

SUMMARY
In eukaryotic genomes, heterochromatin is targeted by RNAi machinery and/or by pathways requiring RNA elimination and transcription termination factors. However, a direct connection between termination machinery and RNA polymerase II (RNAPII) transcriptional activity at heterochromatic loci has remained elusive. Here, we show that, in fission yeast, the conserved cleavage and polyadenylation factor (CPF) is a key component involved in RNAi-in-dependent assembly of constitutive and facultative heterochromatin domains and that CPF is broadly required to silence genes regulated by Clr4SUV39H. Remarkably, CPF is recruited to non-canonical termination sites within the body of genes by the YTH family RNA-binding protein Mmi1 and is required for RNAPII transcription termination and facultative heterochromatin assembly. CPF loading by Mmi1 also promotes the selective termination of long non-coding RNAs that regulate gene expression in cis. These analyses delineate a mechanism in which CPF loaded onto non-canonical termination sites specifies targets of heterochromatin assembly and gene silencing.
Graphical Abstract
In Brief
Vo et al. report that the YTH family RNA-binding protein Mmi1 recruits the cleavage and polyadenylation factor (CPF) to non-canonical transcription termination sites within gene bodies to promote RNA polymerase II termination and RNAi-independent heterochromatin assembly. They show that CPF is required to silence genes that are regulated by the histone methyltransferase Clr4.
INTRODUCTION
The chromatin of eukaryotic genomes is organized into distinct structural domains (Misteli, 2010). DNA is folded with histones and non-histone proteins to form euchromatin and heterochromatin (Grewal and Jia, 2007; Jenuwein and Allis, 2001). Although euchromatin domains are less condensed and relatively accessible, heterochromatin domains are inhibitory to trans-acting factors. Heterochromatin is divided into two types: constitutive and facultative. Constitutive heterochromatin is stably maintained throughout the cell cycle and preferentially targets repetitive DNA elements. On the other hand, facultative heterochromatin targeted to regulated genes is dynamically modulated in response to cellular and environmental signals.
Heterochromatin displays a specific pattern of posttranslational histone modifications (Rando and Winston, 2012) distinguished by histone hypoacetylation and methylation of histone H3 lysine-9 (H3K9me) (Jenuwein and Allis, 2001; Litt et al., 2001; Noma et al., 2001). H3K9me provides binding sites for chromodomain proteins, including HP1 proteins implicated in heterochromatic silencing (Bannister et al., 2001; Lachner et al., 2001; Nakayama et al., 2001; Sadaie et al., 2004). Chromatin-bound HP1 proteins engage RNA processing activities and chromatin modifiers to promote posttranscriptional and transcriptional gene silencing, respectively. Despite numerous studies revealing the diversity of heterochromatin functions (Grewal and Jia, 2007), the mechanisms that specify heterochromatin nucleation across the genome remain poorly understood.
Studies using the fission yeast Schizosaccharomyces pombe have provided insights into conserved heterochromatin assembly pathways. Other than constitutive heterochromatin domains at centromeres, telomeres, and the mat locus, S. pombe contains discrete blocks of facultative heterochromatin, referred to as heterochromatin islands (Cam et al., 2005). The major targets of heterochromatin islands are meiotic genes or genes regulated in response to environmental changes, such as a shift to low temperature (Gallagher et al., 2018; Zofall et al., 2012). In addition, a small number of heterochromatin islands that harbor late replication origins are bound by components of the Shelterin complex (Zofall et al., 2016). Although Shelterin mediates assembly of heterochromatin islands targeting late origins, recruitment of the Clr4SUV39H histone H3K9 methyltransferase complex (ClrC) to other heterochromatin domains is paradoxically linked to RNA polymerase II (RNAPII) transcriptional activity and co-transcriptional processing of RNAs (Chen et al., 2008; Djupedal et al., 2005; Grewal and Elgin, 2007; Kato et al., 2005; Kloc et al., 2008; Reyes-Turcu and Grewal, 2012).
Two distinct mechanisms exploit RNAPII transcription to nucleate heterochromatin: RNAi machinery and RNAi-independent mechanisms involving nuclear RNA processing and degradation factors. Both mechanisms work in parallel to target ClrC to repeat elements within constitutive heterochromatin domains (Chalamcharla et al., 2015; Parsa et al., 2018; Reyes-Turcu et al., 2011; Touat-Todeschini et al., 2017; Tucker et al., 2016). Nuclear RNA processing factors also mediate assembly of facultative heterochromatin islands. At meiotic genes, a YTH family RNA-binding protein, Mmi1, binds transcripts containing a determinant of selective removal (DSR) element (Harigaya et al., 2006; Yamashita et al., 2012) and is implicated in facultative heterochromatin assembly (Hiriart et al., 2012; Tashiro et al., 2013; Zofall et al., 2012). Mmi1 recruits a conserved RNA processing complex called Mtl1-Red1 core (MTREC; pA-tail exosome targeting [PAXT] in mammals) composed of the zinc-finger protein Red1 and the Mtr4-like protein Mtl1, which mediates RNA degradation by the exosome Rrp6 and recruits ClrC to assemble heterochromatin (Lee et al., 2013; Meola et al., 2016; Sugiyama and Sugioka-Sugiyama, 2011; Zhou et al., 2015). However, MTREC also processes RNA transcripts from loci that are not targets of heterochromatin assembly, suggesting that additional factors are required to trigger heterochromatin assembly. Notably, mutations in the RNAPII transcription termination factor Dhp1 (XRN2 in mammals) impair RNAi-independent heterochromatin assembly at meiotic genes and heterochromatin islands detected at low temperature (Chalamcharla et al., 2015; Gallagher et al., 2018; Tucker et al., 2016). However, it was unknown (1) whether the termination machinery directly modulates RNAPII at heterochromatic loci to trigger H3K9me and (2) which factors or pathways couple RNAPII transcription termination to heterochromatic silencing.
We performed an unbiased genetic screen that revealed components of the cleavage and polyadenylation factor (CPF) complex that are involved in transcription-coupled assembly of constitutive and facultative heterochromatin. We find that CPF is recruited to non-canonical sites within the gene body by Mmi1 and is critical for RNAPII transcription termination and assembly of heterochromatin islands via a mechanism involving Dhp1XRN2. Mmi1 also mediates loading of CPF to promote termination of long non-coding RNAs (lncRNAs), which prevents them from invading and repressing downstream genes. Together with our results showing that CPF acts broadly to repress genes regulated by Clr4SUV39H, our findings provide a key mechanistic advance by revealing the role of the evolutionarily conserved CPF complex in specifying targets of heterochromatin assembly and gene silencing.
RESULTS
CPF Is Required for Heterochromatin-Mediated Gene Silencing
We previously described an RNAi-independent mechanism that utilizes RNAPII transcription to trigger facultative heterochromatin formation (Reyes-Turcu et al., 2011). One target locus, SPAC23H3.14, is adjacent to a binding site for the Shelterin complex subunit Taz1 (Figure S1A). Taz1-Shelterin is required for heterochromatic silencing at this region (Zofall et al., 2016), but how RNAPII transcription contributes to assembly of facultative heterochromatin at this locus (referred to as Island 3) remained unclear. To address this, we performed a genetic screen to identify factors affecting the silencing of a ura4+ reporter inserted near the SPAC23H3.14 locus (Is3::ura4+) (Figure 1A). The strain also carried a deletion of the anti-silencing factor Epe1 to reinforce heterochromatic silencing and an ade6+ reporter at the silent mat region (mat2::ade6+) to distinguish mutants specifically defective in Is3::ura4+ silencing from those affecting core heterochromatin factors. Colonies showing growth on medium lacking uracil, but not on counter-selective 5-fluoroorotic acid (5FOA) medium, were examined for mat2::ade6+ expression by replica plating on low-adenine medium. Mutants that maintained mat2::ade6+ silencing formed red colonies, whereas loss of silencing resulted in white colonies. Five mutants specifically defective in silencing of Is3::ura4+ (i.e., Ura+ and red color) were subjected to further analyses. To identify loci harboring mutant alleles, we performed whole-genome sequencing of mutant and wild-type (WT) segregants from tetrad analyses. Mutant alleles exclusively segregating with the silencing phenotype were identified by computational analyses and were further confirmed by conventional DNA sequencing.
Figure 1. Genetic Screen for Factors Required in RNAi-Independent Heterochromatic Silencing.

(A) Genetic screen to identify mutants specifically defective in silencing Is3::ura4+.
(B) Serial dilution spotting assay for expression of Is3::ura4+ and mat2::ade6+. Expression of Is3::ura4+ leads to growth defects on medium containing 5FOA. Expression of mat2::ade6+ results in white colonies on low-adenine medium. Also shown is growth on non-selective medium (N/S).
(C) Graphical representation of Swd22 and Ssu72 protein domains. Nonsense mutations identified in the genetic screen are indicated by red rectangles.
(D) ChIP-qPCR analysis of H3K9me2 enrichment at Is3::ura4+ in CPF mutants. Numbers are enrichment values of Is3::ura4+ relative to the control leu1 locus.
(E and F) H3K9me2 enrichment at (E) ssm4 and (F) mei4 loci as assessed by ChIP-qPCR analysis. Enrichment values are fold changes relative to the leu1 control. n = 2. Error bars are shown as SDs from the mean. Oligonucleotides are listed in Table S5.
Supporting our previous work (Zofall et al., 2016), we found mutant alleles of Taz1 and the Shelterin subunit Ccq1 as well as Rif1, which is known to associate with Taz1 (Figures 1B and S1B). We also identified mutations in Swd22 and Ssu72, which are core subunits of the conserved CPF complex (Figure 1C; Casañal et al., 2017; Roguev et al., 2004). Swd22 and Ssu72 are components of the DPS (Dis2-Ppn1-Swd22) and core phosphatase modules of CPF, respectively (Vanoosthuyse et al., 2014). The swd221–209 truncation mutation eliminates a C-terminal WD40 protein domain required for protein-protein or protein-DNA interactions. The ssu721–86 truncation mutation disrupts the putative phosphatase domain (Reyes-Reyes and Hampsey, 2007).
The discovery of CPF subunits was intriguing, given their direct link to RNAPII transcription. To confirm the results of the screen, we constructed strains in which swd22 and ssu72 loci were replaced with mutant alleles (swd221–209 or ssu721–86). The resulting mutant strains showed de-repression of Is3::ura4+ (Figure 1B). Importantly, deletion of either swd22 or ssu72 caused similar de-repression of Is3::ura4+ (Figure S1C), leading us to wonder whether other CPF subunits might also be required. Indeed, cells lacking another core CPF subunit, Ctf1CSTF2, showed de-repression of Is3::ura4+, similar to swd22Δ and ssu72Δ (Figure S1D). Thus, CPF is functionally required for silencing Is3::ura4+ targeted by heterochromatin machinery.
Mutations in swd22 and ssu72 Affect the Assembly of Heterochromatin Islands
We next investigated whether the defective silencing of Is3::ura4+ observed in CPF mutants was accompanied by changes in heterochromatin. Chromatin immunoprecipitation (ChIP) analyses of H3K9me in swd221–209 and ssu721–86 mutants revealed a marked reduction in H3K9me levels at the reporter locus compared with the WT (Figure 1D).
We also examined H3K9me at other loci where RNAPII transcription is implicated in RNAi-independent facultative heterochromatin assembly, such as meiotic genes (Zofall et al., 2012). Indeed, swd221–209 and ssu721–86 mutants showed a drastic reduction in H3K9me levels at heterochromatin islands coating ssm4 and mei4 meiotic genes (Figures 1E and 1F). Thus, the highly conserved CPF complex is a crucial component of an RNAi-independent pathway for the assembly of facultative heterochromatin.
CPF Acts Broadly to Assemble Facultative Heterochromatin
Besides heterochromatin islands detected under standard growth conditions, RNAi-independent assembly of heterochromatin islands occurs at a number of genes at low temperature (18°C) (Gallagher et al., 2018). To explore the possible role of CPF in this process, we performed a ChIP sequencing (ChIP-seq) analysis of H3K9me in cells grown at 18°C. Loss of Swd22 or Ssu72 resulted in widespread defects in the assembly of heterochromatin islands (Figure 2A; Table S1). In addition to meiotic genes (e.g., ssm4, mei4, mug8, and mbx2) showing reduced H3K9me levels, heterochromatin islands detected at 18°C (e.g., see SPAPB1A11.02 and SPBC428.10) were also affected in swd22Δ and ssu72Δ (Figures 2A-2C). Similarly, loss of the Ctf1CSTF2 subunit of CPF affected H3K9me at both meiotic genes and islands detected at 18°C (Figure S2A), supporting the idea that CPF is involved in facultative heterochromatin assembly.
Figure 2. CPF Is Required for the Assembly of Facultative Heterochromatin Domains.

(A) Genome-wide ChIP-seq analyses of H3K9me2 in WT, swd22Δ, or ssu72Δ cells at 18°C. Representative meiotic or 18°C facultative heterochromatin islands are annotated. Enrichment values are reads per genomic content (RPGC) and represent ratios of immunoprecipitated chromatin over whole-cell extract. Islands are listed in Table S1.
(B) H3K9me2 ChIP-seq enrichment in the indicated strains at 18°C. Shown are meiotic islands at loci ssm4, mei4, mug8, and mbx2. Also shown are islands at SPAPB1A11.02 and SPBC426.10 formed in the WT at 18°C.
(C) H3K9me2 ChIP-qPCR analysis of WT, swd22Δ, or ssu72Δ cells at 18°C. Enrichment values are fold changes relative to the leu1 control. n = 3. Error bars are shown as SDs from the mean.
Although loss of CPF components caused a drastic reduction in H3K9me at meiotic islands and islands detected at 18°C, only a slight reduction in H3K9me was observed at Shelterin-dependent islands (Figure S2B; Table S1). Island 3 was an exceptional case in this analysis because it is targeted by two nucleation mechanisms (Figure S2C). Although Taz1-Shelterin bound to chromosomal-internal telomeric sequences at Island 3 recruits ClrC (Zofall et al., 2016), CPF acts at an adjacent locus to trigger facultative heterochromatin assembly. Together, these results suggest that CPF plays a widespread role in heterochromatin island assembly, specifically targeting meiotic islands and islands detected at 18°C.
CPF and RNAi Act in Parallel to Assemble Centromeric Heterochromatin
In contrast to the broad effects on RNAi-independent facultative heterochromatin islands in swd22Δ and ssu72Δ, RNAi-dependent constitutive heterochromatin domains such as at centromeres appeared to be unaffected (Figures 2A and S2D). However, an increase in centromere repeat-derived small interfering RNAs (siRNAs) was observed in both swd22Δ and ssu72Δ as well as swd221–209 and ssu721–86 mutants (Figure S2E). Such accumulation of siRNAs is reminiscent of cells deficient in heterochromatin silencing factors (such as Rrp6) that act independently of RNAi to assemble centromeric H3K9me (Chalamcharla et al., 2015; Reyes-Turcu et al., 2011). We wondered whether CPF also acts parallel to RNAi. Indeed, cells carrying deletions of CPF and RNAi components (swd22Δ ago1Δ) showed cumulative loss of H3K9me at heterochromatic centromeric repeats (Figure S2F). This result confirmed that CPF is indeed involved in heterochromatin formation at centromeres and that the effects of its loss are masked by the RNAi machinery.
We next addressed whether CPF is required for RNAi-dependent de novo establishment of heterochromatin. Because Clr4SUV39H is the sole H3K9 methyltransferase in S. pombe, clr4Δ cells lack H3K9me (Figure S2G). Establishment of H3K9me occurs upon introduction of a plasmid containing clr4+. To test the requirement for Swd22, we analyzed clr4+-dependent H3K9me establishment in a clr4Δ swd22Δ double mutant strain. We found that H3K9me was established at centromeres in both clr4Δ and clr4Δ swd22Δ (Figure S2G). Moreover, a reporter inserted at a pericentromeric region (otr1::ura4+) was silenced in both strains (Figure S2H). Thus, CPF is dispensable for de novo heterochromatin nucleation at centromeres.
These results further broaden the role of CPF in heterochromatin assembly. In addition to its requirement in facultative heterochromatin assembly at meiotic islands and islands detected at 18°C, CPF also acts in parallel with RNAi to assemble constitutive heterochromatin at centromeres.
CPF Represses Genes that Are Regulated by Clr4SUV39H
Clr4SUV39H moderates upregulated gene expression that occurs genome-wide in response to environmental change (Gallagher et al., 2018). Culturing cells that lack Clr4SUV39H at 18°C results in a wide variety of elevated gene transcripts. We asked whether CPF components, which are required for the assembly of heterochromatin islands detected at 18°C, control expression of Clr4SUV39H target loci. RNA sequencing (RNA-seq) profiles of WT, swd22Δ, and ssu72Δ cultured at 18°C were compared with the WT grown under standard laboratory conditions (30°C). Widespread upregulation of genes occurred in cells lacking Swd22 or Ssu72 compared with WT cells at 18°C (Figure 3A). The expression profiles of cells lacking Swd22 and Ssu72 were highly correlated (Pearson’s correlation coefficient [PCC] = 0.92) (Figure 3B). Among the transcripts that were upregulated 2-fold or more in swd22Δ and ssu72Δ cells (494 and 496, respectively), 413 were upregulated in both mutants (Figure 3C). In addition, transcripts from select loci showed extension of the 3′ ends in swd22Δ and ssu72Δ (Figures S3A and S3B). This indicated possible defects in pre-mRNA processing and readthrough transcription, as also observed in cells lacking Clr4SUV39H (Gullerova and Proudfoot, 2008; Zofall et al., 2009).
Figure 3. CPF and Clr4 Repress Overlapping Gene Sets.

(A) Heatmap of expression changes in WT, clr4Δ, swd22Δ, and ssu72Δ cells at 18°C. Shown are transcripts that are upregulated 2-fold or more (log2 fold change ≥ 1) in clr4Δ at 18°C. Transcripts were clustered based on their upregulation in the WT and all mutants (cluster 1), all 3 mutants and not in the WT (cluster 2), clr4Δ and swd22Δ (cluster 3), clr4Δ and ssu72Δ (cluster 4), or clr4Δ only (cluster 5).
(B) Clustering based on Pearson’s correlation coefficient (PCC) of expression profiles in WT and mutants at 18°C compared with the WT at 30°C.
(C) Euler diagram depicting the overlap of genes that are upregulated 2-fold or more in clr4Δ, swd22Δ, and/or ssu72Δ at 18°C compared with the WT at 30°C. Pairwise intersection p values were assessed using the cumulative hypergeometric distribution.
(D) RNA-seq composite plots showing median expression levels of genes implicated in metabolism, stress response, and/or meiosis that are upregulated in mutants compared with the WT at 18°C. Also shown are RNA-seq expression profiles for representative loci corresponding to metabolic genes (top), stress response genes (center), and meiotic genes (bottom) in WT and mutant cells at 18°C. Genes used to generate composite plots are listed in Table S2. For all RNA-seq expression profiles, normalized reads per kilobase per million (RPKM) values are shown.
We next asked whether any of the loci upregulated in CPF mutants at 18°C are also targets of Clr4SUV39H. Indeed, the expression profiles of swd22Δ and ssu72Δ cells closely resembled the profile of cells lacking Clr4SUV39H (PCC = 0.84 per pairwise comparison) (Figure 3B). Among the 426 transcripts upregulated in clr4Δ, 331 transcripts (77.7%) were also upregulated in swd22Δ or ssu72Δ (Figure 3C; Table S2), suggesting that CPF represses genes that are also controlled by Clr4SUV39H. Because Clr4SUV39H-dependent assembly of constitutive heterochromatin domains was unaffected in the CPF mutants (Figures 2A and S2D), the similarity of the profiles is not due to altered expression of core heterochromatin factors in the CPF mutants. The most notable loci showing upregulation in clr4Δ and CPF deletion mutants included metabolic and stress response genes (Figure 3D). Another major class contained meiotic genes (Figure 3D). Consistent with a critical requirement for CPF and Clr4SUV39H in preventing aberrant expression of genes at low temperature, cells lacking Swd22, Ssu72, or Ctf1 exhibited a cold-sensitive growth phenotype (Figure S3C), analogous to the slow growth phenotype of clr4Δ at 18°C (Gallagher et al., 2018). Collectively, these results support a widespread requirement for CPF components in the regulation of gene expression and show that CPF and Clr4SUV39H repress a common set of genes to properly control their expression.
CPF Localizes within the Gene Body of Facultative Heterochromatin Island Loci
To address whether CPF is directly required for the assembly of facultative heterochromatin islands, we performed ChIP analyses using strains expressing epitope-tagged CPF subunits. Iss1 tagged with GFP (Iss1-GFP) showed the best ChIP enrichment and was chosen for further ChIP-seq analyses. Iss1 is a core subunit of CPF and is homologous to mammalian FIP1 (Roguev et al., 2004; Yamashita et al., 2013). Iss1FIP1 exhibited a unique distribution, localizing to both canonical termination sites as well as non-canonical sites within the gene body at meiotic heterochromatin islands (Figure 4A). The peaks of Iss1FIP1 localization precisely aligned with DSR elements within meiotic genes (Figure 4A). Analyses of recently published ChIP-seq data for CPF accessory complex subunits revealed that Rna14 and Pcf11 (homologs of mammalian CSTF3 and Pcf11, respectively) were also enriched at meiotic heterochromatin islands, in a pattern identical to Iss1FIP1 (Figure S4A). Moreover, we found that cells cultured at 18°C showed elevated levels of Iss1FIP1 at Island 3 that correlated with an increase in H3K9me enrichment (Figure S4B). Altogether, these results are consistent with direct involvement of CPF in facultative heterochromatin assembly.
Figure 4. Mmi1 Recruits CPF to Facultative Heterochromatin Islands.

(A) ChIP-seq analysis of the core CPF subunit Iss1-GFP in the WT (top) or mmi1Δ (bottom). Enrichments at meiotic island loci are shown. DSR motifs are indicated by red triangles.
(B) Chromatin immunoprecipitation combined with microarray (ChIP-chip) enrichment of CFP-Mmi1 in the WT at the indicated meiotic island loci.
(C) Median Iss1-GFP ChIP-seq enrichment over H3K9me island Mmi1 regulon genes (n = 7) versus non-island (n = 21) Mmi1 regulon genes in WT cells as determined by ChIP-seq.
(D) Median Iss1-GFP ChIP-seq enrichment over island Mmi1 regulon genes (n = 7) in WT (green) or mmi1Δ (orange) cells.
(E) Core CPF subunit Iss1FIP1 coimmunoprecipitates with the YTH family protein Mmi1. The purified Iss1-GFP fraction was probed for FLAG-Mmi1.
(F) Colocalization of Mmi1 and CPF in live WT cells expressing CFP-Mmi1 and Iss1-GFP. Also shown are zoomed insets (dashed orange boxes) of 3 representative cells. The scale bar represents 5 μm. PCC was calculated to quantify population-wide colocalization of Mmi1 and Iss1FIP1.
For all median enrichment analyses, the base coordinate ranges of genes were scaled to a common length from the start codon to the stop codon of the open reading frame (ORF). Island and non-island Mmi1-regulon genes are listed in Table S3.
Mmi1 Is Required for CPF Localization at Meiotic Heterochromatin Islands
The presence of Iss1FIP1, Rna14CSTF3, and Pcf11 peaks within DSR-containing genes provided a potential clue to the mechanism of CPF recruitment. DSR-containing RNAs are recognized by Mmi1, which is implicated in facultative heterochromatin assembly (Harigaya et al., 2006; Hiriart et al., 2012; Tashiro et al., 2013; Zofall et al., 2012). Notably, peaks of Mmi1 within specific DSR-containing meiotic heterochromatin islands coincided precisely with CPF components (Figures 4A, 4B, and S4A). Moreover, we found that Iss1FIP1 localization was abolished at these islands in the absence of Mmi1 (Figures 4A and S4C). Thus, CPF localizes to DSR-containing genes in an Mmi1-dependent manner.
Localization of CPF components within the gene body is a general feature of Mmi1-dependent meiotic heterochromatin islands (Figures 4C and S4D). At these loci, CPF enrichment within the gene body was abolished in mmi1Δ, as determined by analysis of Iss1FIP1 peak distributions (Figures 4D and S4E). Thus, not only does CPF colocalize with Mmi1 at meiotic heterochromatin islands, but CPF recruitment to these loci requires Mmi1.
Mmi1 Physically Associates with the CPF Complex
To investigate whether Mmi1 physically associates with CPF, we purified a core CPF subunit, Cft1CPSF1-FLAG, and then performed mass spectrometry analysis. Known subunits of the CPF complex, including Ctf1, Ssu72, Swd22, and Iss1, were present in the purified fraction from the Cft1-FLAG strain but not from the untagged strain (Figure S4F). Additionally, we found a small number of peptides derived from Mmi1, indicating that Mmi1 might physically interact with the CPF complex. Indeed, Mmi1 (FLAG-Mmi1) co-immunoprecipitated with Iss1-GFP (Figure 4E). Moreover, immunofluorescence analyses showed that, in addition to a diffuse nuclear distribution, Iss1FIP1 localized to distinct puncta that overlapped with Mmi1 (Figures 4F and S4G), further supporting a physical association between these factors. Thus, CPF associates with Mmi1, and this association is likely required for loading the CPF complex at heterochromatin islands.
Mmi1 and CPF Collaborate to Control lncRNA-Mediated Gene Silencing
The association of Mmi1 with CPF has another interesting implication. In addition to targeting meiotic genes, Mmi1 is required for terminating regulatory lncRNAs to prevent them from invading and repressing downstream genes (Chatterjee et al., 2016; Lee et al., 2013; Shah et al., 2014; Touat-Todeschini et al., 2017). How Mmi1 controls termination of lncRNAs is unknown. ChIP-seq analysis of CPF (Iss1FIP1) and its accessory complex subunits (Rna14CSTF3 and Pcf11) showed enrichment at cis-acting regulatory lncRNAs (such as lncRNAs located upstream of byr2, phol, and SPCC11E10.01; Figures 5A and S5A) with peaks precisely coinciding with peaks of Mmi1 (Figure 5B). Importantly, loss of Mmi1 abolished CPF localization at these loci (Figures 5A and S5B). Thus, Mmi1 is required to mediate recruitment of CPF to regulatory lncRNAs, similar to its role at DSR-containing meiotic genes.
Figure 5. Mmi1-Mediated Transcription Termination at lncRNAs Requires CPF.

(A) ChIP-seq analysis of the core CPF subunit Iss1-GFP at the lncRNAs adjacent to the genes byr2, pho1, and SPCC11E10.01 in WT or mmi1Δ cells. Enrichment values represent the subtraction of whole-cell extract (WCE, input) from immunoprecipitated chromatin (IP).
(B) ChIP-chip analysis of CFP-Mmi1 in WT cells at the indicated lncRNA loci. Enrichment values represent ratios of IP over input.
(C and D) Northern blot analysis of the (C) byr2 and (D) pho1 loci in WT or CPF mutant cells at 18°C. A schematic of the byr2 and pho1 loci with the adjacent lncRNAs nam1 and prt, respectively, is shown above the blots. The long readthrough transcripts are called nam1-L and prt-L. rRNA was used as a loading control.
See also Figure S5.
To determine whether Mmi1 prevents lncRNAs from invading downstream genes through recruitment of CPF, we studied the effects of nam1 and prt lncRNAs on their adjacent byr2 and pho1 genes, respectively, in CPF mutant cells. Northern blot analyses of byr2 and pho1 revealed the appearance of longer readthrough transcripts (nam1-L and prt-L) in swd22Δ, ssu72Δ, and ctf1Δ, which was coupled to repression of their respective downstream genes (Figures 5C and 5D). Therefore, the recruitment of CPF by Mmi1 is indeed functionally important for lncRNA-mediated control of protein-coding gene expression.
CPF Localizes to Pericentromeric Regions
Mmi1 was recently found to associate with lncRNAs (referred to as nam5, nam6, and nam7) derived from pericentromeric heterochromatic regions (Touat-Todeschini et al., 2017). Because CPF affects heterochromatic silencing at centromeres (Figure S2F), we wondered whether CPF localizes to these lncRNA loci. We compared Iss1FIP1 localization in WT cells, in which pericentromeric repeat elements are tightly repressed and depleted of RNAPII, with Iss1FIP1 localization in cells lacking Clr4SUV39H that show a marked increase in RNAPII levels (Chen et al., 2008). Although Iss1FIP1 enrichment across pericentromeric regions could not be detected in the WT, Iss1FIP1 localized to specific sites within these domains in clr4Δ (Figure S5C). Notably, Iss1FIP1 preferentially localized to the nam5, nam6, and nam7 lncRNAs (Figure S5C). Similarly, Iss1FIP1 was enriched at the centromere-homologous (cenH) heterochromatin nucleation center at the mat locus in clr4Δ (Figure S5D). Together with the results described above (Figure S2F), these findings suggest that CPF directly promotes heterochromatic silencing of these centromeric repeat elements.
Mmi1 and CPF Are Required for RNAPII Termination within the Gene Body
Our finding that Mmi1 mediates CPF localization within the open reading frames of meiotic genes and at lncRNAs suggested that these loci might be sites of non-canonical RNAPII transcription termination. To test this, we mapped the distribution of RNAPII in which the C-terminal domain (CTD) of its largest subunit is phosphorylated at serine-2. Phosphorylation of serine 2 (Ser2-P) peaks near the mRNA cleavage site and facilitates loading of factors involved in pre-mRNA 3′ end processing and RNAPII termination (Buratowski, 2009). Remarkably, RNAPII Ser2-P peaks coincided with CPF enrichment observed within meiotic genes and at lncRNAs (Figures 6A and 6B). The overall binding profile of RNAPII Ser2-P showed that pileups of RNAPII within the open reading frames of genes is a distinctive feature of these Mmi1-regulated loci (Figures 6C and 6D). Notably, peaks of RNAPII Ser2-P at meiotic islands and at lncRNAs were severely affected in cells that lack Mmi1 or that carry deletions of the CPF subunits Swd22 or Ssu72 (Figures 6A and S6A). Moreover, overall gene profiling showed a reduction in RNAPII Ser2-P within the gene body, specifically at Mmi1-regulated islands in mmi1Δ, swd22Δ, and ssu72Δ (Figures 6E, and 6F, and S6B). Thus, Mmi1 and CPF are required for pileups of RNAPII Ser2-P at non-canonical termination sites.
Figure 6. Mmi1 and the CPF Complex Promote RNAPII Pileup at Non-canonical Termination Sites.

(A) ChIP-seq analysis of RNAPII Ser2-P pileup at meiotic (ssm4 and mcp7) and lncRNA loci in the indicated strains. Dashed rectangles highlight regions of RNAPII Ser2-P pileup in the WT. Enrichment values represent the subtraction of WCE from IP.
(B) ChIP-seq localization distributions of the core CPF subunit Iss1FIP1 in the WT and of Dhp1XRN2 in the WT and swd22Δ.
(C) Median enrichment of RNAPII Ser2-P across island Mmi1-regulon genes (n = 7, solid black) or all coding genes (n = 5,144, dashed black) in the WT as determined by ChIP-seq.
(D) Boxplot of the RNAPII Ser2-P peak signals described in (C).
(E) Median enrichment of RNAPII Ser2-P across island Mmi1-regulon genes (n = 7) in the indicated strains.
(F) Boxplot of the RNAPII Ser2-P peak signals described in (E).
(G) Median enrichment of Iss1FIP1 and Dhp1XRN2 across island Mmi1-regulon genes (n = 7) in WT cells.
(H) Boxplot representation of Dhp1XRN2 peak signals in the WT and swd22Δ.
For all median distribution analyses, the base coordinate ranges of genes were scaled to a common length from start codon to stop codon. For boxplots, middle lines indicate median values, box edges indicate the first and third quartiles, and whiskers mark extreme values. p values were calculated using Wilcoxon signed rank test (one-tailed) for paired sets between WT and mutant or Wilcoxon rank sum test (one-tailed) for unpaired sets. Island Mmi1-regulon genes are listed in Table S3.
We next determined whether the RNAPII Ser2-P pileups corresponded to terminating Pol II, as marked by the highly conserved termination factor Dhp1XRN2. Upon cleavage of nascent transcripts by pre-mRNA 3′ end processing factors, the 5′ → 3′ exonuclease activity of Dhp1XRN2 degrades transcripts, leading to release of RNAPII (Porrua and Libri, 2015; Proudfoot, 2016; Richard and Manley, 2009). ChIP-seq analyses showed Dhp1XRN2 enrichment at the 3′ end of genes, and its genome-wide localization pattern matched CPF components (Figure S6C), consistent with tight coupling of RNA 3′ end processing and transcription termination processes (Larochelle et al., 2018; Porrua and Libri, 2015; Proudfoot, 2016; Richard and Manley, 2009). Remarkably, Dhp1XRN2 was enriched at regions showing Mmi1-dependent localization of CPF and RNAPII Ser2-P within meiotic genes and at lncRNAs (Figures 6A and 6B). Average gene profiling confirmed similar distributions of the CPF subunits Iss1FIP1 and Dhp1XRN2 at Mmi1-regulated genes (Figure 6G). Moreover, CPF is required for loading of Dhp1XRN2 to non-canonical termination sites within meiotic heterochromatin islands and lncRNAs targeted by Mmi1 (Figures 6B and 6H). The high degree of colocalization observed among RNAPII Ser2-P, CPF, and Dhp1XRN2 at meiotic heterochromatin islands and lncRNAs (Figure S7) suggests that the RNAPII pileups are indeed sites of transcription termination. At these non-canonical transcription termination sites, Mmi1 recruits CPF, which, in turn, contributes to loading of Dhp1XRN2 to trigger assembly of facultative heterochromatin islands and modulate expression of genes located adjacent to lncRNAs.
DISCUSSION
Heterochromatin assembly paradoxically requires transcription of target sequences by RNAPII (Grewal and Elgin, 2007). Although RNAi-dependent mechanisms exploit RNAPII transcription to generate small RNAs and recruit heterochromatin assembly proteins, it remains unclear how RNAPII transcription supports RNAi-independent nucleation of heterochromatin. We find that the highly conserved CPF complex, implicated in pre-mRNA 3′ end processing and termination of RNAPII, is broadly required for RNAi-independent constitutive and facultative heterochromatin assembly. Loss of CPF causes widespread upregulation of genes that rely on Clr4SUV39H for their regulation during environmental change, highlighting the significance of this complex in adaptive control of the genome. Our analyses suggest that CPF is a key component of a mechanism in which transcription termination machinery specifies targeting of heterochromatin factors to modulate gene expression.
CPF-Mediated Facultative Heterochromatin Assembly and Global Gene Control
Several lines of evidence support the direct involvement of CPF in the assembly of heterochromatin islands and the control of gene expression through collaboration with the YTH family protein Mmi1. First, CPF is enriched at Mmi1-bound sites within the open reading frames of meiotic genes. Second, CPF and Mmi1 physically interact and colocalize within the nucleus. Third, loss of Mmi1 abolishes CPF localization at Mmi1-dependent heterochromatin islands. Thus, in addition to its known role in nuclear RNA elimination mediated by MTREC and Rrp6 (Harigaya et al., 2006; Lee et al., 2013), Mmi1 recruits CPF to promote facultative heterochromatin assembly and meiotic gene silencing.
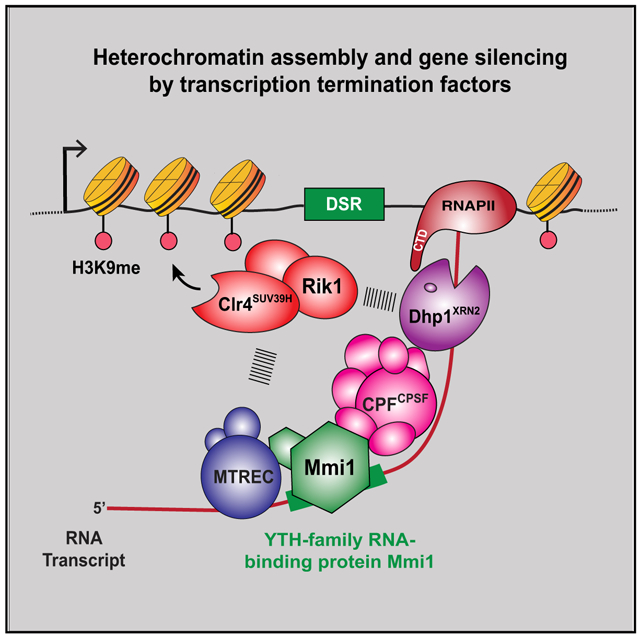
How does CPF recruitment by Mmi1 facilitate heterochromatin formation and gene control? Mmi1 and CPF are required for RNAPII pileups within the gene body and coincide with Dhp1XRN2. The CPF accessory factors Rna14CSTF3 and Pcf11Pcf11, which promote transcription termination (Birse et al., 1998), also colocalize with Mmi1 and Dhp1XRN2 at these sites. Moreover, loss of the CPF components that affect assembly of heterochromatin islands abolish Dhp1XRN2 localization and RNAPII pileups at non-canonical termination sites. Thus, Mmi1-mediated recruitment of CPF within the gene body is linked to non-canonical transcription termination, which determines the fate of transcripts and mediates heterochromatin assembly. We envision that Mmi1-associated termination (e.g., CPF and Dhp1XRN2) and RNA elimination (e.g., MTREC and Rrp6) factors silence target loci by coupling premature RNAPII termination to co-transcriptional RNA degradation and heterochromatin formation (Figure 7). The role of XRN2 in nuclear mRNA decay (Davidson et al., 2012; Miki and Großhans, 2013) and the requirement for Rrp6 in fail-safe termination (Lemay et al., 2014; Shah et al., 2014; Wagschal et al., 2012) are consistent with this model. CPF and other factors, such as MTREC, might coordinate to prepare transcripts for degradation by the 3′ → 5′ exoribonuclease Rrp6 (Lee et al., 2013; Sugiyama and Sugioka-Sugiyama, 2011) and RNAPII termination by the 5′ → 3′ exoribonuclease Dhp1XRN2 (Proudfoot, 2016), which also associate with ClrC to trigger facultative heterochromatin assembly (Chalamcharla et al., 2015; Zofall et al., 2012). In this role, CPF orchestrates the functions of RNA degradation and termination to recruit heterochromatin factors and silence target loci.
Figure 7. Model Showing Facultative Heterochromatin Assembly and Gene Silencing by Transcription Termination and RNA Degradation Factors.

Mmi1 binds target transcripts and recruits factors involved in RNA decay, such as MTREC, as well as 3′ end processing and transcription termination factors, such as CPF, to couple non-canonical termination of RNAPII with RNA degradation and recruitment of heterochromatin assembly factors. RNA cleavage by CPF and other factors may provide an entry site for MTREC to prepare transcripts for degradation by the 3′ →5′ exoribonuclease activity of the nuclear exosome Rrp6 (not shown) and by 5′ → 3′ exoribonuclease Dhp1XRN2, required for RNAPII termination. RNA degradation factors, in particular MTREC, and termination proteins, including Dhp1XRN2, recruit the Clr4SUV39H -containing ClrC complex to trigger RNAi-independent heterochromatin assembly. The ClrC subunit Rik1, which has similarity to a CPSF subunit and is loaded concomitantly with RNAPII transcription of target loci (Zhang et al., 2008), likely plays an important role in linking termination and degradation factors to the recruitment of Clr4 SUV39H.
Beyond meiotic genes, CPF is required for H3K9me and silencing at Island 3, which also requires Taz1-Shelterin for heterochromatin assembly (Zofall et al., 2016). It is unlikely that the two nucleation mechanisms are functionally connected because mutations in CPF have little effect on H3K9me at other islands requiring Taz1-Shelterin, which is known to directly associate with ClrC (van Emden et al., 2019; Wang et al., 2016; Zofall et al., 2016). Particularly exciting is the finding that CPF is required for the assembly of H3K9me peaks at various genes regulated by Clr4 SUV39H in response to low temperature. Thus, CPF might be a key factor in our recently described rheostatlike mechanism, where transcriptional upregulation provides feedback to prevent uncontrolled gene expression through recruitment of Clr4SUV39H (Gallagher et al., 2018).
The Role of CPF in Constitutive Heterochromatin Assembly
Two overlapping pathways mediate constitutive heterochromatin assembly, and both rely on RNAPII transcription. In one pathway, RNAi utilizes RNAPII transcripts to target H3K9me (Reyes-Turcu and Grewal, 2012). In the other, Dhp1XRN2 and Rrp6 promote RNAi-independent transcription-coupled heterochromatin assembly (Chalamcharla et al., 2015; Reyes-Turcu et al., 2011; Tucker et al., 2016). We find that CPF is not required for siRNA production and acts parallel to RNAi machinery. CPF localizes to pericentromeric regions harboring loci encoding the lncRNAs nam5/nam6/nam7 (this study), which are bound by Mmi1 (Touat-Todeschini et al., 2017). Mmi1 likely recruits CPF to the pericentromeric lncRNAs, as it does at meiotic genes. However, it is also possible that additional factors, such as the RNA binding protein Seb1, implicated in heterochromatic silencing (Parsa et al., 2018), are involved. In this regard, we note that Seb1 interacts with both CPF and Ser2-phosphorylated RNAPII (Larochelle et al., 2018; Lemay et al., 2016; Wittmann et al., 2017).
Heterochromatin formation at centromeric repeats also requires splicing factors. Inefficiently spliced cryptic introns within centromeric RNAs likely engage splicing machinery, which, in turn, recruits RNAi factors to trigger heterochromatin assembly (Bayne et al., 2008; Lee et al., 2013; Mutazono et al., 2017). However, splicing factors also interact with mammalian cleavage-polyadenylation specificity factor (CPSF) as well as XRN2 and contribute to pre-mRNA 3′ end processing and termination (Davidson and West, 2013; Kaneko et al., 2007; Kyburz et al., 2006; Millevoi et al., 2002). Therefore, it is possible that CPF provides a link between splicing machinery and factors involved in heterochromatin assembly.
CPF-Mediated Termination of Regulatory lncRNA and Gene Control
Transcription of adjacent lncRNAs has emerged as a major regulatory mechanism for controlling protein-coding genes in diverse eukaryotic species, including S. pombe (Hiriart and Verdel, 2013). Mmi1 has been shown to bind to nascent regulatory lncRNA transcripts to promote termination, preventing them from invading and repressing downstream genes (Lee et al., 2013; Shah et al., 2014; Touat-Todeschini et al., 2017). However, the mechanism by which Mmi1 promotes transcription termination of lncRNAs remained unknown. We find that Mmi1-mediated recruitment of CPF is critical for this process. Indeed, loss of CPF components causes readthrough transcription of lncRNAs, leading to repression of protein-coding genes, similar to mmi1Δ (Chatterjee et al., 2016; Sanchez et al., 2018; Shah et al., 2014; Touat-Todeschini et al., 2017). This function of CPF likely also involves Rrp6 and Dhp1XRN2, which show preferential enrichment at Mmi1 target lncRNAs (Lee et al., 2013; this study).
Our discovery assigns a crucial role to the conserved CPF complex in the control of gene expression in response to environmental and developmental signals. Indeed, lncRNAs have been implicated in this important gene-regulatory process in higher eukaryotes (Hiriart and Verdel, 2013). In this regard, we note that there are several examples in mammals of lncRNA initiating from upstream regions and silencing downstream protein-coding genes (Guil and Esteller, 2012).
The Role of CPF in Heterochromatin Assembly Has Widespread Implications
Although RNAPII transcription plays an important role in heterochromatin assembly in various systems, RNAi is dispensable for heterochromatin formation in mammalian somatic cells and other systems, such as Neurospora crassa (Chan and Wong, 2012; Freitag et al., 2004). Thus, RNAi-independent heterochromatic silencing mechanisms involving CPF are likely applicable to other systems. Indeed, pre-mRNA 3′ end processing factors that promote transcription termination are known to affect gene-regulatory processes (Cui et al., 2008; Liu et al., 2010; Wilusz and Spector, 2010). In budding yeast, the Nrd1/Nab3/Sen1 (NNS) termination complex is linked to heterochromatic silencing (Vasiljeva et al., 2008). However, NNS-like termination is not conserved in fission yeast (Larochelle et al., 2018). Because transcription termination in fission yeast and higher eukaryotes relies predominantly on CPF and Dhp1XRN2, these factors are particularly well positioned to have an evolutionarily conserved role in heterochromatin-mediated gene control.
We propose that a “molecular sensor” comprising pre-mRNA 3′ end processing and termination factors targeted to non-canonical sites specifies targets of heterochromatin assembly. This mechanism likely originated to recognize transposons by exploiting the lack of normal termination signals in these invading foreign elements and silence them through heterochromatin assembly. However, it has been co-opted to regulate gene expression. Termination machinery recruited to non-canonical sites at heterochromatin regions may differ from the machinery assembled at canonical sites. Indeed, transcription termination is complex and involves numerous protein-RNA complex assemblies (Porrua and Libri, 2015; Proudfoot, 2016; Shi and Manley, 2015).
Finally, valuable insight into human diseases such as Friedreich ataxia, in which the expansion of triplet repeats is linked to defects in RNAPII elongation and the appearance of H3K9me (Yandim et al., 2013), may be gained from our findings. Indeed, it would be interesting to explore whether heterochromatin induced by pathogenic repeat expansions also involves termination factors such as CPF.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Correspondence and requests for materials and reagents should be addressed to the Lead Contact, Shiv Grewal (grewals@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. pombe yeast strains used in this study are listed in Table S4. Standard yeast culturing and genetic manipulation methods were used. Strains were generated from crosses or yeast transformations. All experiments were performed in yeast extract rich medium supplemented with adenine (YEA) at 32°C unless otherwise indicated. For 18°C experiments, cells were grown to and maintained at mid-log phase for 3 days at 18°C, as previously described (Gallagher et al., 2018).
METHOD DETAILS
Genetic screen for mutants affecting RNAi-independent heterochromatin assembly
Exponentially growing yeast cells were mutagenized using either UV irradiation (150 J per m2) or N-methyl-N’-nitro-N-nitrosoguanidine (MNNG, M0527, TCI) at 0.5 mg per mL concentration. UV mutagenized cells were recovered on YEA medium at 30°C to obtain 23,702 single colonies. Colonies were replica plated onto minimal media (2% glucose, yeast nitrogen base without amino acid, 0.1 mg/mL adenine, 0.05 mg/mL histidine, 0.05 mg/mL uracil, and 0.05 mg/mL leucine) with 5FOA to initially screen for candidates defective in Is3::ura4+ silencing. Viable MNNG-mutagenized cells from 3,043 single colonies were plated directly onto minimal media with 5FOA. For secondary screening to find candidates with defects in Is3::ura4+ but not mat2::ade6+ silencing, colonies which grew on 5FOA medium were plated onto minimal media, minimal media lacking uracil, minimal media with 5FOA, and low adenine media (3% glucose and 0.5% yeast extract) plates. Colonies that (1) did not grow on minimal media, (2) did not grow on minimal media lacking uracil, and/or (3) grew as white colonies on low adenine media were discarded. The remaining candidates (3 from UV mutagenesis, 26 from MNNG mutagenesis) were backcrossed three times with a non-mutagenized strain. Only candidates which segregated 2:2 based on the secondary screening selection criteria above were considered for mutational analyses by next-generation sequencing. Genomic DNA was isolated from three wild-type and three mutant backcross segregants and sequenced using the NextSeq 500 platform (Illumina). Trimmed reads were aligned using Burrows-Wheeler Aligner (BWA) with default parameters to the S. pombe v2.29 reference genome and duplicated reads were removed using Picard Tools MarkDuplicates. Deduplicate aligned reads were used by Samtools mpileup (Li, 2011) to compute genotype likelihoods and to produce Binary Call Format (BCF) files. Single nucleotide polymorphisms (SNPs), insertions, and deletions were called from the BCF files using Bcftools (Li, 2011) to produce Variant Call Format (VCF) files after which the variants were mapped to features and annotated using SnpEff (Cingolani et al., 2012) to produce annotated VCFs. The annotations produced by SnpEff include the name of the gene affected, the expected amino acid change in the case of variants in coding regions, and an assessment of the probable impact of the variant (HIGH, MODERATE, LOW, MODIFIER). Variants with either HIGH or MODERATE impact include those that introduce premature stop codons, frameshifts or single amino acid changes and were selected for further consideration if they were found in the mutant strain but not in the matched wild-type.
Dilution spotting assays
Mid-log phase yeast cells were serially diluted onto agar medium and incubated at 18°C or 32°C for 2-10 days. 5FOA assays were performed using minimal medium (2% glucose, yeast nitrogen base without amino acid, 0.1 mg/mL adenine, 0.05 mg/mL histidine, 0.05 mg/mL uracil, and 0.05 mg/mL leucine). For experiments using Clr4 expression plasmid (pclr4+), minimal medium lacking leucine was used.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed essentially as described previously (Cam et al., 2005). Briefly, cells were grown in YEA medium to mid-log phase. For H3K9me2 ChIPs, cells were fixed with 1% formaldehyde for 20 min at room temperature. For all other ChIPs, cells were additionally fixed with dimethyl adipimidate (Thermo Fisher Scientific) for 45 min at room temperature. Cell pellets were suspended into 400μL of ChIP lysis buffer (50mM HEPES pH 7.5, 140mM NaCl, 1mM EDTA, 1% Triton, 0.1% deoxycholate) with glass beads and lysed using a bead-beater. Genomic DNA in the lysate was sonicated using a Bioruptor (Diagenode) for 12-cycles on medium power setting (30 s on, 30 s off) at 4°C. Cellular debris was removed by centrifugation at 1,500 g for 5 minutes at 4°C. Lysate supernatant was brought up to 1mL volume using ChIP lysis buffer and was precleared using 20μL of Protein A/G-plus agarose slurry (Santa Cruz) with rotation for 1 hour at 4°C. Precleared lysates were centrifuged at 1,000 g for 1 minute and lysate supernatant was transferred to a new tube for subsequent immunoprecipitation. 50μL of lysate was reserved as whole-cell extract input control. Anti-H3K9me2 (ab115159, Abcam), anti-GFP (ab290, Abcam), and RNAPII phospho S2 (ab5095, Abcam) antibodies were used for immunoprecipitation. Antibodies were recovered using Protein A/G-plus agarose (Santa Cruz). Immunoprecipitated DNA or input DNA was analyzed by performing qPCR, DNA microarray or Illumina sequencing.
ChIP-qPCR analyses were performed using iTaq Universal SYBR Green Supermix (Biorad). Delta-delta Ct normalization used leu1 as the reference gene. Oligonucleotides used for ChIP-qPCR are listed in Table S5. For ChIP-chip analyses, experiments were performed as previously described (Cam et al., 2005). Briefly, DNA was labeled with Cy5/Cy3 and hybridized to a custom 4x44K oligonucleotide microarray according to Agilent’s recommended procedure. Ratios of IP over input were calculated. A moving window average of three probes was assessed for each probe. For ChIP-seq analyses, sequencing libraries were generated using NEBNext Ultra II DNA library prep kit for Illumina (NEB) according to the manufacturer’s protocol. Samples were multiplexed and single-end reads were sequenced on the Illumina MiSeq or NextSeq 500 platform.
For generating H3K9me2 ChIP-seq coverage data, adaptor-trimmed reads were aligned to the S. pombe v2.29 reference genome using BWA-MEM. Correction for GC-content bias and input normalization was performed using the Deeptools suite(Ramírez et al., 2016) functions correctGCbias and bamCompare. Coverage plots were generated using the plotProfile function. For all other ChIP-seq coverage data, reads were trimmed using Trimmomatic (Bolger et al., 2014) and aligned with BWA to the S. pombe v2.29 reference genome with defaults and the “-M” option for compatibility with Picard tools to produce BAM files. Aligned reads were deduplicated using Picard tools and used for normalization to reads per million (RPM) mapped. Normalized samples were then subject to input normalization by input subtraction from the immunoprecipitation sample. Bedtools was used to produce normalized coverage plot bedgraph files. Fold-enrichment bedgraphs were generated using MACS2.
Median distribution profiles and peak density computations were performed by first constructing a profile from deduplicated bedgraphs using a spline function to approximate the ChIP density over the region-of-interest with a uniform resolution of 100 data points regardless of feature length. Next, each profile was input normalized by input subtraction. Median distribution profiles were generated by taking the median of each of the 100 profile points for each set of features-of-interest. Peak density computation per feature were computed by taking the 99% quantile value for each feature profile. In cases where the features were coding sequence (CDS), CDS coordinates were obtained from the PomBase database S. pombe annotations.
The locations of Dhp1 or Iss1FIP1-specific peaks were determined from normalized BedGraphs by identifying peaks in both the ChIP and input samples using an in-house script. Peaks that were found in both the ChIP and input bedGraphs were removed and the positions of the novel peaks that remained, those found only in the ChIP bedGraphs, were deemed to be Dhp1XRN2 or Iss1FIP1-specific. The positions of these novel peaks were used in downstream analyses. Uniformly spaced values (n = 2,000) of the normalized ChIP signal covering a region from 1 kb upstream to 1 kb downstream of each Dhp1XRN2 or Iss1FIP1 peak were plotted as the rows of a heatmap of width 2,000.
Statistical tests were performed using the one-tailed Wilcoxon non-parametric tests (paired or unpaired as indicated in the figure legends). Boxplots show the distribution of the data with horizontal lines marking the median, boxes enclosing the first through third quartiles, and whiskers marking the extreme values. Median distribution and boxplots were generated using R scripts and the ggpubr library. Sequence-based analyses including the intersection of feature coordinates with bedgraphs were performed using the Bioconductor GenomicRanges library. Euler plots were generated using the R Eulerr library. Heatmaps were generated using the R ComplexHeatmap library.
H3K9me Establishment Assay
Strains lacking Clr4 were transformed with either LEU2-based pWH5 (empty vector) or pAK70 (pclr4+) plasmids by lithium acetate transformation. Cells were grown in minimal medium lacking leucine to maintain the plasmids. For H3K9me enrichment analyses, H3K9me2 ChIP was performed as described above using anti-H3K9me2 (ab115159, Abcam) antibody. For spotting assay to determine expression of otr1R::ura4+, cells were grown to mid-log phase and serially diluted, followed by spotting onto minimal medium -Leu or -Leu with counterselection drug 5FOA. Silencing of otr1R::ura4+ leads to cell growth on +5FOA medium.
Northern blot analysis
Total RNA was extracted from mid-log phase cells grown at 18°C for 3 days using the hot phenol method in equal volumes of AES buffer (50mM sodium acetate pH 5.3, 10mM DTA, 1% SDS) and acid-phenol. The mixture was incubated at 65°C and vortexed every minute for 5 minutes. Afterward, the slurry was transferred to Maxtract High Density tubes (QIAGEN) and RNA was purified by chloroform extraction. Purified RNA was precipitated using Glycoblue (Thermo Fisher Scientific), sodium acetate pH 5.3 and isopropanol. 5μg of total RNA per sample was loaded per lane in a 1% formaldehyde agarose gel. T7 MAXIscript kit (Ambion) was used to generate α-P32-UTP (PerkinElmer) labeled RNA probes and hybridizations were carried out using the NorthernMax kit (Ambion).
For probing centromeric small interfering RNAs (siRNAs), siRNAs were purified from exponentially growing cells with mirVana miRNA isolation kit (Thermo Fisher Scientific). 20μg of small RNAs (< 200nt) was resolved on 15% urea-PAGE and electro-transferred to HybondTM-N+ (Thermo Fisher Scientific) membrane in 0.5xTBE for 1 hour at 100V. After UV crosslinking the membrane was probed for siRNAs by hybridization with α-P32-UTP (PerkinElmer) labeled RNA probes (~50nt) corresponding to dg/dh sequence in UltraHyb-oligo hybridization buffer (Thermo Fisher Scientific).
RNA-seq
Total RNA was prepared from cells grown in YEA medium to mid-log phase at 18°C for 3 days using MasterPure Yeast RNA purification kit (Lucigen) and was treated with DNase I (Thermo Fisher Scientific) according to manufacturer recommendations before subsequent manipulations and analyses. Ribosomal RNAs were depleted using the Ribo-Zero Gold rRNA Removal Magnetic Kit (Yeast) (Illumina). Library construction was performed using the ScriptSeq v2 RNA-seq library preparation kit (Epicenter) according to the manufacturer’s instructions. Single-end sequencing was performed on the Illumina NextSeq 500 platform. For generating coverage data, adaptor-trimmed reads were aligned to the S. pombe v2.29 reference genome using Tophat2 (Kim et al., 2013). Minimum and maximum intron sizes were set to 30 and 817, respectively. Normalization by reads per kilobase per million (RPKM) and coverage plots were performed using the Deeptools suite functions bamCoverage and plotProfile (Ramírez et al., 2016). For differential analyses, reads were quality trimmed using Trimmomatic (Bolger et al., 2014) and aligned using the STAR aligner (Dobin et al., 2013) to produce files of uniquely mapping reads, normalized to reads per million (RPM). Aligned reads were assigned to transcripts on the basis of Pombase (Lock et al., 2019) annotations using Rsubread (Liao et al., 2013). DEGseq (Wang et al., 2010) was used to compute the log2 fold-change in transcript expression between experimental strains and previously published WT grown at 30°C (Gallagher et al., 2018). Only transcripts supported by at least 50 mapped reads and corresponding to mRNAs, non-coding RNA, and anti-sense RNA (5,680 total transcripts) were used for analyses.
Composite transcription readthrough analyses
Composite readthrough analyses were performed using 18°C STAR-aligned RNA-seq data. Areas of readthrough transcription were defined as those regions up to 1 kb downstream of an annotated transcript that were covered by reads mapped to the same strand as the upstream transcript but not overlapping with a downstream transcript on the same strand. Further, the junction between the end of the upstream transcript and the putative readthrough region was required to be spanned by at least one read and the trend from the junction to the end of the putative readthrough was required to be one of decreasing read-depth. Finally, the mean read-depth in the readthrough area was required to be less than that of the upstream transcript. From the pool of readthrough candidates, 25 of the best cases were selected by inspection for the construction of the composite plot. The traces in the plot represent the mean normalized read-depth, as reads per million (RPM). The read-depths for all strains are scaled to the level of expression of the upstream WT gene to allow accurate comparison of the degree of readthrough between samples. Readthrough areas that are shorter than 1 kb contribute to the mean trace only to the extent of their length.
Immunoprecipitation and western blotting
Yeast cells expressing epitope tagged proteins under the control of their native gene promoters were used for immunoprecipitation and western blot experiments as described previously (Zofall et al., 2012). Briefly, cells were lysed using a blender in 2X HC lysis buffer (300mM HEPES buffer pH 7.6, 100mM KCl, 2mM EDTA, 0.2% NP-40, 0.2mM DTT) containing protease inhibitor cocktail (11697498001, Roche) and 2mM PMSF. Lysate cleared of cellular debris was incubated with antibody-conjugated beads for immunoprecipitation. Afterward, beads were washed three times with 1X HC lysis buffer (150mM HEPES pH 7.6, 250mM KCl, 1mM EDTA, 1mM PMSF, 0.1% NP-40, 1 tablet protease inhibitor cocktail per 100mL volume) and twice with AC200 wash buffer (20mM HEPES pH 7.6, 1mM EGTA, 200mM KCl, 2mM MgCl2 0.1% NP-40, 1mM PMSF, 1 tablet protease inhibitor cocktail per 100mL volume). Protein elution was performed using 0.2M glycine (pH 2). Eluted protein was recovered by TCA precipitation and dissolved into SDS sample buffer prior to resolution in a polyacrylamide gel. Antibody anti-GFP (gta20, Chromotek) was used for immunoprecipitation. Primary antibodies anti-FLAG M2 (F3165, Sigma) and anti-GFP (7.1 and 13.1, Roche) were used for western blotting analyses. Secondary detection used HRP-conjugated antibody anti-mouse (NA931V, GE).
Fluorescence live microscopy
Strains were grown overnight at 30°C in minimal media to mid-log phase. 1 mL culture was washed and resuspended in 50 μl of fresh minimal media. 4 μL of resuspended cells was mounted on a 2% agarose pad formed on a glass slide. Fluorescent signals corresponding to CFP-Mmi1 (represented in red) and Iss1-GFP (green) in the cells were imaged using SoftWoRx software on a DeltaVision Elite fluorescence microscope (Applied Precision, GE Healthcare) with Olympus 100x/1.40 objective. Optical Z sections were acquired (0.2 μm step size, 20 sections) for each field. Images were deconvolved and all Z-stacks were projected into a single-plane as maximum-intensity projections. Fiji (ImageJ) was used for processing the images and assembling line plot profiles. Intensities were normalized to one of the maximum peaks. Pearson’s correlation coefficient (PCC) of colocalization was calculated using SoftWoRx software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical tests used are described in the figure legends or in the methods section. p values for group overlap of differentially expressed genes from RNA-seq data were calculated from cumulative hypergeometric distribution functions. p values for group comparisons of median ChIP-seq enrichment were calculated from non-parametric Wilcoxon signed rank tests (paired) or rank sum tests (unpaired).
DATA AND CODE AVAILABILITY
The accession numbers for the ChIP-seq and RNA-seq data reported in this paper are GEO: GSE123137 and GSE123138.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3K9me2 | Abcam | ab115159; RRID: AB_10903018 |
| anti-GFP (polyclonal ChIP-grade) | Abcam | ab290; RRID: AB_303395 |
| RNAPII phospho S2 | Abcam | ab5095; RRID: AB_304749 |
| anti-GFP (coupled to agarose beads) | Chromotek | gta20; RRID: AB_2631357 |
| anti-GFP clones 7.1 and 13.1 | Roche | 11814460001; RRID: AB_390913 |
| anti-FLAG M2 | Sigma | F3165: RRID: AB_259529 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ribo-Zero Gold rRNA Removal kit (Yeast) | Illumina | MRZY1324 |
| iTaq Universal SYBR Green Supermix | BioRad | 1725122 |
| Dimethyl adipimidate (DMA) | Thermo Fisher Scientific | 20660 |
| Formaldehyde, 37% solution | VWR | 97064-604 |
| Protein A/G PLUS-Agarose | Santa Cruz | sc-2003 |
| iTaq Universal SYBR Green Supermix | BioRad | 1725122 |
| N-methyl-N’-nitro-N-nitrosoguanidine | TCI | M0527 |
| PureLink RNase A | Thermo Fisher Scientific | 12091039 |
| Proteinase K solution | Thermo Fisher Scientific | AM2548 |
| Masterpure Yeast RNA purification kit | Lucigen | MPY03100 |
| QIAquick PCR Purification kit | QIAGEN | 28106 |
| MAXIscript T7 Transcription kit | Ambion | AM1312 |
| mirVana miRNA isolation kit | Thermo Fisher Scientific | AM1560 |
| 4-12% Bis-Tris PAGE | Thermo Fisher Scientific | NP0321BOX |
| HybondTM-N+ | Fisher Scientific | 45-000850 |
| α-P32-UTP | Perkin Elmer | BLU007H250uc |
| Critical Commercial Assays | ||
| NEBNext Ultra II DNA library prep kit for Illumina | New England Bio Labs | E7645S |
| ScriptSeq v2 RNA-seq library preparation kit | Epicenter | SSV21124 |
| Deposited Data | ||
| ChIP-seq | This study | GEO: GSE123137 |
| RNA-seq | This study | GEO: GSE123138 |
| Figure 3 WT 30°C | Gallagher et al., 2018 | GEO: GSE104547 |
| Figure S4A Rna14, Pcf11 ChIP-seq | Larochelle et al., 2018 | GEO: GSE115595 |
| Figure S5A Rna14, Pcf11 ChIP-seq | Larochelle et al., 2018 | GEO: GSE115595 |
| Figure S7 Rna14, Pcf11 ChIP-seq | Larochelle et al., 2018 | GEO: GSE115595 |
| Experimental Models: Organisms/Strains | ||
| S. pombe | Grewal lab | Table S4 |
| Oligonucleotides | ||
| ChIP-PCR, ChIP-qPCR, Northern blot probing | This study | Table S5 |
| Software and Algorithms | ||
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA-MEM | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/bwa.shtml |
| Bcftools | Li, 2011 | https://samtools.github.io/bcftools/bcftools.html |
| SnpEff | Cingolani et al., 2012 | http://snpeff.sourceforge.net/SnpEff_manual.html |
| Rsubread | Liao et al., 2013 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| ComplexHeatmap | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Deeptools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/content/list_of_tools.html |
| SoftWoRx | Applied Precision, GE Healthcare | http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/6.5.2/SoftWoRx.htm |
| Fiji | ImageJ | https://imagej.net/Using_Fiji |
Highlights.
CPF is required for RNAi-independent assembly of heterochromatin domains
YTH family protein Mmi1 loads CPF at non-canonical termination sites in gene bodies
Mmi1 and CPF promote Dhp1XRN2 and RNAPII pileup to trigger heterochromatin assembly
CPF is globally required for silencing of genes regulated by Clr4SUV39H
ACKNOWLEDGMENTS
We thank members of the Grewal laboratory for valuable discussions and suggestions and J. Barrowman for editing the manuscript. We also thank D. Folco and T. Sugiyama for strains, V. Chalamcharla for strains and helpful contributions, T. Andresson and C. Jenkins for help with analyses of mass spectrometry results, and H. Levin for sharing unpublished results. This study used the Helix Systems and Biowulf Linux cluster at the NIH. This work was supported by a postdoctoral research associate training fellowship from the National Institute of General Medical Sciences (1Fi2GM123947-01 to T.V.V.) and the Intramural Research Program of the NIH, National Cancer Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.107.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, and Kouzarides T (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Bayne EH, Portoso M, Kagansky A, Kos-Braun IC, Urano T, Ekwall K, Alves F, Rappsilber J, and Allshire RC (2008). Splicing factors facilitate RNAi-directed silencing in fission yeast. Science 322, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birse CE, Minvielle-Sebastia L, Lee BA, Keller W, and Proudfoot NJ (1998). Coupling termination of transcription to messenger RNA maturation in yeast. Science 280, 298–301. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S (2009). Progression through the RNA polymerase II CTD cycle. Mol. Cell 36, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, and Grewal SIS (2005). Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet 37, 809–819. [DOI] [PubMed] [Google Scholar]
- Casañal A, Kumar A, Hill CH, Easter AD, Emsley P, Degliesposti G, Gordiyenko Y, Santhanam B, Wolf J, Wiederhold K, et al. (2017). Architecture of eukaryotic mRNA 3′-end processing machinery. Science 358, 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalamcharla VR, Folco HD, Dhakshnamoorthy J, and Grewal SIS (2015). Conserved factor Dhp1/Rat1/Xrn2 triggers premature transcription termination and nucleates heterochromatin to promote gene silencing. Proc. Natl. Acad. Sci. USA 112, 15548–15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FL, and Wong LH (2012). Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 40, 11178–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D, Sanchez AM, Goldgur Y, Shuman S, and Schwer B (2016). Transcription of lncRNA prt, clustered prt RNA sites for Mmi1 binding, and RNA polymerase II CTD phospho-sites govern the repression of pho1 gene expression under phosphate-replete conditions in fission yeast. RNA 22, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, and Grewal SIS (2008). Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451, 734–737. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Allen MA, Larsen A, Macmorris M, Han M, and Blumenthal T (2008). Genes involved in pre-mRNA 3′-end formation and transcription termination revealed by a lin-15 operon Muv suppressor screen. Proc. Natl. Acad. Sci. USA 105, 16665–16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, and West S (2013). Splicing-coupled 3′ end formation requires a terminal splice acceptor site, but not intron excision. Nucleic Acids Res. 41, 7101–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Kerr A, and West S (2012). Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J. 31, 2566–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djupedal I, Portoso M, Spåhr H, Bonilla C, Gustafsson CM, Allshire RC, and Ekwall K (2005). RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 19, 2301–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag M, Lee DW, Kothe GO, Pratt RJ, Aramayo R, and Selker EU (2004). DNA methylation is independent of RNA interference in Neurospora. Science 304, 1939. [DOI] [PubMed] [Google Scholar]
- Gallagher PS, Larkin M, Thillainadesan G, Dhakshnamoorthy J, Balachandran V, Xiao H, Wellman C, Chatterjee R, Wheeler D, and Grewal SIS (2018). Iron homeostasis regulates facultative heterochromatin assembly in adaptive genome control. Nat. Struct. Mol. Biol 25, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, and Elgin SC (2007). Transcription and RNA interference in the formation of heterochromatin. Nature 447, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SIS, and Jia S (2007). Heterochromatin revisited. Nat. Rev. Genet 8, 35–46. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Guil S, and Esteller M (2012). Cis-acting noncoding RNAs: friends and foes. Nat. Struct. Mol. Biol 19, 1068–1075. [DOI] [PubMed] [Google Scholar]
- Gullerova M, and Proudfoot NJ (2008). Cohesin complex promotes transcriptional termination between convergent genes in S. pombe. Cell 132, 983–995. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Tanaka H, Yamanaka S, Tanaka K, Watanabe Y, Tsutsumi C, Chikashige Y, Hiraoka Y, Yamashita A, and Yamamoto M (2006). Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 442, 45–50. [DOI] [PubMed] [Google Scholar]
- Hiriart E, and Verdel A (2013). Long noncoding RNA-based chromatin control of germ cell differentiation: a yeast perspective. Chromosome Res. 21, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiriart E, Vavasseur A, Touat-Todeschini L, Yamashita A, Gilquin B, Lambert E, Perot J, Shichino Y, Nazaret N, Boyault C, et al. (2012). Mmi1 RNA surveillance machinery directs RNAi complex RITS to specific meiotic genes in fission yeast. EMBO J. 31, 2296–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Rozenblatt-Rosen O, Meyerson M, and Manley JL (2007). The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 21, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, and Murakami Y (2005). RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science 309, 467–469. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc A, Zaratiegui M, Nora E, and Martienssen R (2008). RNA interference guides histone modification during the S phase of chromosomal replication. Curr. Biol 18, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyburz A, Friedlein A, Langen H, and Keller W (2006). Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol. Cell 23, 195–205. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, and Jenuwein T (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Larochelle M, Robert MA, Hébert JN, Liu X, Matteau D, Rodrigue S, Tian B, Jacques PE, and Bachand F (2018). Common mechanism of transcription termination at coding and noncoding RNA genes in fission yeast. Nat. Commun 9, 4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, and Grewal SI (2013). Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155, 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bähler J, and Bachand F (2014). The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat. Struct. Mol. Biol 21, 919–926. [DOI] [PubMed] [Google Scholar]
- Lemay JF, Marguerat S, Larochelle M, Liu X, van Nues R, Hunyadkürti J, Hoque M, Tian B, Granneman S, Bähler J, and Bachand F (2016). The Nrd1-like protein Seb1 coordinates cotranscriptional 3′ end processing and polyadenylation site selection. Genes Dev. 30, 1558–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011).A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Gaszner M, Allis CD, and Felsenfeld G (2001). Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 293, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Liu F, Marquardt S, Lister C, Swiezewski S, and Dean C (2010). Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 327, 94–97. [DOI] [PubMed] [Google Scholar]
- Lock A, Rutherford K, Harris MA, Hayles J, Oliver SG, Bähler J, and Wood V (2019). PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 47 (D1), D821–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS, Sandelin A, and Jensen TH (2016). Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell 64, 520–533. [DOI] [PubMed] [Google Scholar]
- Miki TS, and Großhans H (2013). The multifunctional RNase XRN2. Biochem. Soc. Trans 41, 825–830. [DOI] [PubMed] [Google Scholar]
- Millevoi S, Geraghty F, Idowu B, Tam JL, Antoniou M, and Vagner S (2002). A novel function for the U2AF 65 splicing factor in promoting pre-mRNA 3′-end processing. EMBO Rep. 3, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2010). Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol 2, a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutazono M, Morita M, Tsukahara C, Chinen M, Nishioka S, Yumikake T, Dohke K, Sakamoto M, Ideue T, Nakayama JI, et al. (2017). The intron in centromeric noncoding RNA facilitates RNAi-mediated formation of heterochromatin. PLoS Genet. 13, e1006606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, and Grewal SIS (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Noma K, Allis CD, and Grewal SI (2001). Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Parsa JY, Boudoukha S, Burke J, Homer C, and Madhani HD (2018). Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Genes Dev. 32, 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, and Libri D (2015). Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol 16, 190–202. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ (2016). Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 352, aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, and Winston F (2012). Chromatin and transcription in yeast. Genetics 190, 351–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reyes M, and Hampsey M (2007). Role for the Ssu72 C-terminal domain phosphatase in RNA polymerase II transcription elongation. Mol. Cell. Biol 27, 926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, and Grewal SIS (2012). Different means, same end-heterochromatin formation by RNAi and RNAi-independent RNA processing factors in fission yeast. Curr. Opin. Genet. Dev 22, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Zhang K, Zofall M, Chen E, and Grewal SIS (2011). Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat. Struct. Mol. Biol 18, 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Manley JL (2009). Transcription termination by nuclear RNA polymerases. Genes Dev. 23, 1247–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Shevchenko A, Schaft D, Thomas H, Stewart AF, and Shevchenko A (2004). A comparative analysis of an orthologous proteomic environment in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe. Mol. Cell. Proteomics 3, 125–132. [DOI] [PubMed] [Google Scholar]
- Sadaie M, Iida T, Urano T, and Nakayama J (2004). A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J. 23, 3825–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AM, Shuman S, and Schwer B (2018). RNA polymerase II CTD interactome with 3′ processing and termination factors in fission yeast and its impact on phosphate homeostasis. Proc. Natl. Acad. Sci. USA 115, E10652–E10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Wittmann S, Kilchert C, and Vasiljeva L (2014). lncRNA recruits RNAi and the exosome to dynamically regulate pho1 expression in response to phosphate levels in fission yeast. Genes Dev. 28, 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Manley JL (2015). The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 29, 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, and Sugioka-Sugiyama R (2011). Red1 promotes the elimination of meiosis-specific mRNAs in vegetatively growing fission yeast. EMBO J. 30, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro S, Asano T, Kanoh J, and Ishikawa F (2013). Transcription-induced chromatin association of RNA surveillance factors mediates facultative heterochromatin formation in fission yeast. Genes Cells 18, 327–339. [DOI] [PubMed] [Google Scholar]
- Touat-Todeschini L, Shichino Y, Dangin M, Thierry-Mieg N, Gilquin B, Hiriart E, Sachidanandam R, Lambert E, Brettschneider J, Reuter M, et al. (2017). Selective termination of lncRNA transcription promotes heterochromatin silencing and cell differentiation. EMBO J. 36, 2626–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JF, Ohle C, Schermann G, Bendrin K, Zhang W, Fischer T, and Zhang K (2016). A novel epigenetic silencing pathway involving the highly conserved 5′-3′ exoribonuclease Dhp1/Rat1/Xrn2 in Schizosaccharomyces pombe. PLoS Genet. 12, e1005873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Emden TS, Forn M, Forné I, Sarkadi Z, Capella M, Martín Caballero L, Fischer-Burkart S, Brönner C, Simonetta M, Toczyski D, et al. (2019). Shelterin and subtelomeric DNA sequences control nucleosome maintenance and genome stability. EMBO Rep. 20, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V, Legros P, van der Sar SJ, Yvert G, Toda K, Le Bihan T, Watanabe Y, Hardwick K, and Bernard P (2014). CPF-associated phosphatase activity opposes condensin-mediated chromosome condensation. PLoS Genet. 10, e1004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Kim M, Terzi N, Soares LM, and Buratowski S (2008). Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol. Cell 29, 313–323. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, et al. (2012). Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 150, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, and Zhang X (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. [DOI] [PubMed] [Google Scholar]
- Wang J, Cohen AL, Letian A, Tadeo X, Moresco JJ, Liu J, Yates JR 3rd, Qiao F, and Jia S (2016). The proper connection between shelterin components is required for telomeric heterochromatin assembly. Genes Dev. 30, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, and Spector DL (2010). An unexpected ending: noncanonical 3′ end processing mechanisms. RNA 16, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann S, Renner M, Watts BR, Adams O, Huseyin M, Baejen C, El Omari K, Kilchert C, Heo DH, Kecman T, et al. (2017). The conserved protein Seb1 drives transcription termination by binding RNA polymerase II and nascent RNA. Nat. Commun 8, 14861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Shichino Y, Tanaka H, Hiriart E, Touat-Todeschini L, Vavasseur A, Ding DQ, Hiraoka Y, Verdel A, and Yamamoto M (2012). Hexanucleotide motifs mediate recruitment of the RNA elimination machinery to silent meiotic genes. Open Biol. 2, 120014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Takayama T, Iwata R, and Yamamoto M (2013). A novel factor Iss10 regulates Mmi1-mediated selective elimination of meiotic transcripts. Nucleic Acids Res. 41, 9680–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandim C, Natisvili T, and Festenstein R (2013). Gene regulation and epigenetics in Friedreich’s ataxia. J. Neurochem. 126 (Suppl 1), 21–42. [DOI] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, and Grewal SIS (2008). Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol 15, 381–388. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhu J, Schermann G, Ohle C, Bendrin K, Sugioka-Sugiyama R, Sugiyama T, and Fischer T (2015). The fission yeast MTREC complex targets CUTs and unspliced pre-mRNAs to the nuclear exosome. Nat. Commun 6, 7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, and Grewal SIS (2009). Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461, 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, Zhang K, Rubin C, and Grewal SIS (2012). RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science 335, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Smith DR, Mizuguchi T, Dhakshnamoorthy J, and Grewal SIS (2016). Taz1-Shelterin Promotes Facultative Heterochromatin Assembly at Chromosome-Internal Sites Containing Late Replication Origins. Mol. Cell 62, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the ChIP-seq and RNA-seq data reported in this paper are GEO: GSE123137 and GSE123138.