

ABSTRACT
Glucocorticosteroids (GCS) have an established role in oncology and are administered to cancer patients in routine clinical care and in drug development trials as co-medication. Given their strong immune-suppressive activity, GCS may interfere with immune-oncology drugs. We are developing a therapeutic cancer vaccine, which is based on a liposomal formulation of tumor-antigen encoding RNA (RNA-LPX) and induces a strong T-cell response both in mice as well as in humans. In this study, we investigated in vivo in mice and in human PBMCs the effect of the commonly used long-acting GCS Dexamethasone (Dexa) on the efficacy of this vaccine format, with a particular focus on antigen-specific T-cell immune responses.
We show that Dexa, when used as premedication, substantially blunts RNA-LPX vaccine-mediated immune effects. Premedication with Dexa inhibits vaccine-dependent induction of serum cytokines and chemokines and reduces both the number and activation of splenic conventional dendritic cells (cDC) expressing vaccine-encoded antigens. Consequently, priming of functional effector T cells and therapeutic activity is significantly impaired. Interestingly, responses are less impacted when Dexa is administered post-vaccination. Consistent with this observation, although many inflammatory cytokines are reduced, IFNα, a key cytokine in T-cell priming, is less impacted and antigen expression by cDCs is intact. These findings warrant special caution when combining GCS with immune therapies relying on priming and activation of antigen-specific T cells and suggest that careful sequencing of these treatments may preserve T-cell induction.
KEYWORDS: Glucocorticosteroids, dexamethasone, T-cell vaccine, RNA vaccine, cancer immunotherapy, T-cell priming
Introduction
Glucocorticosteroids (GCS) are among the most commonly administered drugs in oncology. The long-acting GCS Dexamethasone (Dexa) is routinely prescribed to patients with advanced cancer for a variety of reasons, including fatigue, loss of appetite, neuropathic and bone pain, night sweats, edema associated with CNS tumors, and to prevent or treat side effects of chemotherapies or targeted drugs such as hypersensitivity, allergic reactions, nausea, and vomiting1.
In addition to their broad application in routine palliative care, GCS are used in clinical trials due to their strong immune-suppressive activity to prevent or to combat immune-related adverse effects of novel drug candidates.
GCS mediate anti-inflammatory effects through binding to the glucocorticoid receptor. This complex translocates to the nucleus and initiates multi-facetted alterations in gene expression. Moreover, direct interactions with a variety of pathways have been described.2-5 As a result, GCS have a plethora of down-stream effects and can modulate the immune system in various ways, which are still not fully understood.
The roll-out of immune checkpoint inhibitors as approved standards of care in many cancer indications and earlier treatment lines has motivated the exploration of therapies such as vaccines, immune-modulators, and adoptive transfer of autologous reprogrammed immune cells.6-8 Any effective cancer immunotherapy is inevitably associated with immune activation, release of proinflammatory cytokines and activation of cytotoxic T cells.
The intended strong immune response can escalate into a life-threatening cytokine release syndrome or result in autoreactive T cells, for which GCS are considered an effective countermeasure.
To which extent GCS-mediated immune suppression may negatively impact the course of a patient's disease or interfere with treatment, in particular with immunotherapy, has not been systematically investigated yet. A limited set of data indicates that Dexa administration is associated with poorer outcome of cancer patients in general9-12 and that it specifically interferes with immune-checkpoint blockade as well as chemotherapy.13-16
We are developing cancer vaccines which are based on tumor-antigen encoding RNA. Being a natural TLR7/8 ligand, RNA has intrinsic adjuvant activity and the antigen is delivered in an HLA-independent manner. RNA is non-integrating and therefore considered as safe. We have optimized the cap structure, the 5ʹ- and 3ʹ-untranslated region and poly(A)-tail of antigen-encoding RNA for high stability and translation efficiency.17-21 The encoded antigen is fused to the MHC class I signal sequence and transmembrane and cytoplasmic domains for routing to the endoplasmic reticulum, resulting in increased presentation efficacy of MHC class I and II epitopes.22 The RNA is formulated as lipoplex (RNA-LPX) which, upon intravenous application, is selectively taken up by dendritic cells (DC) resident in lymphoid compartments throughout the body.23 Induction of a type I IFN response concurrent to presentation of the vaccine antigen promotes efficient priming and expansion of CD8+ and CD4+ T cells against the vaccine-encoded antigen and results in strong anti-tumor immunity in mice.23-25 First clinical data in patients with advanced melanoma indicate execution of the mode-of-action and activity of RNA-LPX vaccines in humans.26-31
To understand potential interference of the long-acting GCS Dexamethasone with our RNA-LPX vaccine, we studied its effects on vaccine-induced innate and adaptive mechanisms when employed under circumstances typically occurring in clinical care.
Materials and methods
Cell lines and mice
Female 8–12 week old BALB/c mice (Janvier Labs) or C57BL/6 mice (Envigo) were kept and treated as approved by the Ethics Committee for animal research of Rhineland-Palatinate, Germany. For experiments shown in Figure 5(e-g), 6(a,b), Supplementary Figure 3, and Supplementary Figure 4(b,c), 6–12 week C57BL/6 mice were purchased from Charles River Labs, and activities and procedures were performed in accordance with a protocol reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Genentech, an AAALAC-accredited animal care and use program. CT26 colon carcinoma cells were purchased in 2011 (ATCC CRL-2638, lot no. 58494154). MC38 cells were a kind gift from R. Offringa, Leiden University Medical Center. Master and working cell banks were generated immediately upon generation or receipt of cells; early passages were used for experiments. Cells were tested for mycoplasma contamination every 3 months.
Figure 5.
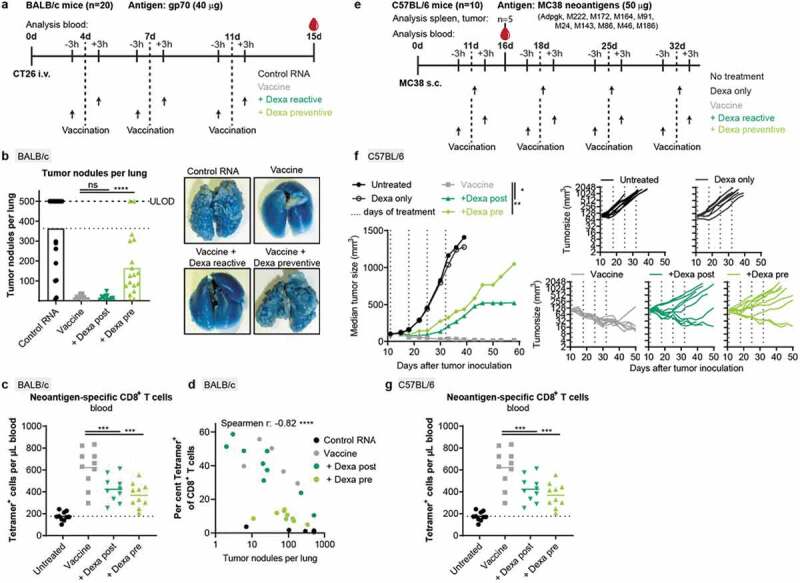
Therapeutic activity of RNA-LPX vaccination is significantly weakened by Dexa co-medication. (a–d), For a hematogenous lung dissemination tumor model, BALB/c mice (n = 20) were injected i.v. with CT26 tumor cells and treated as shown in (a). (b), Tumor nodules per lung of individual mice (left) and representative lung images (right). 500 nodules per lung was the upper limit of detection (ULOD). (c), Fraction of gp70-specific T cells in CD8+ T cells in the blood of mice (n = 10) 15 d after injection of tumor cells. (d), Correlation between tumor nodules per lung and the fraction of gp70-specific (Tetramer+) T cells in the blood. Tumor nodule values of 0 are not depicted but included in the spearman r and corresponding p value. (e-g), C57BL/6 mice (n = 10) were inoculated s.c. with MC38 tumor cells and treated as indicated in (e). (f), Mean tumor growth per group (left) and in individual mice (right). (g), Neoantigen (Adpgk, M86, and M143)-specific CD8+ T cells in the blood (n = 10). Mean (line) and individual values (symbols) are shown.
Figure 6.
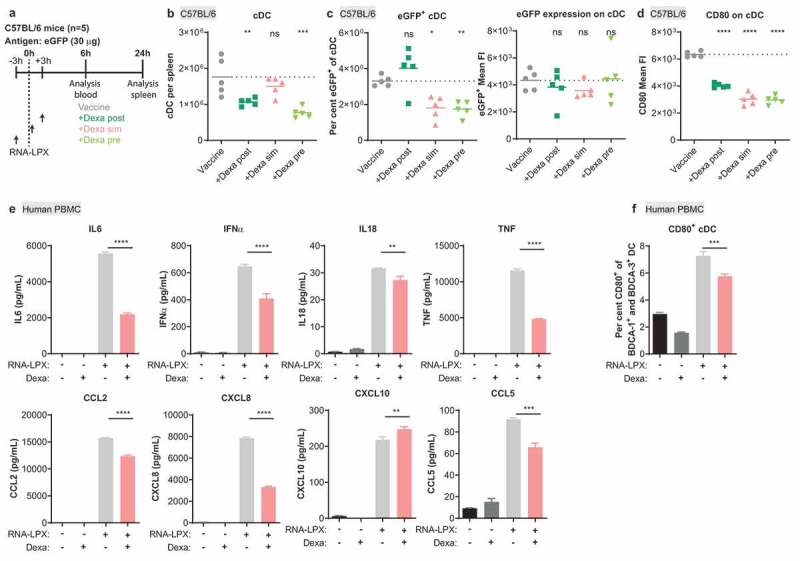
Dexa impairs the stimulatory potential of cDC in mice and of human PBMC. (a), Treatment schedule of C57BL/6 mice. (b), Splenic cDC (MHC class II+, CD11c+, non-T or B cells) as quantified by flow cytometry. (c), Fraction of eGFP+ cells (left) and expression levels of eGFP (right) within cDC. (d), CD80 expression on cDC. FI, fluorescence intensity. (b–d), Mean (line) and individual values (symbols) are shown. The dotted line represents the mean of the control group. (e–f), Human PBMC were stimulated for 17 h with 10 µg/mL RNA-LPX and/or 0.1 µM Dexa as shown. Depicted is the mean of three donors. (e), Cytokine and chemokine secretion. (f), Fraction of CD80+ cells among CD123− BDCA-1+ HLA-DR+ BDCA-3+ cDC. The dotted line represents the mean of the control group.
RNA and peptides
RNAs were synthesized by BioNTech RNA Pharmaceuticals GmbH.32 gp70 RNA codes for the H-2Ld-restricted epitope AH1423-431 derived from the murine leukemia virus envelope glycoprotein 70 (gp70) with a single amino acid substitution at position five (V427A, AH1-A5)33 elongated by three amino acids at the N-terminus and by one amino acid at the C-terminus. H-2 Db-restricted neoepitopes derived from mutations in Adpgk and Reps1 genes identified in the MC38 mouse tumor cell line were described before.34 Therapeutic vaccination of MC38 tumor-bearing mice was conducted with a decatope RNA-LPX encoding the 10 MC38-derived neoantigens Adpgk-M222-M172-M164-M91-M24-M143-M86-M46-M186 described by Capietto et al.35 Neoantigen vaccine RNAs encode 27 amino acids with the mutated amino acid in the center (position 14). Neoantigen sequences on a decatope were separated by 10mer non-immunogenic glycine/serine linkers.32 All epitope sequences were embedded into a construct based on the previously described pST1-Sp-MITD-2hBgUTR-A120 plasmid backbone developed for immuno-pharmacologically optimized gene delivery.22 gp70 AH1-A5 (SPSYAYHQF, gp70423–431, V427A) was synthesized by JPT Peptide Technologies GmbH, Berlin.
Mouse models
For vaccination, RNA was formulated with liposomes consisting of DOTMA and DOPE at a charge ratio (+):(-) of 1.3:2 yielding negatively charged RNA-Lipoplexes (RNA-LPX) as described previously.23 RNA-LPX comprising 20 µg gp70 RNA, 15 µg Reps1 and Adpgk RNA each or 30 µg eGFP RNA was injected i.v. in C57BL/6 or BALB/c mice as described in Figures 1 and 6(a). If not otherwise stated, Dexamethasone (Ratiopharm or Mylan) was injected i.p. at a dose of 4 mg/kg in 200 µL PBS. For lung metastasis experiments, 5 × 105 CT26 cells were injected i.v. in 100 µL PBS and treatment with 40 µg gp70 RNA-LPX and Dexa was performed as depicted in Figure 5(a). CT26 lung tumor burden was quantified after tracheal ink (1:10 diluted in PBS) injection and fixation with Fekete’s solution (5 ml 70% ethanol, 0.5 ml formalin, and 0.25 ml glacial acetic acid). A total of 1 × 105 MC38 tumor cells was implanted subcutaneously in the right hind flank in 100 µL of HBSS + matrigel. MC38 tumor-bearing mice were vaccinated with 50 µg RNA-LPX and treated with 4 mg/kg Dexamethasone as depicted in Figure 5(e), 5 mg/kg Methylprednisolone (Pfizer) or with 0.25 mg/kg Dexamethasone 5 minutes post-vaccination. Tumors were monitored at least twice per week and mice were euthanized if tumors became ulcerated or exceeded the acceptable size limit of 2000 mm3 according to IACUC.
Figure 1.
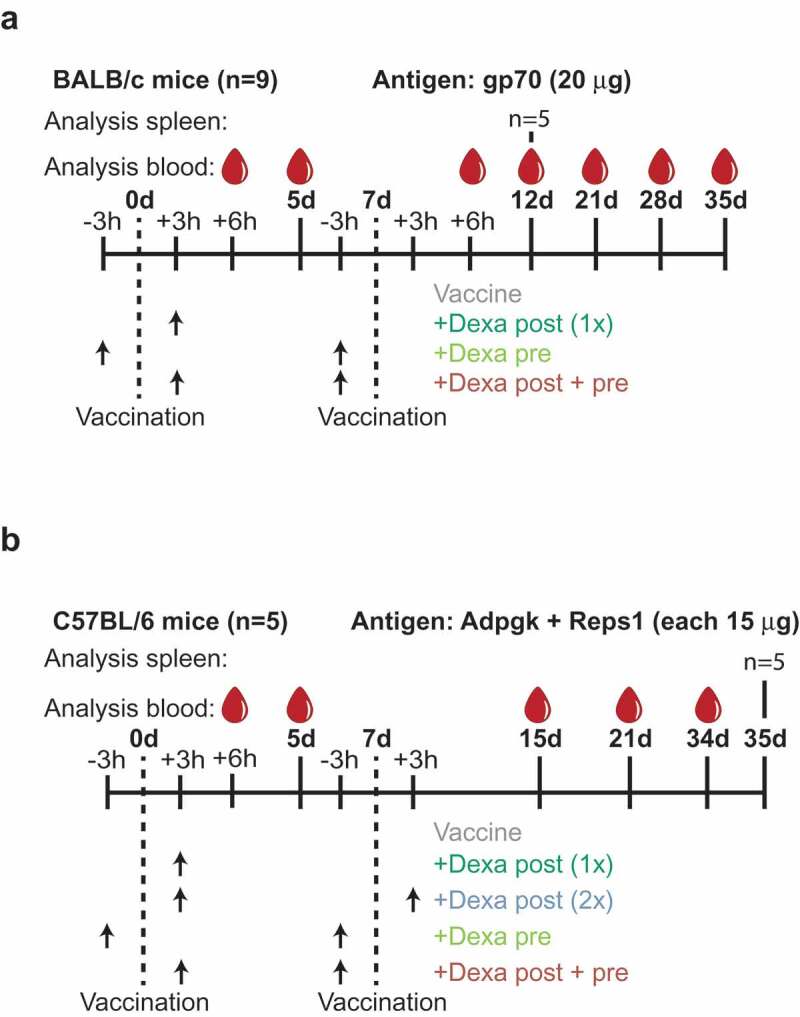
Experimental design. (a), BALB/c mice (n = 9) were vaccinated with 20 µg gp70 RNA-LPX on d 0 and 7. Dexa (4 mg/kg) was injected i.p. 3 h before or after vaccination (arrows). Control mice were vaccinated with RNA-LPX, but not Dexa treated. Blood was taken 6 h after both vaccinations to analyze cytokine release and on d 5, 12, 21, 28 and 35 for immune cell analysis by flow cytometry. Five mice were sacrificed on d 12 for analysis of antigen-specific cytokine release in splenocytes. (b), C57BL/6 mice (n = 5) were vaccinated with 15 µg Adpgk + 15 µg Reps1 RNA-LPX on d 0 and 7 with and without Dexa (4 mg/kg) treatment and underwent a similar analytic regimen as described in (a). On d 35, all mice were sacrificed for analysis of splenocytes for antigen-specific IFNγ release and cytotoxicity.
Protein quantification
Blood from mice was retrieved 6 h after vaccination, serum generated and stored at −20 °C. Serum concentrations of cytokines were determined using a bead-based, custom (Figure 2) or Cytokine & Chemokine Convenience 26-Plex Mouse ProcartaPlex multiplex immunoassay supplemented with murine IFNα (Thermo Fisher Scientific) (Supplementary Figure 5) according to the manufacturer’s instructions. Fluorescence was measured with the Bioplex200 System (Biorad) and analyzed with ProcartaPlex Analyst 1.0 software (Thermo Fischer Scientific).
Figure 2.
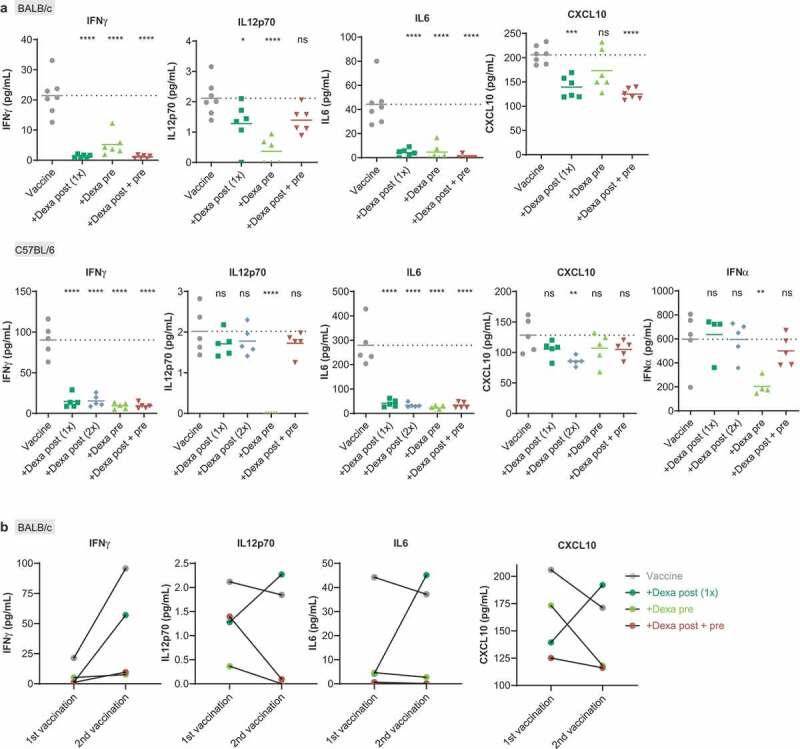
Dexamethasone suppresses RNA-LPX vaccine induced release of immune-response promoting cytokines. (a), Serum cytokine levels were analyzed in the serum of BALB/c mice (upper panel, n = 7 for control, n = 6 for groups treated as described in Figure 1(a)) and of C57BL/6 mice (lower panel, n = 5, treated as described in Figure 1(b)) 6 h post first vaccination. Mean (line) and individual values (symbols) are shown. The dotted line represents the mean of the control group. (b), Comparison of mean serum cytokine levels 6 h after the first and the second gp70 vaccination in BALB/c (n = 7 for control, n = 6 for treated groups, treated as described in Figure 1(a)).
To study cytokine secretion from human PBMC, buffy coats from voluntary healthy donors were obtained as part of the Genentech blood donor program on the day of PBMC isolation. Informed written consent was obtained from all donors. The buffy coats were diluted 1:1 with PBS and isolated by density centrifugation using SepMate-50 tubes and Lymphoprep Medium (both StemCell Technologies). Red blood cells were lysed with ACK lysis buffer and the remaining PBMCs were washed with PBS and passed twice through a 70 µm filter.
In a 96-U-well plate, 3.50 × 105 cells were plated and stimulated with 10 μg/mL eGFP RNA-LPX with or without 0.1 μM Dexamethasone. After 17 hours, the cells were pelleted down, the supernatants were collected and stored at −80 °C until analysis. Supernatant concentrations of human cytokines were determined in thawed samples using a bead-based, custom Bio-Plex group II 4-plex (BioRad) or pre-mix Milliplex panel I 30-Plex (Millipore).
Tissue preparation
Splenocytes were isolated by gently passing spleens through a 70 µm cell strainer followed by erythrocyte lysis using an ACK buffer. For analysis shown in Figure 6(b-d) and Supplementary Figure 3(e) and 5(c-d), spleens were injected with 100 µL of enzymatic solution containing Liberase (Roche Blendzyme-2, 2.5 mg/mL) and DNAse I (Invitrogen) and incubated for 10 minutes before single-cell preparation.
Murine blood was collected into heparinized tubes by retro-orbital bleeding or by puncture of the vena facialis. Following antibody or MHC tetramer staining, blood was lysed using BD FACS™ Lysing Solution (BD Biosciences).
Tumor samples were cut into small fragments and placed in digestion buffer (DMEM, 5% Fetal Bovine Serum, 10 mM HEPES, 2 mg/mL Collagenase D (Roche), 100 µg/mL DNAse I (Roche) and 50 nM desatinib36 (BioVision)) for 15 minutes with gentle mixing at 37 °C.
Flow cytometry
The following antibody clones were used for staining of murine samples: CD8 (53–6.7), CD25 (PC61), FoxP3 (FJK-16s), CD49b (DX5), CD80 (16-10A1), CD45 (30-F11) (all BD Biosciences), CD4 (RM4-5), CD19 (1D3), CD11b (M1/70), CD11 c (N418), F4/80 (BM8), MHC-II (M5/114.15.2), PDCA1 (927), CD4 (GK1.5), CD3 (17A2), CD19 (6D5) (all Biolegend), IFNγ (XMG1.2), granzyme B (GB11) (both Invitrogen) or CD8 (KT-15, MBL).
Antigen-specificity was determined by staining with readily available (gp70, Adpgk, Reps1) or custom (M86; H2-Kb-KILTFDRL, M143; H2-Db-SAIRSYQYV) MHC class I tetramers (MBL International Corporation). Viability was determined using the LIVE/DEAD™ Fixable Yellow Dead Cell Stain Kit or Fixable Aqua Dead Cell Stain Kit (Invitrogen).
Intracellular staining of IFNγ and granzyme B was performed using the cytofix/cytoperm kit (BD Biosciences) after stimulation of 4 × 106 splenocytes with 2 µg/mL gp70 AH1-A5 or no peptide for 1 h, and in the presence of 10 µg/mL brefeldin A (Sigma) and GolgiStop (1:500, BD Biosciences) for 4 hours at 37 °C.
FoxP3 was stained using the eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Invitrogen). Absolute cell numbers were determined with BD Trucount™ Absolute Counting Tubes (BD Biosciences).
Staining of human APC was performed with the following antibody clones: BDCA-3 (AD5-14H12), BDCA-2 (AC144), CD14 (TÜK4), (all Miltenyi Biotec), CD123 (6H6), BDCA-1 (L161) (both Biolegend), HLA-DR (G46-4), CD80 (L307.4) (both BD Biosciences). Viability was determined using the LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit (Invitrogen). On 6-well plates, 3 × 106 PBMC were plated and stimulated with 10 μg/mL eGFP RNA-LPX with or without 0.1 μM Dexamethasone. After 17 hours, the non-adherent cells were collected into a 50 mL tube, the adherent cells were detached using StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific) and pooled together with the non-adherent fraction for a subsequent flow cytometry staining.
Flow cytometry data was acquired on a BD FACSCelesta, BD FACSymphony, or BD Fortessa X20 (BD Biosciences) and analyzed with FlowJo 10.5 software (Tree Star).
IFNγ ELISpot assay
Enzyme-linked ImmunoSpot assays detecting IFNγ release of MC38-specific T cells were performed as previously described.32 Restimulation of 5 × 105 splenocytes was achieved by the addition of 4 × 104 MC38 tumor cells. All samples were tested in duplicates.
Cytotoxicity assay
Splenocytes (3.5x106 in 10 mL medium) were stimulated overnight at 37° C and 5% CO2 in the presence of 100 U/mL IL2 (Proleukin S, Novartis) and 1 × 106 MC38 cells. Lysis of 2 × 105 MC38 cells grown for 24 hours in a 96-well E-Plate (OMNI Life Science) was monitored after the addition of 3 × 105 CD8+ T cells isolated from splenocytes (CD8a (Ly-2) MicroBeads, Miltenyi Biotec) by xCELLigence RTCA MP (ACEA Biosciences).
Statistics
Two-way ANOVA and Dunnett’s multiple comparisons test were used to determine statistically significant differences in Figure 4(b) and Supplementary Figure 4(b). Figure 4(a), Figure 5(f), Supplementary Figure 2, Supplementary Figure 3(b) were analyzed via two-way ANOVA and Sidak’s multiple comparisons test. Kruskal–Wallis and Dunn’s test were applied to analyze Figure 5(b). One-way ANOVA and Sidak’s multiple comparisons test were used in Figure 6(e). One-way ANOVA and Dunnett’s multiple comparisons test were used for all other comparisons. For analyses shown in Figure 5(b,d) and Supplementary Figure 3(a) outliers were removed using a ROUT test (Q = 1%). If not otherwise indicated, all analyses were two-tailed and carried out in comparison to the control group using GraphPad Prism 8. n.s. (not significant), P > .05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Figure 4.
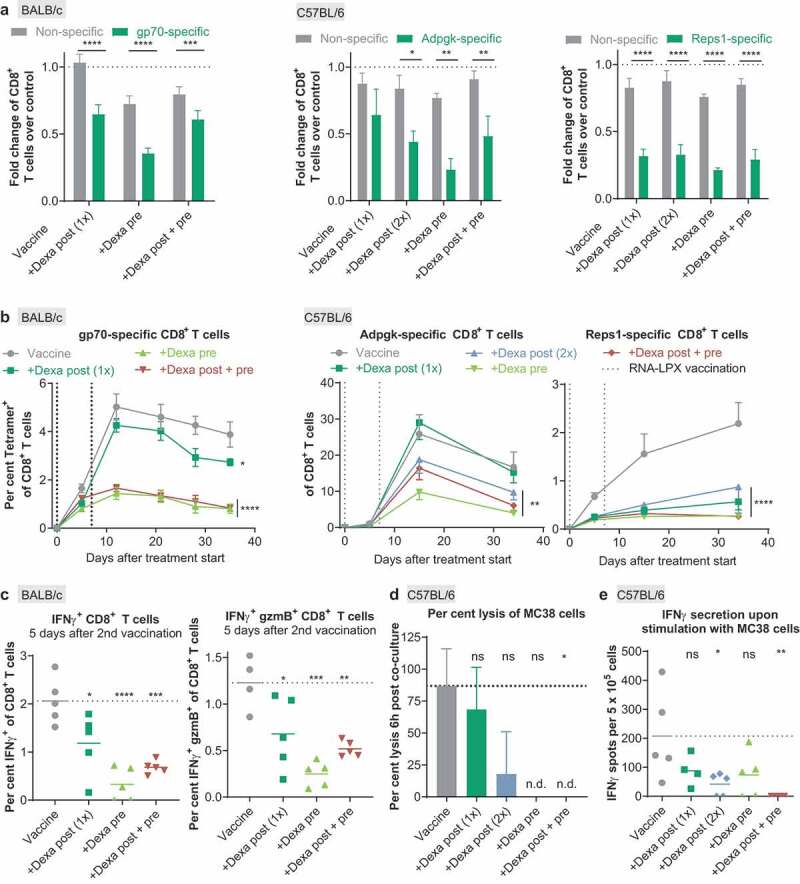
Priming, expansion, and cytotoxicity of antigen-specific T cells induced by RNA-LPX vaccination are severely suppressed by Dexa. (a), The number of nonspecific tetramer-negative or antigen-specific tetramer-positive CD8+ T cells per µL blood of the Dexa-treated groups was divided by the median number of nonspecific tetramer-negative or antigen-specific tetramer-positive T cells per µL blood, respectively, of the vaccine-only group as measured 5 d after the first vaccination. Depicted is the fold change of nonspecific CD8+ T cells or gp70- (left, n = 9), Adpgk- (middle, n = 5) and Reps1- (right, n = 5) specific CD8+ T cells as compared to the control group. Mean+SEM is depicted. (b), Fraction of antigen-specific CD8+ T cells in peripheral blood of RNA-LPX and Dexa-treated mice (gp70-specific (left): n = 9 on d 0 to 11, n = 5 on d 12, n = 4 on d 13 to 35; Adpgk- (middle) and Reps1-specific (right): n = 5). MeanSEM is depicted. Vertical dotted lines represent days of treatment. (c), Splenocytes of BALB/c mice (n = 5) were ex vivo restimulated with gp70 AH1-A5 peptide 12 d after treatment start. IFNγ positive (left) and IFNγ granzyme B (gzmB) double-positive (right) CD8+ T cells were determined by intracellular cytokine staining. Mean (line) and individual values (symbols) are shown. Values from unstimulated samples were subtracted (see also Supplementary Figure 2). (d), Percent lysis of MC38 cells 6 h post-co-culture with isolated CD8+ T cells as measured by xCELLigence RTCA MP. Mean+SEM is depicted (n = 3). (e) IFNγ secretion upon ex vivo restimulation of splenocytes with MC38 tumor cells (n = 5) determined by ELISpot. Mean (line) and individual values (symbols) are shown. The dotted line represents the mean of the control group. n.d., not detectable.
Results
We explored the effect of GCS on vaccination with three antigens expressed in commonly studied mouse tumor models: We immunized BALB/c mice with the H2-Ld-restricted epitope AH1423-431 derived from the murine leukemia virus envelope protein gp70 expressed in the CT26 colon carcinoma cell line33,37 and C57BL/6 mice with the H2-Db-restricted mutant Reps1 and Adpgk-neoepitopes discovered in MC38 colon carcinoma tumor cells.34 The vaccine epitopes were formulated as RNA-LPX and intravenously administered on day 0 and 7. Mice were either premedicated with Dexamethasone (Dexa) 3 hours prior to both vaccinations (Dexa pre) or, to simulate the management of immune-related toxicity, were Dexa-treated 3 hours after the first and in some experiments also after the second vaccination (Dexa post 1x and 2x, respectively). A third regimen combined Dexa 3 hours after the first and 3 hours prior to the second vaccination to simulate a clinical scenario of immune-related adverse-event management resulting in cautionary premedication for the consecutive immunotherapy exposures (Dexa post + pre) (Figure 1).
First, we measured serum levels of IFNγ, IL12p70, IL6, and CXCL10 which are key mediators of the intrinsic adjuvanticity of RNA-LPX vaccines and contribute to the efficient priming and expansion of antigen-specific T cells. As shown in mouse studies23-25 and ongoing clinical trials,23,27-30 these cytokines and chemokines are not detectable at baseline, have a fast, pulsatile, and transient release kinetics with peak levels around 4–6 hours and return to baseline levels within 24 hours after immunization.
In line with this notion, 6 hours after the first immunization of BALB/c mice with gp70 epitope (Figure 2(a), upper panel) and of C57BL/6 mice with two mutant epitopes (Figure 2(a), lower panel), release of these cytokines was detected in the vaccine-only group (Vaccine). In all Dexa-treated groups, however, serum cytokines were very low and IFNγ and IL6 were barely above detection level. Having shown that Dexa interferes with these immune-modulatory factors intended for promoting T-cell priming at first antigen-encounter, we assessed cytokine levels in BALB/c mice after a second vaccination with gp70 epitope (Figure 2(b)). In the group receiving only vaccine, IL12p70 and IL6 levels after the second vaccination were comparable to those after the first vaccination, and IFNγ levels were considerably higher. In contrast, in mice subjected to the pre and the post + pre Dexa regimen, serum IFNγ, IL12p70 and IL6 were hardly measurable after the second vaccination. Mice which had received a single Dexa dose after the first vaccination (Dexa post 1x) demonstrated cytokine levels after the second vaccination that were closer to those observed under nonimmune suppressed conditions after the first vaccination, indicating recovery of the involved immune-cell populations with regard to their capability to secrete these key cytokines.
Immune-cell populations in peripheral blood 5 days after the first vaccination showed the strongest differences in CD4+ T cells. These were significantly lower in the Dexa pre-medicated group as compared to the vaccine-only group. This treatment regimen was also associated with lower CD8+ T- and CD45+ cell numbers. This effect was statistically significant in gp70-vaccinated BALB/c mice and less profound in Reps1- and Adpgk-vaccinated C57BL/6 mice (Figure 3(a)). CD4+ CD25+ FoxP3+ regulatory T cell (Treg) numbers in vaccinated mice with Dexa pre-treatment were lower as compared to the vaccine-only group. However, in particular mice pre-treated with Dexa displayed a significantly higher fraction of Tregs within the CD4+ T cell population (Figure 3(b)). Numbers of B cells and NK cells were not altered by the treatment with Dexa (Supplementary Figure 1).
Figure 3.
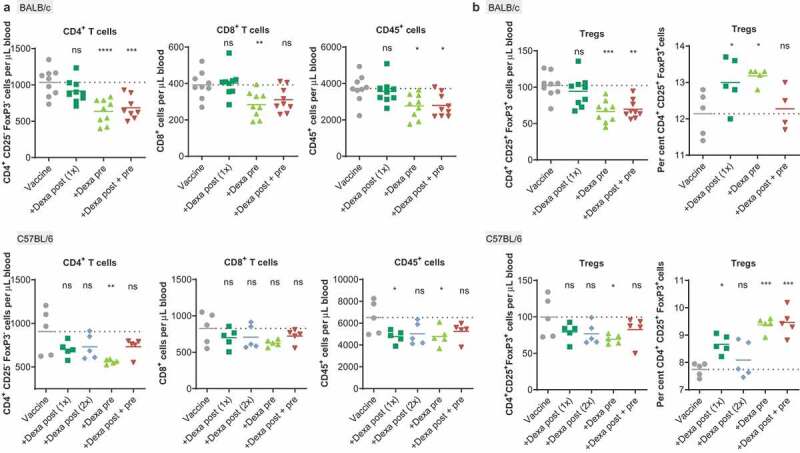
RNA-LPX-vaccinated mice premedicated with Dexa exhibit low CD4+ and CD8+ blood T-cell numbers and elevated Treg fractions. (a), CD4+, CD8+ or CD45+ cells per µL peripheral blood 5 d after the first treatment of BALB/c mice (n = 9, upper panel) with gp70 RNA-LPX or C57BL/6 mice (n = 5, lower panel) with Reps1 + Adpgk RNA-LPX and Dexa co-medication (see Figure 1(a)) as quantified by flow cytometry. (b), CD4+ CD25+ FoxP3+ Tregs per µL peripheral blood 5 d after the first vaccination (left) and fraction of Tregs within the CD4+ T cell population after two vaccinations (right) of BALB/c mice (upper panel, n = 9, left; n = 5, right) treated with gp70 RNA-LPX, or C57BL/6 mice (lower panel, n = 5) treated with Reps1 + Adpgk RNA-LPX and Dexa co-medication. Mean (line) and individual values (symbols) are shown. The dotted line represents the mean of the control group.
Dexa had a more pronounced inhibitory effect on antigen-specific T cells induced with the three different vaccine antigens than on naïve, non-antigen-specific CD8+ T cells (Figure 4(a)).
When we investigated the kinetics of antigen-specific T-cell responses in gp70-vaccinated mice in more detail, we found that the frequency of circulating vaccine-induced antigen-specific T cells of the vaccine-only group increased quickly, peaked at day 10 and was maintained at high levels for the entire observation period of 35 days. Dexa pre-treatment, in contrast, was associated with severely reduced expansion of antigen-specific CD8+ T cells. In the group receiving Dexa only after the first vaccination (Dexa post 1x), expansion kinetics and fractions of gp70-specific CD8+ T cells were more similar to, yet significantly below those of the non-Dexa-treated, vaccine-only group (Figure 4(b), left). Interestingly, kinetics of antigen-specific CD8+ T cells induced with the two other vaccine epitopes differed from what we observed in the gp70 model. Vaccination with the strong and immune-dominant Adpgk epitope resulted in up to 30% of circulating antigen-specific CD8+ T cells in the control group. T-cell expansion was not compromised at all by Dexa post-administered after the first vaccination (Dexa post 1x). All other Dexa regimens, including Dexa post 2x, resulted in significantly impaired expansion and survival of antigen-specific CD8+ T cells, nevertheless reaching low two-digit percentages at peak expansion (Figure 4(b), middle). In contrast, expansion of T cells induced by vaccination with the much weaker Reps1 epitope was heavily suppressed by all tested Dexa regimens, including Dexa post-treatment after the first vaccination (Figure 4(b), right).
As a consequence of Dexa treatment, a significantly lower fraction of CD8+ splenocytes isolated 5 days after the second vaccination was capable of secreting the effector cytokine IFNγ and the cytotoxic effector protein granzyme B (gzmB) upon ex vivo restimulation with the gp70 epitope, further indicating impairment of an effective antigen-specific response (Figure 4(c), Supplementary Figure 2). Again, this effect was most pronounced with Dexa pre-treatment.
For qualitative assessment of the immune response induced against the two MC38-derived neoepitopes, CD8+ T cells were isolated from splenocytes at day 35 of the experiment and tested for killing of MC38 tumor cells. About 80% of tumor cell lysis was observed with T cells from the vaccine-only group, confirming the potency and strong immunogenicity of RNA-LPX vaccination. In contrast, T cells from immunized mice treated with Dexa pre or post + pre were not capable of tumor cell lysis (Figure 4(d)). IFNγ secretion upon costimulation with MC38 cells was significantly reduced in Dexa-treated groups, further indicating that the antigen-specific T cells induced in lower numbers were qualitatively not capable of efficiently exerting a cytotoxic function (Figure 4(e)).
To investigate the impact of Dexa treatment on the therapeutic activity of RNA-LPX vaccination, BALB/c mice were injected i.v. with CT26 tumor cells for hematogenous tumor dissemination into the lung and repetitively treated with gp70 RNA-LPX vaccination (Figure 5(a)). Fifteen days after tumor inoculation, animals treated with irrelevant RNA-LPX presented numerous lung tumor nodules, whereas almost no tumor burden was visible in gp70-vaccinated animals (Figure 5(b), Supplementary Figure 3(a)). Pre-treatment with Dexa in CT26 tumor-bearing mice almost fully reversed the strong anti-tumor activity induced by gp70 RNA-LPX vaccination. In this model, vaccine efficacy was not at all impaired when Dexa was administered post-vaccination. Accordingly, Dexa pre- but not post-treatment significantly affected antigen-specific CD8+ T cells (Figure 5(c), Supplementary Figure 3(b)) and strongly reduced the gp70-specific CD8+ T cell to Treg ratio in the blood (Supplementary Figure 3(c)). Overall, a significant correlation between the fraction of gp70-specific CD8+ T cells in the blood and the number of tumor nodules per lung was found (Figure 5(d)).
To corroborate these findings in a second tumor model, C57BL/6 mice were inoculated subcutaneously with MC38 tumors. Ten previously described mutated tumor neoantigens, including Adpgk, served as vaccine targets.35 Mice received four weekly doses of the vaccine starting 11 days after tumor inoculation, with pre- or post-treatment with Dexa (Figure 5(e)). Tumor growth was significantly inhibited by the neoantigen vaccine, resulting in tumor regression in all mice (Figure 5(f)). Again, Dexa administration prior to vaccination reduced the efficacy of the vaccine significantly and only two of 10 mice showed pronounced tumor regression. Application of steroid treatment after vaccination partially restored anti-tumor activity of the vaccine. Neoantigen-specific CD8+ T cell responses against three out of 10 vaccine targets (Adpgk, M86, M143) were studied with a pool of MHC class I tetramers in the blood, spleen, and tumor of mice 5 days after the first vaccination. Particularly in the blood and spleen, the organs where RNA-LPX primed T cells reside during and shortly after priming, Dexa treatment strongly affected neoantigen-specific (Ki67+, gzmB+) T cell numbers (Figure 5(g), Supplementary Figure 3(d)). Effects were strongest in animals pre-medicated with Dexa, although differences between Dexa regimens were small. Variability of neoantigen-specific T cell numbers in the tumors was large and no significant effect of Dexa was observed (Supplementary Figure 3(e)).
Of note, Dexa pre-treatment with doses as low as 0.25 mg/kg had measurable, although weaker, effects on CD4+ T cells, CD8+ T cells as well as vaccine-induced gp70-specific CD8+ T cells (Supplementary Figure 4(a)). Concomitant administration of 0.25 mg/kg Dexa or Methylprednisolone (MPS), a shorter-acting steroid, however, did not significantly affect therapeutic activity, CD8+ T cell numbers, or the fraction of antigen-specific T cells in vaccinated MC38 tumor-bearing mice (Supplementary Figure 4(b,c)).
To investigate why Dexa treatment particularly prevented the expansion of antigen-specific T cells as opposed to naïve T cells (Figure 4(a), Supplementary Figure 3(b)), we further analyzed its impact on conventional DCs (cDC). C57BL/6 mice were injected with RNA-LPX encoding eGFP and treated with Dexa 3 hours before (Dexa pre), simultaneous with (Dexa sim), or 3 hours after (Dexa post) vaccination (Figure 6(a)). In line with Figure 2(a), proinflammatory cytokines such as IL6, IFNα, or IL18 measured 6 hours post-treatment in the serum were significantly reduced (Supplementary Figure 5(a,b)). IFNα levels were strongly decreased when Dexa was pre- or co-administered but were not reduced after Dexa post-treatment. Notably, IFNα has previously been shown to be crucial for RNA-LPX mediated T-cell priming.23 Dexa treatment significantly curtailed chemokine levels such as CCL4, CCL5, or CXCL10 known to be involved in T cell – DC interaction during priming38 (Supplementary Figure 5(a,b)). At 24 hours after vaccination, no reduction of T cells or B cells was observed within splenocytes yet (Supplementary Figure 5(c)). However, the costimulatory molecule CD80 was slightly reduced on plasmacytoid DCs (pDC), the main producers of IFNα after RNA-LPX treatment, with simultaneous or Dexa pre-treatment (Supplementary Figure 5(d)). In contrast to T and B cells, cDC numbers were significantly diminished in particular when Dexa was pre-administered (Figure 6(b)). Furthermore, the fraction of antigen expressing cDC, as represented by eGFP positivity as well as expression of CD80, was strongly reduced only or primarily when Dexa was administered together with or before vaccination (Figure 6(c left, d)). Antigen expression levels per cDC were not altered (Figure 6(c), right). To corroborate these findings, we further examined the effect of Dexa in human peripheral blood mononuclear cells (PBMC) following simultaneous treatment with RNA-LPX in vitro. Similarly, as in mice, stimulation of PBMC with Dexa resulted in diminished cytokine and chemokine levels and a reduced fraction of CD80+ cDC (Figure 6(e,f)).
In summary, our data show that Dexa co-medication ameliorates RNA-vaccine-mediated release of proinflammatory cytokines and chemokines, significantly affects the number of T lymphocytes, reduces the number and stimulatory potential of cDC and, most importantly, impairs expansion of antigen-specific T cells, leading to impaired effector cytokine release, tumor cell killing, and therapeutic activity. Impairment of vaccine-induced responses by Dexa is dose-dependent and most pronounced if Dexa is administered as pre-medication.
Discussion
In this study, we investigated the effect of Dexamethasone co-medication on the induction of antigen-specific CD8+ T cells by our RNA-LPX vaccine. We observed a large-scale lymphodepletion involving both CD8+ and CD4+ T cells, which is a well-known effect of GCS.39 The key finding of our study is that, in conjunction with a vaccine, antigen-specific T cells are more profoundly affected than naïve T cells. Moreover, vaccine-induced T cells primed in the presence of Dexa are of inferior cytotoxic functionality as shown by their impaired ability to produce effector molecules, their failure to lyse antigen-expressing tumor cells and reject tumors. This is in accordance with the higher dependence of primed T cells on proinflammatory cytokines and a costimulatory environment. We showed that especially Dexa pre-medicated mice respond to vaccination with a considerably lower number of antigen-specific T cells. This coincided with a preferential reduction of IFNα as well as cDC numbers, antigen uptake or translation, and costimulation when Dexa was administered 3 hours before vaccination. Application of Dexa subsequent to the vaccine has a similar but less profound effect.
Potentially contributing to this outcome is that Dexa pre-medication impedes the immediate and transient release of proinflammatory cytokines and chemokines, which we consider relevant for the intrinsic adjuvanticity of our vaccine. For IL12p70 and CXCL10, this effect is cumulative and suppression is stronger after the second vaccination under the Dexa post + pre regimen. Dexa is a long-acting GCS with a half-life of elimination in the range of about 2.5 hours (1 mg/kg dose) in rodents40 and about 3.5 hours (2 mg/kg dose) in humans,41 and therefore the blood concentration in animals pre-treated with Dexa is expected to be still high at the time the vaccine is administered. We reported previously that RNA-LPX induced cytokine release is pulsatile and the decisive immunomodulatory events for induction of antigen-specific T-cell responses are immediate.23 and unpublished results Briefly, within 1 hour after i.v. injection, antigen-encoding RNA is internalized by cDC, pDC, and macrophages in the red pulp and marginal zone of the spleen. DC migrate into the T-cell zones in the white pulp and undergo full maturation with upregulation of costimulatory molecules such as CD86 within the next 2–3 hours.23 IFNα released by pDC and macrophages due to TLR7 triggering by the single-stranded RNA is detectable in the serum of mice within 1–2 hours, which results in transcription of a range of proinflammatory molecules. We showed previously that interference at this early phase, e.g. by inhibition of type I IFN receptor signaling, prevents RNA-mediated adjuvanticity during priming, leads to severe impairment of CD8+ T cell differentiation into KLRG1+, cytotoxic effector T cells, and markedly dampens anti-tumoral activity.23 Altogether, these findings explain our observations with the pre and pre + post regimen.
In contrast, at the time post-administered Dexa was dosed, the relevant immediate immune modulation by the preceding vaccine had most likely already taken place and relevant downstream effects were not compromised by Dexa post-treatment.
Another finding potentially contributing to the impaired antigen-specific T-cell immune response relates to Tregs. As observed for the overall CD4+ T-cell compartment, numbers of CD4+ CD25+ FoxP3+ Tregs in vaccinated mice with pre and pre + post Dexa treatment were lower as compared to vaccine-only controls. However, the fraction of Tregs within the CD4+ T cell population was significantly higher upon Dexa pre-medication, indicating differential sensitivity of CD4+ T-cell subpopulations to this immune-suppressive treatment. GCS are known to have selective effects on Tregs over conventional T cells in mice and humans.42 GCS have been previously described as causing apoptosis in proinflammatory T cells while aiding survival of Tregs.42 This may be attributed to higher proliferation of Tregs in the lymphopenic environment due to a higher affinity for IL2.43 In addition, Tregs were shown to express higher levels of anti-apoptotic Bcl-2 than conventional CD4+ T cells, rendering them more resistant to Dexa-induced cell death.43 A possible additional mechanism may be that TCR-mediated activation of CD4+ T cells in the presence of Dexa has been reported to increase the proportion of IL10-producing cells.44
Another interesting observation is that there are differences between the vaccine antigens. T cells specific for the weaker Reps1 antigen seem to be more sensitive to Dexa than those specific for the stronger antigens gp70 and Adpgk. Dexa post-treatment appears to strongly impair priming and expansion kinetics of Reps1-specific CD8+ T cells, whereas it does only moderately or not at all interfere with the stronger antigens. One may speculate that higher-affinity T cells may be less dependent on proinflammatory cytokines such as IL12,45 which has been shown to augment TCR signaling.46 In addition, T cell survival is directly modulated by TCR affinity, with high-affinity clones exhibiting a lower death rate when proliferating.47 The extent of progressive upregulation and accumulation of Bcl-2 and Bcl-xL during stimulation may depend on the strength of the TCR stimulus,47,48 possibly steeling high-affinity T cells against Dexa-mediated apoptosis.
A recent paper also reported that simultaneous, but not delayed administration of corticosteroids impaired neo-antigen specific anti-tumor responses augmented by checkpoint blockade.15 Reduced CD8+ T-cell proliferation and lack of protection upon rechallenge with tumors lacking the neoantigen were observed. Here, corticosteroids were shown to decrease low- but not high-affinity memory T cells by suppressing fatty acid metabolism essential for memory T cells.
In conclusion, our findings in conjunction with previous data demonstrate that by impacting mechanisms on various levels, corticosteroids, particularly when administered as pre-medication, impair vaccine-induced responses. Our studies indicate that low doses and shorter-acting GCS given preferably several hours or days after vaccination are desirable in clinical practice. Further in-depth analysis of the immunological effects of Dexa alternatives such as short-acting corticosteroids is warranted. The data presented here can inform the development of cancer vaccines and their integration in clinical practice.
Supplementary Material
Acknowledgments
We thank Andres Paler Martinez, Hanna Junginger, Yvonne Feuchter, Bernadette Jesionek, and Nele Brüne for technical assistance.
Abbreviations
- Adpgk
ADP Dependent Glucokinase
- CCL
CC-chemokine ligand
- cDC
Conventional dendritic cell
- CXCL
C-X-C motif chemokine
- COX-2
Cyclooxygenase-2
- Dexa
Dexamethasone
- eGFP
Enhanced green fluorescent protein
- FI
Fluorescence intensity
- GCS
Glucocorticosteroids
- gp70
Murine leukemia virus envelope glycoprotein 70
- i.p.
Intraperitoneal
- i.v.
Intraveneous
- IFN
Interferon
- IL
Interleukin
- MHC
Major histocompatibility complex
- n.d.
Not detectable
- PBS
Phosphate-buffered saline
- pDC
Plasmacytoid dendritic cells
- Reps1
RALBP1 Associated Eps Domain Containing 1
- RNA
Ribonucleic acid
- s.c.
Subcutaneous
- sim
Simultaneous
- TNF
Tumor necrosis factor
- Tregs
Regulatory T cells
- UTR
Untranslated region
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Declaration of interest statement
Some of the authors are inventors on patents and patent applications, which cover RNA-LPX vaccination. Some of the authors have securities from BioNTech or Genentech.
Supplementary material
Supplemental data for this article can be accessed Publisher website.
References
- 1.Nicolaides NC, Pavlaki AN, Maria Alexandra MA, Chrousos GP.. Glucocorticoid Therapy and Adrenal Suppression. Endotext. 2000. [accessed 2020 April 27]. www.endotext.org
- 2.Meijsing SH. Mechanisms of Glucocorticoid-Regulated Gene Transcription. Adv Exp Med Biol. 2015;872:59–12. doi:10.1007/978-1-4939-2895-8_3. [DOI] [PubMed] [Google Scholar]
- 3.Petta I, Dejager L, Ballegeer M, Lievens S, Tavernier J, De BK, Libert C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol Mol Biol Rev. 2016;80(2):495–522. doi: 10.1128/MMBR.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4(10):525–533. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- 5.Shih A, Jackson KC. Role of corticosteroids in palliative care. J Pain Palliat Care Pharmacother. 2007;21(4):69–76. doi: 10.1080/J354v21n04_14. [DOI] [PubMed] [Google Scholar]
- 6.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 7.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019:1. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 8.Vormehr M, Türeci Ö, Sahin U. Harnessing Tumor Mutations for Truly Individualized Cancer Vaccines. Annu Rev Med. 2019;70(1):395–407. doi: 10.1146/annurev-med-042617-101816. [DOI] [PubMed] [Google Scholar]
- 9.Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer. 2015;113(11):1642. doi: 10.1038/bjc.2015.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klement RJ, Champ CE. Corticosteroids compromise survival in glioblastoma in part through their elevation of blood glucose levels. Brain. 2017;140(3):e16. doi: 10.1093/brain/aww324. [DOI] [PubMed] [Google Scholar]
- 11.Shields LBE, Shelton BJ, Shearer AJ, Chen L, Sun DA, Parsons S, Bourne TD, LaRocca R, Spalding AC. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat Oncol. 2015;10(1):222. doi: 10.1186/s13014-015-0527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M, Chan TA, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139(Pt 5):1458–1471. doi: 10.1093/brain/aww046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, Martínez-Bernal G, Ferrara R, Lai WV, Hendriks LEL, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36(28):2872–2878. doi: 10.1200/JCO.2018.79.0006. [DOI] [PubMed] [Google Scholar]
- 14.Maxwell R, Luksik AS, Garzon-Muvdi T, Hung AL, Kim ES, Wu A, Xia Y, Belcaid Z, Gorelick N, Choi J, et al. Contrasting impact of corticosteroids on anti-PD-1 immunotherapy efficacy for tumor histologies located within or outside the central nervous system. Oncoimmunology. 2018;7(12):e1500108. doi: 10.1080/2162402X.2018.1500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokunaga A, Sugiyama D, Maeda Y, Warner AB, Panageas KS, Ito S, Togashi Y, Sakai C, Wolchok JD, Nishikawa H. Selective inhibition of low-affinity memory CD8+ T cells by corticosteroids. J Exp Med. 2019. September 19;216(12):2701–2713. jem.20190738. doi: 10.1084/jem.20190738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Xia L, Chen J, Zhang S, Martin V, Li Q, S L, J C, J C, Lu M, et al. Stress–glucocorticoid–TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med. 2019;25(9):1428–1441. doi: 10.1038/s41591-019-0566-4. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Türeci O, Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17(8):961–971. doi: 10.1038/gt.2010.52. [DOI] [PubMed] [Google Scholar]
- 18.Strenkowska M, Grzela R, Majewski M, Wnek K, Kowalska J, Lukaszewicz M, Zuberek J, Darzynkiewicz E, Kuhn AN, Sahin U, et al. Cap analogs modified with 1,2-dithiodiphosphate moiety protect mRNA from decapping and enhance its translational potential. Nucleic Acids Res. 2016;gkw896. doi: 10.1093/nar/gkw896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warminski M, Kowalska J, Buck J, Zuberek J, Lukaszewicz M, Nicola C, Kuhn AN, Sahin U, Darzynkiewicz E, Jemielity J. The synthesis of isopropylidene mRNA cap analogs modified with phosphorothioate moiety and their evaluation as promoters of mRNA translation. Bioorg Med Chem Lett. 2013;23(13):3753–3758. doi: 10.1016/j.bmcl.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, LM K, Fesser S, Diken M, Löwer M, Vallazza B, Beissert T, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3ʹ UTRs identified by cellular library screening. Mol Ther. 2019;27(4):824–836. doi: 10.1016/j.ymthe.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Türeci O, Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- 22.Kreiter S, Selmi A, Diken M, Türeci Ö, Sahin U, Sebastian M, Osterloh P, Schild H, Huber C, Türeci O, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(5):309–318. 180/1/309 [pii]. doi: 10.4049/jimmunol.180.1.309. [DOI] [PubMed] [Google Scholar]
- 23.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 24.Kranz LM, Diken M, Fritz D, Haas H, Holzmann M, Meng M, Reuter K, Roth R, Selmi A, Kreiter S, et al. Abstract: Novel RNA-lipoplexes with immunostimulatory and targeting properties induce potent anti-tumoral immunity. 43rd Annu. Meet. Ger. Soc. Immunol. (DGfI), Mainz. 2013. [Google Scholar]
- 25.Kranz LM, Diken M, Fritz D, Haas H, Holzmann M, Meng M, Reuter K, Roth R, Selmi A, Kreiter S, et al. Abstract: Characterization of the IFNα response upon systemic administration of targeted mRNA vaccines. 12th Annu. Meet. Cancer Immunother. (CIMT), Mainz. 2014. [Google Scholar]
- 26.Jabulowsky RA, Loquai C, Diken M, Kranz LM, Haas H, Attig S, Britten CM, Buck J, Derhovanessian E, Diekmann J, et al. Abstract B041: A novel nanoparticular formulated tetravalent RNA cancer vaccine for treatment of patients with malignant melanoma. Cancer Immunol Res. 2016;4(1 Supplement):B041–B041. doi:10.1158/2326-6074.CRICIMTEATIAACR15- B041. [Google Scholar]
- 27.Jabulowsky RA, Loquai C, Diken M, Kranz LM, Haas H, Attig S, Bidmon N, Buck J, Derhovanessian E, Diekmann J, et al. Abstract CT032: A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles for potent cancer immunotherapy in patients with malignant melanoma. Cancer Res. 2016;76(14 Supplement):CT032–CT032. doi: 10.1158/1538-7445.AM2016-CT032. [DOI] [Google Scholar]
- 28.Jabulowsky RA, Loquai C, Utikal J, Hassel J, Kaufmann R, Derhovanessian E, Diken M, Kranz LM, Haas H, Attig S, et al. Abstract CT034: A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles for potent melanoma immunotherapy. Cancer Res. 2017;77(13 Supplement):CT034–CT034. doi:10.1158/1538-7445.AM2017- CT034. [Google Scholar]
- 29.Heesen L, Jabulowsky R, Loquai C, Utikal J, Gebhardt C, Hassel J, Kaufmann R, Pinter A, Derhovanessian E, Diken M, et al. 49PA first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for potent melanoma immunotherapy. Ann Oncol. 2017;28(suppl_11):xi14–xi15. doi: 10.1093/annonc/mdx711.030. [DOI] [Google Scholar]
- 30.Jabulowsky RA, Loquai C, Mitzel-Rink H, Utikal J, Gebhardt C, Hassel JC, Kaufmann R, Pinter A, Derhovanessian E, Anft C, et al. Abstract CT156: A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for immunotherapy of malignant melanoma. Cancer Res. 2018;78(13 Supplement):CT156–CT156. doi:10.1158/1538-7445.AM2018- CT156. [Google Scholar]
- 31.Pektor S, Hilscher L, Walzer KC, Miederer I, Bausbacher N, Loquai C, Schreckenberger M, Sahin U, Diken M, Miederer M. In vivo imaging of the immune response upon systemic RNA cancer vaccination by FDG-PET. EJNMMI Res. 2018;8(1):80. doi: 10.1186/s13550-018-0435-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13(4):529–538. doi:10.1016/S1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 34.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 35.Capietto A-H, Jhunjhunwala S, Pollock SB, Lupardus P, Wong J, Hänsch L, Cevallos J, Chestnut Y, Fernandez A, Lounsbury N, et al. Mutation position is an important determinant for predicting cancer neoantigens. J Exp Med. 2020;217(4):4. doi: 10.1084/jem.20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolton G, Tungatt K, Lloyd A, Bianchi V, SM T, Trimby A, CJ H, Donia M, AJ G, DK C, et al. More tricks with tetramers: a practical guide to staining T cells with peptide-MHC multimers. Immunology. 2015;146(1):11–22. doi: 10.1111/imm.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15(1):190. doi: 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32(1):659–702. doi:10.1146/annurev- immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 39.Ashwell JD, Lu FW, Vacchio MS. Glucocorticoids in T cell development and function*. Annu Rev Immunol. 2000;18(1):309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 40.Samtani MN, Jusko WJ. Comparison of dexamethasone pharmacokinetics in female rats after intravenous and intramuscular administration. Biopharm Drug Dispos. 2005;26(3):85–91. doi: 10.1002/bdd.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohdewald P, Möllmann H, Barth J, Rehder J, Derendorf H. Pharmacokinetics of dexamethasone and its phosphate ester. Biopharm Drug Dispos. 1987;8(3):205–212. doi: 10.1002/bdd.2510080302. [DOI] [PubMed] [Google Scholar]
- 42.Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, Heckmann N, Lühder F, Reichardt HM, Arck PC, Gold SM, et al. Glucocorticoid receptor in T cells mediates protection from autoimmunity in pregnancy. Proc Natl Acad Sci U S A. 2017;114(2):E181–E190. doi: 10.1073/pnas.1617115114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Murakami T, Oppenheim JJ, Howard OMZ. Differential response of murine CD4+CD25+ and CD4+CD25- T cells to dexamethasone-induced cell death. Eur J Immunol. 2004;34(3):859–869. doi: 10.1002/eji.200324506. [DOI] [PubMed] [Google Scholar]
- 44.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, de Waal-malefyt R, Coffman RL, Hawrylowicz CM, O’Garra A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195(5):603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, Frucht DM, Chrousos GP, O’Shea JJ. Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol. 2000;164(4):1768–1774. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 46.Richer MJ, Nolz JC, Harty JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity. 2013;38(1):140–152. doi: 10.1016/j.immuni.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hommel M, Hodgkin PD. TCR affinity promotes CD8+ T cell expansion by regulating survival. J Immunol. 2007;179(4):2250–2260. doi: 10.4049/jimmunol.179.4.2250. [DOI] [PubMed] [Google Scholar]
- 48.Broome HE, Dargan CM, Krajewski S, Reed JC. Expression of Bcl-2, Bcl-x, and Bax after T cell activation and IL-2 withdrawal. J Immunol. 1995;155:2311–2317. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Nicolaides NC, Pavlaki AN, Maria Alexandra MA, Chrousos GP.. Glucocorticoid Therapy and Adrenal Suppression. Endotext. 2000. [accessed 2020 April 27]. www.endotext.org