

Abstract
Soluble methane monooxygenase (sMMO) is a multicomponent metalloenzyme that catalyzes the conversion of methane to methanol at ambient temperature using a nonheme, oxygen-bridged dinuclear iron cluster in the active site. Structural changes in the hydroxylase component (sMMOH) containing the diiron cluster caused by complex formation with a regulatory component (MMOB) and by iron reduction are important for the regulation of O2 activation and substrate hydroxylation. Structural studies of metalloenzymes using traditional synchrotron-based X-ray crystallography are often complicated by partial X-ray-induced photoreduction of the metal center, thereby obviating determination of the structure of the enzyme in pure oxidation states. Here microcrystals of the sMMOH:MMOB complex from Methylosinus trichosporium OB3b were serially exposed to X-ray free electron laser (XFEL) pulses, where the [35 fs duration of exposure of an individual crystal yields diffraction data before photoreduction-induced structural changes can manifest. Merging diffraction patterns obtained from thousands of crystals generates radiation damage free, 1.95 Å resolution crystal structures for the fully oxidized and fully reduced states of the sMMOH:MMOB complex for the first time. The results provide new insight into the manner by which the diiron cluster and the active site environment are reorganized by the regulatory protein component in order to enhance the steps of oxygen activation and methane oxidation. This study also emphasizes the value of XFEL and serial femtosecond crystallography (SFX) methods for investigating the structures of metalloenzymes with radiation sensitive metal active sites.
Graphical Abstract
INTRODUCTION
The soluble form of methane monooxygenase (sMMO) isolated from methanotrophic bacteria catalyzes the oxygenation of methane to methanol at room temperature without over-oxidation.1
This enzyme, and a structurally unrelated membrane-bound methane monooxygenase (pMMO) produced by methanotrophs when the solution copper to biomass ratio exceeds ~ 5.7 μmol/g protein, prevent the atmospheric egress of a large fraction of the global production of biogenic methane.2–3 The significant contribution of methane in the atmosphere to global warming is a cause of current concern, highlighting the importance of sMMO and pMMO to economic, health, and societal policies.4–5 The remarkable catalytic prowess of these enzymes also stimulates research into the generation of biomimetic synthetic catalysts for gas-to-liquid conversion, as well as the bioengineering of novel pathways into organisms to make value-added products from methane.6–7
The sMMO enzyme consists of three protein components: a 245 kDa (αβγ)2 hydroxylase (MMOH), a 37 kDa FAD and Fe2S2 cluster-containing reductase (MMOR), and a 15 kDa regulatory protein (MMOB).1, 8–11 The active site is buried deep within sMMOH and contains an oxygen-bridged dinuclear FeIII cluster in which the irons are bridged by two hydroxo moieties and a carboxylate from Glu144.12–15 After reduction, the diiron cluster functions to activate O2 and insert an oxygen atom into a highly stable (105 kcal/mol bond dissociation energy) C-H bond of methane.8, 16
Although chemically reduced sMMOH can carry out the oxygenation chemistry alone,17 the reaction only proceeds at a physiologically relevant rate when sMMOH is complexed with MMOB.8, 18–19 Many regulatory functions of MMOB have been discovered (Scheme 1),20 but its most important effects are to decrease the redox potential of the diiron cluster by 132 mV, accelerate O2 binding by 1000-fold, increase the turnover number 150-fold, and tune sMMOH to selectively bind and oxygenate methane over other more easily oxidized hydrocarbons.1, 10, 19, 21 Indeed, the interaction of sMMOH with MMOB is so central to sMMO catalysis that the chemistry catalyzed by this remarkable enzyme cannot be fully appreciated without also elucidating all of the regulatory functions of MMOB.
Scheme 1.
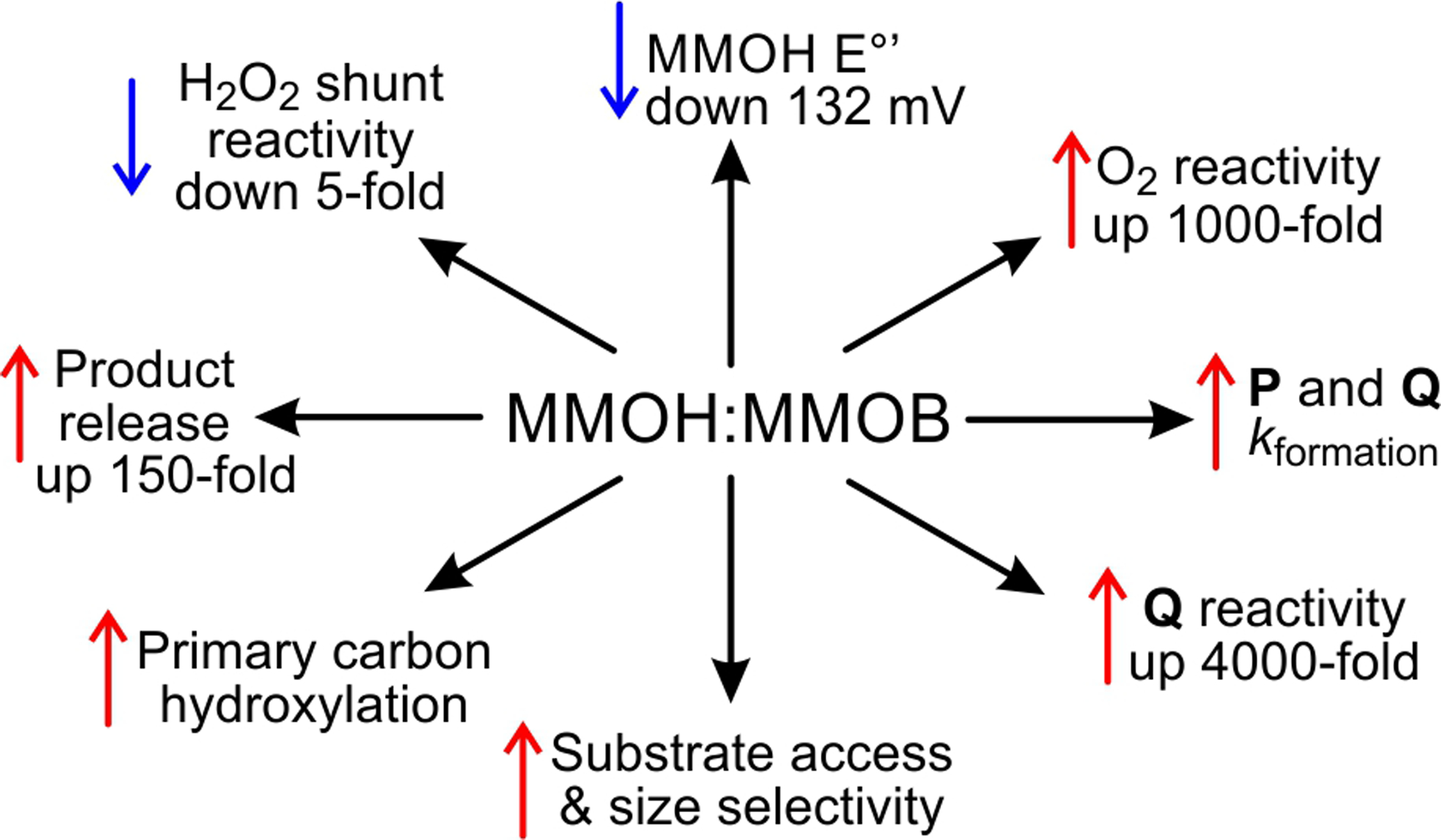
MMOB Serves Many Roles that Affect sMMO Catalysis when Complexed with sMMOH.
The complex between MMOB and sMMOH has been structurally characterized by both spectroscopic and crystallographic approaches in order to understand the mechanism of regulation by MMOB.8, 22–28 The core structural region of MMOB binds to the surface of sMMOH approximately 12 Å above the diiron cluster. An extended 30-residue N-terminal tail region of MMOB is disordered in solution, but it was observed to bind to the sMMOH surface in the structure of the oxidized sMMOH:MMOB complex of sMMO isolated from Methylococcus capsulatus Bath (Mc Bath).24, 26, 28 These structural observations have been supported by transient kinetic and mutagenic studies, which have shown that the core region and both the N- and C-terminal regions of MMOB affect the rate constants of steps throughout the reaction cycle.10, 29–31
Despite the clues provided by the structural and biochemical studies, the mechanisms by which MMOB exerts its many effects on oxygen activation and other aspects of the reaction are largely unknown. The absence of a structure of reduced sMMOH in complex with MMOB has held back our understanding of this regulation, as this is the key enzymatic state that is primed to react with O2. Structural studies of uncomplexed sMMOH have shown that reduction of the diiron cluster causes a shift in the position of Glu243, a monodentate ligand to Fe2 in the diferric cluster as illustrated in Scheme 2.13
Scheme 2.
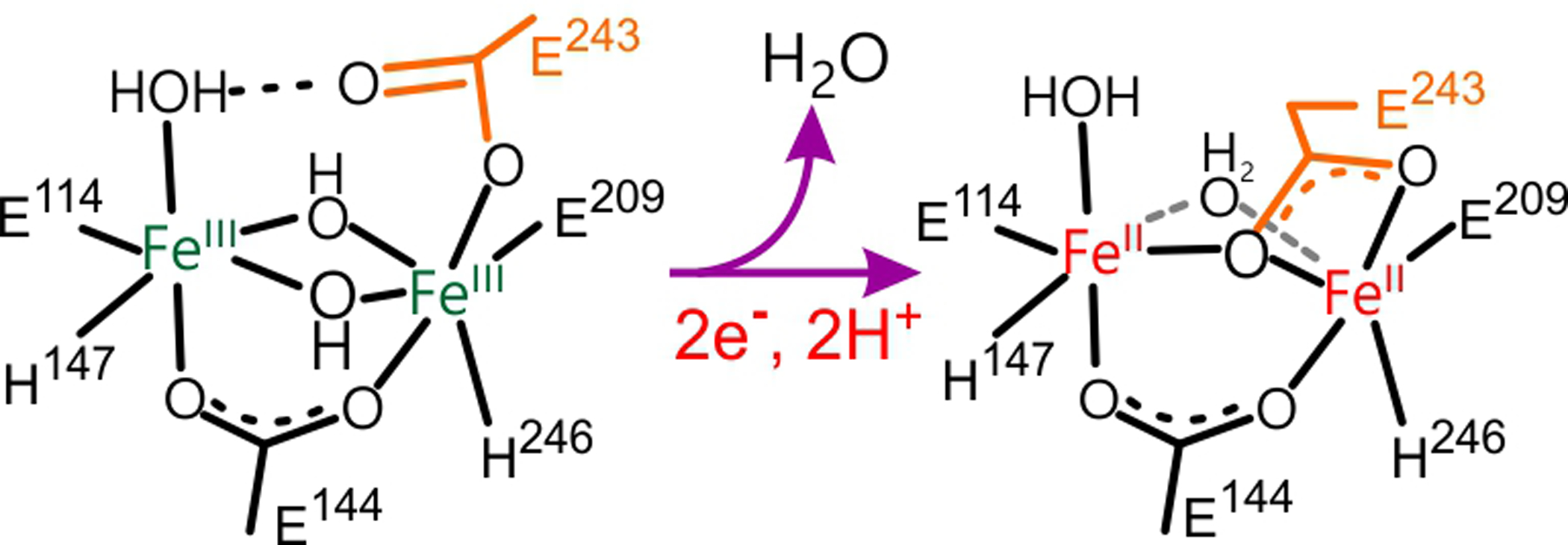
Carboxylate Shift in the Diiron Cluster of sMMOH Upon Reduction
In the shifted position, Glu243 bridges Fe1 and Fe2 via one of its carboxylate oxygens, thereby directly displacing one of the bridging solvents. The second bridging solvent bond is weakened, which presumably allows facile displacement by O2 to begin the oxygen activation process. Surprisingly, in the 2.9 Å resolution X-ray crystal structure of the diferric state of the Mc Bath sMMOH:MMOB complex, the position of Glu243 relative to Fe2 was also found to be shifted to the bridging position.28 The observation of a carboxylate shift in the diferric state of the sMMOH:MMOB complex led to the proposal that one mechanism by which MMOB acts is to cause this critical change in the cluster structure.28 However, a significant problem often encountered in determining the X-ray crystal structure of metalloenzymes is the ease with which the metal is reduced by hydrated electrons generated by the synchrotron X-ray beam.32–37 Partial reduction of the clusters might explain the observed shift in Glu243 without invoking a role for MMOB.
Recently, it has become possible to solve protein crystal structures by injecting a slurry of microcrystals into the beam of an X-ray free electron laser (XFEL).38–45 The instrumentation records a zero-rotation (“still”) diffraction pattern for each micro-crystal exposed to the beam, on the femtosecond time scale (between 10 and 35 fs in this study). The approach has three significant advantages: (a) the diffraction data are generated faster than the rate of reaction with beam-generated solvated electrons, (b) the diffraction data are obtained at physiologically relevant temperature rather than at 100 K as in typical synchrotron protein crystallography experiments, and (c) the potential exists to observe structures of reaction cycle intermediates if the enzyme reaction can be initiated in-crystallo at room temperature.46–52 In addition, concomitant collection of transition metal X-ray emission spectra (XES) has been recently demonstrated for different metalloenzymes at XFELs, enabling a parallel measurement of the metal oxidation state during the crystallographic experiment.41, 53–54
Here, we report new sMMOH:MMOB crystal structures of the highly active sMMO components isolated from Methylosinus trichosporium OB3b (Mt OB3b). Using XFEL data, room temperature structures of the homogeneous diferric and diferrous states of sMMOH:MMOB are reported at 1.95 Å resolution. The structures are used to define the changes effected in sMMOH upon binding of MMOB and to characterize the geometric state of the diiron cluster prepared to bind and activate O2.
EXPERIMENTAL PROCEDURES
Protein Isolation, Crystallization, and Crystal Reduction.
The sMMOH protein was isolated from frozen cell pellets of the native Mt OB3b cultured using methane as the sole carbon source as previously described.55 The MMOB protein was heterologously expressed in E. coli BL21(DE3) cells as previously described.24 sMMOH and MMOB were purified as previously described.56–57 A protein solution of 53 μM sMMOH and 106 μM MMOB in 25 mM MOPS pH 7 was used to screen for suitable crystallization conditions. Swissci MRC-2-drop plates (Molecular Dimensions) were used to setup crystallization experiments using the TTP mosquito® nanoliter pipetting robot. The plates were setup with a reservoir volume of 50 μl and drop volume of 400 nl. Rhombohedral crystals were observed in B7 condition of the Morpheus crystal screen (Molecular Dimensions). The microcrystals were further optimized using the additive screen (Hampton Research), with condition A11 producing 20–30 μm long bipyramidal shaped crystals within 3 days of incubation at room temperature. Larger volume sMMOH:MMOB complex crystals were aerobically grown in Cryschem M sitting drop vapor diffusion plates (Hampton Research), with a 30 μl drop volume (22.5 μl protein solution plus 7.5 μl reservoir solution) against a reservoir volume of 500 μl. The final crystallization condition is as follows: 100 mM HEPES/MOPS pH 7.5, 30 mM NaI, 30 mM NaBr, 30 mM NaF, 20 % (v/v) glycerol, 10 % (w/v) PEG 4000, and 10 mM FeCl3. The crystals were harvested by individually pooling the drops with bipyramidal crystals into a microfuge tube. Chemical reduction of the sMMOH:MMOB crystal slurry was carried out in an anaerobic glove box. The crystal slurry was incubated in reservoir solution containing 46 mM dithionite, 15 mM methyl viologen, and 1 mM phenosafranine for 12 h at room temperature. The crystal slurry primed for in-crystallo re-oxidation experiments was reduced as above before clearing the crystal slurry of any chemical reductants. This was accomplished by pelleting the chemically reduced crystal slurry in a benchtop centrifuge followed by removal of the chemical reductant-containing supernatant solution. The loosely packed crystal pellet was subsequently resuspended in fresh, anaerobic, FeCl3-free reservoir solution. This step was repeated 5 times to dilute the resulting concentration of chemical reductants in the crystal slurry solution used for re-oxidation experiments.
Data Collection and Model Refinement.
The crystals of oxidized sMMOH:MMOB were first tested for XFEL compatibility at SACLA, Japan, using the grease matrix extruder setup.58–59 Crystals of sMMOH:MMOB were mixed with synthetic grease and injected using a 150 μm diameter nozzle and a flow rate of 20–80 μl/min using an HPLC pump. X-ray diffraction was generated by <10 fs long X-ray pulses at 10.9 keV with a pulse energy of around 0.32 mJ, 30 Hz repetition rate and a beam size of 2×2 μm (FWHM). The forward scattering was recorded on a Octal MPCCD detector located 100 mm downstream of the X-ray interaction point. Diffraction images from 7469 crystals were merged to achieve a resolution of 2.24 Å.
Data sets were then collected at the macromolecular femtosecond crystallography (MFX) experimental hutch at LCLS.60 The drop on tape (DOT) method was used to deliver sMMOH:MMOB crystals into the XFEL X-ray interaction region.53 In short, micro-crystal suspensions were loaded in gas-tight Hamilton syringes under inert gas conditions inside a glove box. The syringe was then connected via a single silica capillary to the sample reservoir inside the He-enclosure of the DOT setup. The crystal suspension was dispensed at a flow rate of 8 μl/min using a syringe pump (KD Scientific). The sample was pushed into a 6 μl well underneath and at the focal point of an acoustic transducer, forming droplets of 2.5 – 4 nl volume. The droplets were deposited onto a Kapton tape belt running at a speed of 300 mm/s that transported the droplets into the X-ray interaction region.53 The He-enclosure around the setup ensured anaerobic conditions and limited air scattering of X-rays. The residence time of the droplets inside the He atmosphere (O2 level below 0.1%) was around 0.8 s.
For in-crystallo O2 incubation experiments, an in-situ gas incubation setup was used as previously described.53 In this case, the gas reaction chamber was continuously filled with pure O2 gas at a flow rate of 0.5 – 0.75 liter/min and monitored by an oxygen sensor in the chamber during the experiment. The travel speed of the Kapton belt transporting the sample droplets was reduced to 15 mm/s, thus resulting in a 4 s travel time of individual sample droplets through the O2 reaction chamber and an additional incubation time of 6 s before reaching the X-ray interaction region. To avoid dehydration of the sample droplets, the O2 gas and He gas in the direct environment of the incubation region was first routed through water-filled gas wash bottles, leading to a relative humidity of 35% around the sample.
X-ray diffraction was generated by ~35 fs long X-ray pulses at 9.5 keV with a pulse energy of around 4.0 mJ, 20 Hz repetition rate and a beam size of 4 μm (FWHM). The forward scattering was recorded on a Rayonix MX340 detector at 194 mm downstream of the X-ray interaction point. In addition, diffraction data was collected with the same DOT setup installed at the NCI beam line of the PAL-XFEL facility.61 Here ~35 fs long X-ray pulses at 9.4 keV with a pulse energy of 0.8 mJ, a beam size of 2.5 μm (FWHM) at 15 Hz repetition rate were used and diffraction was measured on a Rayonix MX200 detector located 130 mm downstream of the sample.
X-ray emission data was collected concomitant with diffraction data using a multi-crystal wavelength-dispersive hard X-ray spectrometer based on the von Hamos geometry.53, 62 Four Ge (440) crystals were placed 250 mm from the interaction point, with the center of the crystals at 75.41° with respect to the interaction point covering both Fe Kα lines. The focused X-ray emission signal was collected on an ePIX-100 detector (LCLS) or on a 500k Jungfrau detector (PAL-XFEL). Calibration was performed using aqueous solutions of 10 mM Fe(III)(NO3)3 and Fe(II)Cl2 as reference.
Diffraction images were processed with cctbx.xfel63 and DIALS.64 The MMOH:MMOB structure was solved using molecular replacement by PHASER,65 with truncated coordinates from the Mc Bath MMOH:MMOB complex crystal structure PDB: 4GAM used as the starting model. The structure was initially modeled in space group P41 with two complexes (eight polypeptide chains) per asymmetric unit; however, after running the program labelit.check_pdb_symmetry labelit.check_pdb_symmetry,66 it was realized that the model was consistent with space group P41212 with one complex (four polypeptide chains) per asymmetric unit. Therefore, we used cctbx to merge the P41 reflection file into the higher symmetry, and thus proceeded to refinement in P41212. Refinement of the structure was performed using PHENIX.refine67–68 and model building was done in COOT.69 Individual atomic coordinates, isotropic B factor and TLS parameters were used during refinement. Solvent molecules were added using PHENIX.refine,67–68 followed by manual addition. Table S1 was generated using PHENIX.table_one.67–68
Probing the Active Site Cavity in Protein Structures.
The interior active site cavity in the protein structures was identified with the MOLE 2.5 program.70 Heteroatoms were ignored in the calculation. A probe radius of 3.0 Å, interior threshold of 1.59 Å, and minimum depth of 5.0 Å were used to assess the cavity.
RESULTS
High Resolution Crystal Structures of the sMMOH:MMOB Complex in Diferric and Diferrous Oxidation States.
Crystals of the Mt OB3b sMMOH:MMOB complex in the resting diferric oxidation state were prepared as described in Experimental Procedures and found to diffract to 2.45 Å at the ID30B beamline, European Synchrotron Radiation Facility (data not shown). Microcrystals were then prepared as described in Experimental Procedures and analyzed by XFEL at room temperature. Microcrystals of the diferrous state of the sMMOH:MMOB complex were obtained by chemical reduction of the oxidized sMMOH:MMOB complex crystal slurry under anaerobic conditions. No notable decrease in the diffraction quality of these crystals was observed despite the rigorous reduction procedure. In order to test whether O2 is capable of initiating enzymatic turnover with the reduced sMMOH:MMOB protein complex in crystals, a microcrystal slurry of chemically reduced crystals was washed of excess reductant and then passed through a chamber filled with pure O2 for 4 s before injection into the XFEL beam. The total time from exposure to O2 to diffraction in the beam was 10 s. This sample behaved similarly to the diferric and diferrous crystal slurry. All three structures were refined to a resolution of 1.95 Å (Table S1). As a control, diffraction data were collected from a washed slurry of chemically reduced crystals prior to O2 incubation, and a structure was refined to a resolution of 2.0 Å (Table S1, “t=0 Diferrous”). In parallel, Fe Kα XES was collected for the diferric, diferrous and reoxidized samples.
The overall structure of the Mt OB3b sMMOH:MMOB complex is shown in Figure 1A for the diferric state (PDB: 6YD0). The asymmetric unit contains one molecule of MMOB bound to one αβγ protomer of the (αβγ)2 sMMOH protein. The quaternary structure of the protein complex is unaltered upon reduction of the diiron cluster (PDB: 6YDI) or following reaction of the diferrous complex with O2 (PDB: 6YDU). MMOB binds primarily to the α-subunit of sMMOH in the ‘canyon’ region at the interface of the sMMOH protomers (Figure 1A). This binding site is immediately above the buried active site diiron cluster (Figures 1A and B).
Figure 1.
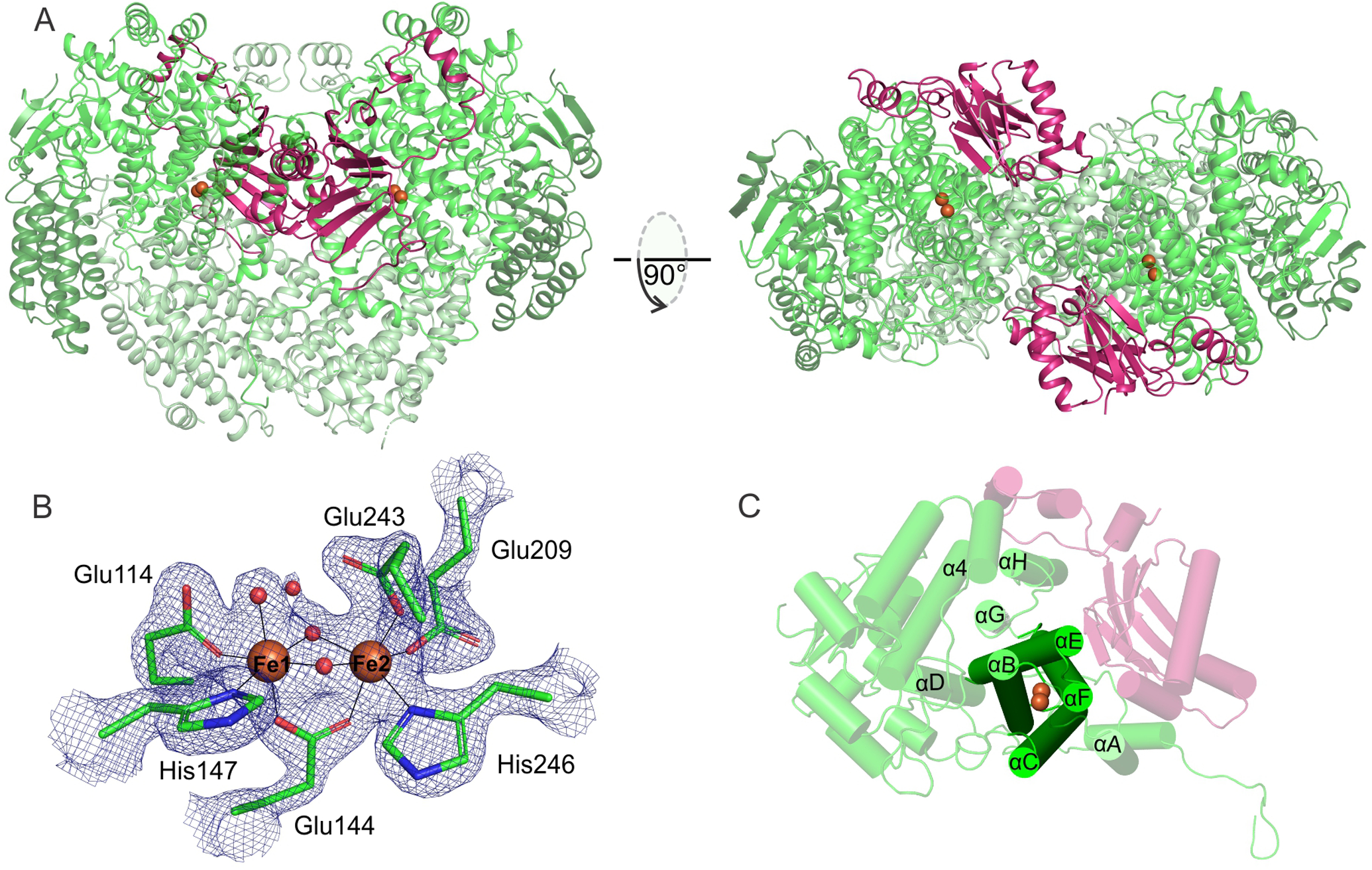
The diferric Mt OB3b sMMOH:MMOB complex (PDB: 6YD0). (A) Two perspectives of the overall complex. The α, β, and γ subunits of the sMMOH protein are shown in different shades of green while MMOB is colored purple. The iron atoms in the sMMOH active site are represented as orange spheres. The dimeric sMMOH:MMOB is shown by generating the symmetry related monomer in PyMOL (B) 2Fo-Fc map of the oxidized diiron cluster contoured to 2 σ. (C) Cartoon of the α-subunit with the helix nomenclature illustrated.
Complex Formation Leads to Localized Structural Reorganization of MMOB as well as the sMMOH Interface.
The availability of a room temperature, high resolution, fully oxidized structure of the Mt OB3b sMMOH:MMOB complex allows a critical evaluation of the changes in both diferric sMMOH and MMOB upon complex formation. A comparison of MMOB in this crystal structure with the NMR structure of Mt OB3b MMOB in isolation (PDB:2MOB)24 shows that there is a significant conformational reorganization of MMOB upon complex formation. These changes are present not only in the well-ordered core region (residues 35 – 125) but also in the N-terminal tail (residues 1 – 35) and C-terminal tail (residues 127 −138) (Figure 2). The reorganization is significant as suggested by the r.m.s.d value of 3.5 Å between isolated MMOB and MMOB in complex with sMMOH. In the absence of sMMOH, the MMOB core region is composed of two domains, where domain 1 (residues 36 – 81) contains a βαββ motif and domain 2 (residues 82 –126) contains a βααββ motif.24 Upon binding to sMMOH, the α-helices on the two domains move closer to each other (Figure 2). In addition, a long helix in domain 2 of MMOB is restructured into a short helix, while an unstructured loop is converted into a β-sheet (Figure 2). This newly-formed β-sheet is involved in hydrogen-bonding interactions with the β-sheets of domain 1. The extent of reorganization of the MMOB core seen in this structure is not observed in the Mc Bath sMMOH:MMOB complex crystal structure.
Figure 2.
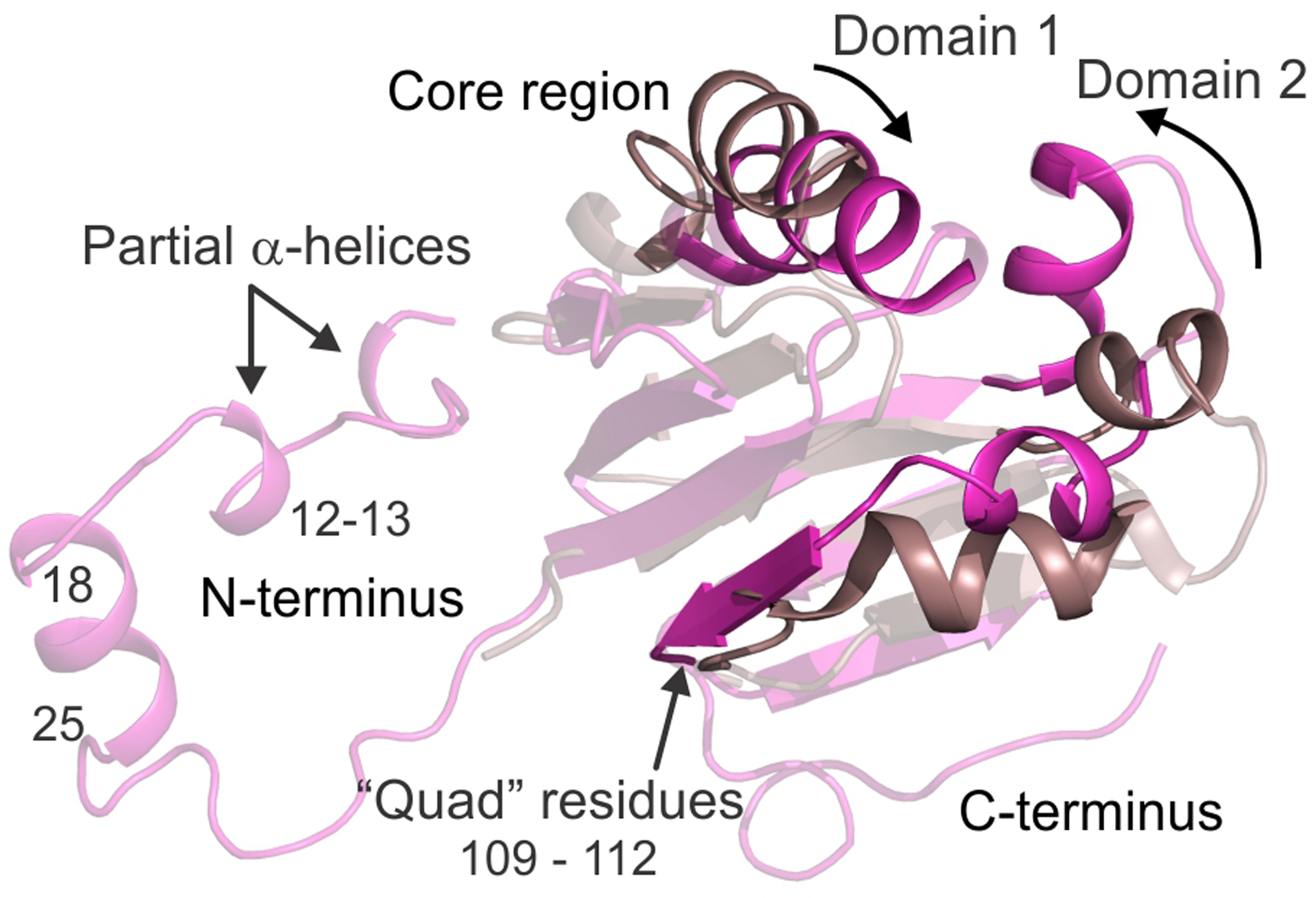
An alignment of the MMOB protein structure in the uncomplexed state (PDB: 2MOB; brown cartoon) and sMMOH bound complex (PDB: 6YD0; magenta cartoon). Both the N-terminal and C-terminal tails of MMOB are ordered in the protein complex with additional secondary structure being generated in the N-terminal tail. The MMOB core also becomes reorganized in the complex as highlighted by the region in focus (opaque cartoon). MMOB domains 1 and 2 reorient closer together upon complex formation with MMOH.
The residues present on the new β-sheet include Ser109, Ser110, Thr111 and Val112. This sequence of MMOB, termed the “Quad region” is important because three of these residues specifically interact with residues on sMMOH and affect catalysis (vide infra).10, 29–30, 71 The N-terminal tail of isolated MMOB is disordered, as it cannot be observed in the NMR structures. However, upon complex formation, this region on MMOB is arranged in a ring-like fashion on the surface of sMMOH (Figures 1A and 3), as observed previously with the 2.9 Å cryo-temperature Mc Bath sMMOH:MMOB complex structure.28 A short α-helix is formed in this N-terminal tail (residues 18 – 25) upon complex formation with sMMOH, which is in agreement with previous solution NMR studies.25 In addition, there are two short regions where a partial helix is formed, which can be seen only in the higher resolution Mt OB3b protein complex structure (Figure 2). These helices appear to form as a result of the insertion of multiple conserved non-polar MMOB residues (Ile12, Met13, Phe20, Phe24, Phe25 and Gln30, Figures 3B and S1) into a predominantly polar region (Helices H and 4, see Figure 1C for helix nomenclature) on the sMMOH surface. Steric clashes, hydrophobic interactions and avoiding non-favorable contacts with the polar residues lead to the stabilization of an α-helix in MMOB. Finally, the C-terminal tail of MMOB, which is disordered in the isolated NMR solution structure, is ordered in the protein complex, although it does not acquire any secondary structure (Figure 2). The higher resolution of the Mt OB3b sMMOH:MMOB crystal structure compared to the Mc Bath structure enables this observation, as the C-terminal tail cannot be observed in the latter structure.
Figure 3.
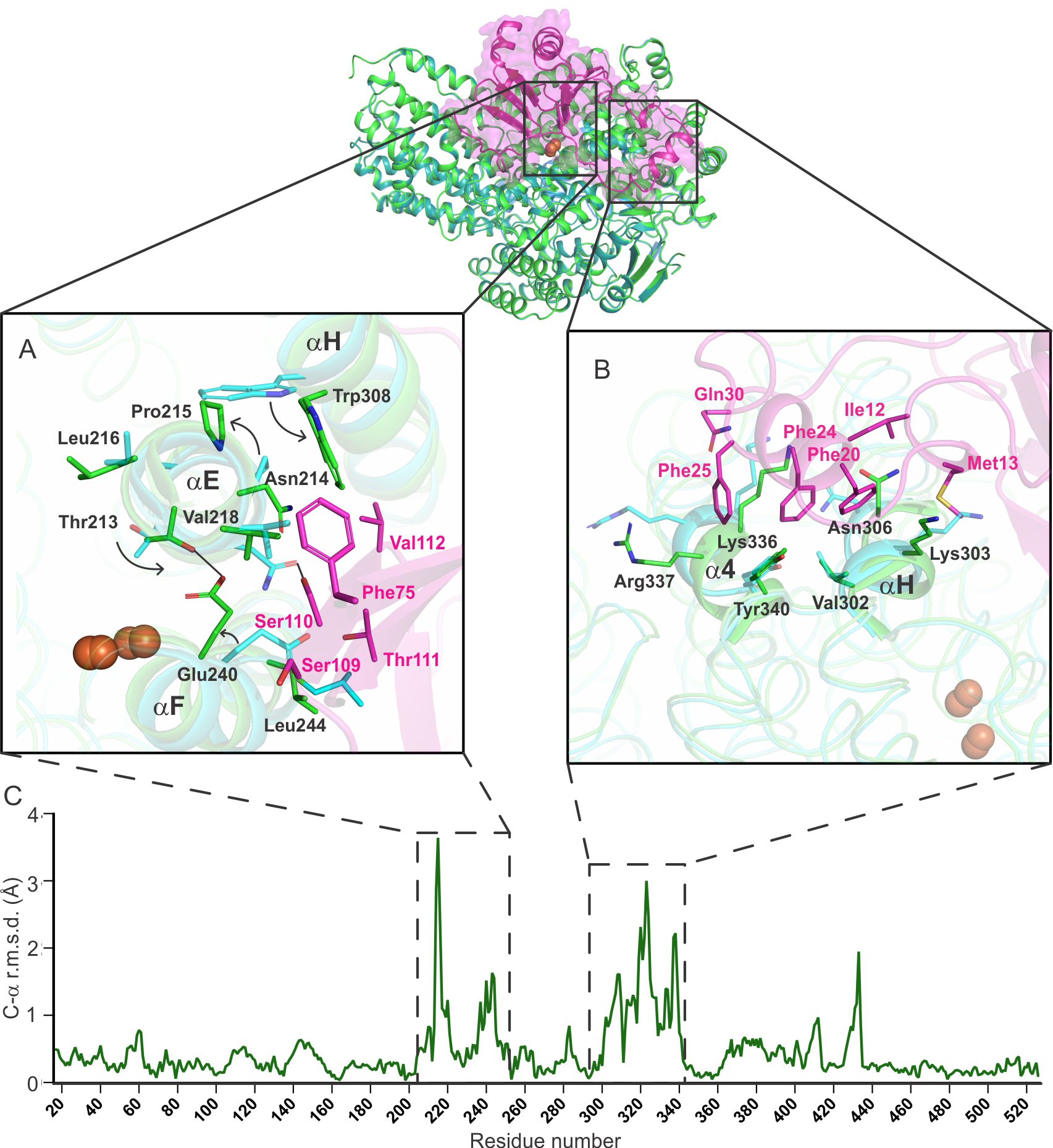
An alignment of the α-subunit of sMMOH in unbound (1MHY) and MMOB-bound (6YD0) states shows that specific regions in sMMOH undergo a large reorganization as depicted by the C-α r.m.s.d. values (panel C). These regions (Helix E – residue 213 – 223; Helix F – residue 237 – 252; Helix H and 4 – residue 302 – 341) are shown in more detail in the square boxes (Panel A: Helices E and F; Panel B: Helices H and 4). MMOB residues responsible for the reorganization are shown in the boxes as magenta sticks. sMMOH in isolation is shown as the cyan cartoon; sMMOHox:MMOB is shown as the green cartoon. Hydrogen bonds are depicted as black lines.
Localized structural reorganization also occurs in the sMMOH protein upon MMOB binding. A comparison of the superposed structures of oxidized sMMOH from Mt OB3b (PDB:1MHY, also solved using data collected at room temperature)14 and the Mt OB3b sMMOH:MMOB complex provides a clear picture of these changes. The reorganization involves two of the four helices of the 4-helix bundle (E and F) and two additional helices on the sMMOH surface (H and 4) (Figures 3C and 1C). All of these regions are located on the sMMOH interface with MMOB.
While a comprehensive list of structural changes in Mt OB3b sMMOH due to MMOB binding is given in Table S2, the most dramatic of these modifications are described here. A long section of sMMOH residues (213 – 223; α-subunit; Helix E) reorganize (Figures 3A and 4) as a result of the formation of a hydrogen bond between Asn214 (MMOH) and Ser110 (MMOB) and steric clashes of Val218 (MMOH) with Phe75 (MMOB). It is important to note that Helix E contains sections that possess the relatively rare π-helical secondary structure. π−helices are evolutionarily conserved features in proteins and their presence is strongly associated with active site residues involved in catalysis.72 The π−helices in the ferritin-like superfamily, of which sMMO is a member along with other bacterial multicomponent monooxygenases (BMM), ribonucleotide reductases (RNR), acyl-ACP desaturases, ferritins and bacterioferritins, are important because they provide some of the ligands to the carboxylate-bridged di-metal cofactors (Glu209 in sMMOH).72 In unbound sMMOH, Helix E contains two small π-helical sections from residues 202 – 205 and 208 – 209 along with residues bereft of intra-main chain hydrogen bonds (206, 210 – 211, 214). MMOB binding reorganizes the second π-helix to 207 – 208 and induces the formation of an additional π-helical section from residues 213 – 215 (Figure 4). The Helix E residues lacking intra-main chain hydrogen bonds in the complex are 206, 209 – 211 and 216 – 217. Such a dramatic reorganization is possible due to a kink in Helix E at Pro215. Prolines in the middle of α-helices are conserved for their structural and functional significance and have also been found to be present at the point of transition from a π-helix to an α-helix.72–73 Upon MMOB binding, Pro215 undergoes the largest r.m.s.d shift of 3.6 Å from its position in the sMMOH alone structure (Figure 3C). Pro215 is a strictly conserved residue in sMMOH (Figure S2), but not in other BMM enzymes.
Figure 4.
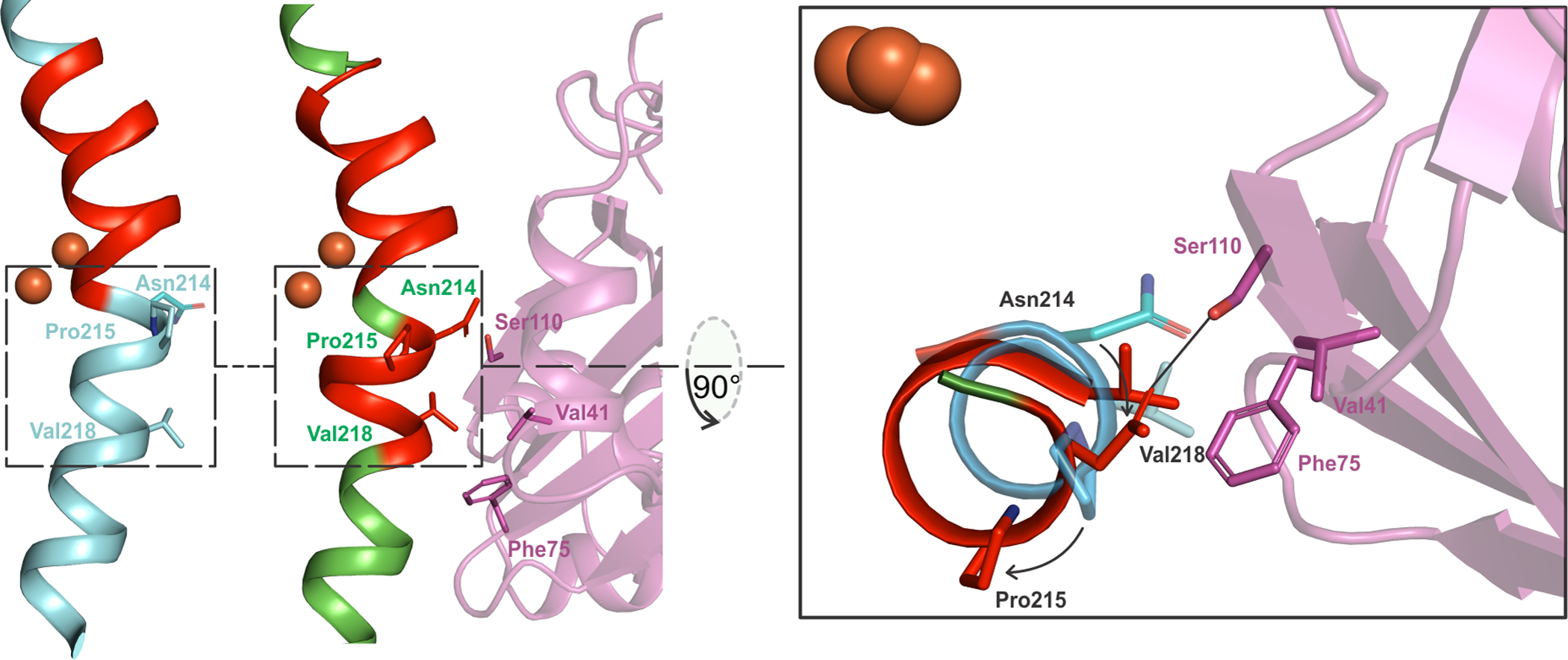
The π-helical portion (red) in Helix E of sMMOH is extended in the sMMOHox:MMOB complex (6YD0, green helix) as opposed to sMMOH in isolation (1MHY, cyan helix). The reorganization is caused primarily by the hydrogen-bond interaction between Ser110 (MMOB) and Asn214 (MMOH) and a steric clash between Val218 (MMOH) and Phe75 (MMOB). This leads to the large movement of Pro215. π-Helix assignment was performed using DSSP74–75 with the DSSP & Stride plugin for PyMOL76 (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Another long chain of residues (236 – 252; α-subunit; Helix F) reorganize upon MMOB binding (Figure 3A) with the largest change occurring at Glu240. The side chain of Glu240 swings away from being solvent exposed to project into the interior of the protein in order to avoid a steric clash with Ser109 of MMOB. In this new position, Glu240 forms a hydrogen bond with Thr213, which has undergone a 180° rotation of the side chain residue. This hydrogen bond formation, and MMOB coverage of the surrounding sMMOH surface, block access to the active site in the diferric sMMOH:MMOB complex. A similar structural change was also observed in the Mc Bath sMMOH:MMOB protein complex.28 The stretch of residues from 237 – 252 is additionally important, because it contains two coordinating ligands, Glu243 and His246 to the Fe2 iron atom. The reorganization of both Helices E and F arise as a result of hydrogen bond interactions and steric clashes with strictly conserved residues Ser109, Ser110 and Thr111 of MMOB (Figures 2 and S1 and Table S2).
A combination of hydrogen bond interactions and steric clashes (Table S2) cause the reorganization of a stretch of 40 residues in sMMOH (302 – 341; α-subunit; Helices H and 4 along with intervening loop) (Figure 3B). Helix H also contains a section with the rare π-helical secondary structure, but unlike Helix E, does not contain either ligands to the diiron cluster or residues in the active site cavity. The π-helical portion of Helix H (residue 306 – 313) is rearranged in the MMOB-bound complex (residues 306 – 309 and 314 – 315). Helix 4 is a short helix in unbound sMMOH (332 – 338) that becomes part of a longer bent helix (331 – 356) in the sMMOH:MMOB protein complex. The extension of this helix in turn rearranges an adjacent unstructured loop region (428 – 435; α-subunit) (Figure 3C). Most of the sMMOH:MMOB interactions leading to the reorganization of Helices H and 4 arise from the N-terminal tail of MMOB (Figures 3B).
It is pertinent to note that specific interactions can be found between all the three regions of MMOB (N-terminal tail, C-terminal tail and core) with sMMOH (Table S2). These discrete interactions of MMOB residues with their counterparts on sMMOH result in very ordered regions of the MMOB protein as indicated by lower B-factors (Figure S3).
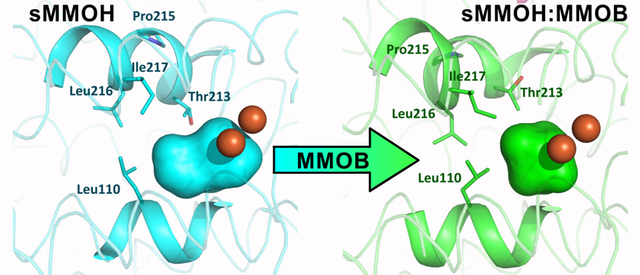
MMOB Reorganizes the Active Site Cavity in sMMOH.
The active site cavity of sMMOH lies along one face of the diiron cluster (Figure 5). It is predominantly lined with non-polar residues on the surface facing the iron atoms (Leu110, Gly113, Ala117, Phe188, Phe192, Leu204, Thr213, Leu216, Ile217) along with the iron ligands Glu209, Glu243, and Glu114. This sMMOH active site cavity is reduced in volume upon MMOB binding (198 Å3 to 156 Å3) primarily due to the projection of the side chains of Leu110, Leu216 and Ile217 further into the cavity (Figure 5). The movement of the latter two residues is due to the reorganization of Helix E upon complex formation with MMOB. The side chain of Leu110 rotates in order to avoid a steric clash with the altered position of Leu216. In addition to size changes, the overall polarity of amino acid side chains that line the cavity is also altered upon MMOB binding (Figure S4). The hydrophobic character is further increased as a result of the rotamer shift of Thr213, which leads to the projection of the methyl group of Thr213 into the active site cavity in place of the hydroxyl moiety. In the unbound sMMOH resting state structure, the side chain hydroxyl group of Thr213 is in hydrogen bonding distance of two water molecules (Figure 6). These water molecules are not found in the active site cavity after MMOB binds sMMOH. Another water molecule that is hydrogen bonded to the backbone carbonyl group of Leu110 and the side-chain carboxylate of Glu114 is also lost. The loss of three crystallographically ordered water molecules and the side-chain hydroxyl group of Thr213 results in a more non-polar active site cavity in the sMMOH:MMOB protein complex. The loss of ordered solvents from the active site cavity not only opens up room for substrates to bind, it also increases the affinity for hydrophobic molecules such as oxygen and methane. The change in hydrophobicity and volume of the active site cavity is a direct result of MMOB binding that induces the extension of the π−helix in Helix E of sMMOH.
Figure 5.
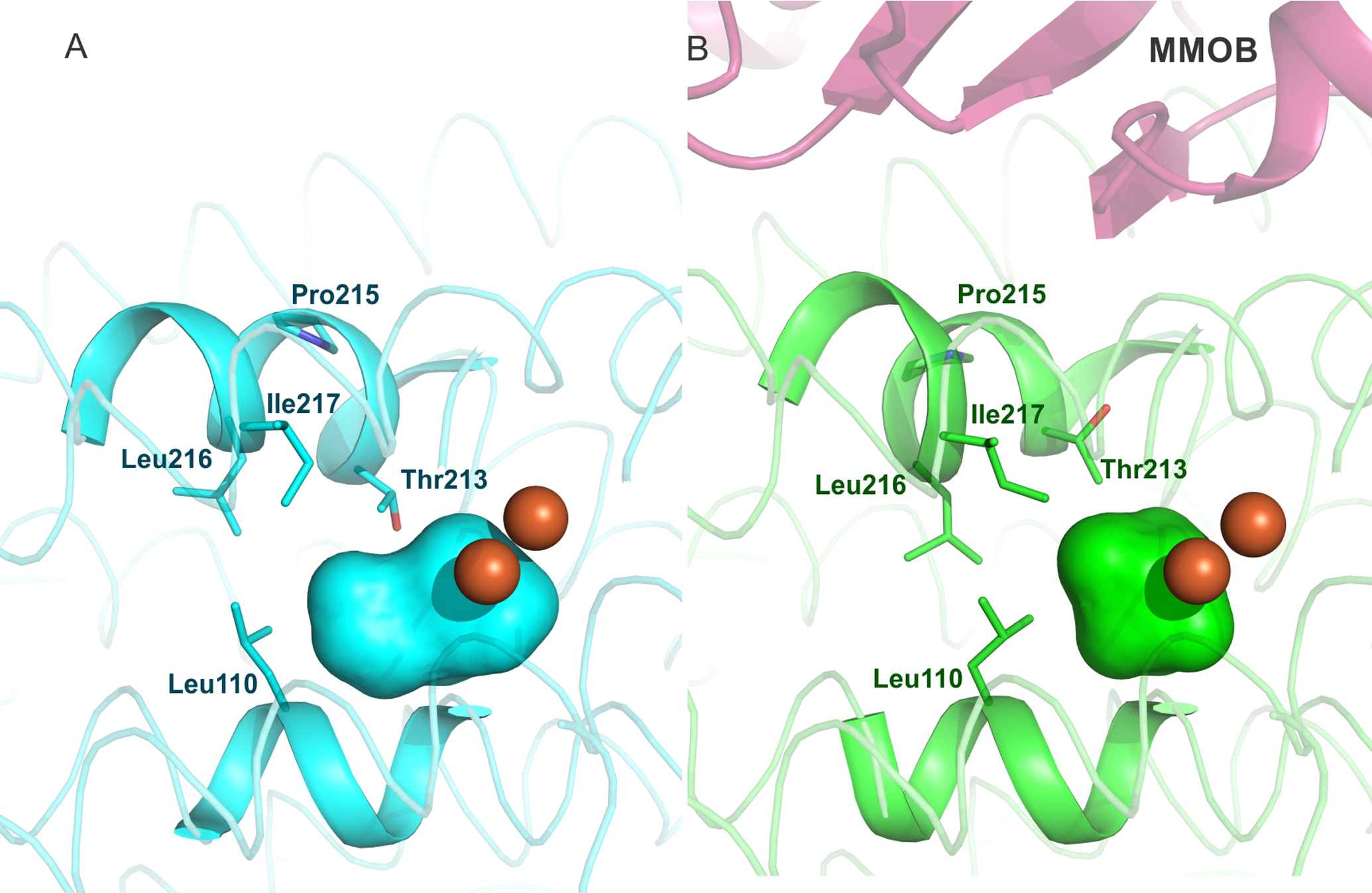
The sMMOH active site cavity (colored surface representation) reduces in size upon MMOB binding, as calculated using HOLLOW.77 Diferric sMMOH in isolation (A) is represented as a cyan colored cartoon while the diferric sMMOH:MMOB complex (B) is shown as a green colored cartoon. MMOB is shown as a magenta colored cartoon. The iron atoms are shown as orange spheres. The active site cavity is indicated by cyan and green surfaces, respectively.
Figure 6.
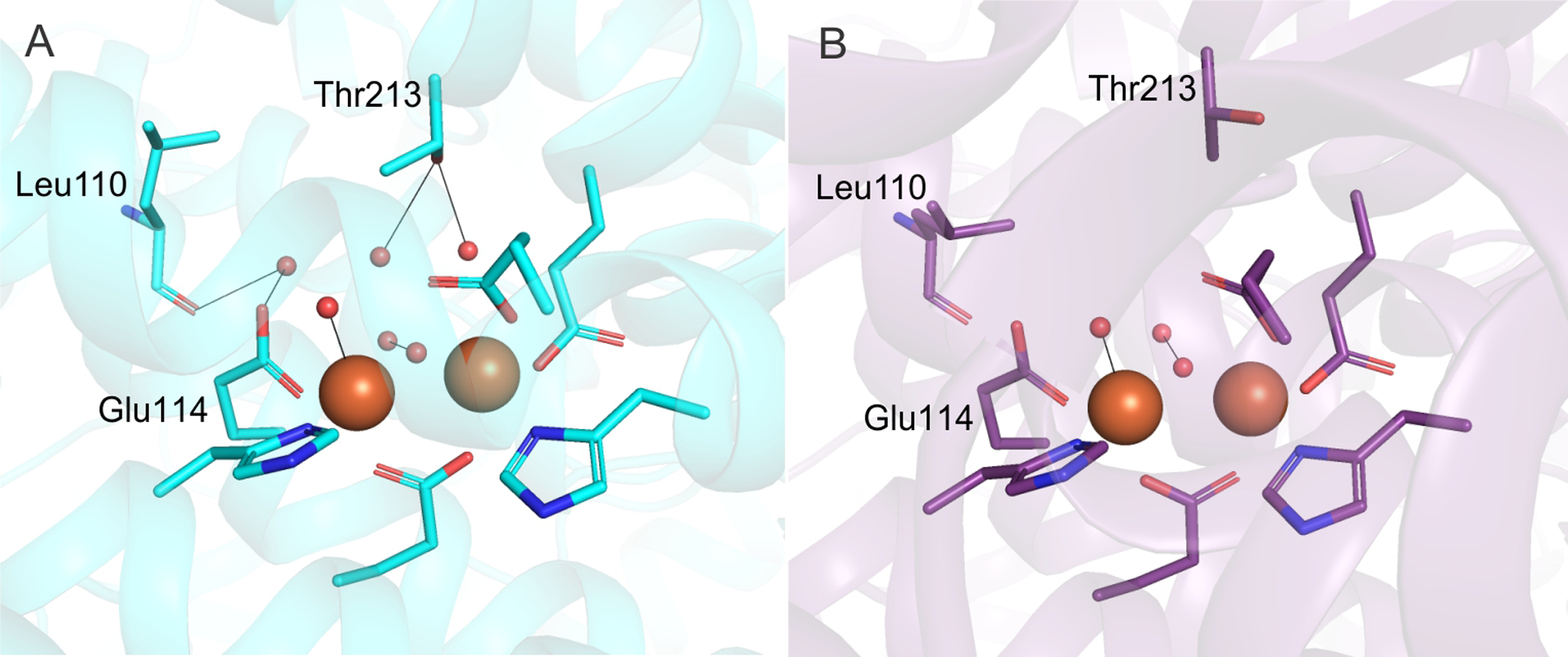
Comparison of active site water molecules between the MMOH (A) and sMMOH:MMOB complex (B) structures. Color scheme – 1MHY (cyan carbon atoms); 6YDU (purple carbon atoms); water molecules and bridging μ-hydroxo groups (red spheres). Hydrogen bonds between residues and water molecules are shown by black lines. The rotamer shift of T213 and L110 leads to the loss of three active site water molecules upon MMOB binding.
The Binding of MMOB Causes Subtle Changes in the Structure of the Oxidized Dinuclear Iron Cluster.
Considering that two of the four diiron cluster-coordinating α-helices (E and F) undergo structural reorganization upon MMOB binding, it is essential to critically evaluate any changes in the metal coordinating environment. Viewed from the superposition of the secondary structure of the α-subunit, MMOB binding leads to a shift of the entire diiron cluster by ~ 0.6 Å. However, the relative arrangement of the ligands with respect to the iron atoms is roughly maintained based upon a superposition of only the metals and ligating residues (r.m.s.d = 0.14 Å) (Figure 7A). Most of the minor reorganization is localized on Fe2 because the E and F helices donating ligands to this iron atom also interact directly with MMOB. There is a small rotameric shift of Glu243 away from the plane of the iron atoms, but the binding mode stays monodentate and non-bridging. The two bridging hydroxo/aquo ligands to the diiron cluster characteristic of oxidized sMMOH are retained.
Figure 7.
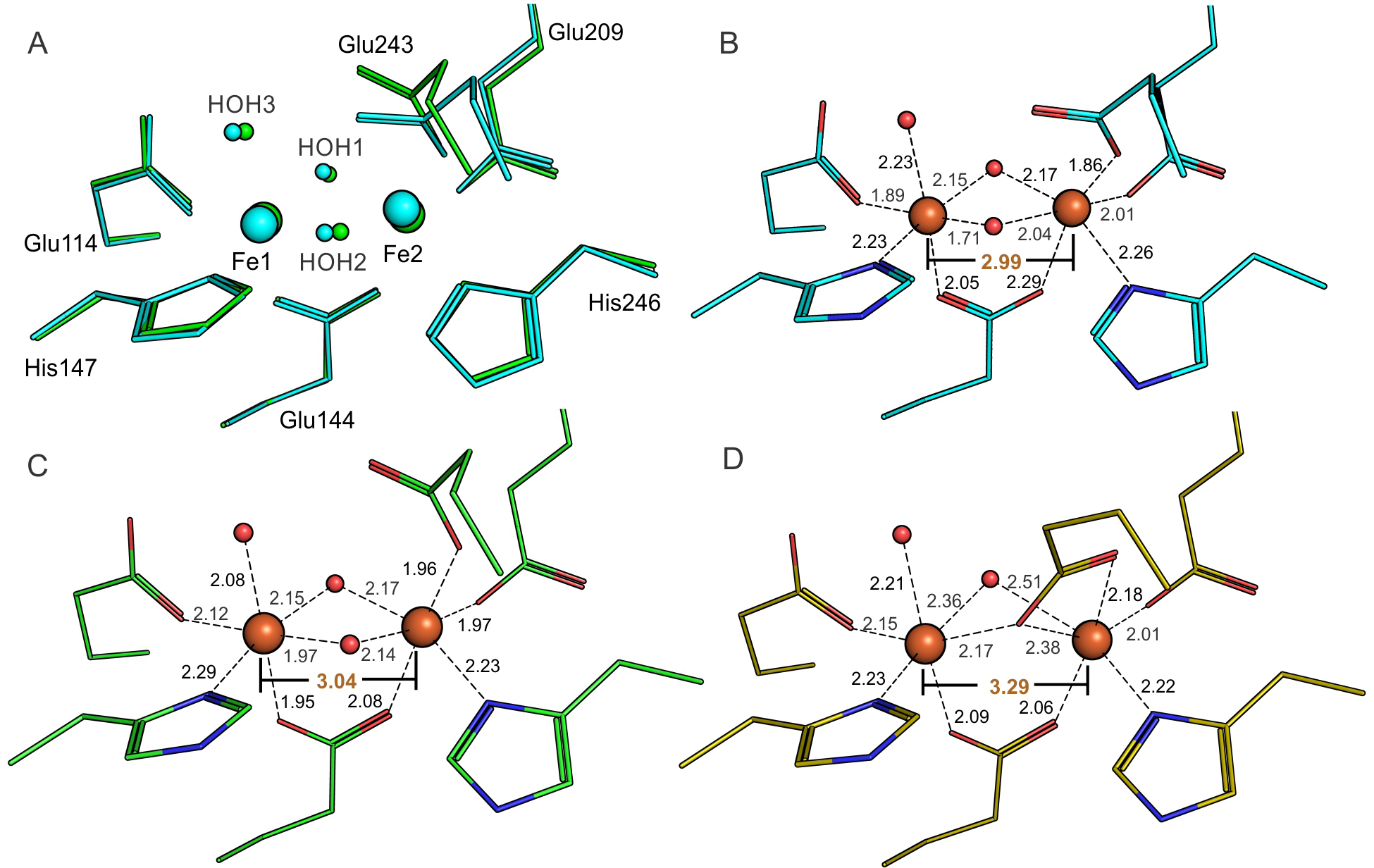
Changes in the diferric sMMOH diiron cluster occur upon MMOB binding and reduction. The ligands and iron atoms are identified as per the labels in panel A. (A) Alignment of the oxidized diiron clusters of unbound sMMOH with the sMMOHox:MMOB complex based on just the first-sphere ligands and iron atoms. Cyan – sMMOH alone (1MHY); Green – diferric sMMOH:MMOB complex (6YD0). (B) Ligand to Fe distances for isolated diferric sMMOH (1MHY). (C) Ligand to Fe distances for diferric sMMOH:MMOB (6YD0). (D) Ligand to Fe distances for anaerobic diferrous sMMOH:MMOB (6YDI). The Fe-Fe distance is labeled in orange text.
The ‘Carboxylate Shift’ of Glu243 Occurs Upon Reduction of the Dinuclear Iron Cluster.
The canonical carboxylate shift of Glu243 to replace one of the bridging hydroxo/aquo ligands (HOH2) is observed upon a chemical reduction of the diiron cluster in the Mt OB3b sMMOH:MMOB complex crystals (Figure 7D). Reduction of the diiron cluster is also evident from the concomitantly measured Fe Kα XES spectra of the microcrystals (Figure 8). Kα XES chemical sensitivity has been reported for some transition metal systems.78–79 Subtle changes in the peak positions and shapes, as well as the ratios between the Kα1 and Kα2 peak height, have been described to be correlated with the number of unpaired d-electrons and hence with the transition metal oxidation state.80–83 The reduced - oxidized difference spectrum (Figure 8, blue line) shows an asymmetric shift in the Kα1 peak as well as a decrease in intensity and a shift in position for the Kα2 peak. This trend is similar to the difference observed between Fe2+ and Fe3+ standards in aqueous buffers collected under the same conditions as the sMMOH:MMOB samples (Figure 8, green line). While ligation environment of the Fe centers are not identical between the two systems, the observation of similar trends in both difference spectra is indicative of a predominant change from Fe3+ to Fe2+ in the sMMOH:MMOB samples. Similar features were also found previously for the Fe/Mn containing ribonucleotide reductase (RNR) R2c, where a transition between Fe oxidation states (orange trace in Figure 8, taken from ref.53) was observed. The observed oxidation is also supported by a change in the full width at half maximum (FWHM) of the Kα1 peak and a change of the intensity ratio of the Kα1 vs. the Kα2 peak, as both have previously been reported as an indicator for the oxidation state.80–81 An increase of the FWHM by 0.17 eV was observed when comparing the diferric with the diferrous sMMOH:MMOB samples. In comparison, an increase of the FWHM by 0.29 eV was observed between the Fe2+ and Fe3+ standards and by 0.1 eV for these states of RNR R2c;53 These numbers are in line with previously reported values for transition metal Kα spectra.80–83
Figure 8.
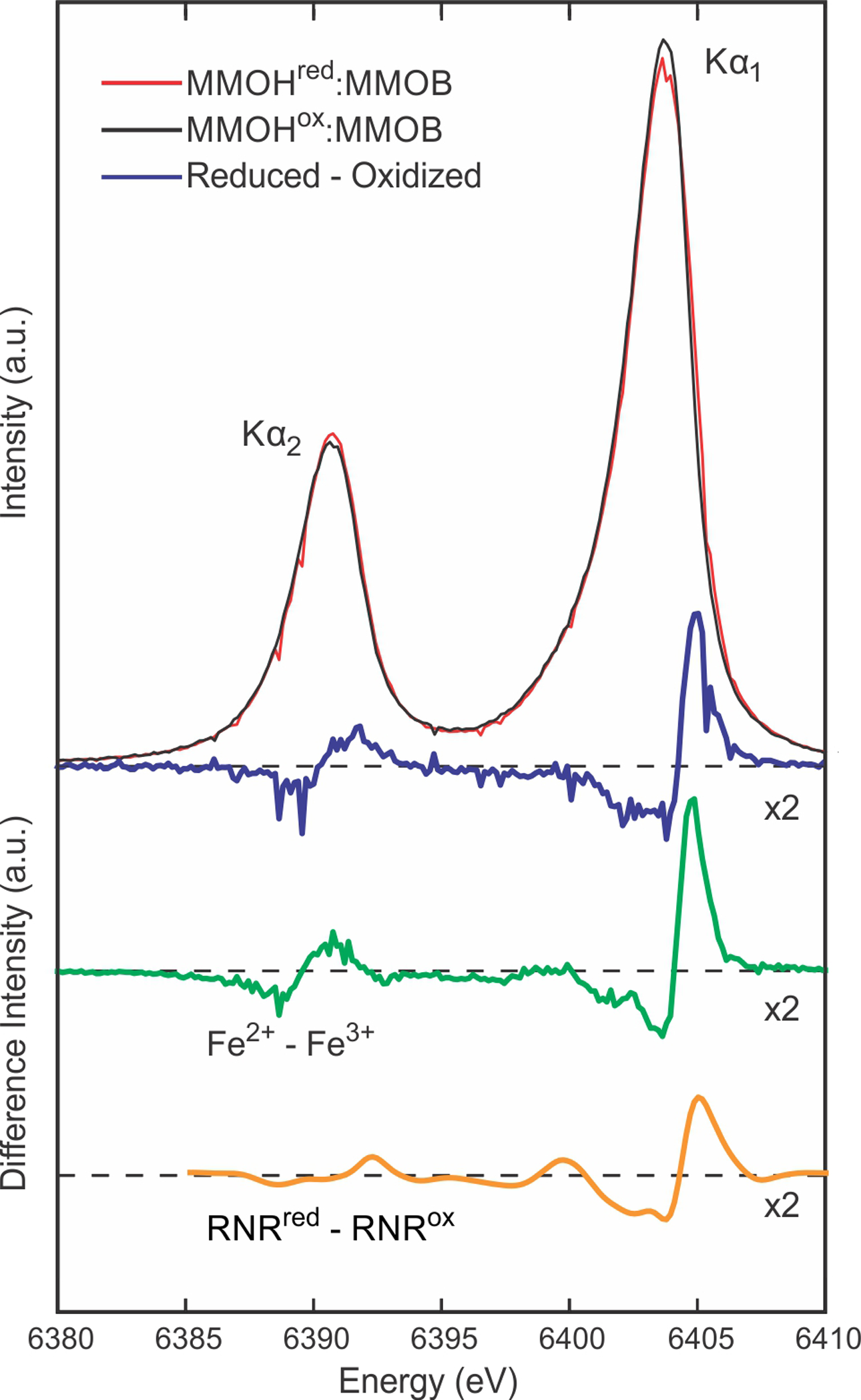
Area-normalized Fe-Kα XES of microcrystals of chemically reduced (red) and oxidized (black) sMMOH:MMOB, and a subtraction (blue, 2× magnification) of the reduced minus oxidized spectra showing a clear asymmetric shift in the Kα1 peak as well as a weakening in intensity and a slight shift in position for the Kα2 peak. For comparison the difference spectra for aqueous Fe2+ - Fe3+ (green, 2× magnification), collected in the same experiment, and for solutions of reduced and oxidized ribonucleotide reductase (RNR) R2c (yellow, smoothed by wavelet denoising and 2× magnification), collected previously at LCLS53 are shown.
The magnitudes of these changes are consistent with the full reduction indicated by the occupancy of the shifted Glu243 in the crystal structure, but the extent of reduction cannot be unequivocally determined from the XES data. In the diferrous state, reorientation of Glu243 is achieved by a rotameric shift at the β-carbon position. The iron-iron distance increases from 3.04 Å in the oxidized state to 3.29 Å in the reduced state, which is in close agreement to previous EXAFS studies (3.056 Å to 3.270 Å).84 In concert, the ligand-to-iron distances increase, especially for HOH3 – Fe1 (2.08 Å to 2.21 Å), Glu144 – Fe1 (1.95 Å to 2.09 Å) and Glu243 – Fe2 (1.96 Å to 2.18 Å). The bridging aquo ligand that faces the active site cavity (HOH1) moves away significantly (~ 0.3 Å) from the iron atoms upon the change of oxidation state. The B-factors for the iron atoms are higher in the reduced state as compared to the resting diferric state (Figure S5). Furthermore, Fe2 specifically has a higher B-factor than Fe1, which is not surprising considering that it is ligated by residues from Helices E and F. The ligands coordinating Fe2 also appear to have higher B-factors, as opposed to the ligands of Fe1. Apart from the carboxylate shift of Glu243 and the lengthening of ligand to iron bond distances, there is no structural reorganization of either sMMOH or MMOB upon diiron cluster reduction. Structural alignments of the sMMOH α-subunits and their respective MMOBs before and after reduction provide r.m.s.d values of 0.08 Å (sMMOH) and 0.13 Å (MMOB). This means that the myriad of the structural changes identified above that occur in oxidized sMMOH upon MMOB binding also pertain in the fully reduced, O2 reactive complex. In the active site, these changes include (i) compression of the active site cavity by shifts in Leu110, Leu216 and Ile217, (ii) closure of an access route from bulk solvent into the active site by reorganization of Glu240 and Thr213, (iii) generation of a more non-polar active site cavity with the loss of water molecules and rotamer shift of Thr213, and (iv) realignment of several of the iron ligands, especially those of Fe2.
Reduced sMMOH:MMOB Complex Reacts Rapidly with O2 in the Microcrystals.
The sMMO catalytic cycle is initiated by the reaction of dioxygen with the diferrous sMMOH:MMOB complex and continues through a well-established series of intermediates to restore the diferric resting state.1, 11, 57, 85–86 While this process occurs readily in solution, it is important to test whether O2 can access and react with the diiron cluster in crystals. This can uniquely be accomplished during the XFEL experiment because the reduced crystals on the tape of the DOT system can be briefly exposed to O2 before being transported into the XFEL interaction region. Moreover, the experiment is conducted at room temperature with very fast XFEL pulses, so structural changes occurring during the reaction can be observed. The crystal structure of the sample after a 4 s exposure to pure O2 atmosphere plus a 6 s travel to the XFEL interaction region (PDB: 6YDU) indicates that the diiron cluster rapidly reacts with O2 to regenerate the diferric complex. The Fe Kα XES difference spectrum for the reduced (t=0) minus10s O2 incubated MMOH:MMOB samples, albeit limited due to a higher noise level, is very similar to that for the reduced minus oxidized MMOH:MMOB samples. This is consistent with the observation from the diffraction data for the in-situ re-oxidized sample, where Glu243 shifts back to the monodentate coordination mode to Fe2 that is characteristic of the diferric cluster (Figure S6). Also, the Fe-Fe distance of the diiron cluster is restored to 3.02 Å and the bridging hydroxo ligand that is lost upon reduction is reacquired. The ligand-to-iron distances are similar to those of the oxidized diiron cluster. Passage of similarly reduced and washed crystals through a chamber filled with helium rather than O2 retained all the structural features of diferrous sMMOH:MMOB (PDB: 6YY3) (Figure S6A and B), showing that the diferrous state is preserved under these conditions in the absence of O2. The 10 s oxidized crystal structure suggests that the reduced sMMOH:MMOB protein complex is indeed capable of binding oxygen, activating it, and completing a single turnover in-crystallo on a similar time scale to that observed in solution.20, 85
During the single turnover in-crystallo reaction, a region of unmodeled electron density disappears from the active site cavity (Figure S7). This density is present in both diferric and diferrous structures and is close to the expected binding location of hydrocarbon substrates. The unmodeled density is too large to be accounted for by methane or methanol, but the sMMO system is known to oxidize a remarkably wide range of other small hydrocarbons and similar molecules.9 One explanation for this density and its loss after O2 incubation is that it may represent an adventitious substrate from the crystallization media, which is oxidized during turnover and diffuses out of the active site. This possibility will be further explored in ongoing studies.
DISCUSSION
This report describes for the first time the crystal structures of the sMMOH:MMOB protein complex in the fully diferric and O2-reactive diferrous oxidation states. The resolutions of the crystal structures of these complexes offer a significant improvement over the previously reported Mc Bath protein complex structure.28 A comparison of the diferric protein complex crystal structures for Mc Bath and Mt OB3b shows that the overall structure of the protein complex is similar. However, the enhanced resolution of the current structures enables detection of important structural reorganization in sMMOH upon binding MMOB not possible in the Mc Bath sMMOH:MMOB crystal structure. These include (i) alteration in the π-helical character of sMMOH Helices E and H, (ii) compression of the active site cavity, (iii) increase in hydrophobicity of the active site cavity due to loss of discrete water molecules, and (iv) MMOB binding-induced modulation of the hydrogen bonding network to the histidine ligands to the diiron cluster (vide infra). A clearer view of the structural reorganization in sMMOH caused by MMOB binding results from these structures and this lies at the heart of the many regulatory roles played by MMOB during catalysis. Furthermore, it is shown that the reduced sMMOH:MMOB protein complex binds and activates O2 in-crystallo to complete a single turnover cycle. This ability, in conjunction with high resolution diffraction and concomitant measurements at 298 K of both crystallographic and Fe Kα XES data, bodes well for future experiments designed to structurally characterize reaction cycle intermediates. Here, the structural changes induced by MMOB binding are discussed in the context of the current understanding of the roles played by MMOB in regulating catalysis.
Structures of sMMOH:MMOB Complexes without Radiation Induced Alterations.
The previously reported comparison of the structures of diferric Mc Bath sMMOH (PDB: 1MTY) and the Mc Bath sMMOH:MMOB protein complex (PDB: 4GAM) led to the conclusion that MMOB binding is sufficient to cause the characteristic ‘carboxylate shift’ of Glu243 in sMMOH without requiring reduction of the diiron cluster.28 In the Mt OB3b sMMOH:MMOB complex structures reported here, this reorientation of Glu243 is only observed in the diferrous oxidation state of sMMOH (Figure 7D). There are several possible explanations for this discrepancy including: (i) slight protein differences, (ii) crystal structure resolution and space group, (iii) temperature (93 K versus 298 K), and (iv) light sources (synchrotron versus XFEL). Of these, the light source is the most likely factor because synchrotron radiation has been demonstrated to cause partial or complete reduction of redox-active metal centers in many different types of proteins.32–33, 87–89 This dose-dependent radiation induced effect arises from solvated electrons generated in situ by the X-ray beam. The serial-femtosecond crystallographic (SFX) approach at XFELs addresses this issue by using significantly shorter and more intense X-ray pulses than produced by synchrotrons. XFEL pulses yield diffraction images from crystals on time scales of 10 – 35 fs, which is much less than the time required for radiation damage to occur by diffusion of solvated electrons or radicals even at room temperature. The concomitantly measured Fe Kα XES spectra in this study showed a clear difference between the oxidized and reduced species, indicating no noticeable photo-induced changes taking place under our experimental conditions. This is in line with previous reports on several proteins and model compounds under conditions similar to the ones used in this study.41, 46, 53–54, 90
Some evidence for radiation-mediated partial reduction of the diiron clusters in the Mc Bath sMMOH:MMOB crystal comes from the wide range of Fe-Fe distances observed in four protomers within the asymmetric unit (2.9 – 3.2 Å).28 The largest of these values are close to that observed for diferrous clusters in the structure of reduced sMMOH.13 In contrast, different distances are observed for the diferric and diferrous Mt OB3b sMMOH:MMOB crystals, and these distances correlate well with those from the more accurate solution EXAFS measurements.84
Another line of evidence suggesting a mixed oxidation state in the Mc Bath sMMOH:MMOB crystal derives from the predicted overall charge of the diiron cluster. It has been observed in structures of diferric sMMOH that the six positive charges of the iron atoms are balanced by the negative charges of the four glutamate residues and two bridging hydroxo ligands.91 This neutral net charge is maintained after reduction of the iron atoms to the diferrous state by either the loss or protonation of the bridging hydroxo ligands, thereby avoiding the thermodynamic penalty of having a charged moiety inside the non-polar environment of the sMMOH active site. This charge-maintenance principle can even be extended to the high-valent intermediates generated during catalysis, wherein the increasing oxidation state of the iron atoms is balanced by the negative charges of the bridging peroxo and bis-μ-oxo (or equivalent) ligands.57, 91 The diiron cluster in the crystal structure of diferric Mc Bath sMMOH:MMOB would have a net 1+ charge due to the loss of a hydroxo ligand without a change in iron oxidation state. In contrast, the structures of the Mt OB3b sMMOH:MMOB complexes show that the dinuclear iron cluster remains charge-neutral during the redox change, as expected. Thus, based upon the range of Fe-Fe distances, the charged active site, and comparison to the Mt OB3b protein complex structures, it is likely that the Mc Bath diferric sMMOH:MMOB protein complex was partially reduced as a result of radiation damage during data collection.
The success of the SFX technique in obtaining accurate representations of the oxidized and reduced diiron cluster in sMMOH:MMOB is especially important towards the future goal of capturing structures of enzymatic reaction cycle intermediates. The reduction potential of the key methane-reactive intermediate Q (dinuclear FeIV) has not been measured, but it is expected to be significantly higher than that of the diferric resting state. A reasonable estimate of ~ +1.4 to +1.6 V can be made based on the measured value of +1.0 V for the isoelectronic Compound I intermediate of heme oxygenases, which is incapable of breaking the strong C-H bond of methane except in the presence of strong chemical oxidants.92–93 The high positive redox potential of Q makes reduction in the X-ray beam very likely, as observed for highly oxidizing intermediates studied in other proteins using synchrotron light sources.32–33, 89 One solution to this problem has been to merge low X-ray radiation dose diffraction data from multiple crystals or multiple positions on one crystal to yield a complete dataset.33, 94 The XFEL-based SFX methodology avoids X-ray induced reduction, and thus it does not have to be tailored to each new protein and unique reaction intermediate. The promise of this technique was recently demonstrated by a study that revealed structures for several intermediate states of the Mn4CaO5 cluster of photosystem II,41, 46 as well as numerous other types of biological molecules.42–45, 47–52
Rationalizing the Regulatory Roles of MMOB.
MMOB exhibits regulatory effects throughout the catalytic cycle of sMMOH (Scheme 1).1, 10 The high-resolution structure of the Mt OB3b sMMOH:MMOB complex allows the basis for some of these effects to be examined. One dramatic effect of MMOB is to control the interaction of substrates with the activated oxygen of the diiron cluster in intermediate Q during the reaction cycle. The most important of these effects is to selectively increase the quantum tunneling component of the reaction with methane, which gives a large kinetic advantage to methane over undesirable substrates such as ethane.30, 71, 95 Reactions of larger adventitious substrates in vitro are also affected. For example in the case of isopentane, the regiospecificity of hydroxylation shifts from the tertiary to the primary carbon upon formation of the sMMOH:MMOB complex.17 Both of these results suggest that MMOB binding must reorganize the sMMOH active site cavity to ensure optimal positioning of methane with respect to the activated oxygen of compound Q in order to enhance tunneling.96 Indeed, the structure of the sMMOH:MMOB complex shows that the volume of the sMMOH active site cavity is decreased by 21 % in the sMMOH:MMOB complex (Figure 5), which would presumably position methane in the site closer to the reactive oxygen species. The same restricted active site architecture may constrain the motion of isopentane so that the primary carbon with the strongest C-H bond, but in the least sterically hindered position, is brought closest to the reactive oxygen of Q and thus is preferentially oxidized. It is interesting to note that a variant of MMOB termed the Quad mutant (N107G/S109A/S110A/T111A) leads to a significant decrease in the methane 2H-KIE for the reaction of compound Q with methane (50 decreased to 6), indicating a loss of the quantum tunneling contribution to the reaction coordinate.30, 71 The Mt OB3b sMMOH:MMOB structure shows that three of the four residues of this MMOB variant, Ser109, Ser110 and Thr111, directly interact with residues on Helices E and F of sMMOH; this interaction causes the reduction in volume of the active site cavity (Figures 3A and 5 and Table S2). A mutation to smaller residues in positions Ser109 and Thr111, which make steric clashes with sMMOH residues, and a removal of the hydrogen-bond interaction of Ser110 with N214 of sMMOH would impair the structural reorganization of sMMOH Leu216, Ile217 and Leu110 (Figures 3A and 5). This would lead to an expanded sMMOH active site cavity, which would be expected to perturb the optimal positioning of methane with respect to Q.
Another effect of MMOB binding to sMMOH is to lower the mid-point reduction potential of the diiron cluster by 132 mV (cf. +48 mV to −84 mV),21 thereby increasing the thermodynamic driving force for the step of O2 binding and activation at the reduced diiron cluster. There are many mechanisms by which structure can affect the redox potential of a metalloprotein, and these primarily involve changes in hydrophobicity of the metal environment and charge donation by the first sphere ligands.97–100 It is shown above that the structural changes evoked by MMOB binding result in a more constricted active site with increased hydrophobicity due to excluding water and introducing more aliphatic residues into the boundary of the substrate binding cavity (Figures 5, 6 and S4). An increase in hydrophobicity would be expected to increase the redox potential of the diiron cluster and decrease its affinity for O2.97, 101 This outcome is opposite to that experimentally observed in solution. However, the increase in hydrophobicity would lead to an increase in the affinity for O2 binding in the active site prior to forming a complex with the diiron cluster. The appropriate balance between binding substrates and activating O2 on the diiron cluster may be achieved through other structural changes revealed by the high resolution structure of Mt OB3b sMMOH:MMOB complex. For example, an increased negative charge density on first sphere ligands that stabilizes the FeIII state over the FeII state will decrease the redox potential. A comparison of the coordination environment of the diferric cluster in sMMOH to that in the sMMOH:MMOB complex shows that the first sphere ligands do not rearrange significantly upon MMOB binding (Figures 7A, B and C). The only reorganization of note is the small change in the rotamer configuration of Glu243 with respect to the diiron cluster, which moves the distal oxygen of the carboxylate function out of hydrogen bonding range of the water ligand (HOH3) on Fe1. However, this change and several additional small second sphere changes cause some of the ligand-to-iron distances to shorten upon complex formation with MMOB, namely HOH3-Fe1, Glu144-Fe1 and Glu144-Fe2 (Figures 7B and C). These shorter bonds reflect a greater charge donation by the ligands to the iron atoms, thereby lowering the reduction potential.
The charge donation by the first sphere ligands may also be affected by hydrogen bonding networks involving second sphere residues. As one example, MMOB binding causes displacement of an active site water that hydrogen bonds to Glu114 in uncomplexed sMMOH, thereby increasing charge density on Glu114 (Figure 6). A potentially much more important example is described in the following section.
Role of Hydrogen Bonding in Second Sphere Active Site Residues.
A set of strictly conserved second-sphere amino acid residues near the diiron cluster histidine ligands has been observed to be present in sMMOH and the entire family of bacterial multicomponent monooxygenases (BMMs) and structurally related enzymes (Figures S2 and 9).102 In these enzyme systems, the second sphere aspartate to histidine hydrogen bonds are strictly conserved such that the typical diiron cluster binding motif [E/D-X~30-E-X2-H] is better described as [E/D-X~29-DE-X-(R)H].103 In the BMM enzyme sub-family, the penultimate Arg is completely conserved [E/D-X~29-DE-X-RH] and the hydrogen bond network is extended further beyond the aspartate residues to the surface of the protein. The function of this network remains undefined in the BMM enzymes. However, the current high resolution structure of Mt OB3b sMMOH:MMOB allows the network of hydrogen bonds to be described in detail for the first time (Figures 9 and S8).
Figure 9.
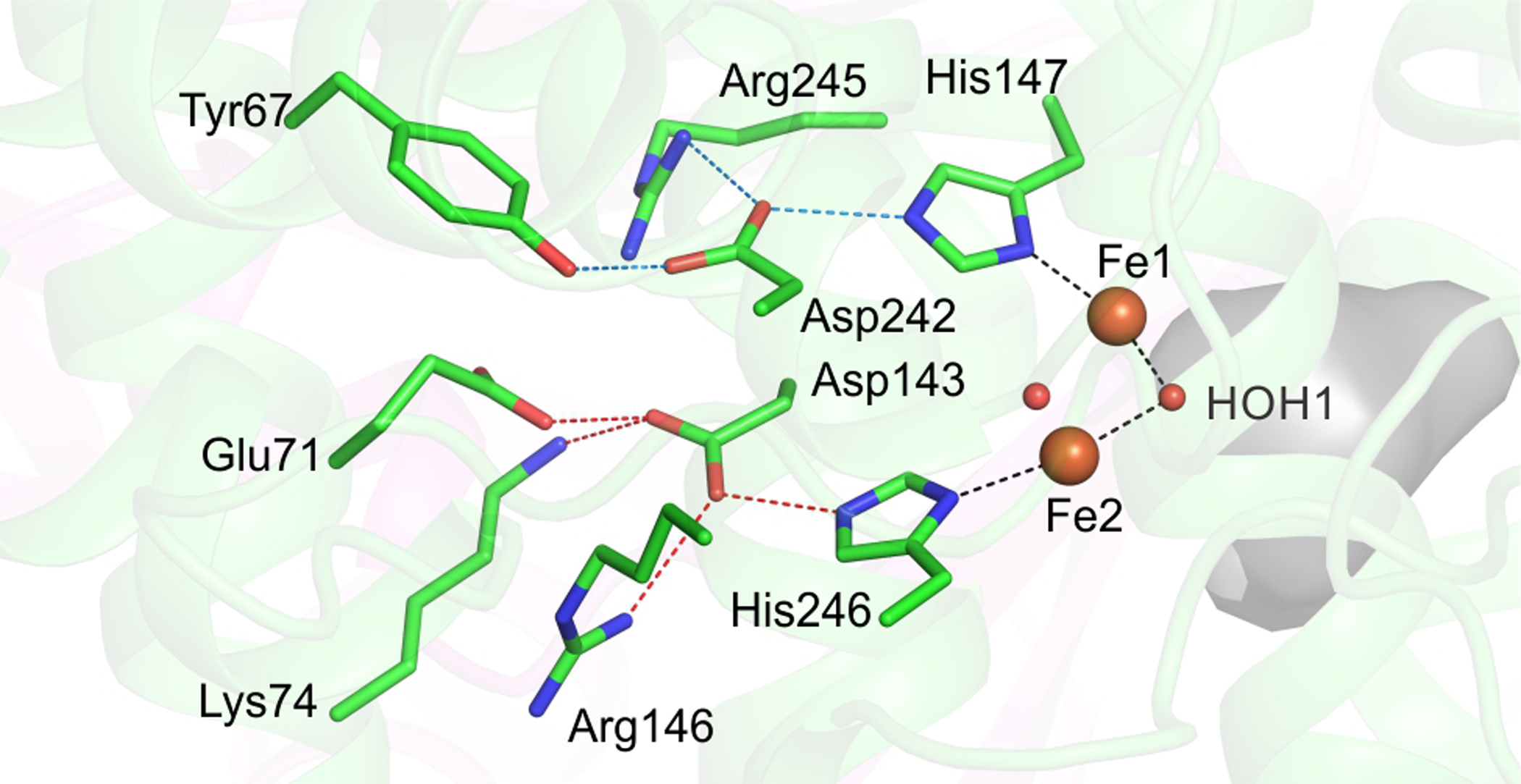
Strictly conserved hydrogen bond network on the distal side of the diiron cluster is shown. The iron atoms are represented as orange spheres while the bridging μ-hydroxo groups are shown as red spheres. The active site cavity is shown as a grey surface. Black dashed lines depict the trans positioning of the histidine ligands with respect to the μ-hydroxo moiety facing the active site. Red and blue dashed lines show the hydrogen bond network to each of the two histidine residues (His147 - blue, His246 - red).
One role that has been suggested for the hydrogen bonding network is electron transfer to the diiron cluster from the reductase MMOR.102 However, this role is unlikely because the exterior sMMOH protein surface from which the hydrogen bond network starts, lies at the dimerization interface between the two sMMOH monomers. As a result, this surface of sMMOH is occluded and cannot be involved in binding MMOR or in electron transfer. A more likely role for this network appears to be a structural stabilization of the diiron cluster, as the hydrogen bonds are arranged in a crisscross manner that would brace the position of the histidine ligands (Figures 9 and S8). Accordingly, the helices containing the residues involved in the hydrogen bond network have the lowest B-factors in the entire sMMOH protein structure (Figure S8).
Another potential role of the hydrogen bond network might be to tune the electronic properties of the diiron cluster by modulating charge donation from the diiron cluster histidine ligands. A comparison of the hydrogen bonding network in diferric Mt OB3b sMMOH alone (PDB:1MHY) to that in sMMOH:MMOB shows an overall shortening of the hydrogen bonds (Table S3). In particular, the two aspartate-to-histidine ligand hydrogen bonds are relatively short as measured in the oxidized Mt OB3b sMMOH:MMOB complex (average donor acceptor distance = 2.7 Å) (Figures 9 and S8 and Table S3). A strong hydrogen bond between the aspartate and histidine residues would partially deprotonate the histidine ligand, thereby increasing its imidazolate character and thus increasing electron donation to the iron atoms and decreasing the redox potential.
Implications of the sMMOH:MMOB Hydrogen Bonding Network for Reactivity.
It is well established that a hydrogen bonding Asp-His motif plays a vital role in increasing the basicity of the active-site histidine residue in both serine proteases and in heme-containing oxygenases and peroxidases.104–105 In the latter enzyme systems, greater charge donation from the axial histidine/cysteine ligand is positively correlated with increased reactivity towards C-H bond activation.106–108 This effect has been rationalized as arising from an increase in the pKa of the FeIV-OH compound II species, resulting in a stronger FeIV-O-H bond in compound II after hydrogen atom abstraction from the substrate C-H bond. This increases the driving force for the compound I mediated hydrogen atom abstraction step and reduces the activation energy barrier.
Similar considerations may apply to sMMO, which must generate an even more potent reactive species to effect the oxygenation of methane. The histidine ligands are positioned opposite to the active site cavity (Figures 9 and S8). In the compound Q species of sMMO, the reactive iron-oxygen moiety is expected to be either a FeIV-O-FeIV group or a terminal FeIV=O group that projects into the active site cavity.84, 109–110 One or both of the histidine ligands are trans to the reactive oxygen species. Thus, modulation in the electron donating capacity of the histidine ligands can have very important effects on the reactivity of Q towards methane oxidation. This modulation is linked in part to the conformational changes induced by MMOB as it binds (Table S3), which may contribute to the dramatic effects MMOB has on the reactivity of Q.10 Unfortunately, it is not currently possible to experimentally examine the role of the conserved hydrogen bonding network by creating variants because efforts to heterologously express sMMOH in numerous laboratories have not been successful.
Correlation of Mt OB3b sMMOH:MMOB Complex Structures with Past Spectroscopic Studies.
The high resolution crystal structures of the sMMOH:MMOB complex also enable a correlation between MMOB binding-induced changes in structure and electronic environment of the diiron cluster. EPR spectroscopic studies of the S = ½ mixed-valent state of sMMOH and the diferric state radiolytically one-electron-reduced at 79K (a probe of the diferric state) have provided evidence for a weakening of the antiferromagnetic coupling between the iron atoms upon MMOB binding.18, 111 The antiferromagnetic coupling constant J for the spin-exchanged mixed-valent diiron cluster is reduced from ~30 cm−1 to ~5 cm−1 in the presence of MMOB. Accordingly, the structure of the sMMOH:MMOB complex shows that the bridging Fe-O bonds (Fe1-HOH2, Fe2-HOH2) are lengthened (Figures 7B and C), which would be expected to moderate the spin-exchange coupling between the iron atoms.
The electronic environment of the reduced diiron cluster of sMMOH has also been probed in the presence and absence of MMOB with circular dichroism (CD) and magnetic circular dichroism (MCD) spectroscopies.112–113 These studies showed that the ligand field environment of only one of the two iron atoms of the diiron cluster changes upon MMOB binding to sMMOH. The crystal structure of the protein complex clearly shows that this insight is correct and identifies the affected iron atom as Fe2 (Figure 10). The ligand field of each iron atom of the reduced diiron cluster was additionally calculated in order to match the experimental ligand field transitions of the sMMOH:MMOB complex.114 A predicted Fe1-Fe2-Glu209 angle of 135° was shown to best match the experimental ligand field transitions. This angle would represent an increase from 120° observed for the diferrous sMMOH alone and would expose a site for O2 binding to Fe2 in the absence of a bridging aquo-ligand (Figure 10). The MCD spectra show that both irons are 5-coordinate in solution, and thus the weak bridging water (HOH1) seen in the crystal structure is lost. In accord with the spectroscopic and computational results, the Fe1-Fe2-Glu209 angle in the reduced sMMOH:MMOB complex is observed to be 143°. It is pertinent to note here that the Glu209 backbone is not anchored by an intra main-chain hydrogen bond upon MMOB binding. Moreover, the B-factors of the first and second sphere residues of Fe2 are large compared to those of Fe1, suggesting that flexibility in this region may be associated with preparing the diiron cluster to bind O2 (Figure S5). Finally, analysis of the CD spectra showed that changes in protein structure occur in the active site away from the diiron cluster upon MMOB binding to diferrous sMMOH that are essential for a rapid reaction with O2.114 The crystal structure of the diferrous complex demonstrates numerous changes in second sphere residue positions and solvent occupancy (Figure 10) that will affect the accessibility to the active site and the geometry of small molecule binding.
Figure 10.
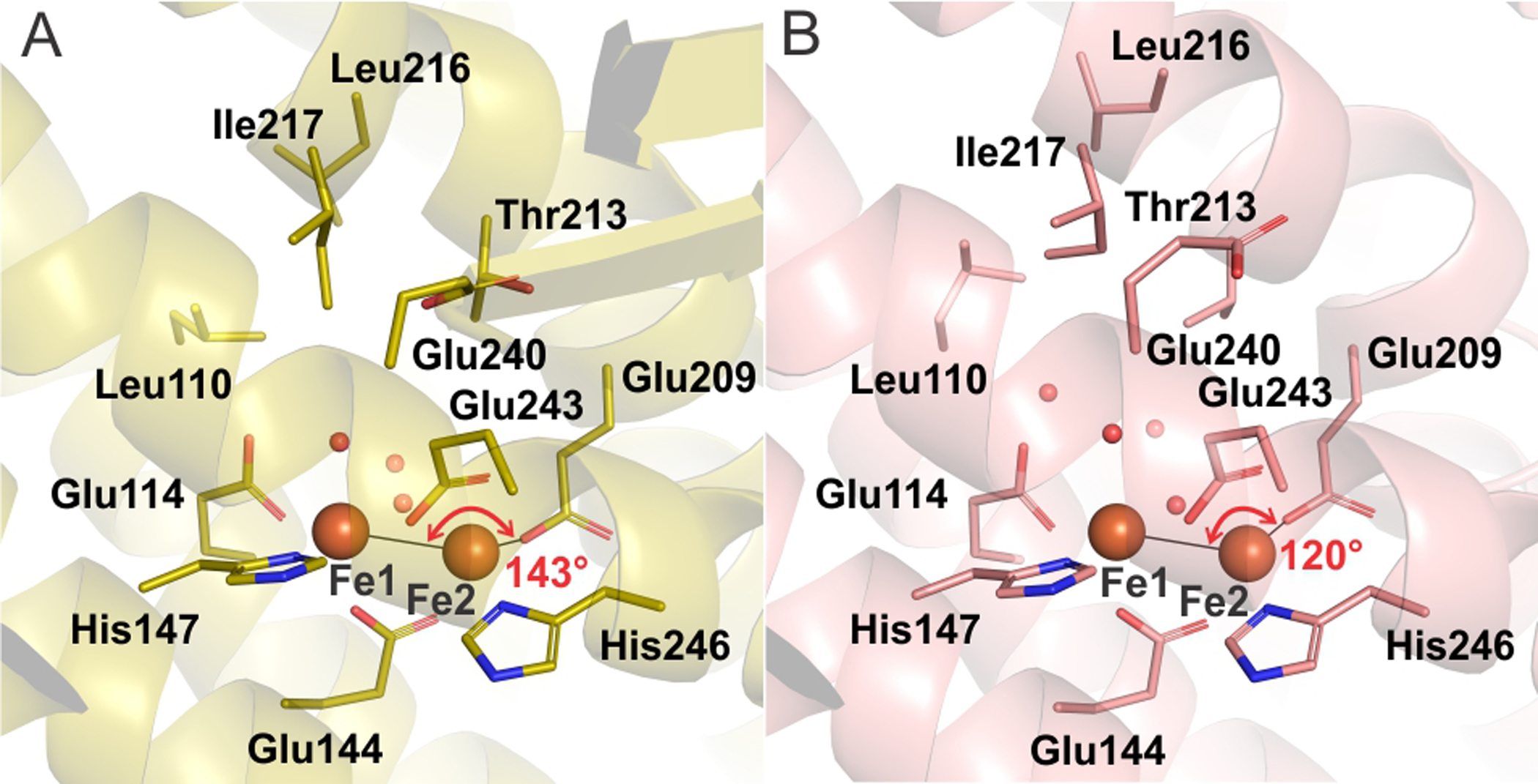
A comparison of the active site cavity of reduced sMMOHred:MMOB complex (PDB: 6YDI, panel A) and reduced sMMOH from M. capsulatus Bath (PDB: 1FYZ, panel B) shows reorganization prior to O2 binding and activation. The iron atoms are shown as orange spheres while red spheres represent water molecules.
CONCLUSION
The use of the XFEL SFX technique in combination with highly diffracting crystals of the Mt OB3b sMMOH:MMOB complex have significantly advanced structural studies of the sMMO enzyme system. This new approach has resulted in a high resolution crystal structure of the fully oxidized resting state of sMMOH:MMOB and the first crystal structure of the O2-reactive diferrous state. The SFX methodology is shown here to circumvent the most common problem in determining the crystal structure of oxidized metalloenzymes, which is reduction during the long duration synchrotron experiment. This conclusion is further supported by the concomitantly collected XES data that indicate a clear change in oxidation state of the Fe site between the oxidized resting state and the diferrous state. The high resolution crystal structures presented here illustrate in detail the extensive structural reorganization of both sMMOH and MMOB upon complexation. Some of these conformational changes shed light on the MMOB mediated regulation of catalysis by providing structural explanations for previous mutation and spectroscopic results. Most importantly, the crystal structures show the manner in which the active site cavity is altered in the protein complex, which is vital for enhancing the reaction with O2 and ultimately methane. Finally, the rapid reoxidation of the diiron cluster in the crystal of reduced sMMOH:MMOB upon exposure to O2 without loss of diffraction quality is very promising for ongoing XFEL SFX experiments designed to capture crystal structures of reaction cycle intermediates such as Q.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the financial support of this work from Grants NIH GM118030 (to J.D.L.), training grant GM08347 (to J.C.J.), training grant GM133081(to K.D.S), GM117126 (to N.K.S), GM55302 (to V.K.Y), GM110501 (to J.Y), GM126289 (to J.K), the Knut and Alice Wallenberg Foundation, and the Swedish Research Council no.2017-04018, and the European Research Council no.HIGH-GEAR 724394 (to M.H.), Biotechnology and Biological Sciences Research Council Grant 102593 (to A.M.O.); Wellcome Investigator Award in Science 210734/Z/18/Z (to A.M.O.); Royal Society Wolfson Fellowship RSWF\R2\182017 (to A.M.O and by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences of the Department of Energy (DOE) (to J.K., J.Y. and V.K.Y.). We thank the synchrotron facilities at the European Synchrotron Radiation facility (proposal no. MX-1948) and Swiss Light Source (proposal no. 20182304). This research used resources of the National Energy Research Scientific Computing Center, a User Facility supported by the Office of Science, DOE, under contract DE-AC02-05CH11231. The structure refinements were performed using PReSTO software stack on resources provided by the Swedish National Infrastructure for Computing (SNIC) at LUNARC Aurora, Lund University; proposal number SNIC 2018/3-251 and SNIC 2019/35-61. XFEL data was collected under proposal LU50 at LCLS/SLAC, Stanford, USA, under proposal 2017B8085 at SACLA, Japan, and under proposal 2019-2nd-NCI-029 at PAL-XFEL, Korea. The Rayonix detector used at LCLS was supported by the NIH grant S10 OD023453. LCLS, SLAC National Accelerator Laboratory, is supported by DOE, OBES under Contract No. DE-AC02-76SF00515. We thank the staff at LCLS, SACLA and PAL-XFEL.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://doi.org/10.1021/jacs.0c05613
UNIPROT ID CODES
Methane monooxygenase hydroxylase component (sMMOH, α, β, γ chains) UniProt P27353, A0A1A6FJQ4 and A0A1A6FHH2; Methane monooxygenase regulatory component B (MMOB) UniProt P27356; Methane monooxygenase reductase component (MMOR) UniProt Q53563.
DATA AVAILABILITY
Structures and crystallographic data have been deposited in the Protein Data Bank with the following accession codes: diferric sMMOH:MMOB, 6YD0; diferrous sMMOH:MMOB, 6YDI; reoxidized sMMOH:MMOB, 6YDU; diferrous sMMOH:MMOB from crystals treated in the same way as used to produce reoxidized sMMOH:MMOB but exposed to helium in the DOT reaction chamber rather than O2, 6YY3. All other data can be obtained from the corresponding authors upon request.
The authors declare no competing financial interest.
REFERENCES
- 1.Banerjee R; Jones JC; Lipscomb JD, Soluble methane monooxygenase. Ann. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- 2.Morton JD; Hayes KF; Semrau JD, Effect of copper speciation on whole-cell soluble methane monooxygenase activity in Methylosinus trichosporium OB3b. Appl. Environ. Microbiol 2000, 66, 1730–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Visscher A; Boeckx P; Van Cleemput O, Artificial methane sinks In Greenhouse Gas Sinks, Reay DS; Hewitt CN; Smith KA; Grace J, Eds. CAB International: Wallingford, Oxfordshire, UK, 2007; pp 184–200. [Google Scholar]
- 4.Reay DS; Smith P; Christensen TR; James RH; Clark H, Methane and global environmental change. Annu. Rev. Environ. Resour 2018, 43, 165–192. [Google Scholar]
- 5.Fletcher SEM; Schaefer H, Rising methane: A new climate challenge. Science 2019, 364, 932–933. [DOI] [PubMed] [Google Scholar]
- 6.Clomburg JM; Crumbley AM; Gonzalez R, Industrial biomanufacturing: The future of chemical production. Science 2017, 355, 38–39. [DOI] [PubMed] [Google Scholar]
- 7.Ravi M; Ranocchiari M; van Bokhoven JA, The direct catalytic oxidation of methane to methanol—A critical assessment. Angew. Chem. Int. Ed 2017, 56, 16464–16483. [DOI] [PubMed] [Google Scholar]
- 8.Fox BG; Froland WA; Dege JE; Lipscomb JD, Methane monooxygenase from Methylosinus trichosporium OB3b. Purification and properties of a three-component system with high specific activity from a type II methanotroph. J. Biol. Chem 1989, 264, 10023–10033. [PubMed] [Google Scholar]
- 9.Pilkington SJ; Dalton H, Soluble methane monooxygenase from Methylococcus capsulatus (Bath). Methods Enzymol 1990, 188, 181–190. [Google Scholar]
- 10.Wallar BJ; Lipscomb JD, Methane monooxygenase component B mutants alter the kinetics of steps throughout the catalytic cycle. Biochemistry 2001, 40, 2220–2233. [DOI] [PubMed] [Google Scholar]
- 11.Tinberg CE; Lippard SJ, Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res 2011, 44, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenzweig AC; Frederick CA; Lippard SJ; Nordlund P, Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [DOI] [PubMed] [Google Scholar]
- 13.Rosenzweig AC; Nordlund P; Takahara PM; Frederick CA; Lippard SJ, Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol 1995, 2, 409–418. [PubMed] [Google Scholar]
- 14.Elango N; Radhakrishnan R; Froland WA; Wallar BJ; Earhart CA; Lipscomb JD; Ohlendorf DH, Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci 1997, 6, 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu LJ; Liu Y; Lipscomb JD; Que L Jr., X-ray absorption spectroscopic studies of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3b. J. Biol. Inorg. Chem 1996, 1, 297–304. [Google Scholar]
- 16.Fox BG; Surerus KK; Münck E; Lipscomb JD, Evidence for a μ-oxo-bridged binuclear iron cluster in the hydroxylase component of methane monooxygenase. Mössbauer and EPR studies. J. Biol. Chem 1988, 263, 10553–10556. [PubMed] [Google Scholar]
- 17.Froland WA; Andersson KK; Lee S-K; Liu Y; Lipscomb JD, Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. A novel role for protein-protein interactions in an oxygenase mechanism. J. Biol. Chem 1992, 267, 17588–17597. [PubMed] [Google Scholar]
- 18.Fox BG; Liu Y; Dege JE; Lipscomb JD, Complex formation between the protein components of methane monooxygenase from Methylosinus trichosporium OB3b. Identification of sites of component interaction. J. Biol. Chem 1991, 266, 540–550. [PubMed] [Google Scholar]
- 19.Liu Y; Nesheim JC; Lee S-K; Lipscomb JD, Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J. Biol. Chem 1995, 270, 24662–246625. [DOI] [PubMed] [Google Scholar]
- 20.Wallar BJ; Lipscomb JD, Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev 1996, 96, 2625–2657. [DOI] [PubMed] [Google Scholar]
- 21.Paulsen KE; Liu Y; Fox BG; Lipscomb JD; Münck E; Stankovich MT, Oxidation-reduction potentials of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3b. Biochemistry 1994, 33, 713–722. [DOI] [PubMed] [Google Scholar]
- 22.Hendrich MP; Münck E; Fox BG; Lipscomb JD, Integer-spin EPR studies of the fully reduced methane monooxygenase hydroxylase component. J. Am. Chem. Soc 1990, 112, 5861–5865. [Google Scholar]
- 23.Zhang J; Wallar BJ; Popescu CV; Renner DB; Thomas DD; Lipscomb JD, Methane monooxygenase hydroxylase and B component interactions. Biochemistry 2006, 45, 2913–2926. [DOI] [PubMed] [Google Scholar]
- 24.Chang SL; Wallar BJ; Lipscomb JD; Mayo KH, Solution structure of component B from methane monooxygenase derived through heteronuclear NMR and molecular modeling. Biochemistry 1999, 38, 5799–5812. [DOI] [PubMed] [Google Scholar]
- 25.Chang SL; Wallar BJ; Lipscomb JD; Mayo KH, Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: a role for the N-terminus. Biochemistry 2001, 40, 9539–9551. [DOI] [PubMed] [Google Scholar]
- 26.Walters KJ; Gassner GT; Lippard SJ; Wagner G, Structure of the soluble methane monooxygenase regulatory protein B. Proc. Natl. Acad. Sci. USA 1999, 96, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W; Lippard SJ, Diiron oxidation state control of substrate access to the active site of soluble methane monooxygenase mediated by the regulatory component. J. Am. Chem. Soc 2014, 136, 2244–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SJ; McCormick MS; Lippard SJ; Cho U-S, Control of substrate access to the active site in methane monooxygenase. Nature 2013, 494, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brazeau BJ; Lipscomb JD, Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B. Biochemistry 2003, 42, 5618–5631. [DOI] [PubMed] [Google Scholar]
- 30.Zheng H; Lipscomb JD, Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling. Biochemistry 2006, 45, 1685–1692. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J; Lipscomb JD, Role of the C-terminal region of the B component of Methylosinus trichosporium OB3b methane monooxygenase in the regulation of oxygen activation. Biochemistry 2006, 45, 1459–1469. [DOI] [PubMed] [Google Scholar]
- 32.Yano J; Kern J; Irrgang K-D; Latimer MJ; Bergmann U; Glatzel P; Pushkar Y; Biesiadka J; Loll B; Sauer K; Messinger J; Zouni A; Yachandra VK, X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA 2005, 102, 12047–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meharenna YT; Doukov T; Li H; Soltis SM; Poulos TL, Crystallographic and single-crystal spectral analysis of the peroxidase ferryl intermediate. Biochemistry 2010, 49, 2984–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebrahim A; Moreno-Chicano T; Appleby MV; Chaplin AK; Beale JH; Sherrell DA; Duyvesteyn HME; Owada S; Tono K; Sugimoto H; Strange RW; Worrall JAR; Axford D; Owen RL; Hough MA, Dose-resolved serial synchrotron and XFEL structures of radiation-sensitive metalloproteins. IUCrJ 2019, 6, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de la Mora E; Coquelle N; Bury CS; Rosenthal M; Holton JM; Carmichael I; Garman EF; Burghammer M; Colletier J-P; Weik M, Radiation damage and dose limits in serial synchrotron crystallography at cryo- and room temperatures. Proc. Natl. Acad. Sci. USA 2020, 117, 4142–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickerson JL; McCubbin PTN; Garman EF, RADDOSE-XFEL: femtosecond time-resolved dose estimates for macromolecular X-ray free-electron laser experiments. J. Appl. Crystallogr 2020, 53, 549–560. [Google Scholar]
- 37.Bury CS; Brooks-Bartlett JC; Walsh SP; Garman EF, Estimate your dose: RADDOSE-3D. Protein Sci 2018, 27, 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue I; Inubushi Y; Sato T; Tono K; Katayama T; Kameshima T; Ogawa K; Togashi T; Owada S; Amemiya Y; Tanaka T; Hara T; Yabashi M, Observation of femtosecond X-ray interactions with matter using an X-ray–X-ray pump–probe scheme. Proc. Natl. Acad. Sci. USA 2016, 113, 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gruner SM, Expanding the femtosecond crystallography toolkit. Proc. Natl. Acad. Sci. USA 2014, 111, 16986–16987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeil BWJ; Thompson NR, X-ray free-electron lasers. Nat. Photonics 2010, 4, 814–821. [Google Scholar]
- 41.Kern J; Alonso-Mori R; Tran R; Hattne J; Gildea RJ; Echols N; Gloeckner C; Hellmich J; Laksmono H; Sierra RG; Lassalle-Kaiser B; Koroidov S; Lampe A; Han G; Gul S; DiFiore D; Milathianaki D; Fry AR; Miahnahri A; Schafer DW; Messerschmidt M; Seibert MM; Koglin JE; Sokaras D; Weng T-C; Sellberg J; Latimer MJ; Grosse-Kunstleve RW; Zwart PH; White WE; Glatzel P; Adams PD; Bogan MJ; Williams GJ; Boutet S; Messinger J; Zouni A; Sauter NK; Yachandra VK; Bergmann U; Yano J, Simultaneous femtosecond x-ray spectroscopy and diffraction of photosystem II at room temperature. Science 2013, 340, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chapman HN, X-Ray free-electron lasers for the structure and dynamics of macromolecules. Annu. Rev. Biochem 2019, 88, 35–58. [DOI] [PubMed] [Google Scholar]
- 43.Schlichting I, Serial femtosecond crystallography: the first five years. IUCrJ 2015, 2, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fromme P, XFELs open a new era in structural chemical biology. Nat. Chem. Biol 2015, 11, 895–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spence J, XFELs for structure and dynamics in biology. IUCrJ 2017, 4, 322–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kern J; Chatterjee R; Young ID; Fuller FD; Lassalle L; Ibrahim M; Gul S; Fransson T; Brewster AS; Alonso-Mori R; Hussein R; Zhang M; Douthit L; de Lichtenberg C; Cheah MH; Shevela D; Wersig J; Seuffert I; Sokaras D; Pastor E; Weninger C; Kroll T; Sierra RG; Aller P; Butryn A; Orville AM; Liang M; Batyuk A; Koglin JE; Carbajo S; Boutet S; Moriarty NW; Holton JM; Dobbek H; Adams PD; Bergmann U; Sauter NK; Zouni A; Messinger J; Yano J; Yachandra VK, Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suga M; Shimada A; Akita F; Shen J-R; Tosha T; Sugimoto H Time-resolved studies of metalloproteins using X-ray free electron laser radiation at SACLA Biochim. Biophys. Acta, 2020,1864, 129466. [DOI] [PubMed] [Google Scholar]
- 48.Stagno JR; Liu Y; Bhandari YR; Conrad CE; Panja S; Swain M; Fan L; Nelson G; Li C; Wendel DR; White TA; Coe JD; Wiedorn MO; Knoska J; Oberthuer D; Tuckey RA; Yu P; Dyba M; Tarasov SG; Weierstall U; Grant TD; Schwieters CD; Zhang J; Ferré-D’Amaré AR; Fromme P; Draper DE; Liang M; Hunter MS; Boutet S; Tan K; Zuo X; Ji X; Barty A; Zatsepin NA; Chapman HN; Spence JCH; Woodson SA; Wang YX, Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kupitz C; Olmos JL Jr.; Holl M; Tremblay L; Pande K; Pandey S; Oberthür D; Hunter M; Liang M; Aquila A; Tenboer J; Calvey G; Katz A; Chen Y; Wiedorn MO; Knoska J; Meents A; Majriani V; Norwood T; Poudyal I; Grant T; Miller MD; Xu W; Tolstikova A; Morgan A; Metz M; Martin-Garcia JM; Zook JD; Roy-Chowdhury S; Coe J; Nagaratnam N; Meza D; Fromme R; Basu S; Frank M; White T; Barty A; Bajt S; Yefanov O; Chapman HN; Zatsepin N; Nelson G; Weierstall U; Spence J; Schwander P; Pollack L; Fromme P; Ourmazd A; Phillips GN Jr.; Schmidt M, Structural enzymology using X-ray free electron lasers. Struct Dyn 2016, 4, 044003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tenboer J; Basu S; Zatsepin N; Pande K; Milathianaki D; Frank M; Hunter M; Boutet S; Williams GJ; Koglin JE; Oberthuer D; Heymann M; Kupitz C; Conrad C; Coe J; Roy-Chowdhury S; Weierstall U; James D; Wang D; Grant T; Barty A; Yefanov O; Scales J; Gati C; Seuring C; Srajer V; Henning R; Schwander P; Fromme R; Ourmazd A; Moffat K; Van Thor JJ; Spence JCH; Fromme P; Chapman HN; Schmidt M, Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2014, 346, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tosha T; Nomura T; Nishida T; Saeki N; Okubayashi K; Yamagiwa R; Sugahara M; Nakane T; Yamashita K; Hirata K; Ueno G; Kimura T; Hisano T; Muramoto K; Sawai H; Takeda H; Mizohata E; Yamashita A; Kanematsu Y; Takano Y; Nango E; Tanaka R; Nureki O; Shoji O; Ikemoto Y; Murakami H; Owada S; Tono K; Yabashi M; Yamamoto M; Ago H; Iwata S; Sugimoto H; Shiro Y; Kubo M Capturing an initial intermediate during the P450nor enzymatic reaction using time-resolved XFEL crystallography and caged-substrate Nat. Commun, 2017,8, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda Y; Tse KM; Nakane T; Nakatsu T; Suzuki M; Sugahara M; Inoue S; Masuda T; Yumoto F; Matsugaki N; Nango E; Tono K; Joti Y; Kameshima T; Song C; Hatsui T; Yabashi M; Nureki O; Murphy MEP; Inoue T; Iwata S; Mizohata E, Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc. Natl. Acad. Sci. USA 2016, 113, 2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuller FD; Gul S; Chatterjee R; Burgie ES; Young ID; Lebrette H; Srinivas V; Brewster AS; Michels-Clark T; Clinger JA; Andi B; Ibrahim M; Pastor E; de Lichtenberg C; Hussein R; Pollock CJ; Zhang M; Stan CA; Kroll T; Fransson T; Weninger C; Kubin M; Aller P; Lassalle L; Bräuer P; Miller MD; Amin M; Koroidov S; Roessler CG; Allaire M; Sierra RG; Docker PT; Glownia JM; Nelson S; Koglin JE; Zhu D; Chollet M; Song S; Lemke H; Liang M; Sokaras D; Alonso-Mori R; Zouni A; Messinger J; Bergmann U; Boal AK; Bollinger JM Jr; Krebs C; Högbom M; Phillips GN Jr; Vierstra RD; Sauter NK; Orville AM; Kern J; Yachandra VK; Yano J, Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nat. Methods 2017, 14, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ibrahim M; Fransson T; Chatterjee R; Cheah MH; Hussein R; Lassalle L; Sutherlin KD; Young ID; Fuller FD; Gul S; Kim I-S; Simon PS; deLichtenberg C; Chernev P; Bogacz I; Pham CC; Orville AM; Saichek N; Northen T; Batyuk A; Carbajo S; Alonso-Mori R; Tono K; Owada S; Bhowmick A; Bolotovsky R; Mendez D; Moriarty NW; Holton JM; Dobbek H; Brewster AS; Adams PD; Sauter NK; Bergmann U; Zoun A; Messinger J; Kern J; Yachandra VK; Yano J, Untangling the sequence of events during the S2 → S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. USA 2020, 117, 12624–12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fox BG; Froland WA; Jollie DR; Lipscomb JD, Methane monooxygenase from Methylosinus trichosporium OB3b. Methods Enzymol 1990, 188, 191–202. [DOI] [PubMed] [Google Scholar]
- 56.Banerjee R; Komor AJ; Lipscomb JD, Use of isotopes and isotope effects for investigations of diiron oxygenase mechanisms. Methods Enzymol 2017, 596, 239–290. [DOI] [PubMed] [Google Scholar]
- 57.Banerjee R; Meier KK; Münck E; Lipscomb JD, Intermediate P* from soluble methane monooxygenase contains a diferrous cluster. Biochemistry 2013, 52, 4331–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugahara M; Mizohata E; Nango E; Suzuki M; Tanaka T; Masuda T; Tanaka R; Shimamura T; Tanaka Y; Suno C; Ihara K; Pan D; Kakinouchi K; Sugiyama S; Murata M; Inoue T; Tono K; Song C; Park J; Kameshima T; Hatsui T; Joti Y; Yabashi M; Iwata S, Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 2015, 12, 61–63. [DOI] [PubMed] [Google Scholar]
- 59.Tono K; Togashi T; Inubushi Y; Sato T; Katayama T; Ogawa K; Ohashi H; Kimura H; Takahashi S; Takeshita K; Tomizawa H; Goto S; Ishikawa T; Yabashi M Beamline, experimental stations and photon beam diagnostics for the hard x-ray free electron laser of SACLA New J. Phys, 2013,15, 083035. [Google Scholar]
- 60.Sierra RG; Batyuk A; Sun Z; Aquila A; Hunter MS; Lane TJ; Liang M; Yoon CH; Alonso-Mori R; Armenta R; Castagna J-C; Hollenbeck M; Osier TO; Hayes M; Aldrich J; Curtis R; Koglin JE; Rendahl T; Rodriguez E; Carbajo S; Guillet S; Paul R; Hart P; Nakahara K; Carini G; DeMirci H; Dao EH; Hayes BM; Rao YP; Chollet M; Feng Y; Fuller FD; Kupitz C; Sato T; Seaberg MH; Song S; van Driel TB; Yavas H; Zhu D; Cohen AE; Wakatsuki S; Boutet S, The macromolecular femtosecond crystallography instrument at the linac coherent light source. J. Synchrotron Rad 2019, 26, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ko I; Kang H-S; Heo H; Kim C; Kim G; Min C-K; Yang H; Baek S; Choi H-J; Mun G; Park B; Suh Y; Shin D; Hu J; Hong J; Jung S; Kim S-H; Kim K; Na D; Lee K, Construction and commissioning of PAL-XFEL facility. Appl. Sci 2017, 7, 479. [Google Scholar]
- 62.Alonso-Mori R; Kern J; Sokaras D; Weng T-C; Nordlund D; Tran R; Montanez P; Delor J; Yachandra VK; Yano J; Bergmann U, A multi-crystal wavelength dispersive x-ray spectrometer. Rev. Sci. Instrum 2012, 83, 073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hattne J; Echols N; Tran R; Kern J; Gildea RJ; Brewster AS; Alonso-Mori R; Glöckner C; Hellmich J; Laksmono H; Sierra RG; Lassalle-Kaiser B; Lampe A; Han G; Gul S; DiFiore D; Milathianaki D; Fry AR; Miahnahri A; White WE; Schafer DW; Seibert MM; Koglin JE; Sokaras D; Weng T-C; Sellberg J; Latimer MJ; Glatzel P; Zwart PH; Grosse-Kunstleve RW; Bogan MJ; Messerschmidt M; Williams GJ; Boutet S; Messinger J; Zouni A; Yano J; Bergmann U; Yachandra VK; Adams PD; Sauter NK, Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat. Methods 2014, 11, 545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winter G; Waterman DG; Parkhurst JM; Brewster AS; Gildea RJ; Gerstel M; Fuentes-Montero L; Vollmar M; Michels-Clark T; Young ID; Sauter NK; Evans G, DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol 2018, 74, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poon BK; Grosse-Kunstleve RW; Zwart PH; Sauter NK, Detection and correction of underassigned rotational symmetry prior to structure deposition. Acta Crystallogr. D 2010, 66, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. E 2010, D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liebschner D; Afonine PV; Baker ML; Bunkóczi G; Chen VB; Croll TI; Hintze B; Hung L-W; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD, Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 2019, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P; Cowtan K, Coot: Model-building tools for molecular graphics. Acta Crystallogr. E 2004, E60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 70.Sehnal D; Svobodová Vařeková R; Berka K; Pravda L; Navrátilová V; Banáš P; Ionescu C-M; Otyepka M; Koča J MOLE 2.0: advanced approach for analysis of biomacromolecular channels J. Cheminformatics, 2013,5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brazeau BJ; Wallar BJ; Lipscomb JD, Unmasking of deuterium kinetic isotope effects on the methane monooxygenase compound Q reaction by site-directed mutagenesis of component B. J. Am. Chem. Soc 2001, 123, 10421–10422. [DOI] [PubMed] [Google Scholar]
- 72.Cooley RB; Arp DJ; Karplus PA, Evolutionary origin of a secondary structure: pi-helices as cryptic but widespread insertional variations of alpha-helices that enhance protein functionality. J. Mol. Biol 2010, 404, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barlow DJ; Thornton JM, Helix geometry in proteins. J. Mol. Biol 1988, 201, 601–619. [DOI] [PubMed] [Google Scholar]
- 74.Joosten RP; te Beek TAH; Krieger E; Hekkelman ML; Hooft RWW; Schneider R; Sander C; Vriend G, A series of PDB related databases for everyday needs. Nucleic Acids Res 2010, 39, D411–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kabsch W; Sander C, Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 76.Zhu H DSSP and Stride plugin for PyMOL, BIOTEC, TU Dresden: Dresden, Germany, 2011. [Google Scholar]
- 77.Ho BK; Gruswitz F HOLLOW: Generating accurate representations of channel and interior surfaces in molecular structures BMC Structural Biology, 2008,8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glatzel P; Bergmann U, High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes-electronic and structural information. Coord. Chem. Rev 2005, 249, 65–95. [Google Scholar]
- 79.Schuth N; Zaharieva I; Chernev P; Berggren G; Anderlund M; Styring S; Dau H; Haumann M, Kα X-ray emission spectroscopy on the photosynthetic oxygen-evolving complex supports manganese oxidation and water binding in the S3 state. Inorg. Chem 2018, 57, 10424–10430. [DOI] [PubMed] [Google Scholar]
- 80.Vankó G; Neisius T; Molnár G; Renz F; Kárpáti S; Shukla A; de Groot FMF, Probing the 3d spin momentum with X-ray emission spectroscopy: The case of molecular-spin transitions. J. Phys. Chem. B 2006, 110, 11647–11653. [DOI] [PubMed] [Google Scholar]
- 81.Svyazhin A; Kurmaev E; Shreder E; Shamin S; Sahle CJ, Local moments and electronic correlations in Fe-based Heusler alloys: Kα x-ray emission spectra measurements. J. Alloys Compd 2016, 679, 268–276. [Google Scholar]
- 82.Baydas E; Oz E, Chemical effects in the Kα and Kβ1,3 of X-ray emission spectra of Fe. J. Electron Spectrosc. Relat. Phenom 2012, 185, 27–31. [Google Scholar]
- 83.Yasuda S; Kakiyama H, X-ray K emission spectra of vanadium in various oxidation states. X-Ray Spectrom 1978, 7, 23–25. [Google Scholar]
- 84.Cutsail GE; Banerjee R; Zhou A; Que L; Lipscomb JD; DeBeer S, High-resolution extended x-ray absorption fine structure analysis provides evidence for a longer Fe···Fe distance in the Q intermediate of methane monooxygenase. J. Am. Chem. Soc 2018, 140, 16807–16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee SK; Nesheim JC; Lipscomb JD, Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem 1993, 268, 21569–21577. [PubMed] [Google Scholar]
- 86.Tinberg CE; Lippard SJ, Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): Evidence for a multi-step, proton-dependent reaction pathway. Biochemistry 2009, 48, 12145–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Corbett MC; Latimer MJ; Poulos TL; Sevrioukova IF; Hodgson KO; Hedman B, Photoreduction of the active site of the metalloprotein putidaredoxin by synchrotron radiation. Acta Crystallogr. D 2007, 63, 951–960. [DOI] [PubMed] [Google Scholar]
- 88.Liu B; Chen Y; Doukov T; Soltis SM; Stout CD; Fee JA, Combined microspectrophotometric and crystallographic examination of chemically reduced and X-ray radiation-reduced forms of cytochrome ba3 oxidase from Thermus thermophilus: Structure of the reduced form of the enzyme. Biochemistry 2009, 48, 820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hersleth H-P; Uchida T; Røhr ÅK; Teschner T; Schünemann V; Kitagawa T; Trautwein AX; Görbitz CH; Andersson KK, Crystallographic and spectroscopic studies of peroxide-derived myoglobin compound II and occurrence of protonated FeIV–O. J. Biol. Chem 2007, 282, 23372–23386. [DOI] [PubMed] [Google Scholar]
- 90.Alonso-Mori R; Kern J; Gildea RJ; Sokaras D; Weng T-C; Lassalle-Kaiser B; Tran R; Hattne J; Laksmono H; Hellmich J; Glockner C; Echols N; Sierra RG; Schafer DW; Sellberg J; Kenney C; Herbst R; Pines J; Hart P; Herrmann S; Grosse-Kunstleve RW; Latimer MJ; Fry AR; Messerschmidt MM; Miahnahri A; Seibert MM; Zwart PH; White WE; Adams PD; Bogan MJ; Boutet S; Williams GJ; Zouni A; Messinger J; Glatzel P; Sauter NK; Yachandra VK; Yano J; Bergmann U, Energy-dispersive X-ray emission spectroscopy using an X-ray free-electron laser in a shot-by-shot mode. Proc. Natl. Acad. Sci. USA 2012, 109, 19103–19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee SK; Lipscomb JD, Oxygen activation catalyzed by methane monooxygenase hydroxylase component: Proton delivery during the O-O bond cleavage steps. Biochemistry 1999, 38, 4423–4432. [DOI] [PubMed] [Google Scholar]
- 92.Hayashi Y; Yamazaki I, The oxidation-reduction potentials of compound I/compound II and compound II/ferric couples of horseradish peroxidases A2 and C. J. Biol. Chem 1979, 254, 9101–9106. [PubMed] [Google Scholar]
- 93.Chen MM; Coelho PS; Arnold FH, Utilizing terminal oxidants to achieve P450-catalyzed oxidation of methane. Adv. Synth. Catal 2012, 354, 964–968. [Google Scholar]
- 94.Berglund GI; Carlsson GH; Smith AT; Szoke H; Henriksen A; Hajdu J, The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [DOI] [PubMed] [Google Scholar]
- 95.Nesheim JC; Lipscomb JD, Large isotope effects in methane oxidation catalyzed by methane monooxygenase: Evidence for C–H bond cleavage in a reaction cycle intermediate. Biochemistry 1996, 35, 10240–10247. [DOI] [PubMed] [Google Scholar]
- 96.Klinman JP, Dynamically achieved active site precision in enzyme catalysis. Acc. Chem. Res 2015, 48, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hosseinzadeh P; Lu Y, Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta 2016, 1857, 557–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tezcan FA; Winkler JR; Gray HB, Effects of ligation and folding on reduction potentials of heme proteins. J. Am. Chem. Soc 1998, 120, 13383–13388. [Google Scholar]
- 99.Moore GR; Pettigrew GW; Rogers NK, Factors influencing redox potentials of electron transfer proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 4998–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DiCarlo CM; Vitello LB; Erman JE, Effect of active site and surface mutations on the reduction potential of yeast cytochrome c peroxidase and spectroscopic properties of the oxidized and reduced enzyme. J. Inorg. Biochem 2007, 101, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y, Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCormick MS; Lippard SJ, Analysis of substrate access to active sites in bacterial multicomponent monooxygenase hydroxylases: X-ray crystal structure of xenon-pressurized phenol hydroxylase from Pseudomonas sp. OX1. Biochemistry 2011, 50, 11058–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pikus JD; Studts JM; Achim C; Kauffmann KE; Münck E; Steffan RJ; McClay K; Fox BG, Recombinant toluene-4-monooxygenase: Catalytic and Mössbauer studies of the purified diiron and Rieske components of a four-protein complex. Biochemistry 1996, 35, 9106–9119. [DOI] [PubMed] [Google Scholar]
- 104.Carter P; Wells JA, Dissecting the catalytic triad of a serine protease. Nature 1988, 332, 564–568. [DOI] [PubMed] [Google Scholar]
- 105.Poulos TL, The role of the proximal ligand in heme enzymes. J. Biol. Inorg. Chem 1996, 1, 356–359. [Google Scholar]
- 106.Krest CM; Silakov A; Rittle J; Yosca TH; Onderko EL; Calixto JC; Green MT, Significantly shorter Fe-S bond in cytochrome P450-I is consistent with greater reactivity relative to chloroperoxidase. Nat. Chem 2015, 7, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Onderko EL; Silakov A; Yosca TH; Green MT, Characterization of a selenocysteine-ligated P450 compound I reveals direct link between electron donation and reactivity. Nat. Chem 2017, 9, 623–628. [DOI] [PubMed] [Google Scholar]
- 108.Poulos TL, Heme enzyme structure and function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Banerjee R; Proshlyakov Y; Lipscomb JD; Proshlyakov DA, Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 2015, 518, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castillo RG; Banerjee R; Allpress CJ; Rohde GT; Bill E; Que L Jr.; Lipscomb JD; DeBeer S, High-energy-resolution fluorescence-detected X-ray absorption of the Q intermediate of soluble methane monooxygenase. J. Am. Chem. Soc 2017, 139, 18024–18033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Davydov A; Davydov R; Gräslund A; Lipscomb JD; Andersson KK, Radiolytic reduction of methane monooxygenase dinuclear iron cluster at 77 K - EPR evidence for conformational change upon reduction or binding of component B to the diferric state. J. Biol. Chem 1997, 272, 7022–7026. [DOI] [PubMed] [Google Scholar]
- 112.Pulver S; Froland WA; Fox BG; Lipscomb JD; Solomon EI, Spectroscopic studies of the coupled binuclear non-heme iron active site in the fully reduced hydroxylase component of methane monooxygenase: Comparison to deoxy and deoxy-azide hemerythrin. J. Am. Chem. Soc 1993, 115, 12409–12422. [Google Scholar]
- 113.Pulver SC; Froland WA; Lipscomb JD; Solomon EI, Ligand field circular dichroism and magnetic circular dichroism studies of component B and substrate binding to the hydroxylase component of methane monooxygenase. J. Am. Chem. Soc 1997, 119, 387–395. [Google Scholar]
- 114.Mitić N; Schwartz JK; Brazeau BJ; Lipscomb JD; Solomon EI, CD and MCD studies of the effects of component B variant binding on the biferrous active site of methane monooxygenase. Biochemistry 2008, 47, 8386–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.