLPs) before and after 6 months of metreleptin administration. Potential mediators of changes in DNL were investigated including measures of glucose disposal (hyperinsulinemic-euglycemic clamp) or insulin exposure (exogenous insulin use) and glycemia. Consequences of changes in DNL were investigated including serum TG and hepatic TG content by magnetic resonance spectroscopy (MRS).
Results
Baseline characteristics and metreleptin treatment.
Eleven patients (3 men, 8 women), 4 with congenital generalized lipodystrophy and 7 with familial partial lipodystrophy, aged 34 ± 17 years, were treated with metreleptin for 7.0 ± 0.8 months at a dose of 8.1 ± 2.7 mg/d. Metreleptin increased serum leptin concentrations from a baseline of 9.5 ± 11.0 to 155.0 ± 71.5 ng/dL (P = 0.002; Table 1).
Table 1. Secondary outcomes before and after 6 months of metreleptin administration.

Metreleptin decreased fasting DNL.
After 6 months of metreleptin treatment, the TG content of TRLP (TRLP-TG) decreased by 38% ± 40%, from 160 to 98 mg/dL (P = 0.02; Figure 1A). The percentage of TRLP-TG that was derived from DNL (%DNL) decreased from a baseline of 20.9% [18.0, 29.7] to 7.3% [5.8, 11.6] (P < 0.001; Figure 1B). Absolute DNL also decreased by 88 ± 7%, from 54.2 ± 32.1 mg/dL to 8.6 ± 6.5 mg/dL (P = 0.003; Figure 1C).
Figure 1. Effects of metreleptin in patients with lipodystrophy.

(A) Triglyceride (TG) in TG-rich lipoproteins (TRLP-TG) (n = 7). (B) Fraction of TG in TRLP derived from de novo lipogenesis (DNL) (%DNL) (n = 10). (C) Absolute DNL as the product of TRLP-TG and %DNL (n = 7). (D) Serum TG (n = 11). (E) Hepatic TG (n = 10). (F) Plasma apolipoprotein B48 (n = 11). Correlation between DNL and peripheral insulin sensitivity before (G) and after (H) metreleptin (n = 9). Gray shaded areas represent normal ranges for healthy individuals. Comparisons were made using paired, 2-tailed Student’s t test or Wilcoxon signed-rank test for normally and nonnormally distributed data, respectively. Univariate analysis was performed using Pearson’s and Spearman’s correlations for normally and nonnormally distributed data, respectively.
Metreleptin improved insulin sensitivity.
As previously reported in an overlapping cohort of subjects (11), metreleptin treatment for 6 months improved multiple measures of insulin sensitivity (Table 1). Peripheral insulin sensitivity, assessed as glucose disposal during a hyperinsulinemic-euglycemic clamp, increased by 101% ± 128% (P = 0.034). Similarly, hepatic insulin sensitivity, assessed as suppression of hepatic glucose production during the clamp, increased by 48% ± 49% (P = 0.012). Fasting insulin and C-peptide decreased by 29% ± 40% (P = 0.049) and 37% ± 28% (P = 0.006), respectively. Insulin total daily dose among insulin users decreased nonsignificantly by 36% ± 52% (P = 0.15). One subject with generalized lipodystrophy was able to completely discontinue insulin treatment after 6 months of metreleptin.
Metreleptin reduced carbon sources for DNL (glucose and branched-chain amino acids).
As previously reported in an overlapping cohort of subjects (11), metreleptin treatment for 6 months lowered hemoglobin A1c (HbA1c) by 15% ± 21% (absolute reduction 1.5%, P = 0.037) and led to nonsignificant reductions in fasting plasma glucose (P = 0.071). Branched-chain amino acids (BCAAs), measured using nuclear magnetic resonance (NMR), decreased by 21% ± 18% after metreleptin treatment (P = 0.005; Table 1).
Metreleptin improved serum lipids and hepatic steatosis.
As previously reported in an overlapping cohort of subjects (11), metreleptin treatment for 6 months improved hepatic steatosis and dyslipidemia (Table 1). Metreleptin treatment decreased total and LDL cholesterol by 18% ± 23% (P = 0.032) and 23% ± 15% (P = 0.028), respectively. Serum TG trended down by 23% ± 58% (P = 0.061; Figure 1D) and hepatic TG decreased by 32% ± 51% after metreleptin (P = 0.016; Figure 1E).
Effects of metreleptin on other potential mediators of serum TG and hepatic TG.
Plasma β-hydroxybutyrate, a marker of hepatic fatty acid oxidation, increased after 6 months of metreleptin treatment (P = 0.009; Table 1). Fasting chylomicrons, as measured by plasma apolipoprotein B48, did not decrease after metreleptin treatment (P = 0.55; Figure 1F). As previously reported in an overlapping cohort of subjects (11), there was a trend toward decreased lipolysis after metreleptin treatment, measured by glycerol and palmitate rate of appearance (P = 0.058 and P = 0.049, respectively; Table 1).
Correlations of DNL with metabolic parameters.
Lower endogenous leptin at baseline did not correlate with baseline DNL (P = 0.65) but did correlate with lower DNL after 6 months of metreleptin treatment (r = 0.81, P = 0.02). Both before and after metreleptin treatment, higher peripheral insulin resistance (Figure 1, G and H) and higher insulin doses were significantly associated with higher levels of DNL (Table 2). Hemoglobin A1c (HbA1c) was associated with higher DNL only after metreleptin treatment (P = 0.604 and P = 0.019; before and after metreleptin treatment, respectively; Table 2). There was a trend toward positive association between BCAAs and DNL both before and after metreleptin treatment (P = 0.097 and P = 0.086; Table 2).
Table 2. Correlations with absolute DNL (mg/dL) at baseline and after 6 months of metreleptin administration.

Correlations of serum TG and hepatic TG with DNL and lipolysis.
Both serum TG and hepatic TG correlated positively with DNL before metreleptin treatment (r = 0.79, P = 0.012, and r = 0.70, P = 0.035 respectively; Table 2). However, DNL did not correlate well with serum TG or hepatic TG after metreleptin treatment (P = 0.17 and P = 0.061, respectively; Table 2). There were no correlations either before or after metreleptin treatment between lipolysis and serum TG or hepatic TG (Table 3).
Table 3. Correlations with lipolysis at baseline and after 6 months of metreleptin administration.

Discussion
This study demonstrates elevated fasting DNL in patients with lipodystrophy that decreased after 6 months of metreleptin treatment. In the current study, we found that subjects with lipodystrophy had fasting DNL of 11% to 35%. In lean, healthy individuals under similar conditions of labeling, DNL contributes to approximately 5%–10% of TRLP-TG in the fasting state, increasing to approximately 10% in the fed state (12, 13). However, increased DNL is thought to play a key role in the pathogenesis of obesity-associated NAFLD, because patients with NAFLD have an increased percentage of hepatic TG and circulating TG derived from DNL, to as much as 20%–40% in the fasted state (14–16). Before metreleptin treatment, subjects with lipodystrophy in this study had %DNL comparable to those with obesity-associated NAFLD. Remarkably, after 6 months of metreleptin treatment, %DNL decreased to the normal range of 5%–10%. Furthermore, absolute DNL decreased by 88% to a mean of only approximately 9 mg/dL (range, 0.4–19.4 mg/dL), which is comparable to the mean level of 4 mg/dL reported in lean, healthy individuals (13).
As carbohydrates and insulin are thought to be the main regulators of DNL (16, 17), we hypothesized that a metreleptin-mediated reduction in DNL would be associated with decreases in hepatic carbohydrate flux and insulin exposure. Carbohydrates, particularly fructose, are sufficient to stimulate DNL; however, insulin signaling is thought to be necessary to drive pathological increases in DNL in insulin-resistant states through increased expression of sterol regulatory element–binding protein-1c, a major lipogenic transcription factor (10, 17). Metreleptin treatment improved insulin sensitivity and glycemia control in our cohort of lipodystrophic patients, consistent with previous studies (9, 11). Both before and after metreleptin treatment, higher peripheral insulin resistance and higher insulin doses were associated with higher levels of DNL, supporting the hypothesis that hyperinsulinemia, whether endogenous or exogenous, stimulates DNL. Consistent with this, administration of diazoxide to a patient with partial lipodystrophy suppressed hyperinsulinemia and lowered VLDL-TG (18).
Increased peripheral tissue glucose disposal after metreleptin might also be expected to reduce DNL by reducing carbohydrate availability in the liver. Glycemia, assessed as HbA1c, improved after metreleptin treatment but was associated with DNL only after metreleptin treatment. These data are consistent with the hypothesis that in the hyperinsulinemic state before metreleptin treatment, insulin is the primary driver of pathologically elevated DNL. Only after insulin resistance and hyperinsulinemia decreased with metreleptin could an association between hyperglycemia and DNL be observed, suggesting that carbohydrate availability is rate-limiting for DNL in the more insulin-sensitive state. Our findings are consistent with a recent study showing strong relationships between insulin sensitivity, 24-hour integrated glycemia and insulinemia, and DNL (16).
Changes in insulin and glucose are not the only mechanisms by which metreleptin might lower DNL. Rodent studies have demonstrated that leptin may also lower DNL via CNS signaling by downregulating enzymes involved in de novo fatty acid synthesis (acetyl-CoA carboxylase, fatty acid synthase, and stearoyl-CoA desaturase–1) (19). A recent study showed that this effect was mediated through vagal signaling to the liver (20). Unfortunately, measures of autonomic nervous system activity in the liver were not available in the current study and are unlikely to be feasible in human studies. In addition to carbohydrates, BCAAs can be a carbon source for DNL. BCAAs decreased with metreleptin therapy and trended toward positive association with DNL before and after metreleptin, suggesting that metreleptin may reduce DNL via reductions in both BCAAs and carbohydrate precursors. However, BCAAs can also be thought of as a measure of positive energy balance that is improved after metreleptin treatment, and thus reductions in BCAAs after metreleptin treatment may not be causal for reductions in DNL.
Consistent with prior studies (9, 21), metreleptin decreased serum TG and hepatic TG. We hypothesized that one way by which metreleptin leads to lower serum TG and hepatic TG levels is through reductions in DNL. Before metreleptin treatment, DNL correlated with both serum TG and hepatic TG, supporting a key pathogenic role of DNL in the development of hepatic steatosis and hypertriglyceridemia in not only obesity-associated NAFLD but also lipodystrophy. However, DNL did not correlate well with serum TG or hepatic TG after metreleptin treatment, reflecting its diminished contribution in the insulin-sensitive state.
Circulating TG and hepatic TG can derive from not only DNL but also chylomicrons from dietary fat, reesterification of free fatty acids (FFAs) from adipocyte lipolysis, and spillover of FFA from lipolysis of TRLP (7, 15). We hypothesized that decreases in circulating TG and hepatic TG after metreleptin would be only partly mediated through decreased DNL, with the potential for additional metreleptin-mediated reductions in circulating TG and hepatic TG resulting from decreased lipolysis (9, 11), decreased chylomicrons (22), and/or increased fatty acid oxidation (23–26). In obesity-associated NAFLD, the majority of fatty acids found in circulating TG and hepatic TG are derived from adipocyte lipolysis (15). Consistent with previous studies (9, 11), there was a trend toward decreased lipolysis after metreleptin treatment, suggesting that decreased glycerol and FFA availability to the liver for TG synthesis is a potential mechanism contributing to reductions in circulating TG and hepatic TG after metreleptin treatment. However, there was no correlation between lipolysis and serum TG or hepatic TG, suggesting that lower lipolysis after metreleptin is not the major driver of reductions in serum TG or hepatic TG. Metreleptin is known to suppress appetite and food intake in states of leptin deficiency including lipodystrophy (27–31), and thus reduction in dietary fat intake is a likely mechanism by which metreleptin lowers serum TG and hepatic TG. Consistent with this, a prior publication from our group showed a reduction in chylomicrons, assessed by lipid NMR, in patients with lipodystrophy after metreleptin treatment (22). The lack of reduction in chylomicrons measured by apolipoprotein B48 after metreleptin treatment in the current study is therefore somewhat surprising. This may be due to measurement of chylomicrons in the fasting state, rather than postprandially, differences in methodology, or the large variance between subjects. Alternatively, this may suggest that metreleptin has more complex effects on chylomicron uptake or turnover independent of its effects on dietary fat intake (32). Prior rodent studies have shown that leptin upregulates hepatic transcription factors involved in fatty acid oxidation (PPARγ coactivator 1α, PPARα, carnitine palmitoyltransferase 1A, and CD36) (23–26). Consistent with this, we observed an increase in plasma β-hydroxybutyrate after metreleptin treatment, suggesting increased hepatic FFA utilization.
Prior studies have shown that metreleptin has greater efficacy to improve metabolic disease in lipodystrophic patients with more severe leptin deficiency (21). Consistent with this, lower endogenous leptin at baseline correlated with lower DNL at 6 months, suggesting greater normalization of DNL in patients who were more leptin deficient. However, metreleptin decreased absolute DNL in all patients, with endogenous leptin ranging from 0.5 to 35.7 ng/mL. Importantly, the metreleptin doses used in this study were pharmacologic, rather than hormone replacement, resulting in supraphysiologic plasma leptin concentrations. This suggests that metreleptin at pharmacologic doses might be effective in reducing hepatic steatosis by lowering DNL even in nonlipodystrophic-, nonleptin-deficient populations with NAFLD. However, the pathophysiology of obesity and lipodystrophy differ not only by endogenous leptin levels but also in other ways such as adipose tissue storage capacity. Therefore, although metreleptin lowered DNL in subjects with lipodystrophy over a wide range of endogenous leptin levels, these findings do not necessarily predict equivalent lowering of DNL in individuals with similar leptin levels without lipodystrophy. In fact, metreleptin has been shown to have only modest effects in its primary action to suppress appetite and cause weight loss in obese subjects with high leptin levels (33). Thus, additional studies are needed to test its effects on DNL and NAFLD in this population. Furthermore, some rodent studies suggest a profibrogenic effect of leptin, suggesting that leptin might be causal for progression of NAFLD to NASH in the context of obesity (5, 6). However, metreleptin treatment has not been shown to increase hepatic fibrosis in humans with lipodystrophy (34). Therefore, the profibrogenic effect in rodents might be due to model-specific pathology.
Limitations.
DNL may be underestimated in this study due to dilutional effects of unlabeled TG from chylomicron remnants; however, apolipoprotein B48 did not change after metreleptin treatment, suggesting that the observed decrease in DNL was primarily due to reductions in VLDL. DNL may also be underestimated due to the relatively short duration of deuterium labeling of body water (11 hours) (16) — although the overnight labeling method has been shown to distinguish populations with markedly different levels of DNL (35). Finally, we speculate that metreleptin-mediated improvements in insulin sensitivity and glycemia were causal for decreased DNL, which in turn was causal for decreased circulating TG and hepatic TG levels. However, this study can only demonstrate association, not causality, between these variables, and there is evidence to support that lower hepatic TG may be causal for lower insulin resistance (36). A demonstration that insulin sensitivity and glycemia improved before changes in DNL would help support the causal role of insulin and glucose in mediating metreleptin-induced reductions in DNL. Although our prior publication showed that insulin sensitivity and glucose improved as early as 2 weeks after metreleptin initiation in an overlapping cohort of subjects (11), DNL data were unfortunately not available at that time point.
Conclusions.
In conclusion, 6 months of metreleptin treatment in very insulin-resistant humans with lipodystrophy led to near normalization of DNL. Improvements in DNL were associated with reductions in glycemia and improved peripheral and hepatic insulin sensitivity, supporting a strong link between the effects of metreleptin to lower insulinemia and increase clearance of blood glucose by peripheral tissues and reduce hepatic carbohydrate flux, and resultant reductions in DNL. This led to lowered hepatic steatosis and dyslipidemia and suggests that treatments targeting multiorgan insulin resistance may improve NAFLD. Importantly, metreleptin-induced improvements in DNL and metabolic disease were observed across all levels of endogenous leptinemia, suggesting that metreleptin may be effective in the broader population with obesity-associated NAFLD who are not leptin-deficient.
Methods
Study design.
Leptin-naive patients with lipodystrophy participated in an open-label study of metreleptin (donated by Aegerion Pharmaceuticals) at the NIH. This analysis includes a subset (9 of 15) of patients described in the primary results of this study (11) plus 2 additional patients who enrolled after the previous publication.
Details of the study design have been published (11). Briefly, patients were admitted and studied for 5 days before metreleptin initiation (5 mg s.c. every 12 hours). Metreleptin was continued for 14 days inpatient, and then patients were discharged to continue metreleptin treatment as outpatients for 6 months. At discharge, the metreleptin dose was decreased in patients with generalized lipodystrophy to prevent excessive weight loss. In all patients, insulin and sulfonylurea doses were reduced as needed to avoid hypoglycemia due to improved insulin sensitivity after metreleptin initiation. No increases in medications for diabetes or dyslipidemia were permitted.
Study procedures.
Study procedures were performed after an 8- to 12-hour fast at baseline (before metreleptin administration) and after approximately 6 months of metreleptin administration. Fasting DNL was measured after oral administration of deuterated water (2H2O) in 4 doses between 2100 and 0300 hours to reach a concentration of 0.3% 2H2O of total body water, measured by IRMS as described previously (37). To isolate the TRLP fraction (density <1.006 g/mL), serum underwent ultracentrifugation for 20 hours with a Beckman Ti rotor at 189,434 g at 4°C and the upper approximately 1 mL was collected by tube slicing. Lipoprotein TGs were separated by thin layer chromatography and fatty acids were transesterified to be analyzed by gas chromatography/mass spectrometry. The fractional contribution of DNL-derived TG palmitate in TRLP was calculated using mass isotopomer analysis as previously described (15). By convention this fraction was multiplied by the TG concentration to estimate absolute DNL (35).
Concentrations of glucose, insulin, and C-peptide were measured every 10 minutes for 30 minutes before the hyperinsulinemic-euglycemic clamp, and the mean of the 4 measurements was reported. Standard methods of NIH Clinical Center laboratory were used to measure glucose, total cholesterol, TG, and HDL-C (Roche Cobas 6000 analyzer); insulin and C-peptide (electrochemiluminescence immunoassay on Roche Cobas e601 analyzer); FFA (colorimetric assay on Roche Cobas C501 analyzer); and HbA1c (high-performance liquid chromatography). LDL-C was calculated using the Friedewald equation if TG was less than 400 mg/dL. Plasma leptin was measured by ELISA (MilliporeSigma, EZHL-80SK). The intra-assay and interassay coefficients of variation were 3.9% and 4.8%, respectively. Plasma BCAA concentration was measured via NMR spectroscopy using the 400-MHz proton Vantera Clinical Analyzer with LP4 deconvolution algorithm as previously described (22). As previously reported, body composition was measured by dual-energy x-ray absorptiometry, and hepatic TG was measured by MRS (11). Apolipoprotein B48 was measured by ELISA (FUJIFILM kit, 637-10641). Both intra-assay and interassay coefficients of variation were 10%.
Glucose, glycerol, and palmitate turnover were measured using the isotope tracer dilution method with [6,6-2H2] glucose, 2H5-glycerol, and [U-13C16] palmitate (Cambridge Isotope Laboratories) as previously reported (11). Hyperinsulinemic-euglycemic clamp studies were performed to measure hepatic and total body insulin sensitivity as previously reported (11). Briefly, patients received a primed insulin infusion for 8 minutes at 240 mU/m2/minute followed by a continuous infusion for approximately 3 hours at 120 mU/m2/minute. The high dose of insulin was chosen to stimulate peripheral glucose uptake with incomplete suppression of hepatic glucose production in this population with severe insulin resistance. Insulin sensitivity (M) was assessed as the mean glucose infusion rate during the final 30 minutes of the clamp, normalized to fat-free mass (mg/kgFFM/minute). Hepatic insulin sensitivity was determined by percentage suppression of endogenous glucose production using [6,6-2H2] glucose.
Statistics.
Outcomes are reported as mean ± SD or median (25th, 75th percentile) based on data distribution. Nonnormally distributed data were log-transformed before analysis. Paired 2-tailed t tests or Wilcoxon signed-rank tests were used to compare outcomes before versus after metreleptin for normally and nonnormally distributed deltas, respectively. Pearson’s or Spearman’s correlations were performed to test associations between DNL and endogenous leptin levels, potential mediators of DNL, and consequences of changes in DNL. Correlations were conducted at baseline and 6-month follow-up. A P value of less than 0.05 represented statistical significance. All P values are 2 sided. Analyses were conducted using GraphPad Prism Software, version 8.1.
Study approval.
This study (NCT01778556) was approved by the institutional review board of the National Institute of Diabetes and Digestive and Kidney Diseases. Patients or legal guardian(s) provided written informed consent before participation; minors provided written assent.
Author contributions
APB analyzed data and wrote the manuscript. EJP conducted experiments, analyzed data, and contributed to manuscript writing. RS, MMSA, SC, EC, MS, AMG, RM, PJW, MW, RO, KZAE, and STC conducted experiments and critically reviewed the manuscript. RJB designed the study, conducted experiments, acquired data, analyzed data, and wrote the manuscript.
Acknowledgments
This work was supported by the intramural research programs of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart Lung and Blood Institute, and the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, Genentech, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors. We acknowledge the services of the NIDDK Clinical Core Laboratory and the companies who donated metreleptin for these studies since 2000, including Amgen, Amylin Pharmaceuticals, Bristol Myers Squibb, AstraZeneca, and Aegerion Pharmaceuticals.
Version 1. 06/23/2020
In-Press Preview
Version 2. 07/23/2020
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: JCI Insight. 2020;5(14):e137180.https://doi.org/10.1172/jci.insight.137180.
Contributor Information
Annah P. Baykal, Email: ANNAH.BAYKAL@NIH.GOV.
Elizabeth J. Parks, Email: parksej@missouri.edu.
Robert Shamburek, Email: bobs@mail.nih.gov.
Majid M. Syed-Abdul, Email: ms9rf@mail.missouri.edu.
Elaine Cochran, Email: elainec@intra.niddk.nih.gov.
Megan Startzell, Email: megan.startzell@nih.gov.
Ahmed M. Gharib, Email: agharib@mail.nih.gov.
Ronald Ouwerkerk, Email: ouwerkerkr@niddk.nih.gov.
Khaled Z. Abd-Elmoniem, Email: abdelmoniemkz@mail.nih.gov.
Peter J. Walter, Email: walterpj@niddk.nih.gov.
Mary Walter, Email: waltermf@niddk.nih.gov.
Ranganath Muniyappa, Email: muniyapr@mail.nih.gov.
Stephanie T. Chung, Email: chungst@niddk.nih.gov.
Rebecca J. Brown, Email: brownrebecca@mail.nih.gov.
References
- 1.Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23(47):8263–8276. doi: 10.3748/wjg.v23.i47.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parikh ND, et al. Projected increase in obesity and non-alcoholic-steatohepatitis-related liver transplantation waitlist additions in the United States. Hepatology. 2019;70(2):487–495. doi: 10.1002/hep.29473. [DOI] [PubMed] [Google Scholar]
- 3.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asilmaz E, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113(3):414–424. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imajo K, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16(1):44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Ikejima K, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122(5):1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 7.Marchesini G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50(8):1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 8.Garg A. Lipodystrophies. Am J Med. 2000;108(2):143–152. doi: 10.1016/S0002-9343(99)00414-3. [DOI] [PubMed] [Google Scholar]
- 9.Petersen KF, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109(10):1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119(2):315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown RJ, et al. Metreleptin-mediated improvements in insulin sensitivity are independent of food intake in humans with lipodystrophy. J Clin Invest. 2018;128(8):3504–3516. doi: 10.1172/JCI95476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timlin MT, Parks EJ. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am J Clin Nutr. 2005;81(1):35–42. doi: 10.1093/ajcn/81.1.35. [DOI] [PubMed] [Google Scholar]
- 13.Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr. 2008;138(6):1039–1046. doi: 10.1093/jn/138.6.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sae-Tan S, Rogers CJ, Lambert JD. Voluntary exercise and green tea enhance the expression of genes related to energy utilization and attenuate metabolic syndrome in high fat fed mice. Mol Nutr Food Res. 2014;58(5):1156–1159. doi: 10.1002/mnfr.201300621. [DOI] [PubMed] [Google Scholar]
- 15.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith GI, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130(3):1453–1460. doi: 10.1172/JCI134165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas JT, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15(6):873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chait A, Janus E, Mason AS, Lewis B. Lipodystrophy with hyperlipidaemia: the role of insulin in very low density lipoprotein over-synthesis. Clin Endocrinol (Oxf) 1979;10(2):173–178. doi: 10.1111/j.1365-2265.1979.tb01363.x. [DOI] [PubMed] [Google Scholar]
- 19.Gallardo N, et al. Tissue-specific effects of central leptin on the expression of genes involved in lipid metabolism in liver and white adipose tissue. Endocrinology. 2007;148(12):5604–5610. doi: 10.1210/en.2007-0933. [DOI] [PubMed] [Google Scholar]
- 20.Hackl MT, et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat Commun. 2019;10(1):2717. doi: 10.1038/s41467-019-10684-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. 2015;100(5):1802–1810. doi: 10.1210/jc.2014-4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinzer AB, Shamburek RD, Lightbourne M, Muniyappa R, Brown RJ. Advanced lipoprotein analysis shows atherogenic lipid profile that improves after metreleptin in patients with lipodystrophy. J Endocr Soc. 2019;3(8):1503–1517. doi: 10.1210/js.2019-00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minokoshi Y, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415(6869):339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 24.Steinberg GR, Dyck DJ. Development of leptin resistance in rat soleus muscle in response to high-fat diets. Am J Physiol Endocrinol Metab. 2000;279(6):E1374–E1382. doi: 10.1152/ajpendo.2000.279.6.E1374. [DOI] [PubMed] [Google Scholar]
- 25.Shimabukuro M, et al. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci U S A. 1997;94(9):4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muoio DM, Dohm GL, Tapscott EB, Coleman RA. Leptin opposes insulin’s effects on fatty acid partitioning in muscles isolated from obese ob/ob mice. Am J Physiol. 1999;276(5):E913–E921. doi: 10.1152/ajpendo.1999.276.5.E913. [DOI] [PubMed] [Google Scholar]
- 27.Farooqi IS, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110(8):1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54(8):690–697. doi: 10.1590/S0004-27302010000800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown RJ, Gorden P. Leptin therapy in patients with lipodystrophy and syndromic insulin resistance. In: Dagogo-Jack S ed. Leptin: Regulation and Clinical Applications. Springer International Publishing; 2015:225–236. [Google Scholar]
- 30.McDuffie JR, et al. Efficacy of orlistat as an adjunct to behavioral treatment in overweight African American and Caucasian adolescents with obesity-related co-morbid conditions. J Pediatr Endocrinol Metab. 2004;17(3):307–319. doi: 10.1515/jpem.2004.17.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oral EA, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 32.Donahoo WT, Stob NR, Ammon S, Levin N, Eckel RH. Leptin increases skeletal muscle lipoprotein lipase and postprandial lipid metabolism in mice. Metab Clin Exp. 2011;60(3):438–443. doi: 10.1016/j.metabol.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 33.Heymsfield SB, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 34.Safar Zadeh E, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol. 2013;59(1):131–137. doi: 10.1016/j.jhep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santoro N, Caprio S, Pierpont B, Van Name M, Savoye M, Parks EJ. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J Clin Endocrinol Metab. 2015;100(8):E1125–E1132. doi: 10.1210/jc.2015-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chacko SK, Sunehag AL. Gluconeogenesis continues in premature infants receiving total parenteral nutrition. Arch Dis Child Fetal Neonatal Ed. 2010;95(6):F413–F418. doi: 10.1136/adc.2009.178020. [DOI] [PubMed] [Google Scholar]