

Abstract
A discovery metabolomic study was performed in a large cohort of adults to identify circulating biomarkers of frailty. The study found that carnitine and vitamin E pathways were dysregulated in frail compared with non-frail participants. These findings point to dysregulated mitochondrial metabolism as a potential root of age-related frailty.
With the ageing of the global population, research on frailty has grown substantially and has become a main focus of the medical quest to find the roots of healthy ageing. The Geroscience hypothesis proposes that susceptibility to multiple chronic diseases, frailty and disability (the main characteristics of ageing) are caused by a progressive discrepancy between the accumulation of damage and resilience. Here, resilience is defined as the capacity to remove and repair damage at the molecular and cellular level to avoid functional decline1. Understanding the mechanisms of declining resilience might enable clinicians to target global susceptibility to disease, therefore extending healthspan and delaying the onset of frailty and disability.
Progress in the science of frailty has been hampered by disagreement about its pathophysiology and operational diagnosis. Dozens of definitions of frailty have appeared in the literature, with those proposed by Ken Rockwood and Linda Fried being the most widely used2. No definition exhaustively defines frailty, but instead all current definitions identify population subsets that are enriched for frail individuals, with substantial misclassification. An appealing alternative is to understand the biological pathways underlying the development of frailty and identify frail individuals based on circulating biomarkers, a diagnostic process that is already used for many diseases. Beyond settling the discussion on the best clinical criteria for frailty, identifying circulating biomarkers would point to specific biological mechanisms and facilitate the discovery of preventive and therapeutic opportunities.
The work published in 2019 by Nicholas J.W Rattray and collaborators is an important step in this direction3. The authors used the Rockwood Frailty Index to classify participants from Wave 4 of the English Longitudinal Study of Aging (ELSA) into frail and non-frail groups. Subsequently, they performed comprehensive, untargeted metabolomics on serum samples and analysed differences of metabolites and metabolic pathways between these two groups. They found that 25 metabolites from the carnitine shuttle, peroxisomal degradation, kynurenine and vitamin E metabolic pathways were significantly dysregulated with frailty3.
Importantly, Rattray and colleagues repeated the same analysis in a subset of the original participants from Wave 6 of ELSA and confirmed an association between frailty and decreased metabolites in the carnitine and vitamin E pathways3. In a Mendelian randomization analysis using four single nucleotide polymorphisms (SNPs) that were previously validated as genetic instruments to assess carnitine, lower predicted serum concentrations of carnitine were associated with increased risk of frailty. The authors of the present study were rightly cautious in interpreting their findings; however, a picture emerges from their analyses where energy metabolism and mitochondrial function take the main stage in the pathogenesis of frailty.
Carnitine is the molecular carrier of long-chain fatty acids (LCFAs) through the inner mitochondrial membrane. A main substrate for oxidative phosphorylation, LCFAs are esterified with Coenzyme A (CoA) to form acyl-CoA and enter the Krebs cycle (FIG. 1. Carnitine also stimulates the mitochondrial respiratory rate and influences the physical properties of the mitochondrial membrane. Thus, a reduction of carnitine is expected to cause reduced availability of LCFAs in the mitochondria and an energetic deficit, especially in the most energy-demanding tissues, such as skeletal muscle.
Fig. 1 |. Proposed mechanisms of mitochondrial dysfunction in individuals with frailty.
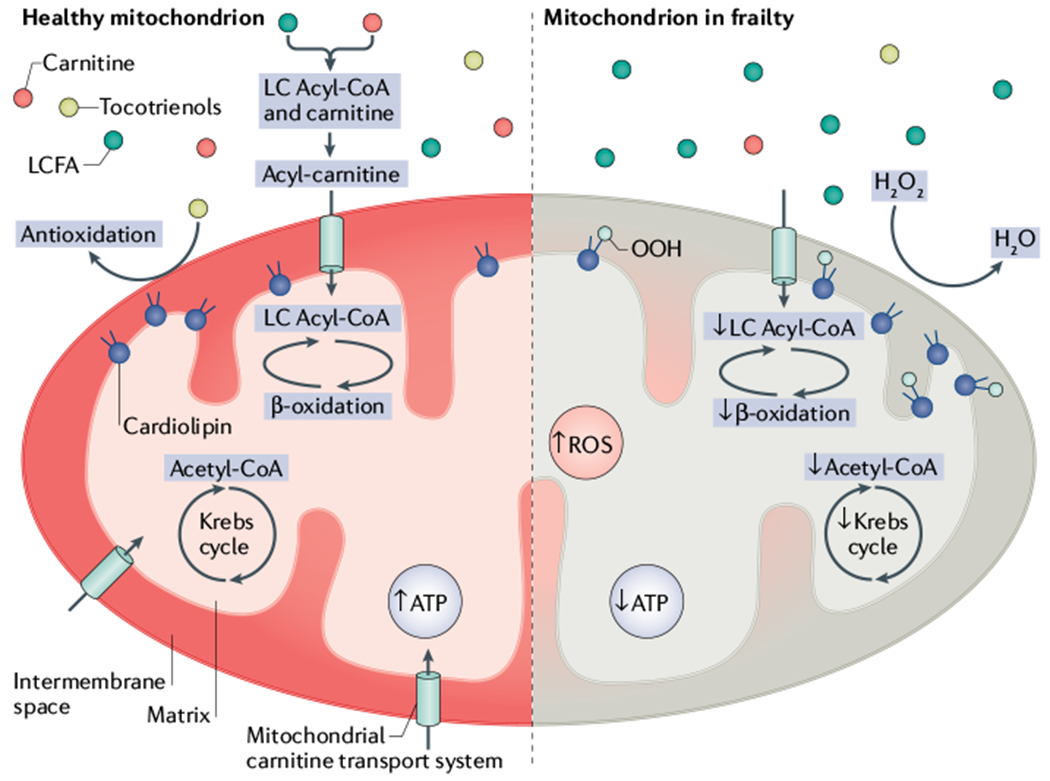
Left, a healthy mitochondrion. Right, putative mechanisms of mitochondrial dysfunction in individuals with frailty. Reduced carnitine content in mitochondria hampers the entrance of long-chain fatty acids (LCFA), causing a decrease in energy production (ATP) and an increase in generation of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2). LC, long-chain.
Genetic defects of the carnitine shuttle are associated with compromised β-oxidation of fatty acids, which leads to clogging of the electron transport chain (ETC), with increased production of free reactive oxygen species (ROS) and myopathy. It is conceivable that scarcity of carnitine occurring in frailty could have similar consequences. During physical activity, reduced availability of LCFAs might lower the threshold of maximal utilization of lipids and shift the metabolism toward utilization of carbohydrates, therefore causing earlier fatigue4. This hypothesis supports the notion that sarcopenia and a sense of exhaustion are early clinical correlates of frailty and is consistent with findings that L-carnitine supplementation favourably affects function and reduces fatigue in older adults5.
The low serum levels of vitamin E associated with frailty in the present study3 might contribute to mitochondrial dysfunction by failing to buffer the excess ROS production induced by carnitine deficiency (FIG. 1). Tocopherols, a class of organic chemical compounds with vitamin E activity, are strong antioxidants that are particularly effective in the mitochondrial environment. Excessive oxidative stress damages proteins, lipids and mitochondrial DNA and RNA, causing substantial mitochondrial dysfunction. Of note, tocopherols are particularly important in protecting against the peroxidation of cardiolipin, which is a mitochondrion-specific phospholipid whose integrity is essential to the structure of the inner mitochondrial membrane. Cardiolipin is also involved in protein transport, signalling, regulation of enzymes of fatty acid β-oxidation and facilitation of the ETC. Furthermore, cardiolipin and carnitine interact synergistically at the inner mitochondrial membrane level6. A scarcity of energy and LCFAs in frail individuals could hamper the synthesis of new cardiolipin. Although cardiolipin is challenging to measure, we argue that carnitine and cardiolipin could influence one another in determining mitochondrial decay and frailty.
The important role of vitamin E in the pathophysiology of frailty is supported by studies showing that low blood levels of vitamin E are cross-sectionally associated with frailty and with future accelerated decline in physical function among community-dwelling older adults7.
Overall, the work of Rattray and colleagues3 suggests that prevention and treatment of frailty should target energy metabolism and mitochondrial function, two of the hallmarks of ageing8. This finding is consistent with studies showing that mitochondrial function, as assessed by in vivo 31phosphorous magnetic resonance spectroscopy, is associated with frailty and mobility performance9. Also, these findings are consistent with data showing that mitochondrial proteins are considerably underrepresented with older age10.
Much work is still needed before the findings of the present study3 can be harnessed for translation. The work of Rattray and collaborators indicates important directions for future research, but methodological problems remain. For example, frailty is a dynamic process and ideally its features should be detected in a longitudinal perspective, as there are time gaps between biological changes and phenotypic manifestations. Rattray and colleagues3 did not identify metabolites that changed with changes in frailty status, but rather performed two cross-sectional analyses at different time points in the same population. As a biomarker of frailty would be particularly valuable when identifying individuals who are still non-frail but have impending risk of frailty, an important analytic opportunity was missed. Finally, the metabolomic assay was performed on serum, which lacks coagulation factors, instead of plasma. Frailty, however, has been associated with changes in coagulation factors and it is known that coagulation removes important metabolites from samples. In spite of these limitations, this work is among the first serious attempts to search for biological roots of frailty and lays out a road map for future research.
Acknowledgements
The authors acknowledge the support of the Intramural Research Program of the National Institute on Aging.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Kennedy BK et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Mao G & Leng SX Frailty syndrome: an overview. Clin. Interv. Aging 9, 433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rattray NJ et al. Metabolic dysregulation in vitamin E and carnitine shuttle energy mechanisms associate with human frailty. Nat. Commun 10, 5027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahlin K, Tonkonogi M & Soderlund K Energy supply and muscle fatigue in humans. Acta Physiol. Scand 162, 261–266 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Pistone G et al. Levocarnitine administration in elderly subjects with rapid muscle fatigue. Drugs Aging 20, 761–767 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Paradies G et al. The effect of aging and acetyl-L-carnitine on the pyruvate transport and oxidation in rat heart mitochondria. FEBS Lett. 454, 207–209 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Balboa-Castillo T et al. Low vitamin intake is associated with risk of frailty in older adults. Age Ageing 47, 872–879 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Fridell Y-W & Sierra F in Progress in molecular biology and translational science Vol. 155 (ed. Ottinger MA) 11–23 (Elsevier, 2018). [DOI] [PubMed] [Google Scholar]
- 9.Adelnia F et al. Moderate-to-vigorous physical activity is associated with higher muscle oxidative capacity in older adults. J. Am. Geriatr. Soc 67, 1695–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Short KR et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl Acad. Sci. USA 102, 5618–5623 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]