

ABSTRACT
Currently, human glioma tumors are mostly modeled in immunodeficient recipients; however, lack of interactions with adaptive immune system is a serious flaw, particularly in the era when immunotherapies dominate treatment strategies. Our group was the first to successfully establish the orthotopic transplantation of human glioblastoma (GBM) in immunocompetent mice by inducing immunological tolerance using a short-term, systemic costimulation blockade strategy (CTLA-4-Ig and MR1). In this study, we further validated the feasibility of this method by modeling pediatric diffuse intrinsic pontine glioma (DIPG) and two types of adult GBM (GBM1, GBM551), in mice with intact immune systems and immunodeficient mice. We found that all three glioma models were successfully established, with distinct difference in tumor growth patterns and morphologies, after orthotopic xenotransplantation in tolerance-induced immunocompetent mice. Long-lasting tolerance that is maintained for up to nearly 200 d in GBM551 confirmed the robustness of this model. Moreover, we found that tumors in immunocompetent mice displayed features more similar to the clinical pathophysiology found in glioma patients, characterized by inflammatory infiltration and strong neovascularization, as compared with tumors in immunodeficient mice. In summary, we have validated the robustness of the costimulatory blockade strategy for tumor modeling and successfully established three human glioma models including the pediatric DIPG whose preclinical study is particularly thwarted by the lack of proper animal models.
KEYWORDS: Glioma, brain tumor, costimulation blockade, immunocompetent, tolerance
Introduction
Malignant gliomas, including glioblastoma (GBM; synonymous with the formerly used term ‘glioblastoma multiforme’) and pediatric diffuse intrinsic pontine glioma (DIPG), have high morbidity and mortality rate. GBM is the most frequently occurring primary central nervous system malignancies in adults.1–3 Decades of concerted effort brought some progress and improvement of 3-y survival rate of GBM patients from 4.4% in the nineties to 10.3% one decade later.4 The prognosis of DIPG in children is even worse: the 3-y survival rate is only 4.3%.5,6 Continuously dismal clinical outcome makes glioma an urgent subject of cancer research. Recently, there was tremendous progress in oncology driven by immunotherapies. Benefits from these strategies are observed across oncology except brain tumors.7 Clearly, the brain with its barriers is a challenging target for all classes of drugs including immunotherapeutics.
The tumor microenvironment plays a large role in cancer progression and therapy responses.8 The glioma model established by human-derived xenograft orthotopic transplantation mostly retains the principal histological and genetic characteristics of their donor tumor, which represents the optimal model in preclinical research.9 To date, immunodeficient 10–12 and immunosuppressed13 animals were typically used in these tumor transplantation experiments. However, due to the important role of immune system in the formation and establishment of tumors, complete elimination of adaptive immunity is a significant drawback.14,15 The interaction between the tumor and the unique immune environment of the central nervous system should be considered when pursuing immune-based therapeutic approaches. Using immunocompetent animal can retain this immune environment, while the lack of effective tumor modeling strategy limited its application. One study reports spontaneous immune tolerance against human GBM in immunocompetent mice. However, no specific intervention was introduced, and the mechanisms for the absence of species-dependent host vs. graft rejection were unclear in their model.16 Humanized mice have been used with some success; however, complexity, cost, and complications such as graft versus host response are serious limitations.17,18
A central element of adaptive immune response is T-cell activation, which has been extensively studied and modulated in organ transplantation and autoimmunity diseases.19,20 Full T-cell activation requires the initial recognition of a specific antigen by a T-cell receptor (TCR), followed by a second costimulatory signal, such as the binding of CD80/CD86 or CD40 on the surface of antigen-presenting cells (APCs) to the CD28 or CD154 receptor on T cells.21,22 Two days after activation, T cells express cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which blocks the CD80/CD86–CD28 interaction and thus functions as a negative regulator of T cell-mediated immune responses.23,24 In addition, the anti-CD154 antibody (MR1) recognizes CD154 and prevents its interaction with CD40 to further limit T-cell stimulation.25
By applying a four-time-point treatment scheme of the selective T-cell costimulation inhibitors (CTLA-4-Ig and MR1) in previous studies, we have successfully not only prevented the rejection of allogeneic glial-restricted progenitors from immunocompetent mouse brains26 but also successfully established the orthotopic transplantation of human GBM1 in immunocompetent mice.27 Here, we further characterized and validated this tolerance induction approach using three distinct brain tumor models: GBM1, DIPG, and GBM551. All these xenograft models in immunocompetent mice recapitulate the histopathological features of corresponding human diseases, and long-term tumor growth is maintained for up to 200 d until the general health of animals deteriorates due to neoplastic progress. Hereby, we provide a new robust and universal approach for modeling brain tumor model in the context of intact immunity that we believe will find broad applications in neurooncology, particularly for studying immunotherapeutics.
Materials and methods
Cell culture
Human GBM551-Luc cell was generously provided by Dr Mihoko Kai (Johns Hopkins University). The human GBM1-Luc cell was kindly provided by Dr Charles Eberhart (Johns Hopkins University).28 Human DIPG-Luc cell was given by Dr Angel Carcaboso (Sant Joan de Deu Hospital).29 The culture medium recipes of all these three cell lines are shown in Supplemental Table 1. All cells were suspension cultured as neurospheres in a humidified atmosphere of 5% CO2 at 37°C.
Quantification of proliferative activity
Cells were seeded and cultured in six-well plates (2 × 105 cells/well). Every other day, cells in one well were collected and dissociated into single cells, incubated with Acridine Orange/Propidium lodide Stain (Logos Biosystems), and then counted with Luna™ Dual Fluorescence Cell Counter (Logos Biosystems). All experiments were conducted in triplicate.
Tumor transplantation
All animal procedures were approved by the Johns Hopkins University Animal Care and Use Committee. Prior to transplantation, GBM1, GBM551, and DIPG neurospheres were harvested, dissociated into single cells, and suspended in phosphate buffered saline (PBS) at a final concentration of 1 × 105/μL. The total 2 × 105 cells were loaded into a 10-μL Hamilton syringe with an attached 31-gauge needle and injected at a rate of 1 μL/min into the right striatum (anteroposterior = 1.0 mm; mediolateral = 2.0 mm; dorsoventral = 2.5 mm) of C57BL/6 J mice (8–10 weeks, n = 5 for each cell line) and SCID mice (8–10 weeks, n = 5 for each cell line, The Jackson Laboratory) through a stereotaxic injector (Stoelting). After the injection was complete, the needle was kept in place for 2 minutes and then withdrawn to avoid backflow of the injected cells through the needle tract.
For T-cell costimulatory blockade experiments, hamster anti-mouse CD154mAb (MR1, BioXcell) and CTLA-4-Ig (Abatacept, Bristol-Myers Squibb) were administered to C57BL/6 J mice (500 μg each) intraperitoneally (i.p.) on d 0, 2, 4, and 6 after tumor inoculation. Mice were monitored twice a week for weight changes and neurological symptoms.
Bioluminescence imaging
For bioluminescence imaging (BLI) of tumor growth rate, mice were injected i.p. with 150 μL D-luciferin substrate (30 mg/mL, Gold Biotechnology), and images were acquired 5–15 minutes after injection by IVIS Spectrum In Vivo Imaging System (PerkinElmer). BLI was quantified (photon flux (p/s)) as previously described.30 Imaging began on d 1 post implantation and then done weekly.
MR imaging
Magnetic resonance imaging (MRI) was performed before animal sacrifice, as previously reported.27 During the MRI, animals were anesthetized using 1–2% isoflurane. Mice were placed on a water-heated animal bed equipped with temperature and respiratory control. Respiration was monitored and maintained at 30–60/min. All MRI experiments were performed on a horizontal bore 11.7 T Bruker Biospec system (Bruker). Baseline T2 (TR/TE = 2,500/30 ms) and T1 (TR/TE = 350/6.7 ms)-weighted images of the brain were acquired. Gadolinium (100 μL) was injected i.p. for contrast-enhanced T1 scans, and T1 post-gadolinium images were acquired.
Immunohistology and immunofluorescence
Mice were sacrificed when they deteriorated neurologically (failure to ambulate, lethargy) or after weight loss >20%.31 Following transcardial perfusion with 5% sucrose and 4% paraformaldehyde in PBS, brain tissues were post-fixed with 4% paraformaldehyde overnight at 4°C and then transferred to 30% sucrose for dehydration. Brains were cryosectioned into 30-μm-thick slices. Hematoxylin and eosin (H&E) staining was performed for histology. For immunofluorescence, sections were blocked using 3% bovine serum albumin (BSA) in 1 × tris buffered saline (TBS) plus with 0.1% Triton and incubated overnight at 4°C with primary antibodies including human-specific Nuclei (HuNu, 1:250; Cat. MAB1281, Millipore); Stem 121 (1:500; Cat. Y40410, Takara); Iba1 (1:250; Cat. 019-19741, Wako); CD45 (1:250; Cat. ab10558, Abcam); CD3 (1:200; Cat. MAB4841, R&D Systems); and Collagen IV (1:300; Cat. ab6586, Abcam). Sections were then incubated for 2 hours with either Alexa-488 or Alexa-594 (1:250; Invitrogen) secondary antibodies at room temperature and mounted with VECTASHIELD® Antifade Mounting Medium with DAPI (Vector Laboratories).
Data processing and statistical analysis
The overall mouse survival time was calculated and expressed by the Kaplan–Meier curve, and the statistical difference was analyzed by log-rank tests. Quantitation of inflammation was based on counting cells immunoreactive to Iba1, CD45, and CD3 per field of view using Image J (Version 1.52p, USA). For the quantitation of tumor diffusion, human cells were detected in the contralateral hemisphere based on immunoreactivity toward the human nuclear antigen. This measure included the contralateral hemisphere area occupied by tumor cells and the farthest distance of tumor cells from tumor center, both done using Image J. Quantitation of blood–brain barrier (BBB) leakage was based on the analysis of the Gd-enhancement area for each mouse. Shapiro–Wilk test was used for normal distribution analysis of data. For normally distributed data, those results were presented as mean ± standard deviation (SD), and an independent t-test was used when comparing two groups. One-way analysis of variance (ANOVA) with least significant difference (LSD) post hoc was used when comparing three groups. Data without normal distribution were presented as median with interquartile range (IQR). Mann–Whitney U test was used when comparing two groups, and Kruskal–Wallis test was used when comparing three groups. All statistical analyses were performed using SPSS software package (Version 22.0, USA). P <0.05 was considered to be statistically significant.
Results
In vitro characterizations of glioma cell lines
DIPG, GBM1, and GBM551 cells were seeded as single-cell suspensions in non-adherent six-well plates, and their proliferative activity and growth characteristics were assessed over 10 d. All cell types grew forming neurospheres, with DIPG (Figure 1a) and GBM551 (Figure 1b) forming compact spheres, while GBM1 exhibited less compacted structures (Figure 1c). When analyzing the growth rate, we found that GBM551 cells were the least proliferative and grew significantly slower than GBM1 (p < 0.05). The GBM1 cells’ doubling time (1.3 d) was shorter than DIPG (1.4 d) or GBM551 (2.7 d). All lines, but particularly DIPG and GBM551, grew slowly during the initial 2 d, and once they established the spheres, the proliferation accelerated (Figure 1d).
Figure 1.
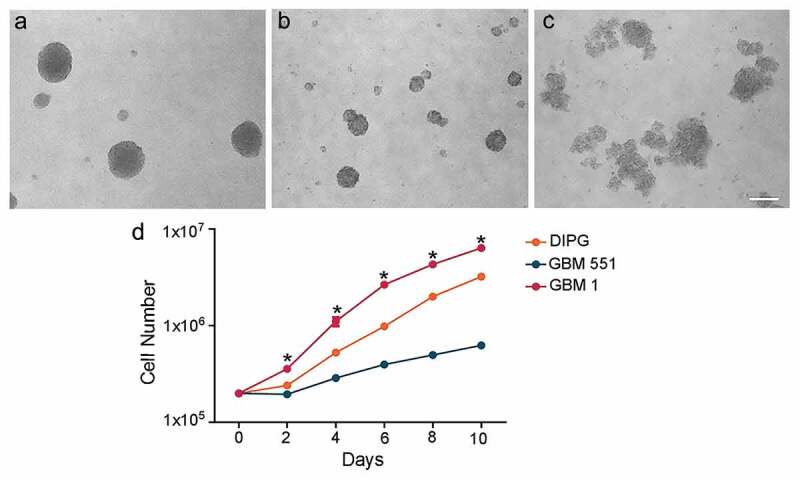
Different in vitro growth patterns and rates in DIPG, GBM1, and GBM551 cells. D 6 in vitro cell culture showed that DIPG (a) and GBM551 (b) exhibited dense spheres, while GBM1 (c) exhibited less compact structures. The scale bar is 200 μm. (d) Cells counting for DIPG, GBM551, and GBM1 at 0, 2, 4, 6, 8, and 10 d after cell seeding (n = 3 wells/condition), mean ± SD, *p < 0.05 compared with GBM551, Kruskal–Wallis test.
Characterization of growth rate of human glioma xenografts in mice
The proliferative activity for individual tumor cell lines observed in vitro was also reflected in the growth rate after inoculation in immune tolerance-induced mice. Adult C57BL/6J mice implanted with tumor cells received the CD154mAb and CTLA-4-Ig injection at d 0, 2, 4, and 6 after tumor inoculation (hereafter termed immunocompetent mice), and adult SCID mice implanted with tumor cells were used as controls (hereafter termed immunodeficient mice). All three tested tumor cell lines successfully established brain tumors as revealed by in vivo BLI (Figure 2). Longitudinal BLI demonstrated that growth rates were different from each cell line, while no significant difference was found between immunocompetent and immunodeficient mice in each cell line (p > 0.05), except for GBM551 which showed a decreased BLI signal in immunocompetent mice as compared with immunodeficient mice at 154 d after tumor inoculation (Figure 2b, p < 0.05). GBM1 displayed an aggressive growth pattern with accelerated progression starting 7 d after inoculation and asymptomatic post-grafting period of about 42 d. DIPG exhibited a relatively slow growth pattern compared to GBM1, with animals deteriorating after 75 d. GBM551 was proved to be characterized by the slowest growth (Figure 2b) and asymptomatic period lasting 150 d. Moreover, we found that two of the GBM551-bearing mice in the immunocompetent group began rejecting tumor starting 140 d after inoculation, as demonstrated by the gradual reduction in bioluminescence signal (Supp. Fig. 1A).
Figure 2.
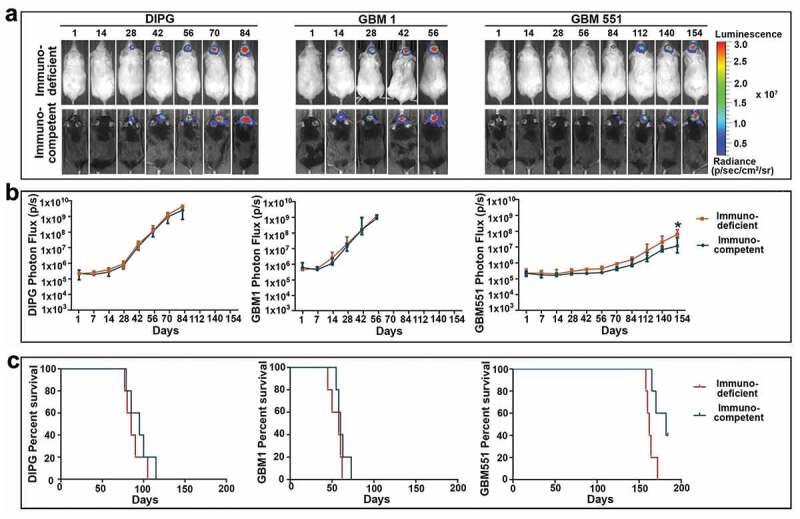
T-cell costimulation blockade facilitates human DIPG, GBM1, and GBM551 tumor cell engraftment in immunocompetent mice. (a) Representative bioluminescent imagings of immunodeficient and immunocompetent mice inoculated with human DIPG, GBM1, and GBM551 xenografts. (b) Longitudinal bioluminescence intensity of each cell line showed stable xenograft survival in both immunocompetent and immunodeficient mice (n = 5 animals/group). Median with IQR, *p < 0.05 compared with immunocompetent mice, Mann–Whitney U test. (c) Kaplan-Meyer curve analysis of survival for immunodeficient and immunocompetent mice (n = 5 animals/group). Two of the five GBM551 mice in the immunocompetent group were sacrificed at 184 d.
Both immunocompetent and immunodeficient recipients displayed similar tumor burdens. The Kaplan–Meyer curve analysis showed that there was no survival difference between immunocompetent and immunodeficient mice (p > 0.05) (Figure 2c). However, the substantial difference was found between these three cell lines, as GBM1 mice have the shortest survival time with a median of 60 d (IQR from 56.5 to 68 d) in immunocompetent group vs. 58 d (IQR from 47.5 to 61 d) in immunodeficient group; DIPG mice have a median survival of 95 d (IQR from 82 to 107.5 d) in immunocompetent group and 85 d (IQR from 78.5 to 97.5 d) in immunodeficient group, while GBM551 mice have the longest survival time, with a median of 170 d (IQR from 165 to 182 d) in immunocompetent group and 162 d (IQR from 159 to 168 d) in immunodeficient group (p < 0.01 between each of these three tumor types; Figure 2c).
Characterization of tumor growth patterns of DIPG, GBM1, and GBM551
Given the different growth rates between these three tumor cell lines, we further examined the tumor growth pattern with MRI and histology staining, focusing on the brain–tumor interface and infiltrative nature. Anatomical T2-weighted imaging revealed that DIPG, GBM1, and GBM551 have different growth patterns and morphologies (Figure 3a). DIPG exhibited highly infiltrative growth with practically no distinguishable tumor mass on T2 scan, while GBM1 and GBM551 showed more restricted growth with large mass effect, visible as compression of the ipsilateral normal brain tissue and midline shift.
Figure 3.
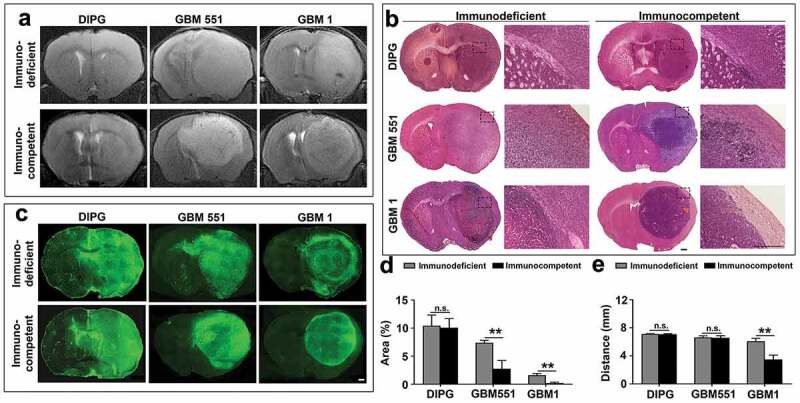
Identification of implanted xenografts and their morphologies with MRI and histology. Anatomical T2-weighted MR imaging (a), H & E staining (b), and Human Nuclear Antigen (HuNu) immunostaining (c) showed the morphologies of human DIPG, GBM551, and GBM1 tumors in immunodeficient and immunocompetent mice. The scale bars are 500 μm. (d) Quantification of contralateral hemisphere HuNu+ area/total contralateral hemisphere area and (e) the farthest distance of tumor cells from tumor center (n = 9 slices from three animals each group). Mean ± SD, **p < 0.01, n.s. is no significant difference, independent t-test.
Consistent with the T2 MRI, H&E staining confirmed the differences in tumor morphologies (Figure 3b). For DIPG, neither the tumor mass nor boundary was found, while GBM551 exhibited a circumscribed growth pattern with a fuzzy boundary. The GBM1 was distinguished by a clear tumor boundary from normal brain tissue. Normal brain structures were destroyed by GBM1 and GBM551 tumors, which was not the case for DIPG. Moreover, after comparing with immunodeficient mice, we found that GBM551 and GBM1 tumors grown in immunocompetent mice have clearer boundaries. No difference was found between DIPG in immunocompetent and immunodeficient animals.
Confirmation of the human origin of the tumors and tumor distribution was further performed by immunostaining with antibodies against the human-specific nuclear antigen (HuNu). All three cell lines (DIPG, GBM551, and GBM1) in immunocompetent and immunodeficient mice showed strong staining of HuNu (Figure 3c). By quantification analysis of the contralateral hemisphere tumor area (Figure 3d) and the farthest tumor infiltration distance (Figure 3e), we found a more restrictive growth in immunocompetent mice of GBM tumors as compared with immunodeficient mice. The percentage of HuNu+ cells in contralateral hemisphere of GBM tumors in immunodeficient recipients (GBM1: 1.60 ± 0.31%; GBM551: 7.39 ± 0.44%) were higher than in immunocompetent recipients (GBM1: 0.19 ± 0.16%; GBM551: 2.75 ± 1.45%; p < 0.01), but not in DIPG tumor (immunodeficient: 10.43 ± 1.89%; immunocompetent: 10.07 ± 1.62%). Moreover, the infiltration distance of GBM1 tumor in immunodeficient mice (6.09 ± 0.40 mm)was farther than in immunocompetent mice (3.47 ± 0.63 mm; p < 0.01), while no difference was found in DIPG (immunodeficient: 7.10 ± 0.09 mm; immunocompetent: 7.08 ± 0.07 mm) and GBM551 (immunodeficient: 6.61 ± 0.24 mm; immunocompetent: 6.57 ± 0.30 mm) tumors.
Infiltration by inflammatory cells in tumors grown in immunocompetent mice
We have previously shown that xenograft rejection could be prevented by inhibiting T-cell costimulation signaling pathway.27 For further analysis of differences in immune cell recruitment to the different tumor types, we performed immunostaining of inflammatory cells. We looked at the status of microglial activation (Iba1), the whole leukocyte population (CD45), and specifically T lymphocyte (CD3) infiltration (Figure 4). While all groups showed microglial activation (Iba1-positive), no significant difference was found in the density of microglia between immunodeficient and immunocompetent mice in any of the tumor groups (Figure 4b, p > 0.05).
Figure 4.
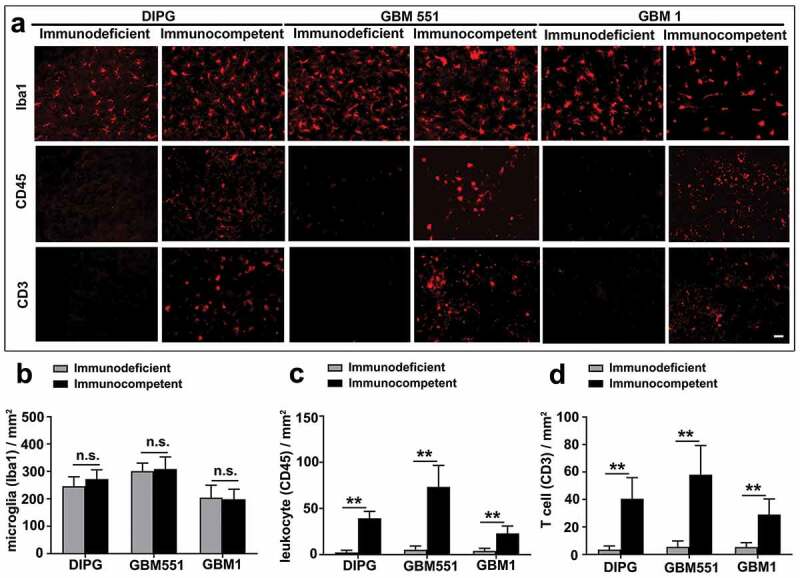
Inflammation infiltration in DIPG, GBM1, and GBM551 tumors in immunocompetent mice. (a) Xenografts in immunocompetent mice engaged an innate and adaptive immune cell surveillance, as revealed by microglia (Iba1), leukocyte (CD45), and T-cell (CD3) infiltration. Immunodeficient mice showed only microglial activation. The scale bar is 50 μm. (b) Quantification of histological assessments for Iba1 immunostaining showed microglial activation in both immunocompetent and immunodeficient mice. Quantification of CD45 (c) and CD3 (d) immunostaining showed leukocyte and T-cell infiltration only in immunocompetent mice. N = 9 slices from three animals/group. Mean ± SD, *p < 0.05, **p < 0.01, n.s. is no significant difference, independent t-test.
Interestingly, we detected a significant increase in the number of leukocytes (CD45) (Figure 4c) and T cells (CD3) (Figure 4d) in immunocompetent mice in comparison to immunodeficient mice (17/14/6 times for CD45, 12/11/6 times for CD3 in DIPG/GBM551/GBM1 tumor, respectively, p < 0.01), indicating active innate and adaptive immune effectors in the immunocompetent mice. It is likely that the rejection of GBM551 xenografts in immunocompetent mice may be partially attributed to a large number of T cell (CD3) and leukocyte (CD45) infiltration (Supp. Fig. 1).
Vascularization and blood–brain barrier status differences in tumors
Gadolinium-enhanced T1-weighted imaging showed the enhancement of GBM1 tumor (both in immunodeficient and in immunocompetent mice) and GBM551 tumor (in immunocompetent mice) signals, indicating the induction of BBB breakdown (Figure 5). However, the BBB in DIPG animals maintained intact. Furthermore, compared to the immunodeficient group, the BBB breakage of GBM1 tumor was more apparent in immunocompetent mice (p < 0.01).
Figure 5.
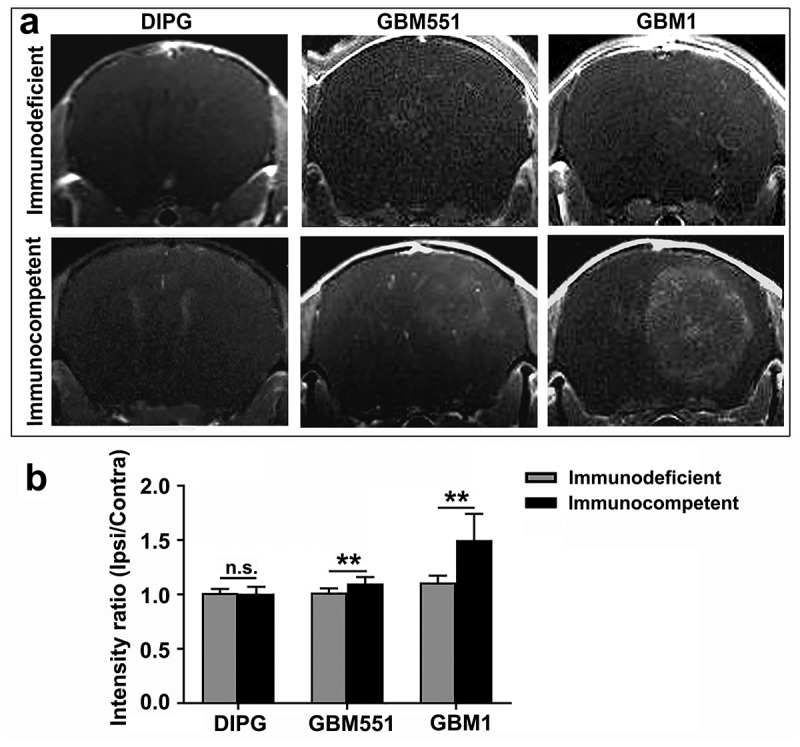
Blood–brain barrier status in immunodeficient and immunocompetent hosts of DIPG, GBM1, and GBM551 tumors. (a) Representative T1-Gd MRI images of the DIPG, GBM551, and GBM1 xenografts in immunodeficient and immunocompetent mice. (b) Quantification of the different tumor graft assessment for T1 intensity. N = 9 slices from three animals/group. Mean ± SD, **p < 0.01, n.s. is no significant difference, independent t-test.
Moreover, all DIPG, GBM551, and GBM1 tumors in immunocompetent mice showed significantly enhanced vascularization in terms of vessel diameter (p < 0.01) (Figure 6), suggesting the contribution of an intact immunity to neovascularization. Of note, the enhanced neovascularization and BBB leakage were most obvious in GBM1 mice with immune tolerance.
Figure 6.
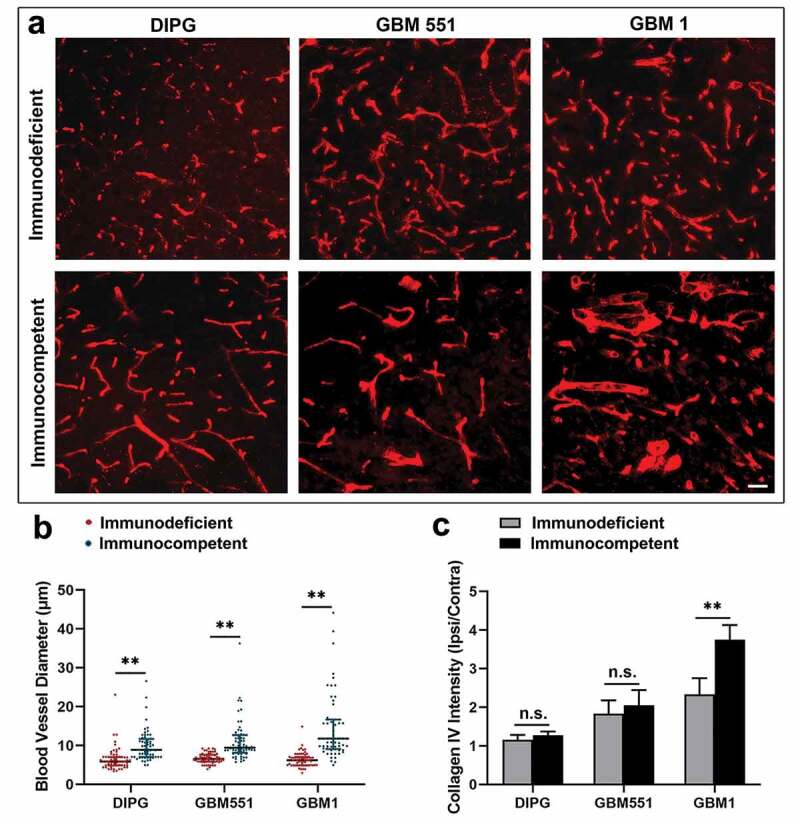
The vascular differences in immunodeficient and immunocompetent hosts of DIPG, GBM1, and GBM551 tumors. (a) Collagen IV staining showed the different blood vessel morphologies in DIPG, GBM1, and GBM551 mice. The scale bar is 50 μm. (b) Quantification analysis of blood vessel diameter showed enhanced vascularization of tumors in immunocompetent mice. N = 180 blood vessels from three animals/group (60 blood vessels for each animal). Median with IQR, ** p< 0.01, Mann–Whitney U test. (c) Quantification analysis of Collagen IV intensity showed more blood supply of GBM1 tumor in immunocompetent mice. N = 9 slices from three animals/group (three brain slices for each animal). Mean ± SD, **p < 0.01, n.s. is no significant difference, independent t-test.
Discussion
A mouse glioma model with high clinical relevance is essential for the development of effective treatments, and the tumor tissue growing in such a model should possess similar features as compared to that in the native human brain in terms of genetic background; epigenetic, phenotypic, and intratumoral heterogeneity; and tumor microenvironment. Besides, the model should be reproducible and stable over time.32 To date, numerous different approaches have been utilized for developing glioma animal models.33 These include chemically induced models, xenograft transplantation models in immunosuppressed recipients13 and immunodeficient hosts, syngeneic transplantation models, and genetically engineered models.34,35 Among them, transplantation of human tumor xenografts into immunodeficient mice is widely used because they retain the advantage of using patient-derived cells. However, full recapitulation of human tumor development is compromised by the absence of immune system.36–38 Genetically engineered mouse models are getting more and more attention with the development of genomic sequencing of human cancers and have the major advantage of studying tumors that arise in their natural microenvironment in immunocompetent animals. But these models lack the intratumor heterogeneity that is observed in human gliomas.39,40 The humanized mouse model, in which immunodeficient mice are engrafted with human hematopoietic cells or tissues, or mice that transgenically express human genes, is another option for studying tumors in clinically relevant setting.41–43 However, this system is complex and costly. Moreover, complications such as graft versus host response and biological constraints that could impair the proper function of its immune response are serious limitations.17 Xenotransplantation of human tumor cells into the brain of an immunocompetent animal represents the optimal strategy for human tumor modeling. In addition to the retention of intratumor heterogeneity, it also has the tumor microenvironment built in the presence of an active immune system. However, the lack of effective and reproducible tumor modeling strategy limited its application.
T cells play an important role in the adaptive immune response toward grafts, which requires both TCR-mediated signals and simultaneously delivered costimulatory signals for their activation.44 These costimulatory signals are provided at least in part by the T cell-based CD28 molecule when bound to its receptors CD80 (B7–1) or CD86 (B7–2) on APCs.44 The activating receptor CD28 engages the same CD80 and CD86 molecules as the inhibitory receptor CTLA-4. Thus, CTLA-4 can compete with CD28 to bind CD80/CD86 or directly inhibit TCR signals leading to suppression of T-cell activation.45 CTLA-4-Ig, which is a selective T-cell costimulation inhibitor that comprises an extracellular domain of CTLA-4 fused with a fragment of human IgG1,46 can mimic the function of CTLA-4. Besides, the interaction of CD40 and its T cell-based ligand, CD40L (CD154), also plays an important role in T-cell activation by upregulating CD80/86 (B7).47,48 In addition, CD40 and CD40L also play a fundamental role in establishing T cell-dependent B-cell activity.49,50 Costimulation blockade of the CD80/86–CD28 and CD40–CD40L (CD154) pathways by CTLA-4-Ig and anti-CD154 mAb has been proven to be a potent inhibitor of T-cell activation, with the combination was 100 times more effective than either alone.51 Apart from prolonging survival of skin allograft,25 renal allograft,51 and islet xenograft,52,53 it was also reported to induce tolerance to stem cell (glial-restricted progenitor) allograft in mouse brain, with further functional integration of stem cells.26
In this study, we showed that costimulation blockade (CTLA-4-Ig and MR1) in immunocompetent mice resulted in long-term different glioma xenografts (human GBM1, GBM551, and DIPG) survival. These results align well with the previous reports on the prevention of rejection of organs25,51-53 or stem cell26 transplantation. All of the animals in GBM1 and DIPG groups, together with three of five animals in GBM551 group treated with both agents, experienced extended (up to 182 d) rejection-free allograft survival. However, two animals in GBM551 group treated with this costimulation inhibitor experienced late, biopsy-proven rejection. Given that the half-life of CTLA-4-Ig is 90 hours and MR1 is 12 d in mouse blood serum,54,55 we speculate that the GBM551 tumor regression may have been triggered in several mechanisms. One possibility is inflammatory/infectious insult triggering leukocyte infiltration and enhancing immune response, as our immunofluorescence results showed substantial leukocyte (CD45) and T-cell (CD3) infiltration and microglial (Iba1) activation in the tumor. Besides, we noticed the disruption of BBB in the immunocompetent mice of GBM551 tumor, which might also contribute to the tumor regression, as peripheral adaptive and innate immune cells, including monocytes, neutrophils, T cells, and B cells, can enter the brain parenchyma and execute distinct cell-mediated effects when the BBB is impaired.56 Finally, it is possible that over the period of several months, new mutations within the tumor cells caused antigenic shift and recognition by immune system.
By further comparing these three tumor cell lines, we found that they exhibit completely different growth patterns and morphologies. DIPG is characterized by extensively diffuse infiltration in the brain parenchyma without obvious tumor mass or clear tumor boundary, which is similar to the findings in DIPG patients,57 while GBM is characterized by comparatively circumscribed growth. Genetic and molecular profile differences are the likely drivers of these distinct infiltrative phenotypes. Previous studies revealed distinct gene expression clusters enriched for different brain tumors;39 for instance, DIPG is genetically distinguished from adult GBM by the high prevalence of the K27 M mutation in the histone H3 variant H3.3 (H3F3A).58 Furthermore, GBM was reported to share expression signatures enriched for extracellular matrix-related genes, which may be related to their restricted growth and reduced infiltration.59 Besides, our glioma models showed similar morphologies and invasion degree with previously reported genetically engineered DIPG or GBM models.35,60-62
Consistent with previous research,16,27 we found that tumors in immunocompetent mice exhibit the characteristics similar to that in glioma patients, including vascular proliferation which is one of the GBM hallmarks seen in patients. We observed that our orthotopic xenotransplant model in immunocompetent mice produced a highly angiogenic tumor with enlarged blood vessels, which is known to be essential to deliver nutrients and oxygen to the tumor.63 Additionally, we observed blood vessels that presented variable calibers, indicating that angiogenesis in the tumor mass is aberrant, as described in GBM patients.64 However, these above characteristics were not found in the tumor transplanted in immunodeficient hosts. Furthermore, we also found microglial activation and substantial recruitment of leukocyte and T cells in the tumor site of immunocompetent mice. This suggests an engagement of both innate and adaptive immune responses during gliomagenesis, which is impossible to obtain in immunodeficient mice. Altogether, these results indicate that the glioma model in immunotolerance mice induced by costimulation blockade is a good candidate for the study of human glioma in vivo.
In summary, we validated the expanded feasibility of brain tumor establishment by using the T-cell costimulation blockade approach, and for the first time, we successfully established human GBM551 and DIPG preclinical orthotopic xenograft models in immunocompetent mice. This will lead a big step forward for glioma study, particularly for DIPG, which poses a real challenge for everyone devoted to the study of pediatric brain tumors. The increased development and availability of these models represent great promise to not only help us gain insights into the biological characteristics and molecular mechanisms of glioma but also provide a suitable platform for in vivo investigations for more effective therapeutic strategies of this devastating disease. It should be noted that, after many passages and expansion in vitro, tumor cells may accumulate genotypic and phenotypic deviations relative to the original cells isolated from the biopsy.65 Thus, for future preclinical tumor studies, transplantation of fresh tumor tissue or cells isolated from patients rather than cultured tumor cell lines would be more meaningful. Such tumors grown in the context of intact immune system would be of particular interest for immunotherapy studies.
Supplementary Material
Acknowledgments
We thank Irina Shats for her immunohistochemical expertise.
Funding Statement
This work was supported by the NIH/NINDS under Grant R01NS102675 and R01NS09111.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
XL, MK, MJ, and PW designed experiments; XL, DK, CC, and AJ performed experiments and analyzed data; XL, SL, YL, MJ, and PW interpreted data and drafted manuscript; and all authors reviewed and edited the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.McNeill KA. Epidemiology of brain tumors. Neurol Clin. 2016;34(4):981–10. doi: 10.1016/j.ncl.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018;20(suppl_4):iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. Epub 2016 May 9. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 4.deSouza RM, Shaweis H, Han C, Sivasubramaniam V, Brazil L, Beaney R, Sadler G, Al-Sarraj S, Hampton T, Logan J, et al. Has the survival of patients with glioblastoma changed over the years? Br J Cancer. 2016;114(2):146–150. Epub Dec 15. doi: 10.1038/bjc.2015.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanan MI, Eisenstat DD. DIPG in children – what can we learn from the past? Front Oncol. 2015;5:237. Epub 2015/ 11/12. doi: 10.3389/fonc.2015.00237. PubMed PMID: 26557503; PubMed Central PMCID: PMCPMC4617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, Lane A, Fuller C, Miles L, Hawkins C, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG registries. J Clin Oncol. 2018;36(19):1963–1972. Epub 2018 May 10. doi: 10.200/JCO.2017.75.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adhikaree J, Moreno-Vicente J, Kaur AP, Jackson AM, Patel PM. Resistance mechanisms and barriers to successful immunotherapy for treating glioblastoma. Cells. 2020;9(2):263. Epub 2020/ 01/25. doi: 10.3390/cells9020263. PubMed PMID: 31973059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyauchi JT, Tsirka SE. Advances in immunotherapeutic research for glioma therapy. J Neurol. 2018;265(4):741–756. doi: 10.1007/s00415-017-8695-5. Epub 2017 Dec 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo GM, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. Epub 2014 Jul 15. doi: 10.158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Hora CC, Schweiger MW, Wurdinger T, Tannous BA. Patient-derived glioma models: from patients to dish to animals. Cells. 2019;8(10):(pii):cells8101177. doi: 10.3390/cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu W, Zhong S, Gong Y, Shan Y, Yuan L, Wang L, Chen J, Zhang Z. A new molecular probe: an NRP-1 targeting probe for the grading diagnosis of glioma in nude mice. Neurosci Lett. 2020;714:134617. Epub 2019 Nov 6. doi: 10.1016/j.neulet.2019.134617. [DOI] [PubMed] [Google Scholar]
- 12.Papachristodoulou A, Silginer M, Weller M, Schneider H, Hasenbach K, Janicot M, Roth P. Therapeutic targeting of TGFbeta ligands in glioblastoma using novel antisense oligonucleotides reduces the growth of experimental gliomas. Clin Cancer Res. 2019;25(23):7189–7201. Epub 2019 Sep 17. doi: 10.1158/078-0432.CCR-17-3024. [DOI] [PubMed] [Google Scholar]
- 13.Qin H, Janowski M, Pearl MS, Malysz-Cymborska I, Li S, Eberhart CG, Walczak P. Rabbit model of human gliomas: implications for intra-arterial drug delivery. PLoS One. 2017;12(1):e0169656. doi: 10.1371/journal.pone. eCollection 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tivnan A, Heilinger T, Lavelle EC, Prehn JH. Advances in immunotherapy for the treatment of glioblastoma. J Neurooncol. 2017;131(1):1–9. doi: 10.1007/s11060-016-2299-2. Epub 2016 Oct 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 16.Garcia C, Dubois LG, Xavier AL, Geraldo LH, da Fonseca AC, Correia AH. The orthotopic xenotransplant of human glioblastoma successfully recapitulates glioblastoma-microenvironment interactions in a non-immunosuppressed mouse model. BMC Cancer. 2014;14(1):923. doi: 10.1186/471-2407-14-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7(2):118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 18.De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39(9):748–763. doi: 10.1016/j.it.2018.07.001. Epub Aug 2 [DOI] [PubMed] [Google Scholar]
- 19.Korhonen R, Moilanen E. Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin Pharmacol Toxicol. 2009;104(4):276–284. doi: 10.1111/j.742-7843.2009.00375.x. Epub 2009 Feb 18 [DOI] [PubMed] [Google Scholar]
- 20.Adams AB, Ford ML, Larsen CP. Costimulation blockade in autoimmunity and transplantation: the CD28 pathway. J Immunol. 2016;197(6):2045–2050. doi: 10.4049/jimmunol.1601135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16(1):111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 23.Graves SS, Stone D, Loretz C, Peterson L, McCune JS, Mielcarek M, Storb R. Establishment of long-term tolerance to SRBC in dogs by recombinant canine CTLA4-Ig. Transplantation. 2009;88(3):317–322. doi: 10.1097/TP.0b013e3181ae285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11(12):852–863. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 25.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381(6581):434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Oh BC, Chu C, Arnold A, Jablonska A, Furtmuller GJ, Qin H-M, Boltze J, Magnus T, Ludewig P, et al. Induction of immunological tolerance to myelinogenic glial-restricted progenitor allografts. Brain. 2019;142(11):3456–3472. doi: 10.1093/brain/awz275. [DOI] [PubMed] [Google Scholar]
- 27.Semenkow S, Li S, Kahlert UD, Raabe EH, Xu J, Arnold A, Janowski M, Oh BC, Brandacher G, Bulte JWM, et al. An immunocompetent mouse model of human glioblastoma. Oncotarget. 2017;8(37):61072–61082. eCollection 2017 Sep 22. doi: 10.18632/oncotarget.7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kahlert UD, Suwala AK, Koch K, Natsumeda M, Orr BA, Hayashi M, Maciaczyk J, Eberhart CG. Pharmacologic Wnt inhibition reduces proliferation, survival, and clonogenicity of glioblastoma cells. J Neuropathol Exp Neurol. 2015;74(9):889–900. doi: 10.1097/NEN.0000000000000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, Yang X, Carrera DA, Downey KM, Shrivastav S, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–157. Epub 2017 Dec 4. doi: 10.1084/jem.20171046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann Oncol. 2017;28(7):1457–1472. doi: 10.093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu CC, Yu CF, Wang SC, Li HY, Lin CM, Wang HH, Abate C, Chiang C-S. Sigma-2 receptor/TMEM97 agonist PB221 as an alternative drug for brain tumor. BMC Cancer. 2019;19(1):473. doi: 10.1186/s12885-019-5700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: experimental models and reality. Acta Neuropathol. 2017;133(2):263–282. doi: 10.1007/s00401-017-1671-4. Epub 2017 Jan 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stylli SS, Luwor RB, Ware TM, Tan F, Kaye AH. Mouse models of glioma. J Clin Neurosci. 2015;22(4):619–626. doi: 10.1016/j.jocn.2014.10.013. Epub 5 Feb 16 [DOI] [PubMed] [Google Scholar]
- 34.Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, Parsa AT, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107. doi: 10.1186/479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misuraca KL, Hu G, Barton KL, Chung A, Becher OJ. A novel mouse model of diffuse intrinsic pontine glioma initiated in Pax3-expressing cells. Neoplasia. 2016;18(1):60–70. doi: 10.1016/j.neo.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Xiao A, diPierro CG, Carpenter JE, Abdel-Fattah R, Redpath GT, Lopes MBS, Hussaini IM. An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am J Pathol. 2010;176(6):3032–3049. doi: 10.2353/ajpath.010.090571. Epub 2010 Apr 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, Nguyen SA, Chan JA, Weljie AM, Cairncross JG, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012;14(2):184–191. Epub 2011 Dec 13. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noorani I. Genetically engineered mouse models of gliomas: technological developments for translational discoveries. Cancers (Basel). 2019;11(9):(pii):cancers11091335. doi: 10.3390/cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashizawa T, Iizuka A, Nonomura C, Kondou R, Maeda C, Miyata H, Sugino T, Mitsuya K, Hayashi N, Nakasu Y, et al. Antitumor effect of programmed death-1 (PD-1) blockade in humanized the NOG-MHC double knockout mouse. Clin Cancer Res. 2017;23(1):149–158. Epub 2016 Jul 25. doi: 10.1158/078-0432.CCR-16-122. [DOI] [PubMed] [Google Scholar]
- 42.Suematsu S, Watanabe T. Generation of a synthetic lymphoid tissue-like organoid in mice. Nat Biotechnol. 2004;22(12):1539–1545. doi: 10.038/nbt039. Epub 2004 Nov 28 [DOI] [PubMed] [Google Scholar]
- 43.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 44.Nagai S, Azuma M. The CD28-B7 family of co-signaling molecules. Adv Exp Med Biol. 2019;1189:25–51. doi: 10.1007/978-981-32-9717-3_2. [DOI] [PubMed] [Google Scholar]
- 45.Alegre ML, Fallarino F. Mechanisms of CTLA-4-Ig in tolerance induction. Curr Pharm Des. 2006;12(2):149–160. doi: 10.2174/138161206775193046. [DOI] [PubMed] [Google Scholar]
- 46.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 1996;273(5283):1862–1864. doi: 10.126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]
- 48.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Flavell RA. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273(5283):1864–1867. doi: 10.126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 49.Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L. Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help). J Exp Med. 1992;175(4):1091–1101. doi: 10.84/jem.175.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lederman S, Yellin MJ, Inghirami G, Lee JJ, Knowles DM, Chess L. Molecular interactions mediating T-B lymphocyte collaboration in human lymphoid follicles. Roles of T cell-B-cell-activating molecule (5c8 antigen) and CD40 in contact-dependent help. J Immunol. 1992;149:3817–3826. [PubMed] [Google Scholar]
- 51.Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, Hong X, Thomas D, Fechner JH, Knechtle SJ, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci U S A. 1997;94(16):8789–8794. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehnert AM, Mottram PL, Han W, Walters SN, Patel AT, Hawthorne WJ, Cowan PJ, d'Apice AJ, O'Connell PJ. Blockade of the CD28 and CD40 pathways result in the acceptance of pig and rat islet xenografts but not rat cardiac grafts in mice. Transpl Immunol. 2001;9(1):51–56. doi: 10.1016/s0966-3274(01)00040-5. [DOI] [PubMed] [Google Scholar]
- 53.Wu J, Hu M, Qian YW, Hawthorne WJ, Burns H, Liuwantara D, Alexander SI, Yi S, O’Connell PJ. In vivo costimulation blockade-induced regulatory T cells demonstrate dominant and specific tolerance to porcine islet xenografts. Transplantation. 2017;101(7):1587–1599. doi: 10.097/TP.0000000000001482. [DOI] [PubMed] [Google Scholar]
- 54.Srinivas NR, Shyu WC, Weiner RS, Tay LK, Greene DS, Barbhaiya RH. Pharmacokinetics of CTLA4Ig (BMS-188667), a novel immunosuppressive agent, following intravenous and subcutaneous administration to mice. J Pharm Sci. 1995;84(12):1488–1489. doi: 10.002/jps.2600841217. [DOI] [PubMed] [Google Scholar]
- 55.Foy TM, Shepherd DM, Durie FH, Aruffo A, Ledbetter JA, Noelle RJ. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. II. Prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med. 1993;178(5):1567–1575. doi: 10.084/jem.178.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20(2):136–144. doi: 10.1038/nn.4475. Epub 2017 Jan 16 [DOI] [PubMed] [Google Scholar]
- 57.Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. 2014;128(4):573–581. doi: 10.1007/s00401-014-1319-6. Epub 2014 Jul 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE. Histone H3.3K27M Represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017;15(9):1243–1254. Epub 2017 May 18. doi: 10.158/541-7786.MCR-16-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, Modrusan Z, Meer H, Westphal M, Lamszus K, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008;27(20):2897–2909. Epub 2007 Nov 26. doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- 60.Koschmann C, Calinescu -A-A, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M, Mulpuri L, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med. 2016;8(328):328ra28. doi: 10.1126/scitranslmed.aac8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oldrini B, Curiel-Garcia A, Marques C, Matia V, Uluckan O, Grana-Castro O, Torres-Ruiz R, Rodriguez-Perales S, Huse JT, Squatrito M, et al. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat Commun. 2018;9(1):1466. doi: 10.038/s41467-018-03731-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346(6216):1529–1533. doi: 10.126/science.1253799. Epub 2014 Nov 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1(1):97–117. doi: 10.1146/annurev.pathol.1.110304.00043. [DOI] [PubMed] [Google Scholar]
- 64.Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. 2013;369(16):1561–1563. doi: 10.056/NEJMcibr1309402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res. 2008;6(1):21–30. Epub 2008 Jan 9. doi: 10.1158/541-7786.MCR-07-0280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.