

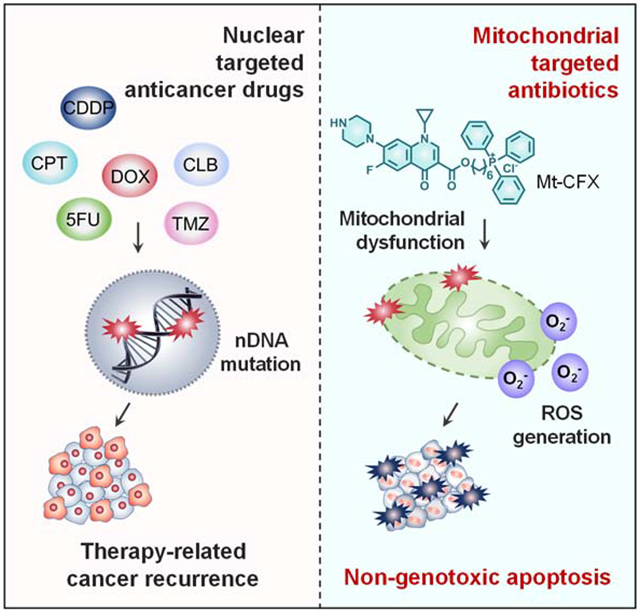
SUMMARY
Tumor recurrence as a result of therapy-induced nuclear DNA lesions is a major issue in cancer treatment. Currently, only a few examples of potentially non-genotoxic drugs have been reported. Mitochondrial re-localization of ciprofloxacin, one of the most commonly prescribed synthetic antibiotics, is reported here as a new approach. Conjugating ciprofloxacin to a triphenyl phosphonium group (giving lead Mt-CFX), is used to enhance the concentration of ciprofloxacin in the mitochondria of cancer cells. The localization of Mt-CFX to the mitochondria induces oxidative damage to proteins, mtDNA, and lipids. A large bias in favor of mtDNA damage over nDNA was seen with Mt-CFX, contrary to classic cancer chemotherapeutics. Mt-CFX was found to reduce cancer growth in a xenograft mouse model and proved to be well tolerated. Mitochondrial relocalization of antibiotics could emerge as a useful approach to generating anticancer leads that promote cell death via the selective induction of mitochondrially-mediated oxidative damage.
Keywords: Non-genotoxic cancer therapy, Mitochondria, Targeted therapeutics, Prodrug, DNA damage, Reactive oxygen species, Ciprofloxacin
Graphical Abstract
eTOC blurb
Therapy-associated nuclear DNA damage is a key driver for secondary malignancies. It represents a major problem in clinical practice that has yet to be overcome. The mitochondria-targeted ciprofloxacin conjugate reported here eliminates therapy-induced nuclear DNA mutagenicity and induces cancer cell death as the result of oxidative damage. These findings could provide a new framework for the development of non-genotoxic therapeutics that overcome the conceptual limitations of the inherently mutagenic chemotherapeutics that define the current standard of care in cancer chemotherapy.
INTRODUCTION
Tumor recurrence remains the greatest challenge in cancer therapy and is responsible for up to 90% of cancer mortality.1 Paradoxically, recurrence is often elicited by chemotherapeutics due to the accumulation of nuclear genomic mutations during the treatment regime.2 Antineoplastic agents relying on direct nuclear DNA damage, such as alkylating agents, are common chemical mutagens and have been implicated in tumor recurrence, e.g., therapy-related acute myeloid leukemia (t-AML).3–6 Additionally, drug-induced genomic alterations are carried over into the next generation, detectable up to two generations in mice.7 Given the steady increase in pediatric malignancy survival rates,8 prevention of these long-term effects might warrant a paradigm shift towards rationally designed non-genotoxic chemotherapeutics.
Mitochondria lie at the nexus of cellular signaling events, including the induction of apoptosis via the caspase cascade. As such, mitochondrial damage-induced programmed cell death represents an appealing pathway for cancer chemotherapy. Agents that target the mitochondria could bypass direct nuclear DNA damage mechanisms, thus preventing the accumulation of nuclear DNA mutations. Mitochondria are widely believed to have a bacterial evolutionary origin and share many similarities with the bacterial genome and biosynthetic machinery.9 While operating via disparate mechanisms in bacteria and eukaryotic cells, common antibiotics such as fluoroquinolones, aminopenicillins, aminoglycosides ,and tetracyclines inhibit the mitochondrial electron transfer chain (ETC).10 The associated reduction in mitochondrial electron transfer efficiency leads to the production of ROS,10 resulting in mitochondrial damage. This damage can trigger apoptosis. We thus thought that by targeting antibiotics to the mitochondria it might be possible to discover new non-genotoxic drugs for cancer chemotherapy. The present study was designed to test this hypothesis.
Efforts to apply antibiotics to cancer chemotherapy have a long history.11,12 For instance, bacteria-derived bactericides, such as doxorubicin and mitomycin C, have found broad clinical applications in chemotherapy; however, their mode of action is thought to involve direct nuclear DNA damage, which can lead to drug-induced genomic alterations.13,14 In contrast, many commonly prescribed synthetic bactericidal drugs were developed with a goal of maintaining low toxicity levels in eukaryotic cells. This has made the standard repurposing of such FDA-approved drugs as potential anticancer agents attractive. However, difficulties in achieving effective sub-cellular localization of such antibiotics has, in part, prevented success. While large doses of synthetic antibiotics have been shown to reduce cancer cell viability, typically non-cancerous cells have proved more susceptible to these antimicrobiocidal drugs than cancer cells.15 Nonetheless, the potential role of antibiotics as chemotherapy adjuvants has been noted.16 Below, we show that redirection of the antibiotic ciprofloxacin (CFX) to the mitochondria via chemical modification results in a new agent with cancer cell selective antiproliferative activity. A reduction in vivo is also seen as inferred from tumor regrowth experiments in murine models. The approach illustrated here could overcome the difficulties encountered in the standard repurposing of antibiotics for cancer treatment.
RESULTS AND DISCUSSION
Mt-CFX induces cell death in a mitochondrial membrane potential (MMP)-dependent way
Ciprofloxacin (CFX) is one of the most commonly prescribed synthetic antibiotics worldwide. It is thus an attractive target for repurposing as a potential anticancer agent. However, we hypothesized that in order to be effective in this latter regard CFX would need to be directed to the mitochondrial. To test this hypothesis, CFX was linked to a triphenyl phosphonium subunit, a group known to promote mitochondria localization.17,18 The resulting conjugate, Mt-CFX (Figure 1A; for synthesis, see the Supplemental Information), was found to reduce cell viability in a dose-dependent manner in MDA-MB-231 cancer cells (Figure 1B). An IC50 value of 31 μM was recorded for Mt-CFX, while CFX or the phosphonium targeting group alone proved several orders of magnitude less effective (Figure 1B). An associated BrdU incorporation assay revealed a statistically significant reduction in cell proliferation in the presence of Mt-CFX as compared to CFX or vehicle only (Figure S1). Relative to MDA-MB-231, a reduced cytotoxicity profile was seen for Mt-CFX in the non-cancerous MCF10A mammary gland epithelial cell line (Figure 1C), thus illustrating cancer selectivity in vitro. The antiproliferative effect of Mt-CFX was found to be on the same order of magnitude across several cancerous cell lines (e.g., MDA-MB-231, SW620, DU145, A549, and PC3). The toxicity of Mt-CFX was at least an order of magnitude lower in the normal human fibroblast cell line (BJ) as compared to the cancer cell lines. Moreover, as noted above, virtually no toxicity was observed in the MCF 10A cell line (Figure 1F).
Figure 1. Mt-CFX induces cell death in a mitochondrial membrane potential (MMP)-dependent manner.
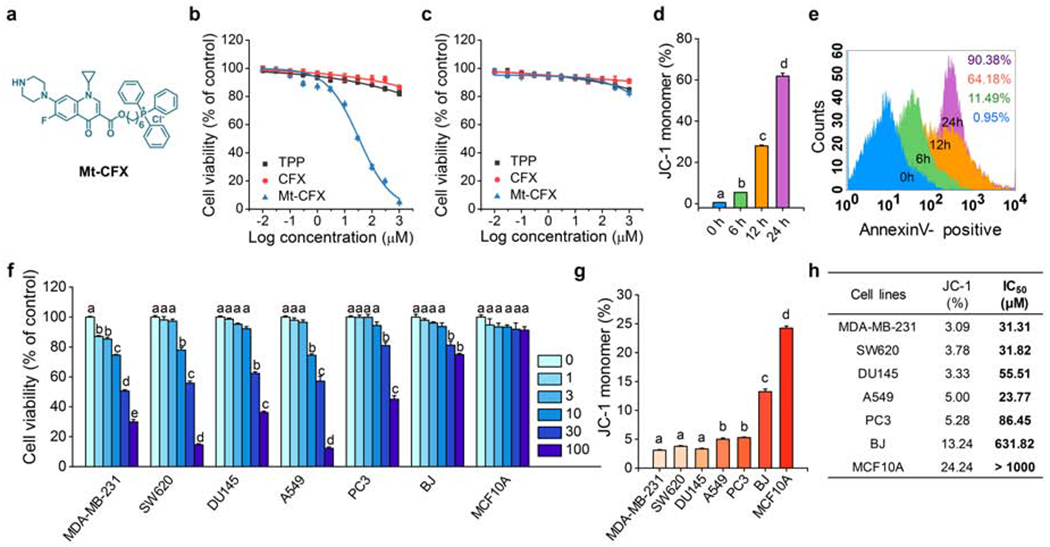
(A) Chemical structure of Mt-CFX.
(B) Concentration-dependent cell-viability of MDA-MB-231 cells incubated with Mt-CFX, CFX, and TPP, as determined by a Cyto-Tox96 Assay (72h).
(C) Concentration-dependent cell-viability of MCF10A cells incubated with Mt-CFX, CFX, and TPP, as determined by a CytoTox96 Assay (72h).
(D) Proportion of JC-1 monomer fluorescence in a flow cytometry assay of MDA-MB-231 pretreated with 30 μM Mt-CFX with incubation times as indicated in the figure.*
(E) Time depended Annexin V-FITC flow cytometry analysis of MDA-MB-231 pretreated with 30 μM Mt-CFX per the incubation times indicated in the figure.
(F) Concentration-dependent cell-viability of different cell lines incubated with Mt-CFX, as indicated in the figure, as determined by a CytoTox96 Assay (72h).*
(G) Different proportions of JC-1 monomer fluorescence in a flow cytometry assay of untreated cells.
(H) Summarized JC-1 monomer fractions from panel G and IC50 values of Mt-CFX-treated cell lines from panel F.*
All cytotoxicity experiments were carried out three times and in triplicate wells. Data are represented as mean ± SEM. Statistical significance was determined using a one-way ANOVA test with post-hoc Bonferroni test.
*Different letters (e.g., a–d) signify data which are statistically different (p < 0.05).
JC-1 monomer/aggregate flow cytometry assays were used in this study to assess the mitochondrial membrane potentials (MMP), with high proportions of monomer fluorescence corresponding to a decreased MMP. The addition of Mt-CFX to MDA-MB-231 cells resulted in a time-dependent MMP depolarization (Figures 1D and S2) with a concurrent increase in Annexin-V binding (Figures 1E and S3). Such findings are consistent with an apoptosis-induced cell death mechanism. We thus sought to investigate the relationship, if any, between cell viability and the mitochondrial membrane potential. A population of each cell line was subjected to a JC-1 assay (Figures 1G and S4). A strong general trend towards lower cell viability with stronger MMP hyperpolarization was found (Figure 1H).
Localization of the Mt-CFX system to the mitochondria as evidenced by studies of a fluorescent probe, Bo-Mt-CFX
To obtain further support for the suggestion that Mt-CFX localizes to the mitochondria, we synthesized a related conjugate incorporating the fluorophore resorufin. This system, termed Bo-Mt-CFX, was designed to allow for an accurate spatiotemporal analysis of the drug action in vitro and in vivo (see the Supplemental Information for synthetic details).19–22 A phenylboronate moiety was used to prepare Bo-Mt-CFX since it is a known trigger that will undergo peroxide-induced cleavage, thus allowing release of both the fluorophore and putative Mt-CFX drug under conditions of ROS over-production (i.e., in cancerous tissues).23 As can be seen in Figure 2A, peroxide-triggered drug release results in the liberation of the CFX and the fluorophore resorufin. This self-immolative release relies on the formation and subsequent nucleophilic attack of various quinone methide moieties which are produced throughout the release mechanism.
Figure 2. Bo-Mt-CFX, a ROS-triggered DDS system incorporating Mt-CFX.
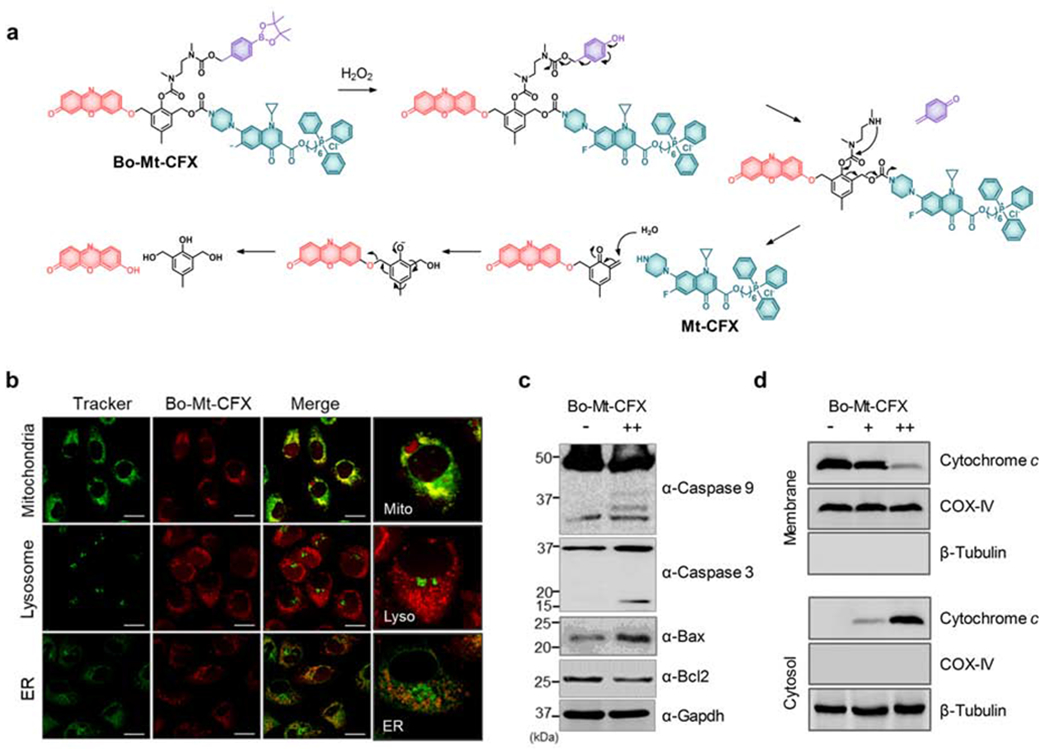
(A) Peroxide-induced self-immolative activation mechanism of Bo-Mt-CFX
(B) Localization of resorufin release from 10 μM Bo-Mt-CFX in MDA-MB-231 cells, compared with MitoTracker Green FM, LysoTracker Green DND-26, and ER-Tracker Green.
(C) Western blotting of apoptosis-related proteins in MDA-MB-231 cells treated with 10 μM Bo-Mt-CFX or a control.
(D) Western blotting of cytochrome c release from mitochondria to the cytosol seen in MDA-MB-231 cells treated with 3 μM or 10 μM Bo-Mt-CFX, as well as a control.
We validated the peroxide-triggered self-immolative release of the resorufin fluorophore from Bo-Mt-CFX in aqueous solution (5 μM) by observing the characteristic fluorescent band centered at 580 nm. We observed a clear time-dependent response with a half-life of approximately 6,000 s in the presence of 50 equiv. of H2O2 (Figure S5A). A concentration-dependent effect was also observed (Figure S5B). Moreover, the identity of the fluorescent species was confirmed to be resorufin via a time-dependent HPLC assay. On this basis, we conclude that under these test conditions the self-immolative reaction is complete within 8 hours (Figure S6). Mt-CFX release was also confirmed by HPLC/MS (Figure S7). The influence of Bo-Mt-CFX on intracellular cellular ROS and its effects on cellular activity (i.e., cell viability across a panel cell lines, glutathione homeostasis, ROS dependence) was further evaluated in vitro (Figures S8–S14), revealing the expected correlation between cancer cell inhibition and cellular oxidative stress.
Confocal microscopy studies confirmed that in MDA-MB-231 cells release of resorufin from Bo-Mt-CFX occurs in the mitochondria (Figure 2B). The Mt-CFX released from Bo-Mt-CFX was found to be largely retained in the mitochondria for at least 8 hours (Figure S15). The addition of Bo-Mt-CFX to MDA-MB-231 cells also resulted in the activation of the caspase pathway (Figures 2C and S16), while engendering a dose-dependent cytoplasmic relocalization of cytochrome c (Figures 2D and S16). An in-depth study of several pro-apoptotic (BAX, BAC, BAD, PUMA, Noxa, and BID) and anti-apoptotic (Bcl-2, Bcl-w, Mcl-1, Bcl-xl and A1) members of the Bcl-2 family was conducted in the presence of Mt-CFX (Figure S17). A lowered or constant protein expression level was found for the anti-apoptotic proteins, while the pro-apoptotic BAX levels were increased. The PUMA and Noxa levels were scarcely elevated, which is consistent with a lack of (nuclear) genotoxicity.24 Finally, a comparison between Mt-HCT116 WT and HCT116 Bax−/− cells revealed the requirement of BAX for apoptosis induction following Mt-CFX treatment (Figure S18), a finding that supports mitochondrial involvement in the cell death process. Taken together, these results lead us to conclude that the activity of Mt-CFX is strongly dependent on the mitochondrial accumulation of the drug, a phenomenon that generally tracks with the MMP.
Mt-CFX induces mitochondrial ROS production with minimal nuclear DNA damage
Several antibacterial drugs have been reported to cause mitochondrial damage as a result of ROS production.10 Therefore, we investigated the influence of Mt-CFX on ROS production in MDA-MB-231 cells. As can be seen in Figure 3A, the intracellular Amplex red fluorescence was elevated in cells treated with Mt-CFX and this indicator. The fluorescence intensity ascribed to Mito-SOX, a mitochondria-selective superoxide (O2• −) fluorescent dye, is likewise enhanced upon treatment with Mt-CFX. Such findings are consistent with an increase in ROS production within the mitochondria (Figures 3B and S19A). The use of the non-selective fluorescein-based oxidative stress indicator, CM-H2DCFDA, provided further support for the contention that ROS are being generated in the presence of Mt-CFX (cf. Figures 3C and S19B). Furthermore, cells pre-incubated with N-acetyl cysteine (NAC) (cf. Figures S20–S21) or other antioxidants (Figure S21) not only decreased the cellular ROS burden but rescued cells from Mt-CFX-induced cytotoxicity.
Figure 3. Mt-CFX induces mitochondrial ROS production with minimal nuclear DNA damage.
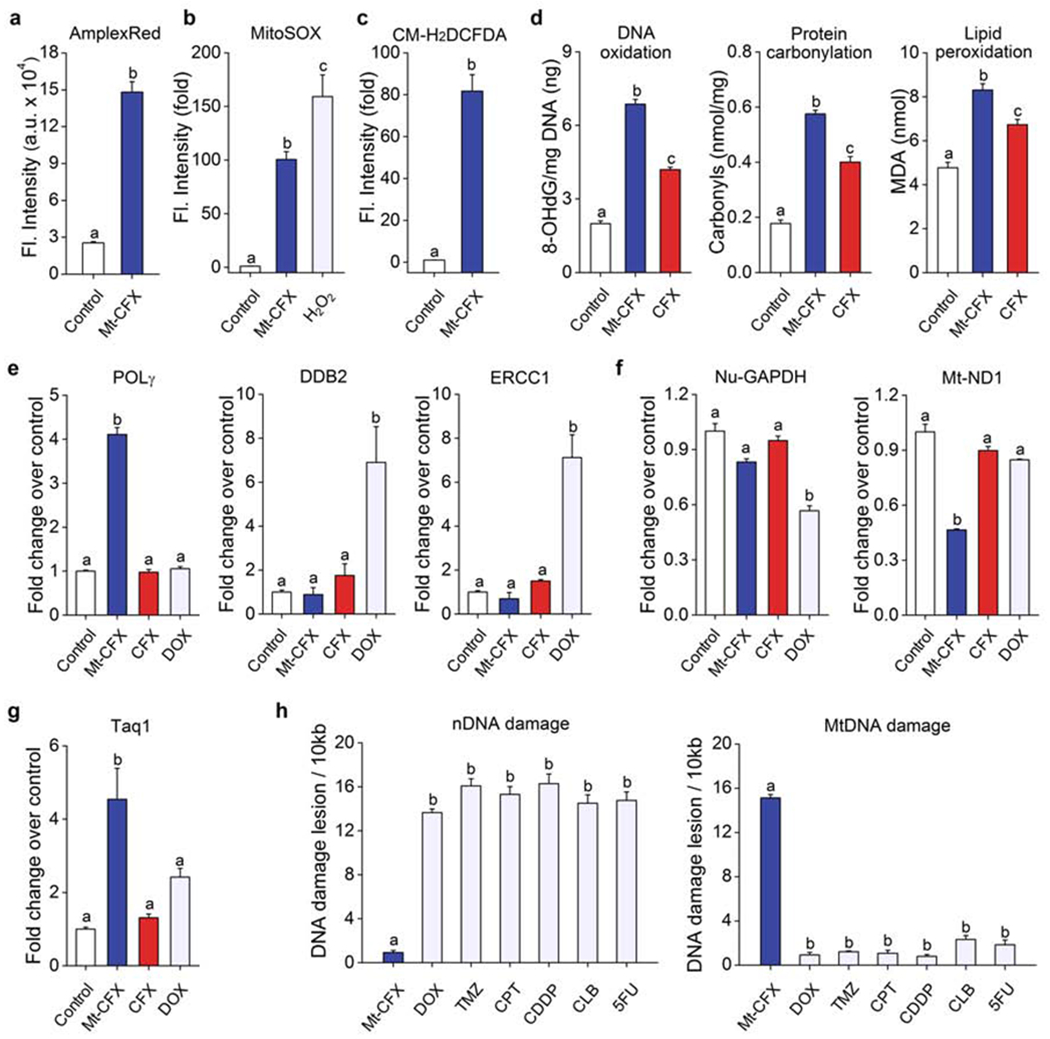
(A) Confocal microscopic fluorescence intensity of MDA-MB-231 cells treated with Mt-CFX (30 μM) or a control co-incubated with 400 μM Amplex Red® for 30 min (n = 6).
(B) Confocal microscopic fluorescence intensity of Mt-CFX (30 μM) or control-treated MDA-MB-231 cells incubated with 5 μM Mito-Sox for 15 min (n = 5).
(C) Confocal microscopic fluorescence intensity of Mt-CFX (30 μM) or control treated MDA-MB-231 cells incubated with 10 μM CM-H2DCFDA for 30 min (n = 15).
(D) ELISA DNA oxidation and protein carbonylation assay results of MDA-MB-231 cells treated with 30 μM Mt-CFX, CFX as well as control samples. Colorimetric malondialdehyde (MDA) assay results, as a measure of lipid peroxidation, of similarly treated MDA-MB-231 cells.
(E) q-PCR analysis of DNA repair genes in MDA-MB-231 cells treated with 30 μM Mt-CFX, CFX, DOX, and control samples.
(F) q-PCR analysis of housekeeping genes in MDA-MB-231 cells treated with 30 μM Mt-CFX, CFX, DOX, and control samples.
(G) Taq1 restriction site mutation assay using MDA-MB-231 cells treated with 30 μM Mt-CFX, CFX, DOX, and control samples.
(H) Long-range q-PCR DNA lesion assay using MDA-MB-231 cells treated with Mt-CFX (30 μM), temozolomide (TMZ) (1 μM), camptothecin (CPT) (250 nM), cis-platin (CDDP) (3 μM), chlorambucil (CLB) (50 μM), and fluorouracil (5-FU) (30 μM), respectively. Concentrations were selected on the basis of their respective IC50 values.
Data are represented as mean ± SEM. (n = 3, unless specified otherwise). Statistical significance was determined using a one-way ANOVA test with post-hoc Bonferroni test. Different letters (e.g., a–d) signify data which are statistically different (p < 0.05).
We next investigated whether damage to cellular components ascribable to ROS was seen upon incubation of MDA-MB-231 cells with Mt-CFX or CFX (Figure 3D and Figure S22). We found elevated 8-hydroxy-2′-deoxyguanosine (8-OHdG) levels, a classic marker for radical-induced oxidative damage to nuclear and mitochondrial DNA. Both protein carbonylation and elevated malondialdehyde (MDA) levels were seen. These are indicators of protein oxidation and lipid peroxidation, respectively. Thus, considered in concert, these results lead us to suggest that Mt-CFX interferes with the mitochondrial ETC process, inducing a significantly enhanced level of oxidative stress. TEM (transmission electron microscopy) revealed noticeable morphology changes, including the loss of cristae, deformation and mitochondrial swelling, when treated with Mt-CFX alone, while NAC pre-treatment also prevented discernable mitochondrial damage (Figure S23). Mitochondrial de-energization was found to mirror these observations, as the cellular ATP content was reduced in the case of Mt-CFX treated cells, but not for those pre-treated with NAC (Figure S24)
The nature and extent of Mt-CFX-mediated DNA was then explored. Oxidative DNA damage induces the expression of base-excision repair proteins in both the nucleus and mitochondria, whereas nucleotide-excision repair (NER) expression is localized to the nucleus.25 We thus employed quantitative PCR (q-PCR) to probe the expression levels of organelle-specific repair enzymes upon Mt-CFX treatment. Doxorubicin (DOX) was also studied since this clinically approved drug is known to induce ROS production.8 POLG, encoding a mitochondria-selective base excision repair protein, was found to be significantly increased following incubation with Mt-CFX only (Figure 3E). DOX, on the other hand, elicited a clear upregulation of NER, as observed for both ERCC1 and DDB2, while Mt-CFX did not cause significantly increased expression levels. Under these conditions treatment with CFX alone did not lead to an upregulation in any of these repair enzymes.
The impact of Mt-CFX on cellular protein metabolism was investigated by monitoring the expression levels of a nuclear (GAPDH, glyceraldehyde 3-phosphate dehydrogenase) and mitochondrial (ND1, NADH-ubiquinone oxidoreductase chain 1) housekeeping proteins. While DOX treatment produced a reduction in GAPDH expression levels, no statistically alterations in GAPDH were seen upon exposure to either Mt-CFX or CFX. Conversely, Mt-CFX was found to interfere with mitochondrial function (Figure 3F). Furthermore, Mt-CFX was found to reduce the ND1/GAPDH expression level ratio in both a dose- and time-dependent manner. On the other hand, a much smaller reduction in the mitochondrial/nuclear expression level ratio was observed for CFX (Figure S25).
The extent of drug-induced mitochondrial DNA mutation was then measured using a TaqI restriction site mutation assay.26 This restriction enzyme cleaves the double stranded 5′-TCGA-3′ DNA sequence leading to DNA digestion. In contrast, restriction enzyme-resistant sites undergo PCR amplification. Mt-CFX-treated cells showed an approximately 4.5-fold increase in resistance to TaqI restriction, whereas no significant difference in the amount of TaqI resistance was observed in the case of DOX-treated cells (Figure 3G).
We also investigated the location of predominant DNA mutagenicity for Mt-CFX, as well as that for several conventional chemotherapeutic drugs, including doxorubicin (DOX), temozolomide (TMZ), camptothecin (CPT), cis-platin (CDDP), chlorambucil (CLB), and fluorouracil (5-FU). Long range qPCR (3129 bp and 3723 bp for nuclear and mitochondrial targets, respectively)27 revealed that all the conventional drugs induced a considerable amount of nuclear DNA mutations, while having relatively little effect on the mitochondrial genome. In contrast, Mt-CFX was found to promote mitochondrial DNA damage while engendering little in the way of nuclear DNA damage (Fig 3H). This mirrors the lack of increase in the PUMA and Noxa protein levels (cf. Figure S17). Taken in aggregate, these results are consistent with our core hypothesis, namely that Mt-CFX localizes to the mitochondria and induces levels of ROS that are increased to the point where mitochondrial (but not nuclear) DNA damage is seen.
Identification of cellular processes affected by Mt-CFX
A DNA microarray study, comprising 197 genes, was conducted to determine the impact of Mt-CFX on protein expression and pathway activation in MDA-MB-231 cells (Figure 4A,B). Several pathways related to programmed cell death, such as apoptosis, ferroptosis and necroptosis, were altered in a statistically significant manner. Genes corresponding to the biosynthesis and metabolism of amino acids, fatty acids, and pyruvate were also affected.
Figure 4. Identification of cellular processes affected by Mt-CFX.
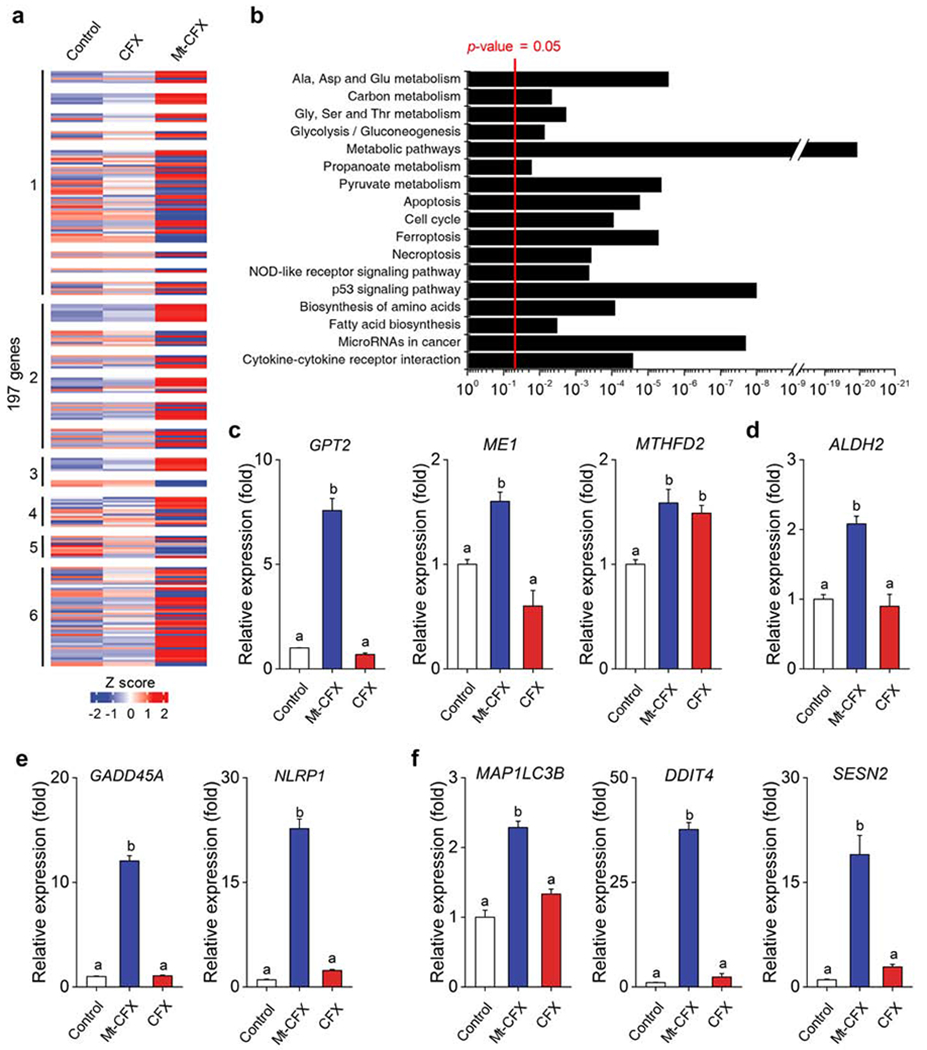
(A) Heatmap representing relative expression levels of the DEGs (differentially expressed genes) to control versus 10 μM CFX and Mt-CFX.
(B) KEGG pathway enrichment analysis of the 197 DEGs.
(C-F) q-PCR gene expression levels of selected genes in cells MDA-MB-231 cells treated with 10 μM Mt-CFX, CFX and a control sample.
(C) Cellular metabolism.*
(D) Mitochondrial detoxification.*
(E) Cellular repair and inflammasome induction.*
(F) Autophagy and mitophagy.*
Data are represented as mean ± SEM. (n = 3, in panels C-F). Statistical significance in panels c-f was determined using a one-way ANOVA test with post-hoc Bonferroni test. *Different letters (e.g., a–d) signify data which are statistically different (p < 0.05).
Selected examples of gene expression levels were assayed using q-PCR (Figure 4C). On this basis it is inferred that cellular metabolism is strongly affected by Mt-CFX, as reflected in an increased expression of the transaminase, pyruvate synthesis, and folate metabolism pathways (GPT2, ME1 and MTHFD2). Importantly, we also found that several ROS-induced cellular responses were upregulated in the presence of Mt-CFX. A mitochondrial aldehyde dehydrogenase (ALDH2) gene was significantly overexpressed. Presumably, this overexpression reflects a need to mitigate the effects of increased protein carbonylation, which is a recognized form of ROS damage (Figure 4D). GADD45A, encoding for a protein involved in stimulating DNA excision repair and inducing cell cycle arrest, was also overexpressed by ~12-fold (Figure 4E). An approximately 20-fold overexpression of NLRP1 was also seen, which is taken as evidence of cellular structure damage and the induction of the NLRP1 inflammasome (Figure 4E). DDIT4 and SESN2, involved in inhibiting mTORC1 and thus enabling autophagy, were overexpressed by ~40-fold and ~23-fold, respectively, indicating a need for the degradation of damaged cellular components. Overexpression of MAP1LC3B was also detected; this result suggests that damaged mitochondria are being degraded via mitophagy and may indicate onset of ferroptosis (Figure 4F).
In vivo tumor growth reduction by Mt-CFX and Bo-Mt-CFX
Finally, we investigated the in vivo activity of Mt-CFX in MDA-MB-231 tumor xenograft-bearing mice. All agents (e.g., vehicle, CFX, Mt-CFX, and Bo-Mt-CFX) were administered intravenously to these mice via tail vein injection. To assess biolocalization in vivo, Bo-Mt-CFX was administered to mice and its fluorescence was monitored. Fluorescence was detected at the tumor site and was found to persist >48 hours (Figure 5A). We confirmed that the inferred selectivity was indeed cancer specific by ex vivo analyses of organs harvested from mice euthanized 48 hours after a single Bo-Mt-CFX treatment. Nearly all fluorescence was confined to the tumor site, whereas other organs, such as the heart and reticuloendothelial system, remained unaffected (Figure 5B).
Figure 5. In vivo tumor growth reduction by Mt-CFX and Bo-Mt-CFX.
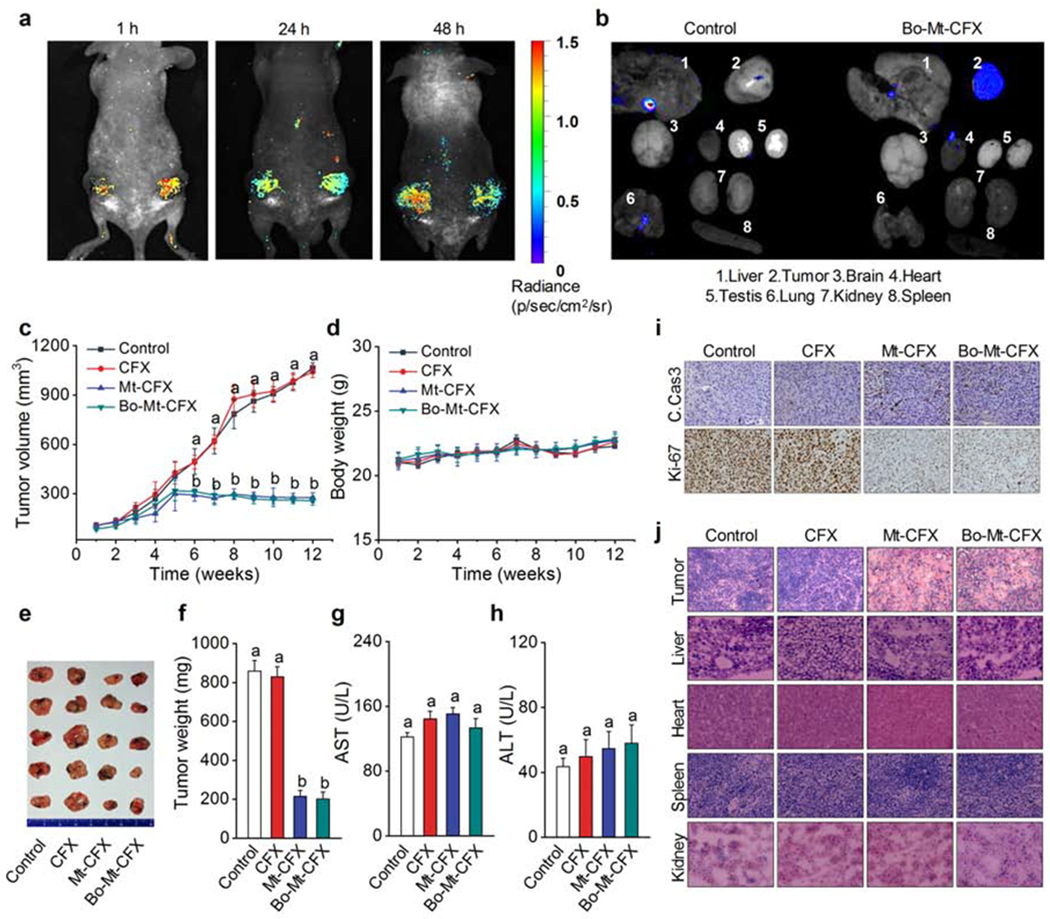
(A) In vivo images of MDA-MB-231 xenograft mouse 1 h, 24 h, and 48 h post intravenous tail vein injection of a single dose of 0.5 μmol kg−1 Bo-Mt-CFX. Excitation at 550 nm. Emission at 650 nm.
(B) Ex vivo imaging of excised tumors and organs 48 h post injection of a single dose of 0.5 μmol kg−1 Bo-Mt-CFX or vehicle only. Excitation at 550 nm. Emission at 650 nm.
(C) In vivo tumor volume determination (1/2 × length × width2) of mice treated with 0.5 μmol kg−1 CFX, Mt-CFX, Bo-Mt-CFX, or vehicle alone, once a week for 3 weeks.*
(D) Body weight of mice during the treatment regime.
(E) Excised tumors at the treatment endpoint.
(F) Excised tumor weight per treatment group.*
(G-H) Blood serum AST and ALT activity levels, as determined using a colorimetric assay.*
(I) Immunohistochemistry (cleaved caspase 3 and Ki-67) of representative tumor tissue slices of the different treatment groups.
(J) H&E staining of representative tissue slices of the different treatment groups.
Data are represented as mean ± SEM. Panels C, D, F: n = 4 mice and n =8 tumors per group, Panels G, H: n = 4. Statistical significance was determined using a one-way ANOVA test with post-hoc Bonferroni test.
*Different letters (e.g., a–d) signify data sets that are statistically different (p < 0.05).
Chemotherapeutic intervention was investigated using a 3-week regimen involving 1-dose per week treatment with 5 mmol kg−1 CFX, Mt-CFX, Bo-Mt-CFX, or vehicle alone. The study revealed a statistically significantly reduction in tumor growth in mice treated with Bo-Mt-CFX or Mt-CFX from the sixth week onwards (Figure 5C). The experiment was terminated at the twelfth week, when the tumors in the control group reached 1,000 mm3. The reduction in tumor burden produced by Bo-Mt-CFX or Mt-CFX was mirrored in the excised tumor weights (Figure 5E,F). This treatment regime was well-tolerated by the mice and no significant differences in animal body weights were observed during the course of treatment (Figure 5D). Liver function tests at the experimental endpoint revealed no significant differences relative to the outset, with the overall AST and ALT ratios of all mice being within the normal range.28
Tissue samples from the 4 treatment groups were subjected to immunohistochemistry. Cells with cleaved caspase were clearly more prevalent in the Mt-CFX and Bo-Mt-CFX groups, while a Ki-67 cell proliferation also revealed a dramatic reduction in cell proliferation in these two treatment groups (Figure 5E). Finally, tissue samples from tumor, liver, heart, spleen and kidneys were subject to H & E (haemotoxylin and eosin) staining. These studies revealed statistically significant differences in the histology for the tumor samples obtained from the mice treated with Mt-CFX or Bo-Mt-CFX relative to controls. Importantly no obvious signs of tissue damage were found in any of the other samples subject to analysis (Figure 5E).
CONCLUSION
Based on the findings presented here, we propose that Mt-CFX acts as a mitochondrially targeted chemotherapeutic drug. Mt-CFX was found to have inherent selectivity towards cancer cells. The use of the fluorescent conjugate Bo-Mt-CFX revealed mitochondrial localization, whereas an oxidative stress response was seen when MDA-MB-231 cells were treated with either Mt-CFX system. Mt-CFX was found to interfere with the mitochondrial ETC, inducing significantly enhanced levels of ROS. This was reflected in increased protein carbonylation, lipid peroxidation, and oxidative DNA damage in vitro. Moreover, Mt-CFX was found to cause a dramatic decrease in nuclear DNA damage as compared to DOX, TMZ, CPT, CDDP, CLB and 5-FU. Bo-Mt-CFX was retained in tumor xenografts as inferred from fluorescent imaging, while treatment of xenograft-bearing mice with well-supported doses of Mt-CFX and Bo-Mt-CFX resulted in reduced cancer burden. We thus suggest that rerouting CFX and potentially other approved antibiotics to the mitochondria could provide a new approach to generating small molecule chemotherapeutics. Support for this notion comes from the finding that the present system appears to be free of many of the genotypic-based side-effects that plague the majority of chemotherapeutics in current use.
EXPERIMENTAL PROCEDURES
The full experimental procedures are provided in the Supplemental Information.
Supplementary Material
The Bigger Picture.
Cancer recurrence is a major concern in clinical practice, severely limiting the life expectancy of cancer patients. Recurrence is often the result of therapy, which can also lead to drug-induced secondary tumorigenesis. In many cases, the resulting secondary malignancies are the result of nuclear DNA lesions induced by traditional genotoxic chemotherapeutics. In this study we show that by rerouting a commonly used microbiocidal, ciprofloxacin, to the mitochondria, it is possible to produce a highly selective and well-tolerated anticancer agent. Mitochondrial targeting not only induces a high degree of cancer specificity, it also minimizes nuclear DNA damage. On the other hand, it promotes mitochondrial damage-associated cancer cell death and tumor growth reduction. The present targeted ciprofloxacin derivative thus illustrates what could emerge as a new approach to generating less genotoxic and more effective chemotherapeutics.
Highlights.
Mitochondrial re-localization of ciprofloxacin reduces nuclear genotoxicity
Mitochondrial localization and spatiotemporal drug activation were confirmed
qPCR analyses provide support for the proposed mode of action
In vivo experiments support the efficacy and target-specificity
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2018R1A2A1A05020236, S.-G.C. and CRI project no. 2018R1A3B1052702, J.S.K.) and the Basic Science Research Program (2017R1D1A1B03032561, P.V. and 2017R1D1A1B03030062, M.W.) funded by the Ministry of Education as well as the Korea Research Fellowship Program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (2016H1D3A1938052, P.V.). K.S. is a recipient of the global PhD fellowship (GPF) program of the NRF funded by the Ministry of Science and ICT, Korea (2014H1A2A1020978, K.S.). Funding from the National Cancer Institute (RO1 CA68682 for J.L.S. and R15 CA232765 for J.F.A.) is acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
The micro array data was deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE137679.
SUPPLEMENTAL INFORMATION
The supplemental Information includes experimental procedures, synthetic procedures, and characterization data for of new compounds, as well as 18 supplemental figures (Figures S1-S18), and 2 Tables (Tables S1-S2). The associated files and can be found linked to this article online.
DECLARATION OF INTERESTS
Mt-CFX and analogues as non-genotoxic anticancer agents are the subject of a pending patent application, filed by Korea University, with J.S.K, K.S., P.V. and M.W. named as inventors. J.L.S. holds a part-time summer position at Shanghai University.
REFERENCES AND NOTES
- 1.Mehlen P, and Puisieux A (2006) Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458. [DOI] [PubMed] [Google Scholar]
- 2.Grosse Y, Baan R, Straif K, Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Galichet L, and Cogliano V (2009) A review of human carcinogens—Part A: pharmaceuticals. Lancet Oncol. 10, 13–14. [DOI] [PubMed] [Google Scholar]
- 3.Eichenauer DA, Thielen I, Haverkamp H, Franklin J, Behringer K, Halbsguth T, Klimm B, Diehl V, Sasse S, Rothe A, et al. (2014) Therapy-related acute myeloid leukemia and myelodysplastic syndromes in patients with Hodgkin lymphoma: a report from the German Hodgkin Study Group. Blood 123, 1658–1664. [DOI] [PubMed] [Google Scholar]
- 4.Pui CH, Ribeiro RC, Hancock ML, Rivera GK, Evans WE, Raimondi SC, Head DR, Behm FG, Mahmoud MH, Sandlund JT, et al. (1991) Acute Myeloid-Leukemia in Children Treated with Epipodophyllotoxins for Acute Lymphoblastic-Leukemia. New Engl. J. Med 325, 1682–1687. [DOI] [PubMed] [Google Scholar]
- 5.Travis LB, Holowaty EJ, Bergfeldt K, Lynch CF, Kohler BA, Wiklund T, Curtis RE, Hall P, Andersson M, Pukkala E, et al. (1999) Risk of leukemia after platinum-based chemotherapy for ovarian cancer. New Engl. J. Med 340, 351–357. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia S, Robison LL, Oberlin O, Greenberg M, Bunin G, Fossati-Bellani F, and Meadows AT (1996) Breast cancer and other second neo-plasms after childhood Hodgkin’s disease. New Engl. J. Med 334, 745–751. [DOI] [PubMed] [Google Scholar]
- 7.Glen CD, and Dubrova YE (2012) Exposure to anticancer drugs can result in transgenerational genomic instability in mice. P. Natl. Acad. Sci. USA 109, 2984–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith MA, Seibel NL, Altekruse SF, Ries LAG, Melbert DL, O’Leary M, Smith FO, and Reaman GH (2010) Outcomes for Children and Adolescents With Cancer: Challenges for the Twenty-First Century. J. Clin. Oncol 28, 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyall SD, Brown MT, and Johnson PJ (2004) Ancient invasions: From endosymbionts to organelles. Science 304, 253–257. [DOI] [PubMed] [Google Scholar]
- 10.Kalghatgi S, Spina CS, Costello JC, Liesa M, Morones-Ramirez JR, Slomovic S, Molina A, Shirihai OS, and Collins JJ (2013) Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci. Transl. Med 5, 192ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeVita VT Jr., and Chu E (2008) A History of Cancer Chemotherapy. Cancer Res. 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- 12.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, and Shen B (2005) Antitumor Antibiotics: Bleomycin, Enediynes, and Mitomycin. Chem. Rev 105, 739–758. [DOI] [PubMed] [Google Scholar]
- 13.Tacar O, Sriamornsak P, and Dass CR (2013) Doxorubicin: an update on anti-cancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol 65, 157–170. [DOI] [PubMed] [Google Scholar]
- 14.Fu D, Calvo JA, and Samson LD (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 12, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esner M, Graifer D, Lleonart ME and Lyakhovich A (2017) Targeting cancer cells through antibiotics-induced mitochondrial dysfunction requires autophagy inhibition. Cancer Lett. 384, 60–69. [DOI] [PubMed] [Google Scholar]
- 16.Alibek K, Bekmurzayeva A, Mussabekova A, and Sultankulov B (2012) Using antimicrobial adjuvant therapy in cancer treatment: a review. Infect. Agent Cancer 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, and Kalyanaraman B (2017) Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev 117, 10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samanta S, He Y, Sharma A, Kim J, Pan WH, Yang ZG, Li J, Yan W, Liu LW, Qu JL, and Kim JS (2019) Fluorescent Probes for Nanoscopic Imaging of Mitochondria. Chem 5, 1697–1726. [Google Scholar]
- 19.Lee MH, Sharma A, Chang MJ, Lee J, Son S, Sessler JL, Kang C and Kim JS (2018) Fluorogenic reaction-based prodrug conjugates as targeted cancer theranostics. Chem. Soc. Rev 47, 28–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang R, Xu Y, Zhang Y, Kim HS, Sharma A, Gao J, Yang G, Kim JS and Sun Y (2019) Rational design of a multifunctional molecular dye for dual-modal NIR-II/photoacoustic imaging and photothermal therapy. Chem. Sci 10, 8348–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Ding F, Zhou Z, Li C, Pu M, Xu Y, Zhan Y, Lu X, Li H, Yang G, et al. (2019) Rhomboidal Pt(II) metallacycle-based NIR-II theranostic nanoprobe for tumor diagnosis and image-guided therapy. Proc. Natl. Acad. Sci. USA 116, 1968–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Ding F, Chen Z, Zhang R, Li C, Xu Y, Zhang Y, Ni R, Li X, Yang G, et al. (2019) Melanin-dot-mediated delivery of metallacycle for NIR-II/photoacoustic dual-modal imaging-guided chemo-photothermal synergistic therapy. Proc. Natl. Acad. Sci. USA 116, 16729–16735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liou GY, and Storz P (2010) Reactive oxygen species in cancer. Free Radical Res. 44, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu J, and Zhang L (2008) PUMA, a potent killer with or without p53. Oncogene 27, S71–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dianov GL, Souza-Pinto N, Nyaga SG, Thybo T, Stevnsner T, and Bohr VA (2001) Base excision repair in nuclear and mitochondrial DNA. Prog. Nucleic Acid Res. Mol. Biol 68, 285–297. [DOI] [PubMed] [Google Scholar]
- 26.Vermulst M, Bielas JH, and Loeb LA (2008) Quantification of random mutations in the mitochondrial genome. Methods 46, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans SO, Jameson MB, Cursons RT, Peters LM, Bird S, and Jacobson GM (2016) Development of a qPCR Method to Measure Mitochondrial and Genomic DNA Damage with Application to Chemotherapy-Induced DNA Damage and Cryopreserved Cells. Biology (Basel) 5, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sher Y and Hung M (2013) Blood AST, ALT and UREA/BUN Level Analysis. Bio-protocol 3, e931. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.