

Abstract
Long-lived somatic stem cells regenerate adult tissues throughout our lifetime. However, with aging, there is a significant deterioration in the function of stem and progenitor cells, which contribute to diseases of aging. The decision for a long-lived somatic stem cell to become activated and subsequently to undergo either a symmetric or an asymmetric division is a critical cellular decision process. The decision to preferentially divide symmetrically or asymmetrically may be the major fundamental intrinsic difference between normal somatic stem cells and cancer stem cells. Based upon work done primarily in our laboratory over the past 15 years, this article provides a perspective on the critical role of somatic stem cells in aging. In particular, we discuss the importance of symmetric versus asymmetric divisions in somatic stem cells and the role of the differential usage of the highly similar Kat3 coactivators, CREB-binding protein (CBP) and p300, in stem cells. We describe and propose a more complete model for the biological mechanism and roles of these two coactivators, their evolution, and unique roles and importance in stem cell biology. Finally, we discuss the potential to pharmacologically manipulate Kat3 coactivator interactions in endogenous stem cells (both normal and cancer stem cells) to potentially ameliorate the aging process and common diseases of aging.
Keywords: Kat3 coactivator, Somatic stem cells, Cancer stem cells, Pharmacology, CBP, p300
Introduction
“He who knows nothing is closer to the truth than he whose mind is filled with falsehoods and errors.”
―Thomas Jefferson
Throughout our lifetime, long-lived somatic stem cells (SSCs) regenerate adult tissues both during homeostatic processes and repair after injury. However, with aging, there is a significant deterioration in stem cell function in a wide array of tissues including blood (Chen et al. 2000), forebrain (Molofsky et al. 2006), skeletal muscle (Conboy et al. 2005), and skin (Nishimura et al. 2005). This decline in SSC functionality with age is associated with reduced (e.g., thinning of the epidermis and dermis) and/or aberrant tissue regeneration (e.g., fibrosis), increased degenerative diseases (e.g., Alzheimer’s disease), and cancer (Sharpless and DePinho 2007). It appears that age-related decline in the function of stem and progenitor cells contributes to most diseases of aging, particularly in regenerative tissues. This perspective will discuss the potential to pharmacologically manipulate our endogenous stem cell populations (both normal and cancer stem cells) to reverse the deterioration of somatic stem cell functionality with age, with emphasis on the biological roles and evolution of the two human Kat3 coactivators, p300 and CBP.
Stem cell definitions
What is a stem cell? In the lay press, stem cells have been touted as a panacea, potentially able to cure a wide range of diseases (Parkinson’s, AD, diabetes, etc.) or produce new organs for replacement after damage via disease or injury (spinal cord, heart, etc.). However, the term stem cell is in fact quite broad and covers a diverse array of cell types. The defining feature of all stem cells is their capacity to both self-renew (i.e., make at least one identical copy of itself at each division) as well as differentiate into more mature, albeit less potent, specialized cells. Stem cells can be embryonic (ES), induced pluripotent cells (iPS), or adult/SSC if derived from tissues. The enormous interest elicited by ES and more recently iPS cells is based on their key property of pluripotency. ES and iPS cells possess the rare and precious capacity to generate all the cell types found in embryos, as well as adult organisms. This unique property has been lost in SSCs. Many excellent recent reviews on ES and iPS cells have been published, and the interested reader is referred to these (Tanabe et al. 2014; Grabel 2012). However, there are still significant hurdles to overcome to effectively utilize either ES or iPS cells for therapeutic benefit (Barker and de Beaufort 2013).
SSCs
SSCs are multipotent, i.e., capable of generating multiple differentiated cell types but generally restricted to that of a particular tissue, organ, or physiological system (e.g., hematopoietic stem cells, neural stem cells, etc.) in which they reside, as opposed to pluripotent. One enormous advantage of endogenous SSCs over ES or iPS cells is that they already reside in the proper environment/location or “niche” for effective regeneration of endogenous tissues. Additionally, if it is possible to pharmacologically manipulate endogenous stem cells, by definition, they would be autologous and concerns about histo-incompatibility therefore absent.
The first type of SSC to be isolated and utilized therapeutically was the hematopoietic stem cell (HSC) in the form of bone marrow for transplantation therapy (Weissman and Shizuru 2008). Subsequent studies have demonstrated the existence of a large variety of additional SSC populations. Adult SSCs, although generally present in quite limited numbers, are believed to be essentially immortal (i.e., they are with us for our entire lives) and the source of endogenous tissue regeneration, both homeostatic and repair after injury, in adult tissues. This has already been demonstrated in the lung (Warburton et al. 2008), in the heart after myocardial infarction (Yacoub et al. 2006), or in the adult CNS for therapeutic approaches to stroke and neurodegenerative disorders (Taupin 2006). However, pinpointing exactly what is/are the adult somatic stem cell population(s) for each organ system is a matter of great contention (De Mey and Freund 2013), which we will not discuss in this perspective. We will more generically refer to the concept of SSCs, their potential different states (i.e., quiescent versus activated), and their plasticity, rather than try to define exactly which cells constitute the adult SSC population.
The “dark side” of the immortality of SSCs is their capacity to be corrupted into so-termed cancer stem cells (CSCs). Like their normal counterpart SSCs, CSCs exhibit self-renewal capacity and differentiation potential, albeit with aberrant and incomplete differentiation potential, and have the capacity to maintain or renew and propagate a tumor. Since the initial isolation of CSCs in leukemia, their existence in a wide variety of other cancers has been successfully demonstrated (Zhang and Rosen 2006). The focus of this perspective will be on SSCs and CSCs and whether we can safely and appropriately pharmacologically manipulate these cell types in vivo for long-term health benefits.
Did Count Dracula have the answer?
What constitutes immortality? One definition is, “endless life, the condition of living forever, of never dying.” Vampires are mythical creatures that feed on their victim’s life essence, the blood of living creatures. Immortality is often cited as one of the key characteristics of the vampire, which likely contributes to the tremendous appeal of the vampire in western popular culture. Furthermore, for many, the lure of the vampire lies in the ideal of eternal youth. Recently, a surgical procedure called parabiosis, which connects the circulatory systems of aged and young mice, has demonstrated the presence of factors in young blood that can restore many features of youth (e.g., faster recovery and decreased fibrosis after injury, increased neurogenesis, and memory consolidation) in aged mice (Laviano 2014; Sinha et al. 2014). These studies highlight that the problem of “aging” is not purely intrinsic, i.e., a deficiency of endogenous stem cells, but rather extrinsic via a gain of deleterious and/or a decrease of beneficial circulating factors that control the activation and normal division of our immortal SSCs.
In that regard, perhaps Count Dracula’s methods already highlighted the exciting and distinct possibility that many deleterious consequences of aging may be reversible and effectively treated. However, rather than advocate vampirism, our goal is to understand the mechanisms whereby our aged stem cell population no longer maintains its youthful regenerative capacity and subsequently to find safe and efficacious pharmacologic therapies that can restore it. As discussed in this perspective, our investigations over the past 15 years point to the possibility that we can pharmacologically modulate the decision processes of our endogenous SSC populations that deteriorate with age and at the same time eliminate the CSC population that drives tumorigenesis.
Symmetric or asymmetric that is the question
Long-term SSCs appear to remain relatively quiescent for the majority of their lifetime during normal tissue homeostasis, perhaps dividing only once every few months (Foudi et al. 2009) or even less frequently (Baker et al. 2014). However, quiescent SSCs can enter the cell cycle and undergo mitosis to give rise to two daughter cells, both under normal homeostatic conditions as well as more frequently after insult or injury to replace and repair damaged organs and tissues. Stem cells when they divide can do so in two basic modes, i.e., symmetrically or asymmetrically (Fig. 1). In the ideal situation, an asymmetric balance is maintained, whereby one of the daughter cells remains in its niche as a stem cell, while the other daughter proceeds forward in the differentiation process to maintain tissue homeostasis (Fig. 1, upper panel). However, this asymmetric balance of fates between the two daughter cells is not always maintained. SSCs can also undergo symmetric divisions. There are two modes of symmetric division. In symmetric non-differentiative divisions, both daughter cells remain as stem cells in their niche. Alternatively, SSCs can undergo symmetric differentiative divisions, where both cells leave the niche and go on to differentiate, thereby losing their “stemness” (Fig. 1, lower panel). Both of these modes of symmetric division are presumed to be deleterious to the normal long-lived stem cell population, as they can either lead to premature exhaustion of the stem cell pool (via symmetric differentiative divisions) or alternatively increase the number of DNA mutations accumulated in the SSC pool (via symmetric non-differentiative divisions). A potential, although still controversial, rationale for the preference of SSCs to undergo asymmetric versus symmetric cell divisions stems from the “immortal strand hypothesis” described almost 40 years ago by John Cairns (1975). Stated simply, when SSCs undergo mitosis, the stem cell desires to retain the original uncopied strands of DNA and pass on the duplicated strands that contain multiple copy errors, inherent in the DNA replication process, to the differentiated daughter cell that will continue on a path towards terminal differentiation. In this way, the total number of DNA mutations that accumulate in the long-lived SSC population in the niche can be minimized.
Fig. 1.
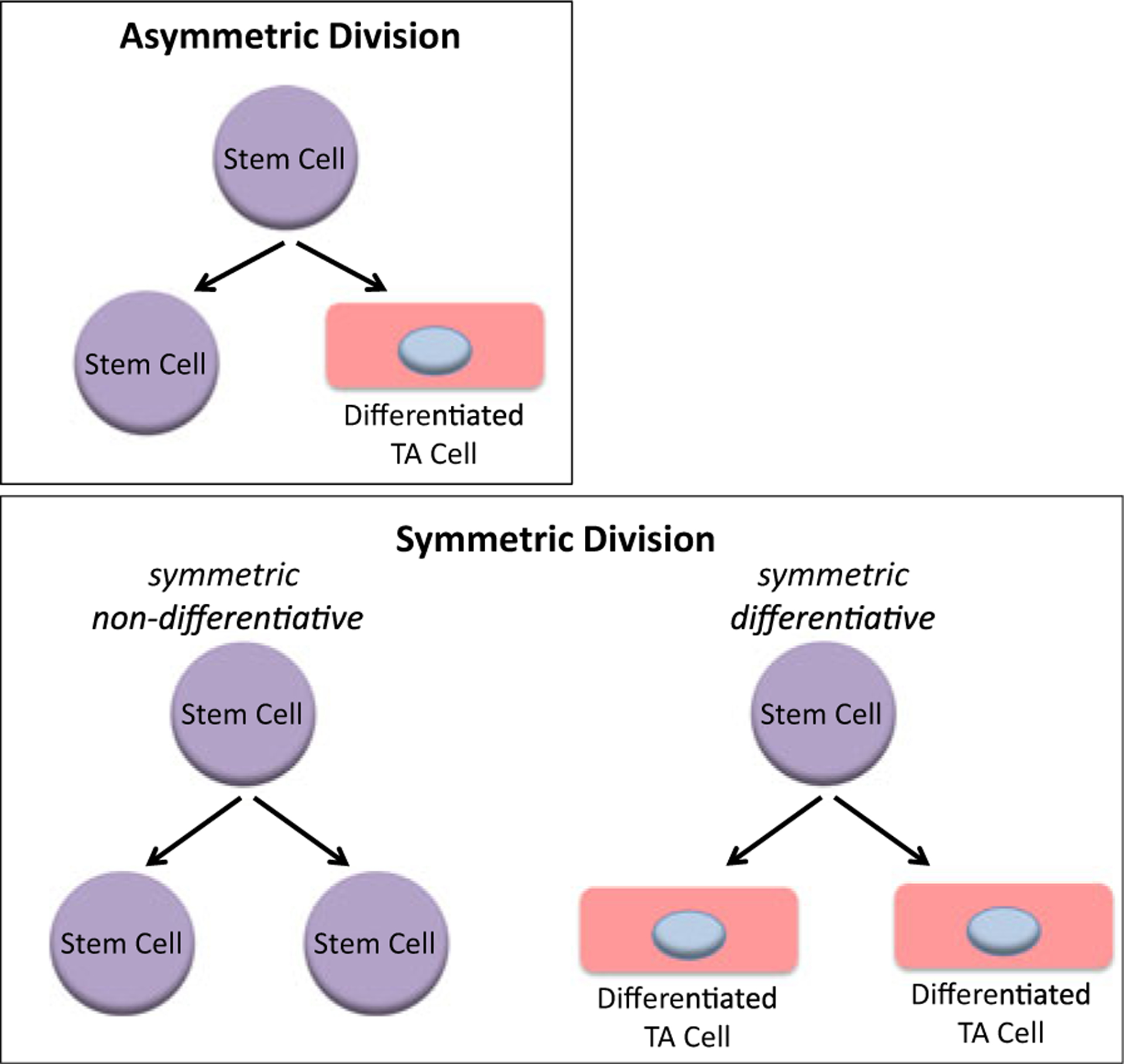
An asymmetric division results in the production of two daughter cells with different cell fates—one a stem cell and the other a differentiated transient amplifying (TA) cell (upper panel). A symmetric non-differentiative division occurs when the two daughter cells remain as stem cells. A symmetric differentiative division gives rise to two daughter cells, both of which are differentiated TA cells (lower panel)
The timely activation and asymmetric division of the SSC pool are critical decisions required for both normal homeostasis and repair after injury. We propose and will discuss in more detail that this decision process becomes corrupted with age due to a variety of factors including accumulating mutations in the SSC pool, chronic or acute injury, reversion of differentiated daughters to SSCs, serum factors, and SSC senescence. The critical questions to address then are (a) how can we entice quiescent SSCs to become activated and enter the cell cycle, and (b) once the SSC has committed to enter cycle and undergo mitosis, how is the decision for that stem cell to undergo a symmetric versus an asymmetric division made and can we pharmacologically control it? These are probably the most critical cellular decisions in adult organisms and likely underlie the aging problem and a host of diseases of aging, including cancer, neurodegeneration, and decreased tissue maintenance/homeostasis (as in wound healing, sarcopenia, fibrosis, and osteoporosis).
Somatic stem cell and cancer stem cells
Drug resistance, disease relapse, and metastasis constitute the central challenges in the management of advanced malignancies. Recently, cancer initiation, metastasis, and disease progression have been attributed to subpopulations of self-renewing, highly tumorigenic, drug-resistant tumor cells termed CSCs (Reya et al. 2001). The concept of CSCs is not new though, as almost 150 years ago Cohnheim et al. proposed that cancer might arise from a rare population of cells with stem cell-like properties (Cohnheim 1867). CSCs have now been proven to exist in a wide array of tumor types including leukemias, brain, breast, bladder, prostate, colon, etc. and are associated with disease recurrence, multidrug resistance, and metastasis (Holland et al. 2013). A major focus in cancer research over the past decade has been to both prospectively identify CSCs and even more critically to develop therapeutic strategies to safely eliminate this cell population without deleterious effects to the normal SSC populations.
Somatic stem cells and cancer stem cells; more similarities than differences
Unfortunately, from the standpoint of safely targeting CSCs, it appears that the similarities between normal adult SSCs and CSCs far outweigh the differences between them. This is not all that surprising in that CSCs likely arise from SSCs in many instances. Importantly, by the definition of “stemness,” they both have the ability to self-renew and also proceed on to more differentiated cell types. CSCs express similar “stemness” markers and exhibit cellular behaviors highly reminiscent of SSCs. SSCs appear to reside in specialized niches within tissues or organs (e.g., hematopoietic stem cells, neuronal stem cells, intestinal stem cells, etc.) and are critical for both normal tissue homeostasis and regeneration after injury (Gage and Temple 2013; Cullen et al. 2014; Clevers 2013). Long-lived quiescent SSCs infrequently enter cell cycle to maintain homeostasis but more frequently upon injury to repair damaged tissue. This process seems to be degraded with aging, and we will return to this subject later in the perspective.
Similarly, CSCs appear to reside in similar niches to SSCs and in fact can compete with one another for the limited space within the niche. The same signaling pathways involved in regulating SSC maintenance (i.e., Wnt, Notch, Hedeghog, TGFβ/BMP, JAK/Stat, Hippo, FGF/MAPK/PI3K) are also involved in the regulation of CSCs (LaBarge 2010; Merchant and Matsui 2010; Pannuti et al. 2010). Aberrant regulation of these same pathways leads to neoplastic proliferation in the same tissues. For both CSCs and SSCs, there are multiple points of intersection and crosstalk, including feedback and feedforward loops, connecting the various signaling cascades that modulate “stemness.” The question then becomes, how is all of this information integrated to decide a stem cell’s fate, i.e., to exit quiescence and subsequently divide either asymmetrically or symmetrically, be it a normal SSC or a CSC? Furthermore, can we safely pharmacologically manipulate our endogenous stem cell population, both normal SSCs and CSCs, as this would provide a breakthrough in treating diseases of malignancy as well as to ameliorate the aging process?
Decisions, decisions
Although symmetry versus asymmetry is essentially a simple binary decision process (a 0/1 decision as in computer logic), the stem cell in the niche undergoing mitosis must read an enormously complex array of information from its environment (e.g., oxygen levels, nutrient levels, circadian cycles, nervous system innervation, growth factors, adhesion molecules, kinase cascades, cell–cell contacts, and so on) to arrive at this eventual binary decision. The question then is: how does a stem cell (either normal SSC or CSC) in the niche read this plethora of information and distill it down into a simple molecular binary decision? Interestingly, a preference for symmetric over asymmetric divisions appears to be one of the fundamental differences between CSCs and normal SSCs. For example, breast cancer stem cells with mutations in the gene p53 preferentially undergo symmetric divisions (Cicalese et al. 2009). Loss of function of the tumor suppressor PTEN leads to premature exhaustion of the normal HSC population (presumably due to increased symmetric differentiative divisions), whereas there is an expansion of the leukemic stem cell (LSC) population (presumably due to increased symmetric non-differentiative divisions) (Lee et al. 2010). Similarly, genetic activation of Hedgehog signaling involving indirect perturbation of Notch signaling causes an increase in neural stem cell (NSC) symmetric divisions (Ferent et al. 2014). The decision to preferentially undergo symmetric non-differentiative versus symmetric differentiative divisions appears to be another intrinsic difference between CSCs and SSCs carrying critical mutations (i.e., p53, p73, PTEN, etc.), respectively. Presumably, this provides a potential mechanism to stochastically eliminate mutated defective SSCs prior to the accumulation of additional deleterious mutations. Clearly, multiple signaling cascades can affect a stem cell’s decision to divide symmetrically or asymmetrically and to remain quiescent or become activated. The key then is to understand how this diverse array of signals and crosstalk are integrated and processed into the stem cell’s ultimate decision to become activated and divide either asymmetrically or symmetrically.
Aging, injury, and repair and SSCs
A recent report estimated that 50 % or more of somatic mutations found in tumors likely occur prior to tumor initiation and a majority of these occur in utero (Tomasetti et al. 2013) during the rapid expansion of the fertilized egg to the developed fetus. This is logical in that during this explosive period of growth and development more symmetric stem cell divisions must occur than under normal homeostasis in the adult. However, if the fundamental preference for asymmetric over symmetric divisions in adult normal SSCs was absolute, in principle, our SSC pool would never add any additional mutations with aging without intrinsic damage to the SSC. Unfortunately, this is clearly not the case as epidemiologic data demonstrate that the risk of developing cancer, fibrosis, or neurodegeneration increases significantly with age, starting at approximately age 50, and DNA damage has been shown to specifically accumulate in SSC compartments with age (Mandal et al. 2011). There also appears to be a correlation between the total number of divisions of normal SSC populations and the total lifetime risk of developing cancer in a particular tissue (Tomasetti and Vogelstein 2015). What are some of the factors that potentially lead to accumulating DNA damage in the SSC pool with age?
DNA damage and repair in SSCs
Quiescent SSCs primarily exhibit properties that apparently limit their propensity to acquire DNA damage. The quiescent nature of adult SSCs combined with their low metabolic activity and decreased production of reactive oxygen species (ROS), albeit with decreased expression of anti-oxidant defense genes (Chen et al. 2010), presumably is beneficial with regard to reducing DNA damage. As opposed to rapidly proliferating differentiated somatic cells, the metabolic requirements for quiescent SSCs are relatively modest and they are predominantly reliant on the glycolytic pathway (Rehman 2010). This confers an advantage to adult SSCs in terms of minimizing DNA damage, as mitochondrial respiration utilized by differentiated somatic cells generates large amounts of ROS, which is believed to contribute to DNA damage (Naka et al. 2008). Lower ROS levels and a reducing redox microenvironment have been shown to be crucial for SSC maintenance and function (Ito et al. 2004). The maintenance of a hypoxic environment in the SSC niche also helps to reduce accumulating DNA damage and thereby helps to maintain the self-renewing potential of SSCs. Interestingly, subsets of CSCs from human and mouse breast tumors have lower levels of ROS and express higher levels of free radical scavengers than their non-tumorigenic counterparts (Diehn et al. 2009), although the functional significance of this is not yet understood. Finally, SSCs, as well as their CSC counterparts, intrinsically express high levels of ATP-binding cassette (ABC) transporter/multidrug resistance gene family members. This leads to more efficient efflux of toxic metabolites and xenobiotics thereby minimizing potential DNA damage by these agents (Huls et al. 2009).
However, due to their quiescence, the primary mechanism utilized in non-replicating SSCs to repair DNA damage, non-homologous end joining (NHEJ), is significantly more error prone than the homologous recombination mechanism utilized by their more differentiated cycling progeny and can introduce small deletions or insertions into repaired regions (Delacote and Lopez 2008; Mao et al. 2008). NHEJ is therefore thought to contribute to the acquisition of mutations during repair in SSCs over time (Mohrin et al. 2010). NHEJ plays a key role in DNA repair in SSCs, as mice deficient in components of the NHEJ pathway, including DNA ligase 4 (LIG4) and Ku80, show impaired HSC repopulating potential (Rossi et al. 2007; Nijnik et al. 2007).
Steady state homeostasis versus injury and stem cell plasticity
Accumulating evidence indicates that somatic stem cells exist in minimally two distinct states based upon their ease of activation. Very recently, Rando and colleagues demonstrated that highly quiescent SSCs in G0 actively and reversibly transition into a so-called Galert phase before becoming fully activated (Rodgers et al. 2014). Often, it is critical for organismal survival after injury to rapidly repair tissue in whatever manner possible and strict stem cell hierarchy may be compromised under these conditions. Under these conditions, cellular plasticity, fate conversion, and reacquisition of “stem cell” characteristics or “de-differentiation” can occur (Blanpain and Fuchs 2014). For example, in the hair follicle, both cells in the bulge and in the neighboring hair germ (HG) possess many features of “stemness,” and both bulge and HG cells can generate the seven different lineages in the hair follicle. Although under normal homeostatic settings bulge cells normally generate HG cells, HG can also replenish depleted laser-ablated bulge cells (Rompolas et al. 2013). Similarly, in the intestine, two intestinal stem cell (ISC) populations with multi-lineage regenerating capacity have been identified: (1) the Lgr5+ columnar basal cell (CBCs) and the highly quiescent, asymmetrically dividing, less radiosensitive +4 Bmi1+ ISCs (Barker et al. 2007; Sangiorgi and Capecchi 2008; Montgomery et al. 2011; Takeda et al. 2011; Ritsma et al. 2014). The +4 populations, because of the traits listed above and their mode of chromosomal segregation (Potten et al. 1978), appear to behave more like bona fide long-lived SSCs. However, these populations have been shown to inter-convert and both possess the capacity to generate all lineages of the intestinal epithelium (Ritsma et al. 2014) and the identity of bona fide long-lived SSCs remains a hotly debated issue (De Mey and Freund 2013). Similar situations exist in numerous other stem cell populations including the mammary and lung epithelium (Blanpain and Fuchs 2014) as well as the hematopoietic system (Oh and Humphries 2012).
Although the mechanism(s) regulating cellular plasticity and the regaining of a “stem-like” state by partially committed or differentiated cells after injury remain incompletely understood, this plasticity may also have important implications in regards to tumorigenesis and aging. This concept was first postulated in 1990 based upon experiments where oncogenic H-ras was targeted to the differentiated epidermis in mice using a Keratin-10 promoter. Papillomas developed preferentially at sites of irritation and wounding (Bailleul et al. 1990) that are associated with increased stem cell activation. Subsequently, it was demonstrated that a single oncogenic mutation will only initiate intestinal tumor formation if it is introduced into one of the putative ISC populations (i.e., Lgr5/prominin/Bmi1 positive populations) (Sangiorgi and Capecchi 2008; Barker et al. 2009; Zhu et al. 2009), whereas targeting transient amplifying (TA) cells had either no effect or generated only microadenomas (Barker et al. 2009). Therefore, the plasticity associated with the response to tissue injury may allow more differentiated progeny, which carry more significant levels of DNA damage from previous rounds of replication, to revert to a “stem-like” state, and potentially become CSCs. In mice, activation of the critical NF-kappa B (NF-κB) inflammatory pathway, in conjunction with Wnt activation in the intestine, can lead to the reversion of normally differentiated TA cells to a “stem-like” state and tumor formation (Schwitalla et al. 2013). Of the multiple signaling pathways involved in colonic inflammation, the NF-κB pathway is dominant and epidemiological studies have demonstrated that chronic inflammation predisposes patients to cancers of many organs and that administration of non-steroidal anti-inflammatory drugs decreases the incidence of colorectal cancer (Mantovani et al. 2008; Chan et al. 2007). A number of human tumor DNA viruses including Epstein-Barr virus (EBV), Kaposi sarcoma-associated herpesvirus (KSHV), human papillomaviruses (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), human T lymphotropic virus (HTLV-1), and more recently Merkel cell polyomavirus (MCPyV) and RNA viruses such as HCV and HTLV-1 have been shown to be able to induce the immortalization of differentiated infected human host cells (Saha et al. 2010). The virally infected cells can persist in a state of chronic infection, which can lead to oncogenesis, demonstrating that viral infection can induce de-differentiation thereby generating CSCs. The underlying correlation between inflammation, viral infection, and increased cancer risk is likely caused by the “corruption” of the stem cell pool via the mechanisms discussed above.
Wnt/catenin-dependent transcription and “stemness”
Wnt signaling constitutes an ancient pathway that dates back to the early metazoans. The Wnt/catenin pathway is critical in virtually every tissue and organ system throughout normal embryonic development and the life of the organism. It is an extremely complex signal transduction pathway involving 19 mammalian Wnt ligands, with at least 10 already present in the metazoan common ancestor (Mi et al. 2013) that trigger a variety of intracellular responses broadly classified as either canonical (increase in nuclear β-catenin) or non-canonical (planar cell polarity, Ca2+/PKC activation) (Niehrs 2012; Moon 2005). Although a gross oversimplification, the former is often associated with proliferation and lack of differentiation (for example, as a hallmark of dysregulated Wnt signaling in cancer), whereas the latter is often associated with cell, tissue, and organ differentiation. β-catenin plays a key role in both canonical and non-canonical Wnt signaling through its nuclear functions and cytoskeletal/cytoplasmic membrane interactions, respectively. However, rather than being thought of as two completely distinct signal transduction systems, we believe that a continuum exists that coordinates β-catenin-dependent gene expression and cytoplasmic/cytoskeletal β-catenin to affect key developmental and regulatory processes.
The entry of β-catenin, although other catenins, such as γ-catenin/plakoglobin, may also play a critical role under particular circumstances (Kim et al. 2011), into the nucleus and the subsequent transcriptional processes that ensue are controlled by the so-termed “canonical Wnt” or “Wnt/β-catenin” signaling cascade. Alternative signaling pathways can also induce the nuclear translocation of β-catenin and its subsequent participation in transcription. The process of epithelial to mesenchymal transition (EMT) involves down-regulation of E-cadherin, which normally binds cytoplasmic β-catenin (Onder et al. 2008), leading to the subsequent nuclear translocation of β-catenin (Kim et al. 1998). Receptor tyrosine kinases (Wagh et al. 2011) and non-receptor tyrosine kinases including Src (Coluccia et al. 2006) and Abl (Ress and Moelling 2006) can disrupt the E-cadherin/β-catenin interaction, thereby enhancing β-catenin-mediated transcription. Additionally, prostaglandins (Ishimoto et al. 2010), hypoxia (Mazumdar et al. 2010; Kida and Kahn 2013), high glucose levels (Chocarro-Calvo et al. 2013), and cholinergic innervation (Zhao et al. 2014) can also activate Wnt/β-catenin signaling. It is clear that a wide range of inputs influence β-catenin dynamics and β-catenin-dependent transcription (Brembeck et al. 2006; van Veelen et al. 2011; Kawabata 2011).
These signals must be successfully integrated with signals from a number of other key pathways (e.g., Notch, Hedgehog, JAK/Stat, BMP, Hippo, FGF/MAPK), for nuclear β-catenin to play an essential role in balancing self-renewal versus differentiation in adult stem cells (Fig. 2). Wnt signaling plays important roles throughout development (Komiya and Habas 2008) and although most would agree that Wnt signaling is critical in stem cell biology, there is no consensus as to whether Wnt signaling is important for either maintenance of potency (Reya et al. 2001; Sato et al. 2004) or differentiation of stem cells (Otero et al. 2004). Wnt/β-catenin signaling is important for the maintenance of ES cell pluripotency (Sato et al. 2004) and in the expansion of neural stem/progenitors, thereby increasing brain size (Chenn and Walsh 2002). However, Wnt/β-catenin signaling also induces the differentiation of ES cells (Otero et al. 2004) and controls fate determination in neural crest stem cells (Hari et al. 2002). Wnt/β-catenin signaling clearly plays dichotomous roles in stem cell biology.
Fig. 2.
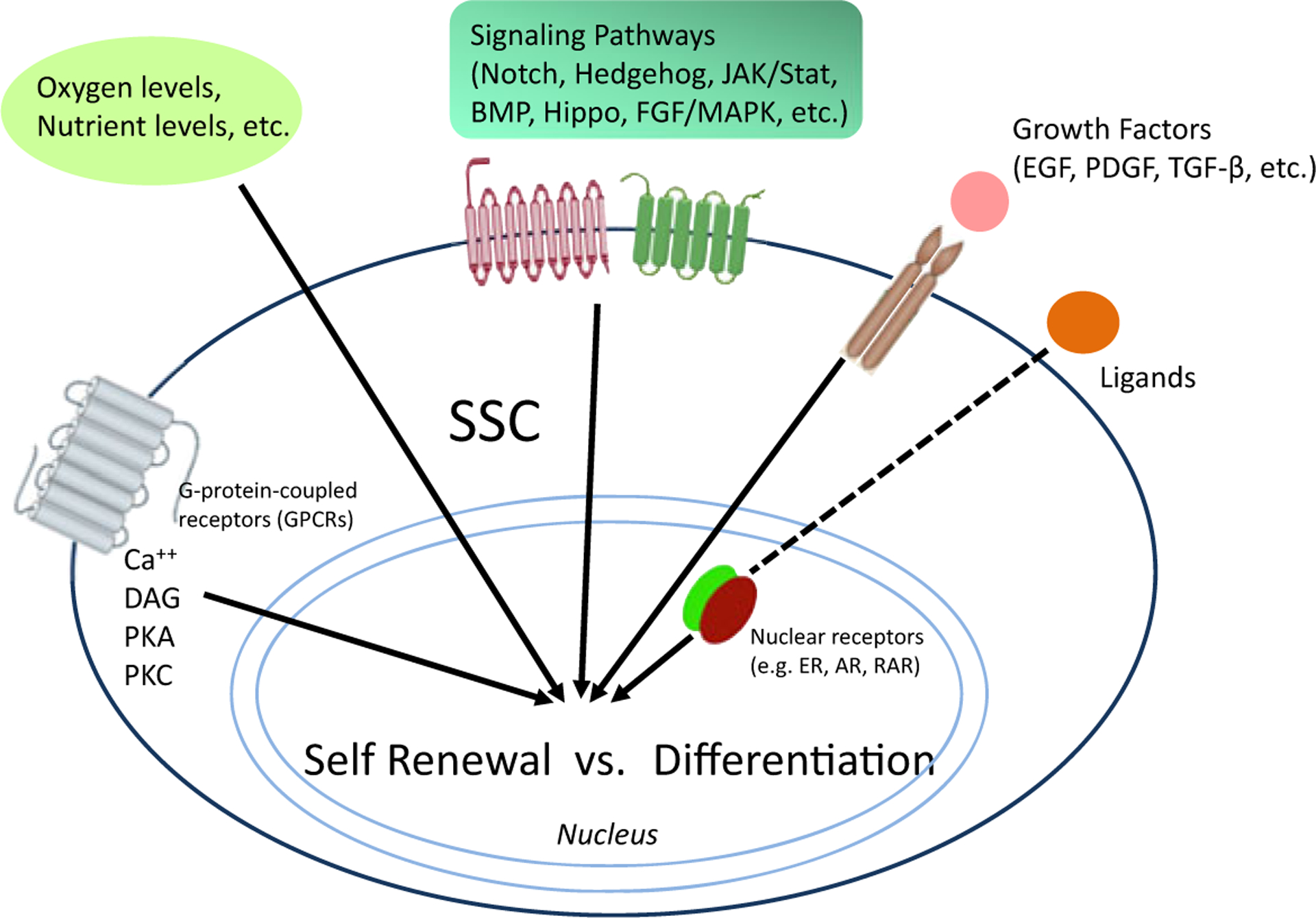
Integration and coordination of multiple signaling pathways are necessary to regulate a stem cells’ response to these inputs to decide to either self-renew or differentiate
Wnt/catenin, cancer, and cancer stem cells
The Wnt pathway has emerged as a pivotal player in the specification and maintenance of SSC in multiple stem cell niches, in a wide array of tissues and organs including the intestines, heart, blood, brain, and mammary gland (Kühl and Kühl 2013). It is therefore not surprising that a recurrent theme in cancer biology is aberrant regulation of Wnt signaling (Polakis 2012; Anastas and Moon 2013). This has engendered substantial efforts into the development of therapeutic approaches to target the Wnt pathway. However, a number of factors have thwarted progress in this field, including the enormous complexity of the pathway (Niehrs 2012). However, this represents only the tip of the iceberg in regards to the difficulty in attempting to develop safe and effective specific Wnt pathway therapeutics. Further complexity is encountered when targeting transcriptionally competent β-catenin, as β-catenin can bind to a broad spectrum of transcription factors beyond members of the TCF/LEF family (Le et al. 2008). Transcriptionally active β-catenin modulates a plethora of downstream biological processes including pluripotency, EMT, oxidative stress, and lineage commitment (Le et al. 2008). Although the successful therapeutic manipulation of endogenous “stemness” (normal or cancerous) via modulation of aberrant catenin-regulated transcription offers enormous promise, it requires significant precision to affect the desired transformations without deleterious effects (e.g., depletion of or increases in somatic mutations) in normal SSC populations (Takahashi-Yanaga and Kahn 2010).
Differential coactivator modulation
β-catenin must recruit one of the two Kat3 transcriptional coactivators, cAMP response element binding protein (CREB-binding protein (CBP) or its closely related homolog, p300 (E1A-binding protein, 300 KDa) as well as other components of the basal transcriptional apparatus to generate a transcriptionally active complex (Moon 2005; Teo and Kahn 2010). These coactivators interact with hundreds of proteins in their roles as master orchestrators of transcription, and due to their high degree of protein sequence identity and even higher similarity, they have long been considered as largely redundant. However, accumulating evidence indicates that CBP and p300 are not redundant and play definitive and unique roles both in vitro and in vivo (Kung et al. 2000; Yamauchi et al. 2002; Roth et al. 2003).
Fifteen years ago, from a library of 5000 secondary structure mimetics, our lab identified ICG-001 in a cell-based TopFlash reporter gene assay in SW480 colon carcinoma cells. In this assay, ICG-001 had an IC50 value of 3 μM. We subsequently demonstrated that ICG-001 binds specifically and with high affinity (~1 nM) to the N-terminus of the coactivator CBP (McMillan and Kahn 2005; Emami et al. 2004). Subsequently, we found that selectively blocking the interaction between CBP and β-catenin with ICG-001 leads to the initiation of a differentiation program in a wide variety of stem/progenitor cells (Fig. 3a) (Hasegawa et al. 2012; Banerjee et al. 2012). This led to the development of our model of differential coactivator usage, which highlights the distinct roles of the coactivators CBP and p300 in catenin-mediated transcription (Fig. 3b) (Miyabayashi et al. 2007). In our model, the differential utilization of either CBP or p300 as the catenin coactivator is the first decision that guides a stem cell to either maintain potency or initiate a differentiative transcriptional program, respectively (Fig. 3b). Subsequently, we have identified several small molecules (IQ-1, ID-8, and most recently the specific direct p300/catenin antagonists YH249/250) that selectively block the p300/catenin interaction, thereby increasing the CBP/catenin interaction, resulting in enhancement of symmetric divisions and the maintenance of potency (pluri- or multipotency) in a variety of stem cell populations (ES, iPS, and SSC), both in mouse and human cells (Hasegawa et al. 2012; Miyabayashi et al. 2007; Marson et al. 2008; Higuchi et al. 2015) (Fig. 3c).
Fig. 3.

ICG-001 selectively blocks the interaction between β-catenin and CBP. This results in biasing towards p300 usage and thereby initiates a differentiation transcriptional program (with the loss of self-renewal capacity). a Differential coactivator usage by β-catenin regulates transcriptional programs of differentiation versus self-renewal. Depending on its usage of the two Kat3 coactivators, β-catenin will lead to transcriptional activation of sets of genes that are either implicated in self-renewal (CBP) or differentiation (p300). b IQ-1, ID8 (both indirectly), and YH 249/250 (directly) block the interaction between β-catenin and p300. By selectively blocking this interaction, CBP usage is increased, and consequently the initiation of a self-renewal transcriptional program is favored
Pharmacologically manipulating stem cells
Over the years, we have extensively examined the therapeutic potential of selectively antagonizing the CBP/β-catenin interaction in a variety of preclinical tumor models, where we have demonstrated the ability to safely eliminate drug-resistant CSCs, via forced differentiation, without deleterious effects on the normal endogenous stem cell populations (Wend et al. 2013; Gang et al. 2014; Zhao et al. 2015). CBP/catenin antagonists have also demonstrated efficacy in a variety of injury models including pulmonary and renal fibrosis (Henderson et al. 2010; Hao et al. 2011), myocardial infarction (Sasaki et al. 2013), and neurodegeneration (Teo et al. 2005). The beneficial effects of CBP/catenin antagonists in these models are at least in part due to enhanced asymmetric differentiation of SSCs and accelerated repair (Ring et al. 2014). The differential effects of CBP/catenin antagonists on CSCs versus normal SSCs (i.e., forced differentiation and elimination versus differentiation and enhanced repair without depletion) must therefore be cell intrinsic. CBP/catenin antagonists apparently take advantage of the intrinsic propensity of CSCs to increase the number of symmetric divisions at the expense of asymmetric divisions due to various mutations (e.g., p53, PTEN, etc.) (Cicalese et al. 2009; Lee et al. 2010). As discussed previously, normal long-term repopulating SSCs preferentially divide asymmetrically with one daughter cell remaining in the niche and the other going on to a transient amplifying cell required for generating the new tissue involved in repair processes (Fig. 4a, upper panel) (Ress and Moelling 2006), whereas CSCs undergo more symmetric divisions (Fig. 4a, lower panel). However, when treated with CBP/catenin antagonists, CSCs will eventually be cleared out of the niche via symmetric differentiative divisions (Fig. 4b), whereas normal SSCs that divide asymmetrically will always retain one of the dividing daughter cells in the stem cell niche (Fig. 4a). This fundamental cell intrinsic difference between SSCs and CSCs provides a unique opportunity to therapeutically target and eliminate CSCs as well as enhance the repair potential of normal SSCs, without damaging the normal endogenous stem cell populations (Teo et al. 2005).
Fig. 4.
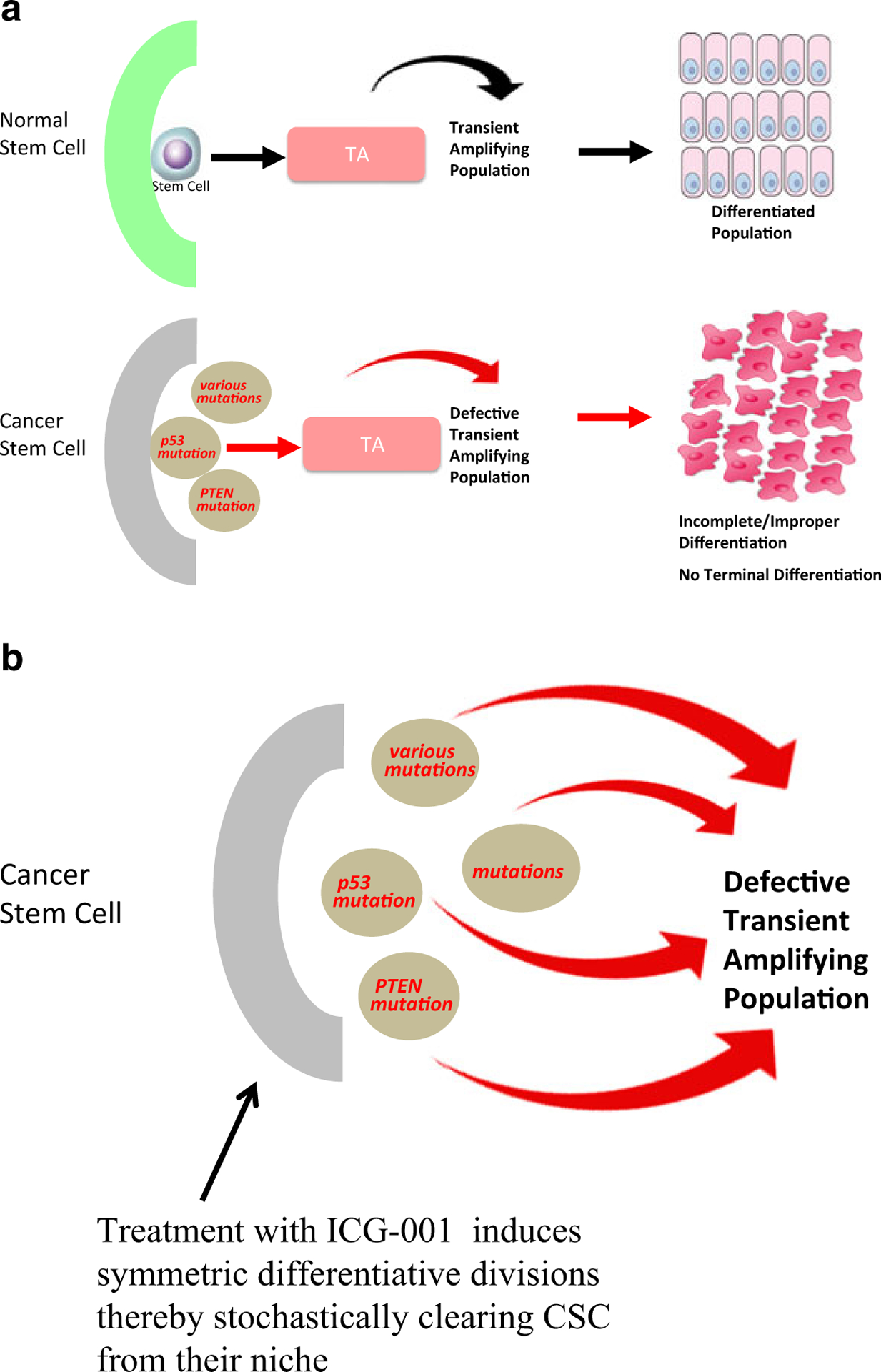
a In normal somatic stem cells (SSCs), asymmetric division is favored. Cancer stem cells (CSCs) undergo both symmetric and asymmetric divisions, leading to an increase in CSCs over time. b Treatment of CSCs with CBP/catenin antagonists (e.g., ICG-001) induces symmetric differentiative divisions of the CSC population thereby eventually clearing the CSC population from the niche. Normal SSCs continue to undergo asymmetric divisions upon treatment with CBP/catenin antagonists and are thus not depleted
To the clinic
In principle, significant concerns about specificity could be raised using small molecule inhibitors that target the coactivator protein CBP, as it has as many as 500 molecular partners, including a vast number of transcription factors (Ring et al. 2014). However, these concerns have not been borne out either preclinically or even more importantly clinically, utilizing specific CBP/catenin antagonists (i.e., ICG-001 or the second-generation CBP/catenin antagonist PRI-724). This is perhaps at first very surprising. A few salient features are worth mentioning though: the first is the extremely high biochemical selectivity of ICG-001/PRI-724 for its molecular target CBP; second, these small molecules only disrupt a small subset of all CBP interactions; and third the unique properties, based upon specific differences, of the two Kat3 coactivators CBP and p300, which will be discussed in more detail later in the perspective.
The second-generation specific CBP/catenin antagonist PRI-724 (IC50 ~150 nM) was developed by Prism Pharma. PRI-724 proved to be extremely safe in preclinical IND enabling toxicology studies, with the no adverse event level for PRI-724 being 120 mg/kg/day in dogs given 28-day continuous infusion. An open label phase Ia safety study in subjects with solid tumors was initiated at USC in March 2011, and the results of this trial were reported at ASCO in June 2013 (El-Khoueiry et al. 2013). As had been observed in preclinical studies, PRI-724 had a very acceptable toxicity profile with dose escalation from 40 to 1280 mg/m2/day with 7 days of continuous i.v. infusion. Downregulation of the biomarker survivin/BIRC5 with upregulation of the differentiation antigen CK20 in CTCs strongly correlated with increasing plasma concentrations of drug (El-Khoueiry et al. 2013). Additional oncology trials and a trial for HCV-induced hepatic fibrosis with PRI-724 are currently underway (https://clinicaltrials.gov).
As previously discussed, CBP/catenin antagonists have demonstrated efficacy in a wide variety of preclinical injury models including pulmonary and renal fibrosis (Henderson et al. 2010; Hao et al. 2011) and myocardial infarction (Sasaki et al. 2013). Given the apparent safety of these agents, additional clinical trials with CBP/catenin antagonists are anticipated in the future.
Aging and aging stem cells
Although not a disease per se, aging and an aging population are creating major health and economic issues for our society. As we age, both the fidelity and the efficiency of our bodies homeostatic and repair processes decrease. This in principle could be due to a decline in the size of SSC populations; however, it appears that rather than a decline, at least in many SSC populations (HSC, skin/hair, etc.), there is often an increase in the number of SSCs. Yet, the “effectiveness” of SSCs to serve as a regenerative pool during homeostasis and repair decreases with age. Several mouse models of premature aging and decreased effectiveness of repair after injury (i.e., increased fibrosis) have demonstrated an increase in Wnt signaling (Hernandez et al. 2010; Liu et al. 2007; Brack et al. 2007). Furthermore, serum complement factor C1q, whose concentration increases with aging, augments Wnt signaling and decreases skeletal muscle regeneration in wild-type mice (Naito et al. 2012). We believe that this increase in the number of SSCs, albeit with decreased efficiency in homeostasis and repair, arises from increased SSC quiescence along with an increase in the number of symmetric renewing divisions at the expense of asymmetric divisions in the SSC pool. We propose that this arises from an increase in the CBP/catenin interaction/transcription at the expense of the p300/catenin interaction/transcription with aging. This also fits with epidemiologic data demonstrating that the risk of developing cancer, fibrosis, or neurodegeneration increases significantly with age after the age of 50 (Tomasetti and Vogelstein 2015). The increase in stem cell symmetric versus asymmetric divisions and increased quiescence with age could be engendered and/or influenced by a variety of factors including genetics (both germline and somatic variants), various insults, or life style decisions, including infection, xenobiotics and pollutants, diet/caloric intake/metabolism, and radiation, e.g., UV and X-ray. In combination, these factors could bias the equilibrium between CBP/catenin and p300/catenin-driven transcription leading to an increase in CBP/catenin-driven processes and an increase in symmetric versus asymmetric divisions in SSCs populations (Fig. 5). In general, replicative stress in an aged SSC population may be associated with the declining functionality of SSCs with aging (Flach et al. 2014). If the underlying problem with aging is due to an imbalance in Kat3/catenin coactivator usage and a bias towards the CBP/catenin side, then selective small molecule CBP/catenin antagonists by correcting this bias could thereby provide a more optimal (youthful) balance in asymmetric versus symmetric divisions. This could potentially ameliorate the aging process and/or manifestations thereof and also provide potential prophylaxis against many of the common diseases of aging (e.g., cancer, fibrosis, neurodegeneration, etc.).
Fig. 5.
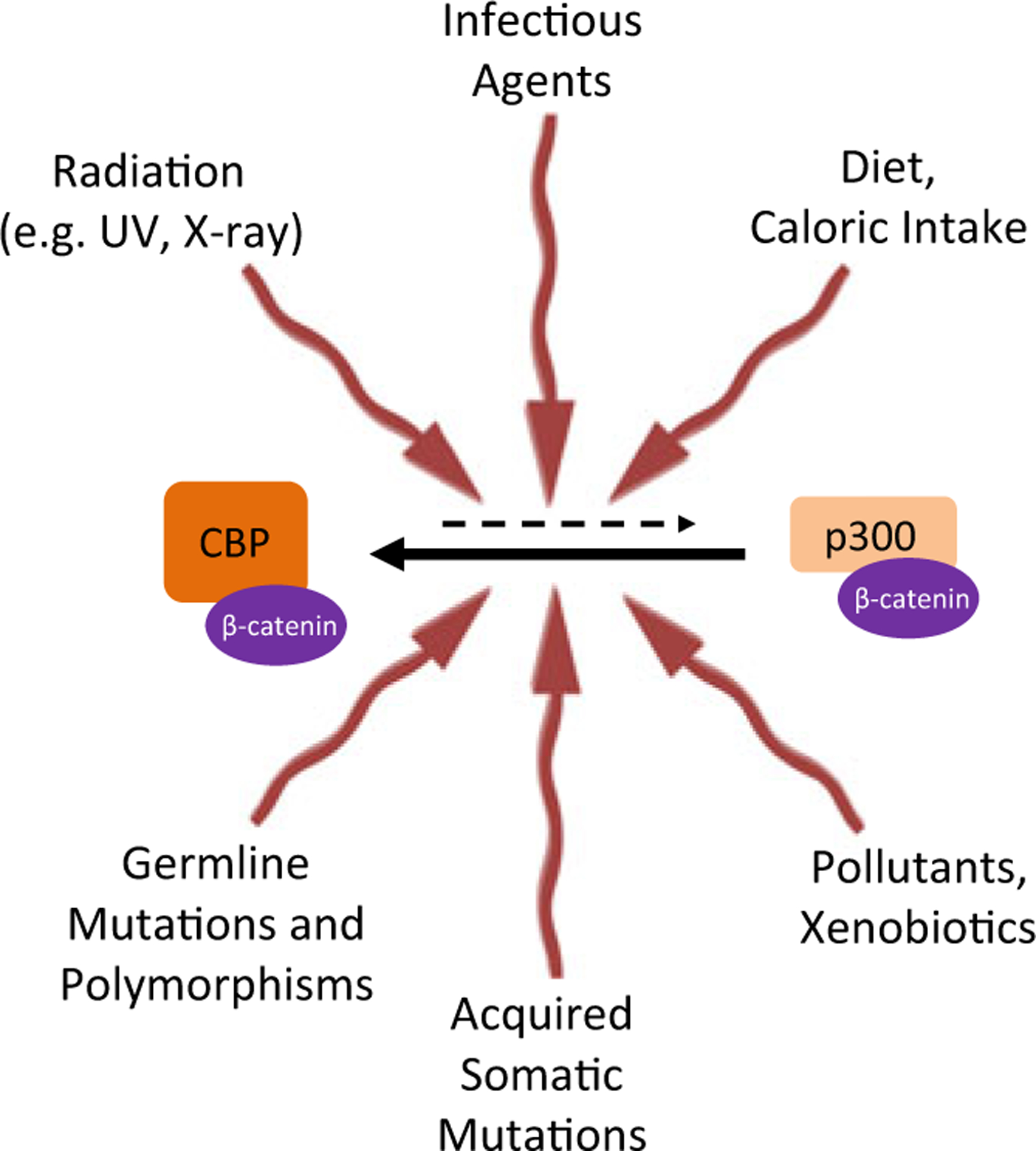
As we age, various physiological and pathological inputs that SSC receive bias the equilibrium between CBP/catenin and p300/catenin, favoring the CBP/catenin interaction and thereby symmetric stem cell divisions
Quiescence, aging, and metabolism
The survival, quiescence, and activation of long-lived SSCs is dependent on a plethora of signals both intrinsic and extrinsic, i.e., from the microenvironment or “niche.” As described previously, most organs appear to contain minimally two populations of stem cells, a highly quiescent slow cycling population as well as an activated cycling population that is required for both homeostatic control in organs with rapid turnover (intestine, hematopoietic system) or after injury in essentially all organ systems (Li and Clevers 2010). The inputs that control quiescence or activation are either cellular from the SSC itself or from differentiating daughter cells through paracrine signaling (e.g., lateral inhibition) or from stromal or mesenchymal cells in the niche. The extracellular matrix as well as more distant external cues (i.e., neuronal, hematologic or immunological input) also play critical roles in this decision process. In long-lived SSCs, quiescence provides a safeguard to preserve the function of SSCs by limiting damage to the cell caused by mitochondrial respiration, i.e., oxidative phosphorylation, and by uncontrolled cell cycle entry and exhaustion of the stem cell pool via symmetric differentiative divisions (Orford and Scadden 2008; Bakker and Passegué 2013). Accumulating evidence suggests that SSCs (as well as CSCs and cancer cells more generally) utilize glycolysis rather than oxidative phosphorylation despite the inefficiency of glycolysis compared to oxidative phosphorylation in regards to ATP generation (Kohli and Passegué 2014). Interestingly, a switch from glycolysis to oxidative phosphorylation is associated with activation of quiescent SSCs and the initiation of differentiation, whereas reprogramming to pluripotency is associated with “anaerobicizing” (Panopoulos and Izpisua Belmonte 2011). mTor plays a critical role in integrating growth factor signals and nutrient/energy levels and is a key regulator of protein translation, mitochondrial biogenesis, and autophagy (Laplante and Sabatini 2009). mTORC1, which partners with mTor as part of the RAPTOR complex, has been demonstrated to be critical in the control of activation of quiescent stem cells (Rodgers et al. 2014).
Kat3A/CBP and Kat3B/p300
“Nothing in Biology Makes Sense Except in the Light of Evolution”
—Theodosius Dobzhansky.
Roughly 450 million years ago, the evolution of the vertebrate lineage initiated a new lifestyle having a relatively long-lived adult stage. One of the critical adaptations that had to occur to successfully accommodate this “life style” change was a mechanism for long-term homeostatic maintenance (cell turnover) and tissue repair. This necessitated the advent of SSCs and their corresponding niches, to maintain a relatively quiescent “anaerobic” metabolic state as opposed to their more proliferative aerobic-differentiated daughter cells, in order to protect the integrity of the genetic material in SSCs (Trosko and Kang 2012). The advent of two different populations generated via asymmetric stem cell division, i.e., one a long-lived anaerobically metabolizing, relatively quiescent stem cell population and the other a differentiating aerobically metabolizing, cycling expanding daughter cell population, further required a robust, high fidelity mechanism to ensure the proper maintenance of “stemness” in one daughter cell, while on the one hand, the execution of a proliferative and differentiative program in the other daughter cell. Intriguingly, the Kat3 coactivator family CBP and p300 diverged via a gene duplication apparently just prior to the vertebrate radiation over 450 million years ago, as these two paralogs are found in virtually all vertebrate genomes sequenced to date but not in other chordates (Mi et al. 2013). CBP and p300 are extremely large proteins with molecular weights of ~300 kd that are encoded over 33 exons. Despite having diverged over 450 million years ago, CBP and p300 retain an extremely high degree of identity, up to 93 %, particularly over a large central core that includes the CH1, KIX, Bromodomain, and CH2 and CH3 regions (Fig. 6) (Arany et al. 1994; Eckner et al. 1994).
Fig. 6.
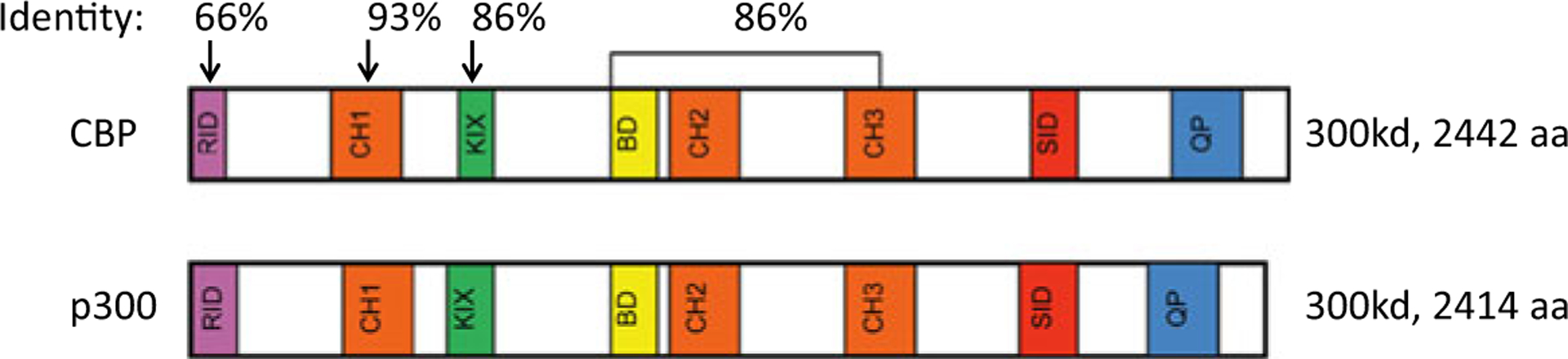
Schematic representation of CBP and p300 and the high percentage of identity at the amino acid level between various regions of these large Kat3 coactivators, despite their divergence more than 450 million years ago. The very amino terminal of CBP, to which CBP/catenin antagonists bind, is by far the most divergent region between these two Kat3 coactivators
Interestingly, the extreme N-terminal regions of CBP and p300, which bind to both β-catenin and the nuclear receptor family through a highly conserved LXXLL sequence and also to the small molecules ICG-001/PRI-724 and YH 249/250 (Higuchi et al. 2015), are the least conserved region with only 66 % identity between the two Kat3 coactivators. Despite this, the N-terminal regions within each orthologous group are highly conserved; for example, human and mouse CBP are 98 % identical at the amino acid level within this region of the proteins. What drove the rapid divergence of these two Kat3 coactivators from one another within the N-terminal regions of these coactivators prior to the vertebrate radiation and their subsequent conservation?
Both the Wnt signaling pathway and the nuclear receptor family appear to be inventions of the first multicellular animals (metazoans) (Holstein 2012; Markov and Laudet 2011) and are found in all animals including nematodes, flies, and vertebrates. We would like to propose that the gene duplication that provided the two Kat3 coactivators CBP and p300 occurred in conjunction with the expansion of the nuclear receptor family prior to vertebrate radiation. This evolutionary event solved the problem of the requirement for a high fidelity control mechanism to guide differential cell fates following asymmetric stem cell division, thereby generating and maintaining two inherently different cell populations: one a long-lived anaerobic metabolizing, relatively quiescent stem cell population and the other a differentiating aerobically metabolizing, rapidly cycling and expanding daughter cell population that could be further committed to generate multiple cell types. Importantly, this provided a mechanism for the protection of the genetic material in SSCs, especially critical in longer lived organisms, which was not required in shorter-lived invertebrates, from corruption via DNA copy errors as well as other mechanisms of insult and the ability to “read” multiple inputs that can affect the stability of the genetic material. For example, the ozone layer that was already present at this time in evolution absorbs the most dangerous part of the ultraviolet light spectrum; however, ultraviolet light can still induce a huge number of lesions in DNA (Jackson and Bartek 2009). Presumably for this reason, the circadian cycle also plays a role in the control of normal SSC activation and division (Brown 2014). Aberrant circadian regulation is associated with the increased risk for cancer development in shift workers (Davis et al. 2001; Schernhammer et al. 2001). More generally, the regulation of the timing and mode of SSC division appear to be regulated by both metabolic and circadian control (Chen and McKnight 2007).
It would also seem logical that over millions of years of evolution that a “naturally occurring” CBP/catenin antagonist, analogous to ICG-001, would have evolved that could enhance SSC asymmetric differentiation. In fact, we believe that there are numerous naturally occurring CBP/catenin antagonists. Acute promyelocytic leukemia (APL) is uniquely sensitive to all-trans retinoic acid (ATRA), a derivative of vitamin A. Unlike most other chemotherapies, ATRA does not directly kill the malignant cells but induces them to differentiate, similarly to ICG-001. Vitamin D occupies a prominent position in cancer prevention. Interestingly, both ATRA and vitamin D, via their respective transcriptional complexes (RAR/RXR and VDR/RXR), have been shown at least in certain settings (e.g., colorectal cancer cells) to antagonize aberrant Wnt signaling (Dillard and Lane 2007) and thereby phenotypically behave as CBP/catenin antagonists via competition for binding to the N-terminus of CBP. However, there are also reports of synergistic effects on the activation of gene expression by ATRA and Wnt for example (Szeto et al. 2001) and both vitamin D and ATRA drive the expression of distinct cassettes of genes. Clearly, ATRA and vitamin D therefore are not simply “pure antagonists” of CBP/catenin signaling and, in that sense, not identical to ICG-001. The highly conserved LXXLL sequence that is present in the very amino termini of both CBP and p300 can recruit these as well as potentially all other nuclear receptor signaling complexes (e.g., AR, PPAR, etc.), to this region of the Kat3 coactivators that binds both catenin (beta and gamma) and the CBP/catenin antagonists (e.g., ICG-001, PRI-724). Interestingly, a number of nuclear receptors, liganded or not, also demonstrate the ability to either maintain potency or initiate differentiation in stem cell populations, similarly to what we have observed with specific CBP/catenin or p300/catenin antagonists (Mullen et al. 2007). Vitamins A and D are required during normal development and have many beneficial health effects in adulthood. However, at high levels, they can have deleterious effects especially during development. For example ATRA is highly teratogenic. One of the most surprising findings during our investigations of mouse development was that selectively antagonizing the CBP/catenin interaction with ICG-001, even at very high levels, is extremely safe and has no deleterious effects. Mice born from mothers treated topically or orally with high doses of ICG-001 throughout pregnancy gave birth to normal litters, and at 6 weeks of age, the mice exhibited normal weight and size compared to their control littermates and could breed a second generation, testifying to the fact that there were also no deleterious effects on germ stem cell populations (data not shown). In dramatic contrast, selective antagonism of the p300/catenin interaction in utero causes dramatic defects in development in virtually every organ system investigated (i.e., vasculature, heart, lung, CNS, limbs, etc.) (Sasaki et al. 2013; Sasaki and Kahn 2014). In fact, the safety of CBP/catenin antagonists during embryonic development and their ability to increase differentiation prompted us to investigate the possibility of pharmacologically correcting genetic defects in utero. In two mouse genetic knockout models (Axin2 and p73), we have demonstrated that in utero administration of ICG-001 corrects the defects observed (Axin 2 (−/−), absence of Hardarian gland development and p73 (−/−), premature neuronal differentiation) (data not shown).
Although nuclear receptor ligands can be considered CBP/catenin antagonists and thus naturally occurring analogs of ICG-001 in a sense, there are several important differences. CBP/catenin antagonists (ICG-001 and PRI-724) are direct inhibitors (i.e., they bind directly to CBP and do not require any protein cofactors) and are pure CBP antagonists (i.e., they have no agonistic activity per se). Furthermore, they allow for stochastic differentiation (i.e., non-deterministic), whereas ATRA or vitamin D, after antagonizing the CBP/catenin interaction, presumably via their p300-dependent interaction agonistic properties, bias lineage commitment and thereby have deleterious effects at high dose levels on embryonic development (Laplante and Sabatini 2009). Striking differential coactivator usage by the nuclear receptor family was also recently highlighted in a publication from the Stallcup lab. They demonstrated that in a prostate cancer cell line, 47 % of androgen-regulated genes are p300-dependent, whereas only 0.3 % are CBP-dependent (Ianculescu et al. 2012). Nuclear receptors via their corresponding ligands also appear to influence stem cell senescence and aging. For example, PPARδ agonists improved HSC maintenance via increased asymmetric division (Ito et al. 2012) and bile acids, products of cholesterol metabolism, likely control aging and longevity, at least in part via their nuclear receptors (LXR, FXR) and their effects on stem cells (Groen and Kuipers 2013).
That being said, symmetric versus asymmetric mitosis predates vertebrate radiation and was already present in the bilaterian common ancestor. The role of the Wnt signaling cascade in symmetric versus asymmetric division has been demonstrated in both flies and nematodes (Lu et al. 1998). Thus, it predates the gene duplication of the Kat3 coactivators in the vertebrate lineages. The vertebrate Wnt cascade is not the only one to have diverged from its ancestral state. In Caenorhabditis elegans, multiple gene duplication events have led to at least four β-catenin homologs: bar-1, hmp-2, wrm-1, and sys-1. The β-catenin homolog bar-1 is utilized in a canonical Wnt/catenin (Wβ) pathway that utilizes the TCF homolog pop-1 (Lin et al. 1995; Thorpe et al. 1997), generally conserved among animals including vertebrates. Unlike vertebrates and flies however, C. elegans has a Wnt/catenin asymmetry (WβA) pathway that also utilizes pop-1 in conjunction with the divergent (by sequence but not function) β-catenin-like molecule sys-1 (Phillips and Kimble 2009). Sys-1 does not have any known orthologs outside the nematodes, so it seems likely that this WβA asymmetry pathway evolved specifically in that lineage. There are two additional β-catenin-like molecules in C. elegans, hmp-2 and wrm-1, whose major roles are in adhesion and regulation of TCF export, respectively (Phillips and Kimble 2009). The WβA pathway controls key asymmetric divisions at multiple stages of development in C. elegans (Phillips and Kimble 2009). It has been proposed that the role of sys-1 in asymmetric cell division is an example of specialization (i.e., sub-functionalization), in which the multiple roles of β-catenin/armadillo in different processes in other animals have been divided among the different catenins in nematodes (Liu et al. 2008). Thus, the ancestral bilaterian asymmetric division was likely controlled via a Wnt/catenin/TCF-like pathway (as still found today in flies and vertebrates), via a single Kat3 coactivator gene (still found today as the Drosophila Nejire gene).
We propose that the evolution of long-lived populations of SSCs in vertebrates was enabled by the duplication of this ancestral coactivator gene to generate CBP and p300, in concert with the expansion of the nuclear receptor family. This provided a robust mechanism to maintain the “stemness” of one daughter cell (associated with anaerobic metabolism and CBP/β-catenin signaling) while allowing the other daughter cell to be modulated by nuclear receptor ligands via their corresponding receptors (many of which are associated with metabolism, including PPAR, FXR, and others) to drive specific lineage commitment to accommodate longer lived, more complex organism development. How was this accomplished? We propose that the unique difference in the N-termini of CBP and p300 enabled this process. The increased fidelity afforded by this new mechanism may have been important to enhance the maintenance of somatic stem cell genetic material that would be critical for increasing longevity (C. elegans a few weeks versus vertebrates that can live up to 100 years or more) in vertebrate populations.
Interestingly, one significant difference that has been highly evolutionarily conserved between the N-termini of CBP and p300 is a 27 bp/9aa deletion in CBP between the β-catenin-binding region (DELI-sequence) and the nuclear receptor binding sequence (LXXLL). We propose that this deletion provided a mechanism for nuclear receptors, via steric inhibition, to cleanly antagonize CBP/β-catenin signaling, thereby either maintaining SSC quiescence or initiating asymmetric divisions. Utilizing p300 in asymmetric divisions, β-catenin and nuclear receptor signaling have the ability to synergize to affect a feedforward mechanism to drive differentiation and lineage commitment, as steric constraints do not preclude this synergy (Fig. 7). As one validation of the importance of this synergy in p300, we have recently performed CRISPR/Cas9 editing of the 27-bp insertion in p300 in murine P19 cells and demonstrated its effect on cell differentiation and the ability for synergistic β-catenin and nuclear receptor interactions to occur. Further details will be reported in a separate manuscript. As the closest orthologs of the Kat3 coactivators in other species (including Nejire in Drosophila and cbp-1 in C. elegans) have an even longer “spacer” sequence between the β-catenin and nuclear receptor binding motifs than either p300 or CBP, we propose that vertebrate p300 conserves the ancestral state most closely. Thus, the ancestral Kat3 coactivator could bind β-catenin (with or without nuclear receptor binding) to drive proliferation and differentiation following either symmetric or asymmetric cell division, with the possibility for either antagonistic or synergistic interactions between the two domains. In this view, the evolution of CBP in vertebrates, in which CBP nuclear receptor binding can purely antagonize the β-catenin interaction and not synergize with it, was a key innovation that provided a high fidelity mechanism for asymmetric cell fates of SSC daughter cells, wherein one daughter preserves (using a CBP-regulated program) its quiescent, pluripotent stem cell status, while the other daughter subsequently generates (using the ancestral p300-regulated proliferation and differentiation program) a rapidly symmetrically dividing TA population, enabling both normal tissue homeostasis and repair in longer lived organisms. Simply stated, the Kat3 coactivator gene duplication generated a robust switch to tightly regulate asymmetric fate commitment of daughter cells in long-lived SSCs in vertebrates (Fig. 7b).
Fig. 7.
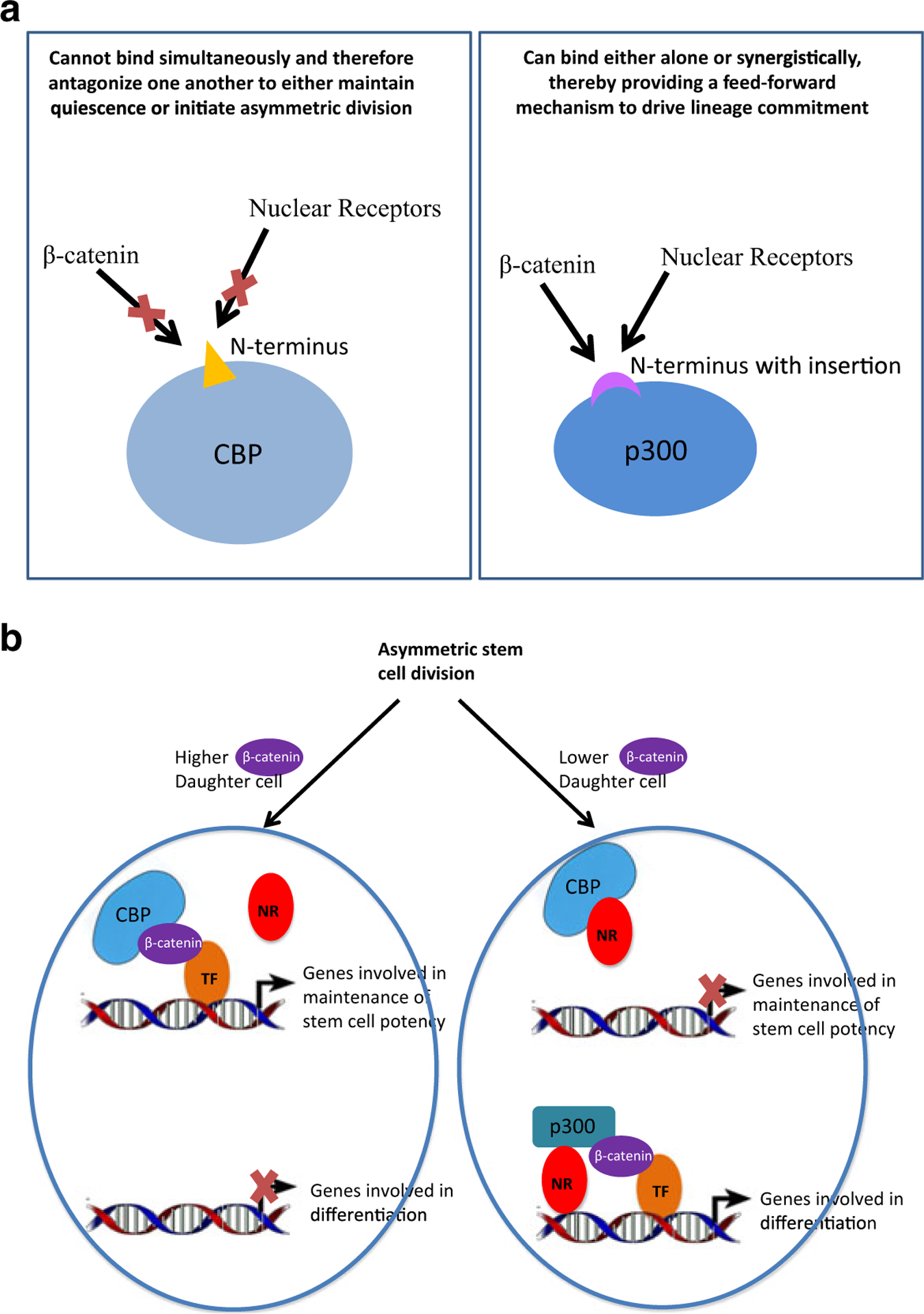
A model depicting the distinct modes of interaction (i.e., antagonism or potential synergy) between β-catenin and members of the nuclear receptor family to maintain asymmetric differentiation of the SSC population and to generate a feedforward mechanism to drive differentiation. a CBP shows antagonistic, competitive binding of β-catenin and nuclear receptors, while p300 can bind both simultaneously. b A simple model to show how competitive binding of CBP can act as a cell fate switch driven by differential coactivator usage
Conclusion and future prospects
The Spanish Explorer and Conquistador Ponce De Leon according to legend went searching for the Fountain of Youth. The initial motive for our investigations was to find a way to target aberrant activation of the Wnt signaling pathway in colorectal cancer. However, we believe that our discoveries have now provided us with a way to safely pharmacologically manipulate the balance of differential catenin coactivator usage (i.e., catenin/CBP versus catenin/p300) in SSC populations. If stem cell aging is intimately connected with longevity and diseases of aging, we may not have discovered the “fountain of youth”; however, a safe therapeutic approach to ameliorate the effects of aging and in particular to target diseases of aging, i.e., cancer, fibrosis, and neurodegeneration, may be available in the not too distant future.
However, we have only begun to scratch the surface of understanding how this balance is endogenously controlled. Stem cells in the niche receive an enormously complex array of information from its environment (e.g., oxygen levels, nutrient levels, light/dark, i.e., circadian cycles, growth factors, adhesion molecules, cell/cell contacts, etc.) to arrive at the eventual decision to remain quiescent or enter cycle and then to divide either symmetrically or asymmetrically. Beyond containing conserved catenin and nuclear receptor binding regions in their N-terminal domains, within the first 111 amino acid residues of CBP and p300, the interferon responsive transcription factor Stat1 binds within this region and there are approximately 20 serine and threonine residues that can be post-translationally modified (Zhang et al. 1996). Figure 8 depicts our model for the “funneling down” of information through various kinase cascades that can assist in coordinating this flood of information, to determine the binary decision to symmetrically or asymmetrically divide via controlling the balance between the CBP/catenin interaction and the p300/catenin interaction. The rapid and reversible (via phosphatases) ability of kinase cascades to modulate protein/protein interactions offers a very versatile and facile mechanism to incorporate numerous inputs to modulate this critical binary switch. We have only begun to address some of these questions of how important kinase cascades (e.g., PKC, MAPK, etc.) can affect the CBP/catenin versus p300/catenin equilibrium using genetic, proteomic, and pharmacologic approaches, which will be the subject of further investigations and communications.
Fig. 8.
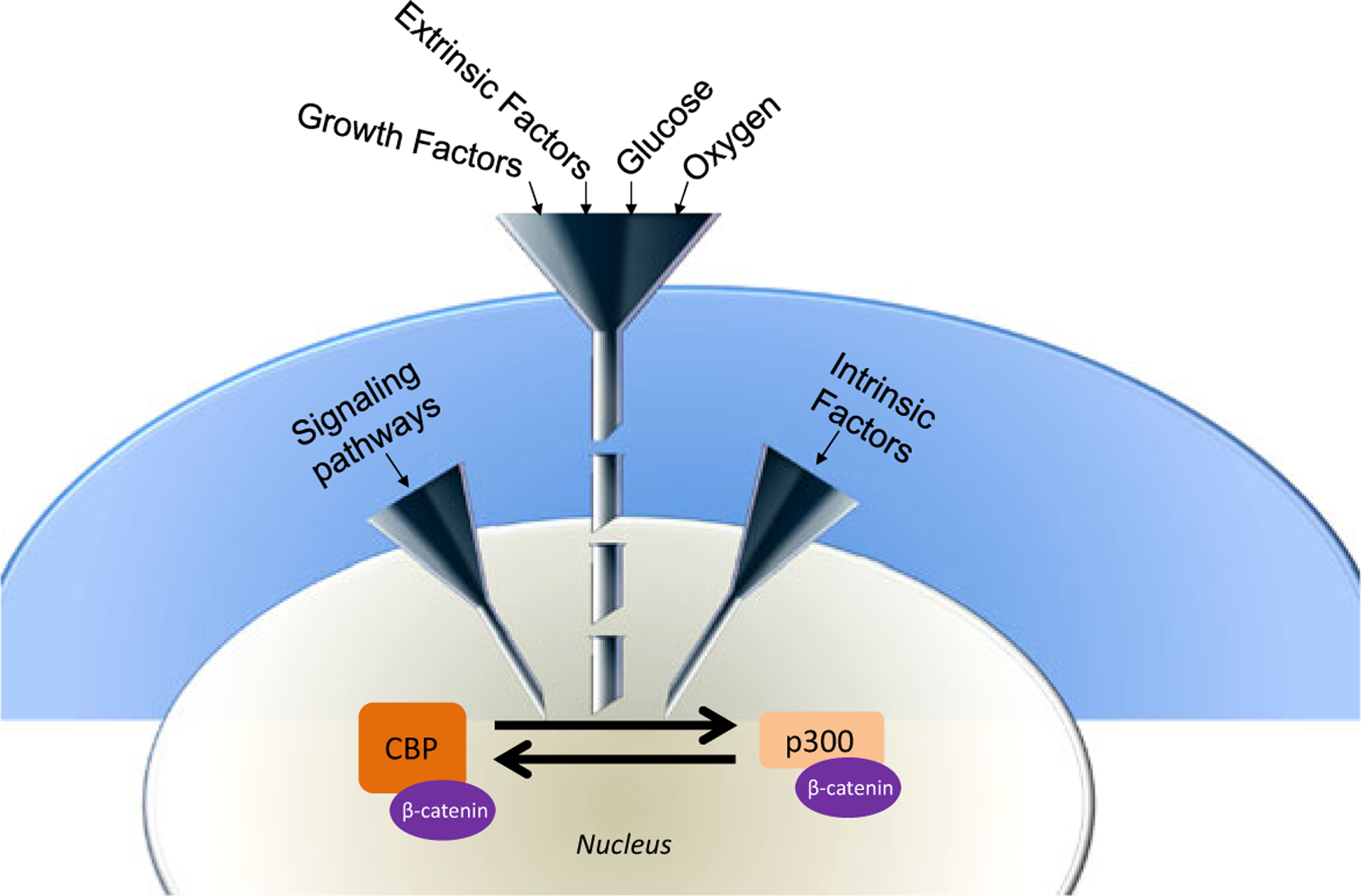
Model depicting the integration of a host of inputs to regulate the binary decision of symmetric versus asymmetric division in stem cells via differential β-catenin/Kat3 coactivator usage
These studies, in conjunction with preclinical models to evaluate the role of this critical switch in a range of devastating diseases (e.g., AD, Parkinson’s, MS, inflammatory bowel disease, etc.) and to pharmacologically intervene with small molecule CBP/catenin antagonists, as well as with more generic health problems such as metabolic syndrome and aging should provide the rationale for future clinical investigations.
“Vision is the art of seeing what is invisible to others.”
Jonathan Swift
Acknowledgments
Support from USC Norris Comprehensive Cancer Center Support Grant P30 CA014089 (PDTand MK), NIH 1R01CA166161-01A1, 1R21NS074392-01, 1R21AI105057-01, and NIH 1R01 HL112638-01 (MK) is gratefully acknowledged. We would also like to thank Dr. Jia-Ling Teo for critical review and assistance with preparation of this manuscript.
Footnotes
Conflict of interest PDT declares no conflict of interest.
M. K. was a consultant for Prism Pharma and has an equity interest in Makucell.
References
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. [DOI] [PubMed] [Google Scholar]
- Arany Z, Sellers WR, Livingston DM, et al. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77(6):799–800. [DOI] [PubMed] [Google Scholar]
- Bailleul B, Surani MA, White S, et al. Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell. 1990;62(4):697–708. [DOI] [PubMed] [Google Scholar]
- Baker AM, Cereser B, Melton S, et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014;8(4):940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker ST, Passegué E. Resilient and resourceful: genome maintenance strategies in hematopoietic stem cells. Exp Hematol. 2013;41(11):915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee ER, Laflamme MA, Papayannopoulou T, et al. Human embryonic stem cells differentiated to lung lineage-specific cells ameliorate pulmonary fibrosis in a xenograft transplant mouse model. PLoS ONE. 2012;7(3):e33165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker RA, de Beaufort I. Scientific and ethical issues related to stem cell research and interventions in neurodegenerative disorders of the brain. Prog Neurobiol. 2013;110:63–73. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229): 608–11. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344(6189): 1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–10. [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Rosário M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16(1):51–9. [DOI] [PubMed] [Google Scholar]
- Brown SA. Circadian clock-mediated control of stem cell division and differentiation: beyond night and day. Development. 2014;141(16):3105–11. [DOI] [PubMed] [Google Scholar]
- Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255(5505):197–200. [DOI] [PubMed] [Google Scholar]
- Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356(21):2131–42. [DOI] [PubMed] [Google Scholar]
- Chen Z, McKnight SL. A conserved DNA damage response pathway responsible for coupling the cell division cycle to the circadian and metabolic cycles. Cell Cycle. 2007;6(23): 2906–12. [DOI] [PubMed] [Google Scholar]
- Chen J, Astle CM, Harrison DE. Genetic regulation of primitive hematopoietic stem cell senescence. Exp Hematol. 2000;28(4):442–50. [DOI] [PubMed] [Google Scholar]
- Chen CT, Hsu SH, Wei YH. Upregulation of mitochondrial function and antioxidant defense in the differentiation of stem cells. Biochim Biophys Acta. 2010;1800(3):257–63. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297(5580):365–9. [DOI] [PubMed] [Google Scholar]
- Chocarro-Calvo A, García-Martínez JM, Ardila-González S, et al. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol Cell. 2013;49(3):474–86. [DOI] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138(6):1083–95. [DOI] [PubMed] [Google Scholar]
- Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154(2):274–84. [DOI] [PubMed] [Google Scholar]
- Cohnheim J. Ueber entzuendung und eiterung [About inflammation and suppuration]. Path Anat Physiol Klin Med. 1867;40(1–2):1–79. [Google Scholar]
- Coluccia AM, Benati D, Dekhil H, et al. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp 60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006;66(4):2279–86. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433(7027):760–4. [DOI] [PubMed] [Google Scholar]
- Cullen SM, Mayle A, Rossi L, et al. Hematopoietic stem cell development: an epigenetic journey. Curr Top Dev Biol. 2014;107:39–75. [DOI] [PubMed] [Google Scholar]
- Davis S, Mirick DK, Stevens RG. Night-shift work, light at night, and risk of breast cancer. J Natl Cancer Inst. 2001;93(20): 1557–62. [DOI] [PubMed] [Google Scholar]
- De Mey JR, Freund JN. Understanding epithelial homeostasis in the intestine: an old battlefield of ideas, recent breakthroughs and remaining controversies. Tissue Barriers. 2013;1(2):e24965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacote F, Lopez BS. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle. 2008;7(1):33–8. [DOI] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard AC, Lane MA. Retinol decreases beta-catenin protein levels in retinoic acid-resistant colon cancer cell lines. Mol Carcinog. 2007;46(4):315–29. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8(8):869–84. [DOI] [PubMed] [Google Scholar]
- El-Khoueiry AB, Ning Y, Yang D et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol 31(Suppl), 2013. Abstract 2501. [Google Scholar]
- Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004;101(34):12682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferent J, Cochard L, Faure H, et al. Genetic activation of Hedgehog signaling unbalances the rate of neural stem cell renewal by increasing symmetric divisions. Stem Cell Reports. 2014;3(2):312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Hochedlinger K, Van Buren D, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27(1):84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Temple S. Neural stem cells: generating and regenerating the brain. Neuron. 2013;80(3):588–601. [DOI] [PubMed] [Google Scholar]
- Gang EJ, Hsieh YT, Pham J, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33(17): 2169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabel L. Prospects for pluripotent stem cell therapies: into the clinic and back to the bench. J Cell Biochem. 2012;113(2): 381–7. [DOI] [PubMed] [Google Scholar]
- Groen AK, Kuipers F. Bile acid look-alike controls life span in C. elegans. Cell Metab. 2013;18(2):151–2. [DOI] [PubMed] [Google Scholar]
- Hao S, He W, Li Y, et al. Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol. 2011;22(9):1642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari L, Brault V, Kleber M, et al. Lineage-specific requirements of beta-catenin in neural crest development. J Cell Biol. 2002;159(5):867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Yasuda SY, Teo JL, et al. Wnt signaling orchestration with a small molecule DYRK inhibitor provides long-term xeno-free human pluripotent cell expansion. Stem Cells Transl Med. 2012;1(1):18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson WR Jr, Chi EY, Ye X, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107(32): 14309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Roux KJ, Wong ES, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19(3):413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi Y, Nguyen C, Yasuda S, et al. Specific direct small molecule p300/β-Catenin antagonists maintain stem cell potency. Curr Mol Pharm. 2015;8:1–8. [DOI] [PubMed] [Google Scholar]
- Holland JD, Klaus A, Garratt AN, et al. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25(2):254–64. [DOI] [PubMed] [Google Scholar]
- Holstein TW. The evolution of the Wnt pathway. Cold Spring Harb Perspect Biol. 2012;4(7):a007922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huls M, Russel FG, Masereeuw R. The role of ATP binding cassette transporters in tissue defense and organ regeneration. J Pharmacol Exp Ther. 2009;328(1):3–9. [DOI] [PubMed] [Google Scholar]
- Ianculescu I, Wu DY, Siegmund KD, et al. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J Biol Chem. 2012;287(6):4000–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Oshima H, Oshima M, et al. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010;101(3):673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. [DOI] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A. Prostaglandin E2 and pain—an update. Biol Pharm Bull. 2011;34(8):1170–3. [DOI] [PubMed] [Google Scholar]
- Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6(3): 204–10. [DOI] [PubMed] [Google Scholar]
- Kim K, Daniels KJ, Ha ED. Tissue-specific expression of beta-catenin in normal mesenchyme and uveal melanomas and its effect on invasiveness. Exp Cell Res. 1998;245(1):79–90. [DOI] [PubMed] [Google Scholar]
- Kim YM, Ma H, Oehler V, et al. The gamma catenin/CBP complex maintains survivin transcription in β-catenin deficient/depleted cancer cells. Curr Cancer Drug Targets. 2011;11(2): 213–25. [DOI] [PubMed] [Google Scholar]
- Kohli L, Passegué E. Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014;24(8): 479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl SJ, Kühl M. On the role of Wnt/β-catenin signaling in stem cells. Biochim Biophys Acta. 2013;1830(2):2297–306. [DOI] [PubMed] [Google Scholar]
- Kung AL, Rebel VI, Bronson RT, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14(3):272–7. [PMC free article] [PubMed] [Google Scholar]
- LaBarge M. The difficulty of targeting cancer stem cell niches. Clin Cancer Res. 2010;16(12):3121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviano A. Young blood. N Engl J Med. 2014;371(6):573–5. [DOI] [PubMed] [Google Scholar]
- Le NH, Franken P, Fodde R. Tumour-stroma interactions in colorectal cancer: converging on beta-catenin activation and cancer stemness. Br J Cancer. 2008;98(12):1886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nakada D, Yilmaz OH, et al. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell. 2010;7(5):593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327(5965):542–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Thompson S, Priess JR. Pop-1 encodes an HMG box protein required for the specification of a mesoderm precursor in early C. elegans embryos. Cell. 1995;83(4):599–609. [DOI] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317(5839):803–6. [DOI] [PubMed] [Google Scholar]
- Liu J, Phillips BT, Amaya MF, et al. The C. elegans SYS-1 protein is a bona fide β-catenin. Dev Cell. 2008;14(5):751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Jan LY, Jan YN. Asymmetric cell division: lessons from flies and worms. Curr Opin Genet Dev. 1998;8(4):392–9. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Blanpain C, Rossi DJ. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12(3):198–202. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–44. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, et al. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst). 2008;7(10):1765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markov GV, Laudet V. Origin and evolution of the ligand-binding ability of nuclear receptors. Mol Cell Endocrinol. 2011;334(1–2):21–30. [DOI] [PubMed] [Google Scholar]
- Marson A, Foreman R, Chevalier B, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3(2):132–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar J, Obrien WT, Johnson RS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12(10):1007–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan M, Kahn M. Investigating Wnt signaling: a chemogenomic safari. Drug Discov Today. 2005;10(21): 1467–74. [DOI] [PubMed] [Google Scholar]
- Merchant AA, Matsui W. Targeting Hedgehog—a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(Database issue):D377–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Teo JL, Yamamoto M, et al. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 2007;104(13): 5668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Bourke E, Alexander D, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443(7110): 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RK, Carlone DL, Richmond CA, et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108(1):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT. Wnt/beta-catenin pathway. Sci. STKE 2005(271), cm1 2005. [DOI] [PubMed] [Google Scholar]
- Mullen EM, Gu P, Cooney AJ. Nuclear receptors in regulation of mouse ES cell pluripotency and differentiation. PPAR Res. 2007;2007:61563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito AT, Sumida T, Nomura S, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012;149(6):1298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K, Muraguchi T, Hoshii T, et al. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal. 2008;10(11):1883–94. [DOI] [PubMed] [Google Scholar]
- Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767–79. [DOI] [PubMed] [Google Scholar]
- Nijnik A, Woodbine L, Marchetti C, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447(7145):686–90. [DOI] [PubMed] [Google Scholar]
- Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307(5710):720–4. [DOI] [PubMed] [Google Scholar]
- Oh IH, Humphries RK. Concise review: multidimensional regulation of the hematopoietic stem cell state. Stem Cells. 2012;30(1):82–8. [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–54. [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115–28. [DOI] [PubMed] [Google Scholar]
- Otero JJ, Fu W, Kan L, et al. Beta-catenin signaling is required for neural differentiation of embryonic stem cells. Development. 2004;131(15):3545–57. [DOI] [PubMed] [Google Scholar]
- Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16(12):3141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, Izpisua Belmonte JC. Anaerobicizing into pluripotency. Cell Metab. 2011;14(2):143–4. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Kimble J. A new look at TCF and beta-catenin through the lens of a divergent C. elegans Wnt pathway. Dev Cell. 2009;17(1):27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31(15):2737–46. Erratum in: EMBO J. 31(15), 3375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten CS, Hume WJ, Reid P, et al. The segregation of DNA in epithelial stem cells. Cell. 1978;15(3):899–906. [DOI] [PubMed] [Google Scholar]
- Rehman J. Empowering self-renewal and differentiation: the role of mitochondria in stem cells. J Mol Med. 2010;88(10):981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ress A, Moelling K. Bcr interferes with beta-catenin-Tcf1 interaction. FEBS Lett. 2006;580(5):1227–30. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. [DOI] [PubMed] [Google Scholar]
- Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014;10(4): 512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritsma L, Ellenbroek SI, Zomer A, et al. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507(7492):362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. 2014;510(7505):393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502(7472):513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Seita J, et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9. [DOI] [PubMed] [Google Scholar]
- Roth JF, Shikama N, Henzen C, et al. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J. 2003;22(19):5186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Kaul R, Murakami M, et al. Tumor viruses and cancer biology: modulating signaling pathways for therapeutic intervention. Cancer Biol Ther. 2010;10(10):961–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40(7):915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Kahn M. Inhibition of β-catenin/p300 interaction proximalizes mouse embryonic lung epithelium. Transl Respir Med. 2014;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Hwang H, Nguyen C, et al. The small molecule Wnt signaling modulator ICG-001 improves contractile function in chronically infarcted rat myocardium. PLoS ONE. 2013;8(9):e75010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, et al. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10(1):55–63. [DOI] [PubMed] [Google Scholar]
- Schernhammer ES, Laden F, Speizer FE, et al. Rotating night shifts and risk of breast cancer in women participating in the Nurses’ Health Study. J Natl Cancer Inst. 2001;93(20): 1563–8. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1–2):25–38. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. Cancer biology: gone but not forgotten. Nature. 2007;445(7128):606–7. [DOI] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344(6184):649–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto W, Jiang W, Tice DA, et al. Overexpression of the retinoic acid-responsive gene Stra6 in human cancers and its synergistic induction by Wnt-1 and retinoic acid. Cancer Res. 2001;61(10):4197–205. [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin. Cancer Res 2010;16(12):3153–62. [DOI] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, et al. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334(6061):1420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Takahashi K, Yamanaka S. Induction of pluripotency by defined factors. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(3):83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taupin P. Stroke-induced neurogenesis: physiopathology and mechanisms. Curr Neurovasc Res. 2006;3(1):67–72. [DOI] [PubMed] [Google Scholar]
- Teo J-L, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Deliv Rev. 2010;62(12):1149–55. [DOI] [PubMed] [Google Scholar]
- Teo JL, Ma H, Nguyen C, et al. Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc Natl Acad Sci U S A. 2005;102(34):12171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe CJ, Schlesinger A, Carter JC, et al. Wnt signaling polarizes an early C. elegans blastomere to distinguish endoderm from mesoderm. Cell. 1997;90(4):695–705. [DOI] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A. 2013;110(6):1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosko JE, Kang KS. Evolution of energy metabolism, stem cells and cancer stem cells: how the Warburg and Barker hypotheses might be linked. Int J Stem Cells. 2012;5(1):39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veelen W, Le NH, Helvensteijn W, et al. β-catenin tyrosine 654 phosphorylation increases Wnt signalling and intestinal tumorigenesis. Gut. 2011;60(9):1204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh PK, Gary JK, Zinser GM, et al. β-Catenin is required for Ron receptor-induced mammary tumorigenesis. Oncogene. 2011;30(34):3694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton D, Perin L, Defilippo R, et al. Stem/progenitor cells in lung development, injury repair, and regeneration. Proc Am Thorac Soc. 2008;5(6):703–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112(9):3543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wend P, Fang L, Zhu Q, et al. Wnt/β-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013;32(14):1977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoub M, Suzuki K, Rosenthal N. The future of regenerative therapy in patients with chronic heart failure. Nat Clin Pract Cardiovasc Med. 2006;3 Suppl 1:S133–5. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Oike Y, Kamon J, et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet. 2002;30(2):221–6. [DOI] [PubMed] [Google Scholar]
- Zhang M, Rosen JM. Stem cells in the etiology and treatment of cancer. Curr Opin Genet Dev. 2006;16(1):60–4. [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Vinkemeier U, Gu W, et al. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc Natl Acad Sci U S A. 1996;93(26):15092–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Masiello D, McMillan M et al. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene. 2015. doi: 10.1038/onc.2015.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CM, Hayakawa Y, Kodama Y, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6(250): 250ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457(7229):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]