

Abstract
Cystic fibrosis (CF) is caused by loss-of-function mutations in the CFTR (CF transmembrane regulator) gene. Pharmacologic therapies directed at CFTR have been developed but are not effective for mutations that result in little or no mRNA or protein expression. Cell therapy is a potential mutation-agnostic approach to treatment. One strategy is to harvest human bronchial epithelial cells (HBECs) for gene addition or genetic correction, followed by expansion and engraftment. This approach will require cells to grow extensively while retaining their ability to reconstitute CFTR activity. We hypothesized that conditionally reprogrammed cell (CRC) technology, namely growth in the presence of irradiated feeder cells and a Rho kinase inhibitor, would enable expansion while maintaining cell capacity to express functional CFTR. Our goal was to compare expression of the basal cell marker NGFR (nerve growth factor receptor) and three-dimensional bronchosphere colony-forming efficiency (CFE) in early- and later-passage HBECs grown using nonproprietary bronchial epithelial growth medium or the CRC method. Cell number and CFTR activity were determined in a competitive repopulation assay employing chimeric air–liquid interface cultures. HBECs expanded using the CRC method expressed the highest NGFR levels, had the greatest 3D colony-forming efficiency at later passage, generated greater cell numbers in chimeric cultures, and most effectively reconstituted CFTR activity. In our study, the HBEC air–liquid interface model, an informative testing platform proven vital for the development of other CF therapies, illustrated that cells grown by CRC technology or equivalent methods may be useful for cell therapy of CF.
Keywords: airway epithelial progenitors, cell therapy, colony-forming efficiency, competitive repopulation, cystic fibrosis
Clinical Relevance
Cell therapy has the potential to be a mutation-blind treatment for people with cystic fibrosis. Effective cell therapy will require therapeutic cells that proliferate, compete with native stem cells, and reconstitute CFTR (cystic fibrosis transmembrane regulator) ion transport. This work represents a step toward identifying the optimal cell population.
Cystic fibrosis (CF) is a life-limiting genetic disease that affects over 70,000 people worldwide (1, 2). Caused by mutations in the CFTR (CF transmembrane regulator) gene (1, 3), pathology develops in the lungs, where absent or dysfunctional CFTR protein leads to insufficient airway surface hydration, thick airway mucus, chronic infection and inflammation, airway remodeling, bronchiectasis, and eventual functional decline. Mainstay CF treatments, such as chest physical therapy, nebulized medications to loosen mucus, and antibiotics, alleviate symptoms (4–6) but do not treat the root cause. Newer pharmacologic therapies target specific CFTR variants (7–9), but these drugs are not approved for mutations that cause low mRNA or protein levels (10). Thus, mutation-agnostic approaches, such as gene or cell therapy, are being sought.
Clinical trials investigating CF gene therapy have largely been disappointing. Modest effects were seen in a study of cationic DNA–liposome complex delivered directly to the lungs of individuals with CF (11). However, clinical trials using adeno-associated virus did not improve forced expiratory volume in 1 second, decrease sputum disease markers, or reduce days of antibiotic use (12). Despite improvements in gene delivery vectors (13), significant obstacles to sustainable CFTR correction of the airway epithelium remain, including mucus, epithelial tight junction barriers (14), and adaptive immunity to viral vectors (15). Cell therapy is another potential treatment approach, but the optimal cell type has yet to be determined. Engrafted cells must express a normal copy of CFTR, self-renew, compete effectively with native CFTR-deficient cells, and differentiate into columnar cells, reconstituting clinically significant levels of CFTR ion transport. Two candidate cell types include heterologous embryonic stem cells (ESCs) and autologous induced pluripotent stem cells (iPSCs). However, there are ethical quandaries about ESCs and safety concerns about both (16, 17). In addition, uniform directed differentiation of either ESCs or iPSCs into airway epithelial cells remains a challenge, requiring prolonged culture (18, 19), cell sorting, and temporal regulation of Wnt signaling (20, 21).
Endogenous airway epithelial progenitor cells may be viable candidates for CF cell therapy. In theory, these cells could be obtained from an individual, gene corrected, expanded ex vivo, and readministered for airway engraftment. The remarkable success of transgenic keratinocyte cell therapy for the blistering skin disease, junctional epidermolysis bullosa (22), provides hope that such an approach may be successful. Like transgenic keratinocytes, airway epithelial cells have a long history of being expanded in vitro (23, 24). However, human transplantation of airway epithelial cells has only been reported in two recipients with bronchiectasis (25), and this, to our knowledge, has not been replicated.
Cell therapy will likely require large numbers of cells to be effective (23, 26), but airway epithelial progenitors have a limited lifespan ex vivo. One approach known to increase cell yield is the conditionally reprogrammed cell (CRC) culture method, in which epithelial cells are grown in the presence of irradiated NIH3T3J2 embryonic fibroblasts and Y-27632, a Rho kinase inhibitor (27). The CRC technique enables extensive cell proliferation while maintaining organ-specific differentiation potential (27). Thus, we hypothesized that the CRC method would allow airway epithelial cells to overcome the loss of growth capacity that occurs during conventional in vitro expansion, thereby increasing the feasibility of using airway epithelial progenitors for CF cell therapy.
Airway epithelial progenitors are heterogeneous, with varying clonal capacity (28–30), and effective engraftment may require the use of markers to identify and isolate populations with the highest growth capacity. A potential marker is the low-affinity NGFR (nerve growth factor receptor), which has been identified in a highly proliferative subset of airway basal cells by lineage tracing and whole-transcriptome sequencing (28). Harnessing proliferative subpopulations of NGFR+ airway epithelial cells could be a step toward making cell therapy possible. To test this hypothesis, we evaluated NGFR expression and bronchosphere colony-forming efficiency (CFE) in five distinct populations of human bronchial epithelial cells (HBECs): 1) cells grown in nonproprietary bronchial epithelial growth media (BEGM) for 1 day to passage 1 (D1P1); 2) cells grown in BEGM to 70–90% confluence at passage 1 (P1); 3) cells grown in BEGM to passage 3 (P3); 4) cells grown to CRC passage 1 (CRC P1); and 5) cells grown to CRC passage 3 (CRC P3). We employed a competitive repopulation assay in which different proportions of non-CF donor cells were coseeded with CF cells to create a mature, chimeric air–liquid interface (ALI) culture. Ussing chamber studies, digital droplet PCR (ddPCR), and whole-mount immunostaining were performed for functional, proliferative, and morphological evaluation. Finally, we examined the relationship between the proportion of CFTR-expressing cells and functional reconstitution of ion transport. A preliminary version has previously been reported in abstract form (31).
Methods
Cell Isolation and Tissue Culture
HBECs obtained under University of North Carolina’s Office of Human Research Ethics/Institutional Review Board study no. 03-1396 were isolated and cultured immediately or after cryopreservation using conventional (32) or CRC (33, 34) culture methods. NIH3T3J2 mouse fibroblasts were replaced with MRC5 (Medical Research Council cell strain 5) human fibroblasts (ATCC) where indicated. Donor demographics are given in Table E1 in the data supplement. The five cell populations examined are illustrated in Figure E1.
Flow Cytometry
HBECs (0.5–1 × 106 cells) were stained with FITC-labeled anti-epithelial cell adhesion molecule (EpCAM), phycoerythrin-labeled anti-NGFR, Pacific blue–labeled annexin V, and sytox blue (Methods E1 and Figure E2). Analysis and sorting were performed using a Sony SH800 cytometer. FlowJo V10 (Becton Dickinson) was used to obtain fluorescence intensity and population density data.
Colony-Forming Assay
HBECs (1 × 103 cells) were suspended in 50 μl of ALI media, mixed 1:1 with 50 μl of Matrigel (356237; Corning), and seeded on 6.5-mm Transwell inserts (CLS3422; Corning), as previously described (28). Bronchospheres were stained between Days 14–18 with Calcein AM (C3100MP, 1 ng/ml in ALI media; Life Technologies) and imaged on an Olympus IX-81 inverted wide-field microscope using a 2× objective. Spheres were counted to calculate CFE, defined as the proportion of spheres generated to the number of cells seeded.
Competitive Repopulation Assay
Non-CF cells were coseeded with the CF cell line UNCCF7T (35) or the parent primary CF cells in proportions of 0%, 1%, 5%, 10%, or 100% non-CF cells at a total seeding density of 1 × 105 cells in 12-mm Millicell CM inserts (PICM01250; Millipore) coated with human placental collagen (C7510; Sigma) (Figure E3). Ussing chamber analysis was performed between Days 24–32 with the addition of amiloride, forskolin, CFTRinh-172 (CFTR inhibitor-172), and UTP, as previously described (33, 36).
ddPCR
DNA was isolated using 1% SDS/50 mM EDTA/50 mM Tris/100 mM NaCl/5 mM DTT/500 μM spermidine in H2O and purified by chloroform:phenol:isoamyl alcohol extraction. The Taqman SNP genotyping assay (4351379; SNP ID, rs113993959; Life Technologies) and the Bio-Rad QX200 system were used to distinguish CF from non-CF cells. Thresholds were set at 3,000 for the fluorescein channel (G542X CFTR mutation) and 2,000 for the victoria fluorescent dye channel (wild-type CFTR) in QuantaSoft 1.7.4.0917 (Bio-Rad) to denote positive reads.
Whole-Mount Immunostaining and Imaging
ALI cultures were fixed in 4% formaldehyde in PBS and stained for α-tubulin, MUC5AC, phalloidin (F-actin), and DNA (Methods E1) using species-specific secondary antibodies as previously described (37). Subsets of cultures were stained with forkhead box I1 (FOXI1) and CFTR where indicated (Methods E1). Cultures were imaged using a Leica TCS SP8 with a 40× objective, systematically sampling nine fields (291 × 291 μm) per condition. Fluorescence intensity of MUC5AC (goblet cells) and α-tubulin (ciliated cells) was obtained using Volocity imaging software (Perkin Elmer).
Statistical Analysis
All data were analyzed using linear mixed-effects models with the donor as a random-effect factor using the R packages lme4 and lmerTest. Post hoc comparisons were performed using the general linear hypothesis test from the multcomp R package. All results are presented as the median (interquartile range). Graphs were created using GraphPad Prism 5 (GraphPad Software Inc.).
Results
NGFR Protein Expression in HBEC Populations
Mouse or human airway epithelial cells that express the basal cell marker NGFR form three-dimensional bronchospheres in vitro, but extended passage is associated with loss of both NGFR and CFE (28). NGFR protein expression was examined using flow cytometry in five candidate EpCAM+ HBEC populations (Figure E1) and also at P5 after BEGM or CRC culture (Figure 1 and Table E2). D1P1 HBECs were 50.3 (44.4–56.5)% NGFR+, which increased slightly at P1 in BEGM to 68.5 (34.5–72.5)% but decreased by P5 in BEGM to 35.6 (21.7–38.5)%. In contrast, HBECs grown using the CRC method more homogeneously expressed NGFR at P1, with 90.2 (81.7–93.2)% positive for NGFR, and maintained NGFR expression in 86.7 (85.5–87.1)% of cells at P5. The median NGFR fluorescence intensity of CRC HBECs was approximately fivefold greater at P1, eightfold greater at P3, and twofold greater at P5 than that of conventionally cultured HBECs (P < 0.001). Thus, the CRC culture method generated cells with higher NGFR protein levels than did conventionally grown cells at all passages.
Figure 1.
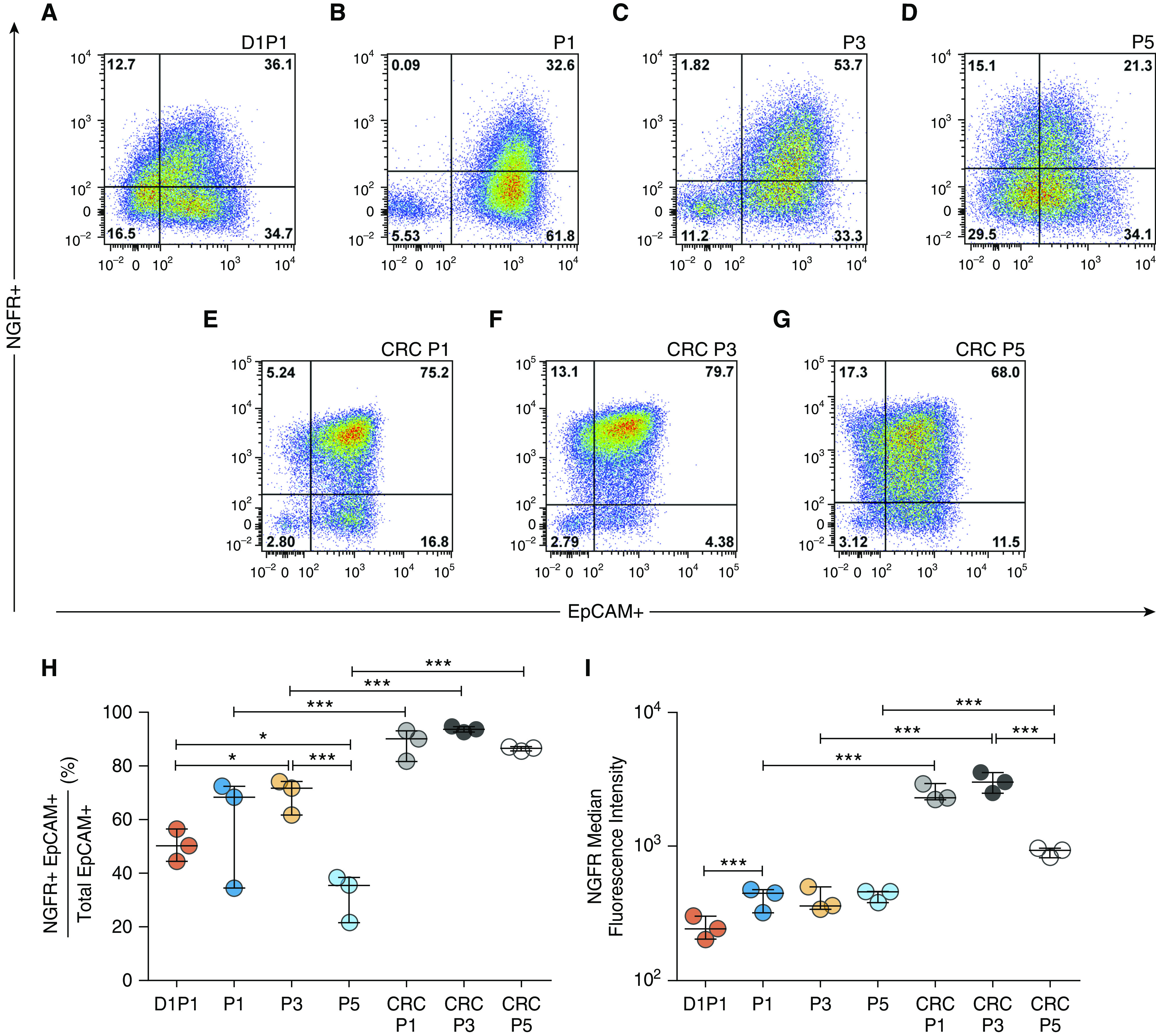
Assessment of NGFR (nerve growth factor receptor) protein in airway epithelial progenitor populations. (A–G) Representative flow cytometry plots from a single human donor. Shown are (A) 1 day to passage 1 (D1P1), (B) P1, (C) P3, (D) P5, (E) conditionally reprogrammed cell (CRC) P1, (F) CRC P3, and (G) CRC P5. The gating strategy for each population can be seen in Figure E2. (H) NGFR median fluorescence intensity. (I) Percentage of NGFR+EpCAM+ cells out of total EpCAM+ cells (Table E2). Biological n = 3 donors; one replicate per donor. *P < 0.05 and ***P < 0.001. Data are presented as median and interquartile range. EpCAM = epithelial cell adhesion molecule.
Growth of NGFR-Positive and -Negative Cells and Induction and Maintenance of NGFR Expression by the CRC Method
To assess the relationship between NGFR expression and growth capacity, EpCAM+ D1P1 cells were sorted into NGFR+ and NGFR− populations and compared in CFE and competitive repopulation assays. Sorted cells were then cultured in the CRC method and assayed for changes in growth capacity (Figure 2A). The CFE of NGFR+ D1P1 cells was higher than the CFE of NGFR− cells (P < 0.01; Figure 2B and Table E3). However, NGFR− cells converted to CRC (cCRC) significantly rescued the colony-forming ability (P < 0.01), bringing the CFE to levels equivalent to NGFR+ cells. In a competitive repopulation assay, NGFR+ cells mixed in as 1% of the initial seeding density expanded to 14.0 (12.4–18.6)% after 24–32 days, whereas NGFR− cells expanded to only 3.7 (1.8–4.7)% (P < 0.001; Figure 2C and Table E3). CFTR activity was also significantly greater in chimeric ALI cultures seeded with 1% NGFR+ cells than in cultures seeded with 1% NGFR− cells (P < 0.001; Figure 2D and Table E4). Notably, CFTR function and competitive repopulation by NGFR− populations were also rescued by CRC culture conditions.
Figure 2.
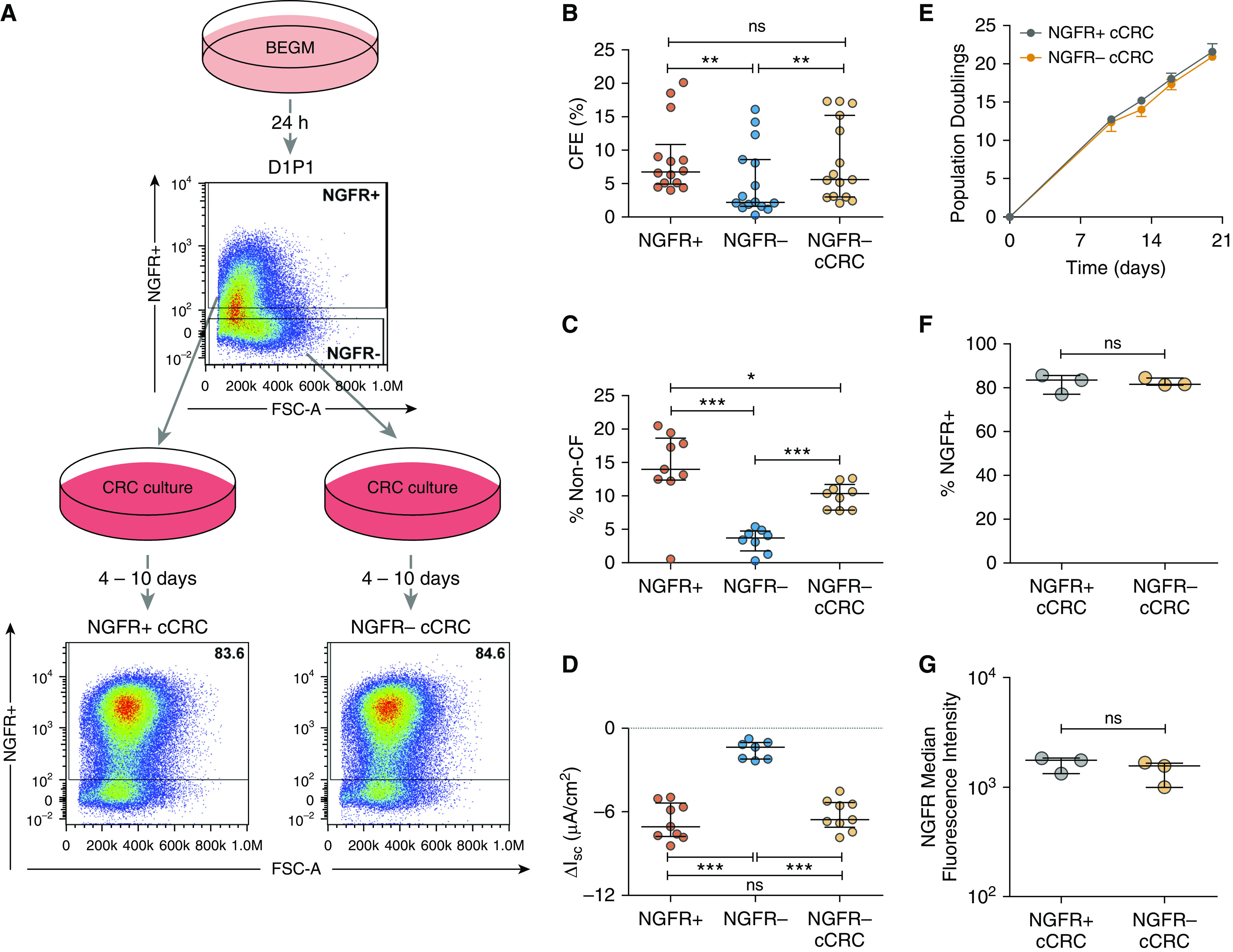
CRC culture rescues growth capacity of NGFR− human bronchial epithelial cells (HBECs). (A) Schematic outline. Primary HBECs were cultured in bronchial epithelial growth media (BEGM) for 1 day (D1P1), dissociated, stained for NGFR, and sorted based on NGFR expression. Colony-forming efficiency (CFE) and competitive repopulation of NGFR+ and NGFR− cells were analyzed. D1P1 NGFR+ and NGFR− subpopulations were converted to the CRC method (cCRCs), reanalyzed by flow cytometry, and colony forming and competitive repopulation were analyzed. FSC-A = forward scatter area. (B) CFE of NGFR+, NGFR−, and NGFR− cCRCs. Biological n = 5; two to three technical replicates for each donor. Competitive repopulation results: (C) percent non–cystic fibrosis (CF) cells between Days 24–32 (also see Table E3) and (D) short-circuit current response to CFTRinh-172 (CFTR inhibitor-172) in Ussing chambers (also see Table E4). Biological n = 4; one to three replicates per donor. *P < 0.05, **P < 0.01, and ***P < 0.001. (E) Growth curves for NGFR+ and NGFR− cCRCs. Analysis of NGFR expression in cCRCs: (F) percentage of NGFR+ cells and (G) median fluorescence intensity of NGFR in NGFR+ and NGFR− cCRCs. Biological n = 3. Data are presented as median and interquartile range. ΔIsc = change in short-circuit current; ns = not significant.
To assess how the CRC method might be rescuing clonal growth and competitive repopulation capacities, we compared the growth trajectories and NGFR expression of NGFR+ and NGFR− cCRCs. Both NGFR+ cCRCs and NGFR− cCRCs grew rapidly, with a small, but distinguishable, difference in population doublings over time (P < 0.05; Figure 2E). Cells that were previously NGFR− exhibited a significant rescue in NGFR expression upon conversion to CRC, becoming 81.7 (81.5–84.6)% positive (Figure 2F and Table E5) and with a median fluorescence intensity equivalent to NGFR+ cCRCs (Figure 2G and Table E5). NGFR+, NGFR−, NGFR+ cCRC, and NGFR− cCRCs were also analyzed by immunofluorescence for the canonical basal cell markers TP63 (28) and K5 (38) (Figure E4). TP63 expression was significantly diminished in NGFR− cells compared with NGFR+ cells (P < 0.001) but was rescued upon conversion to CRC. In contrast, K5 was only slightly lower in the NGFR− populations and remained unchanged upon conversion to CRC. These findings indicate that the CRC culture method rescues growth capacity, competitive repopulation, and markers of stemness, such as NGFR and TP63.
CFE and Growth of HBEC Populations
Colony-forming ability in vitro has historically been considered a measure of stemness (39–41). The baseline CFE of D1P1 cells was 13.5 (5.1–18.1)%, which increased to 23.0 (11.2–34.7)% when expanded to P1 in BEGM and to 19.1 (8.9–27.5)% using the CRC method (Figure 3 and Table E6). Notably, CFE declined significantly by P3 in BEGM, whereas it was maintained in CRC P3 HBECs. In addition, although the growth rate plateaued in BEGM, it increased linearly in CRC culture, which generated 10 population doublings by P3 versus two doublings in BEGM (Figure 3G).
Figure 3.

CFE and growth curve of five candidate lung progenitor populations. Representative images of bronchospheres from: (A) D1P1, (B) P1, (C) P3, (D) CRC P1, and (E) CRC P3. Scale bar, 1 mm. (F) Average CFE of each candidate lung progenitor population (Table E5). Biological n = 8–10 donors; two to three replicates per donor. ***P < 0.001. (G) Growth curves comparing BEGM (solid line) versus CRC (dashed line) growth methods. Biological n = 3; one replicate for each donor. Data are presented as median and interquartile range.
Competitive Repopulation Reveals Functional, Proliferative, and Morphological Properties of Candidate Populations
To assess the competitive growth capacity of the five candidate populations, HBECs were mixed at varying ratios, with the UNCCF7T cell line at a low total seeding density. The resulting chimeric cultures were analyzed between Days 24–32 for CFTR function and the proportion of non-CF cells. Consistent with prior reports that Bmi-1/hTERT cell lines generate abundant goblet cells and few ciliated cells (36), ALI cultures consisting of 100% UNCCF7T cells formed a thin, nonciliated, MUC5AC-expressing cell layer (Figure 4). As the proportion of non-CF cells increased, a thicker pseudostratified columnar epithelium with more ciliated cells and fewer MUC5AC+ cells was observed. Furthermore, seeding as few as 1% non-CF cells measurably recovered CFTR function from baseline (Figures 5A and 5B and Tables 1 and 2 and Table E7). At a seeding density of 1% non-CF cells, CRC P1 HBECs generated the most CFTR function, whereas BEGM P3 HBECs generated the least. CFTR activity did not increase linearly in proportion to seeding density but plateaued, with relatively small increases between the 5% and 10% non-CF seeding densities. In parallel, quantitation of relative cell number between Days 24–32 by ddPCR demonstrated that CRC P1 HBECs consistently generated the most cells in chimeric cultures at all seeding densities (Figure 5C, Tables 1 and 2, and Table E8). When primary G542X homozygous CF cells replaced the UNCCF7T cell line in the competitive repopulation assay, similar results were obtained (Figures 5D and 5E).
Figure 4.

Cell morphology in the competitive repopulation assay. (A–E) Representative images of mature air–liquid interface (ALI) cultures coseeded with UNCCF7T cells and varying percentages of CRC P1 non-CF cells. White, α-tubulin (ciliated cells); green, MUC5AC (mucin 5AC); blue, Hoescht; and red, phalloidin (F actin). Scale bar, 50 μm. (A) 0% non-CF, (B) 1% non-CF, (C) 5% non-CF, (D) 10% non-CF, and (E) 100% non-CF. (F) Orthogonal view of the same representative images. Note the change from a single monolayer, produced by the CF cell line, to a pseudostratified epithelium with more ciliated cells and fewer MUC5AC-expressing cells as more non-CF cells are added. Scale bar, 20 μm. (G) Number of ciliated cells present in the mature ALI culture. (H) Number of MUC5AC+ cells present in the mature ALI culture. Biological n = 3 donors; one replicate per donor. Nine representative fields (291 × 291 μm) per ALI culture were quantified. *P < 0.05 and ***P < 0.001. Data are presented as median and interquartile range.
Figure 5.
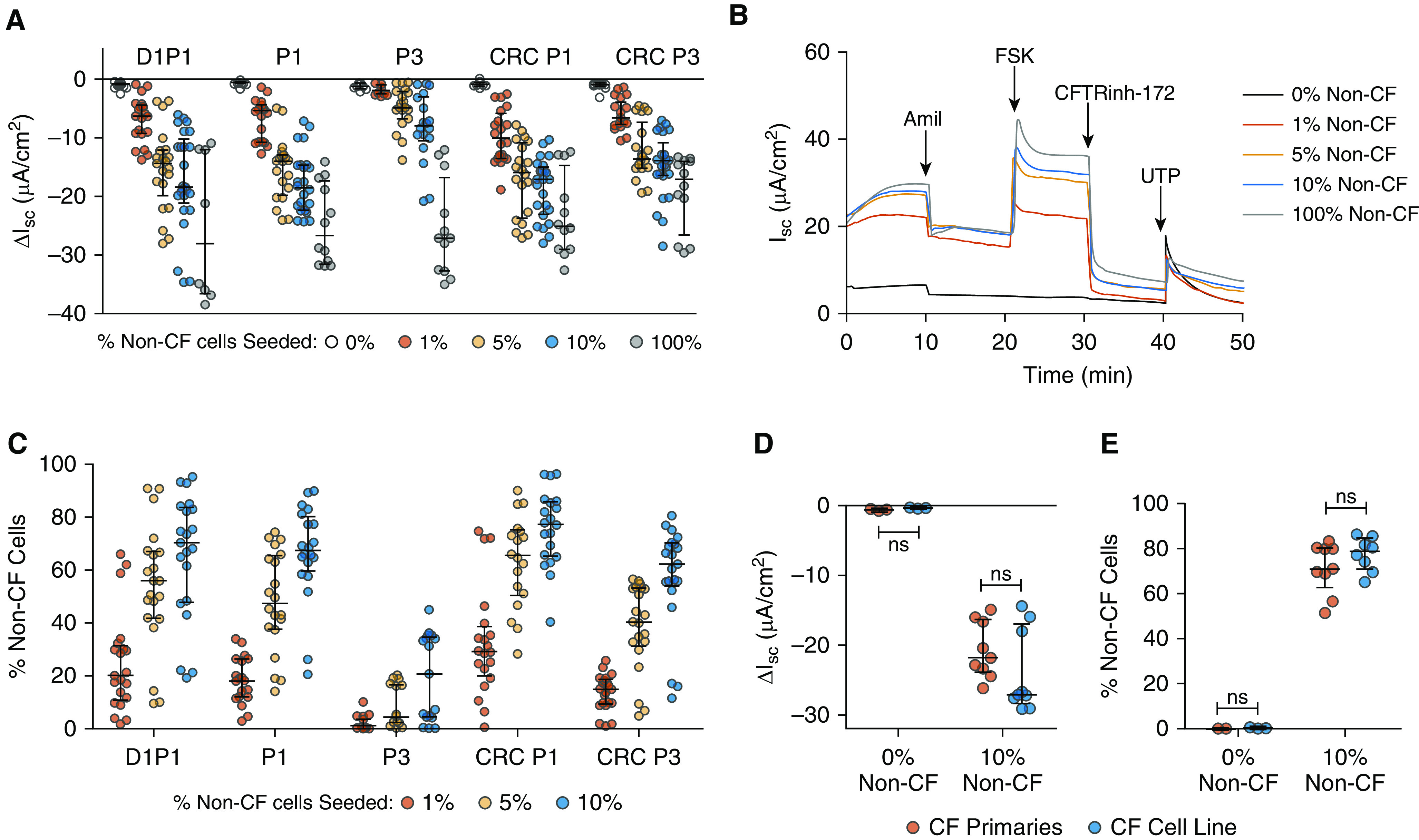
Competitive repopulation by five candidate lung progenitor populations. (A and B) Comparison of the five populations seeded at 0%, 1%, 5%, 10%, and 100% non-CF cells in the competitive repopulation assay. (A) Average short-circuit current response to CFTRinh-172 (Table E6). Biological n = 6–7 donors; two to three replicates per donor. (B) Representative Ussing tracings of the CRC P1 population depicting a dose–response relationship between the percent non-CF cells seeded and the Ussing response. Amil = amiloride; FSK = forskolin; UTP = uridine-5′-triphoshpate. (C) Percent non-CF cells between Days 24–32 in ALI cultures, measured by digital droplet PCR (ddPCR; see Table E7). (D and E) Comparison of CRC P1 cells seeded at 10% non-CF cells in the competitive repopulation assay with either the UNCCF7T cell line (blue) or the primary cells from which the cell line was derived (red). (D) Short-circuit current response to CFTRinh-172. (E) Percent non-CF cells present in ALI cultures between Days 24–32. Biological n = 3 donors; two to three replicates per donor. P values for (A and C) are given in Tables 1 and 2. Data are presented as median and interquartile range.
Table 1.
Statistical Significance of CFTRinh-172 ΔIsc and Relative Cell Number by ddPCR Analysis
| Population |
P Values |
||||||
|---|---|---|---|---|---|---|---|
| 1% Non-CF Cells Seeded |
5% Non-CF Cells Seeded |
10% Non-CF Cells Seeded |
100% Non-CF Cells Seeded | ||||
| CFTRinh-172 ΔIsc | Relative Cell No. | CFTRinh-172 ΔIsc | Relative Cell No. | CFTRinh-172 ΔIsc | Relative Cell No. | CFTRinh-172 ΔIsc | |
| D1P1 vs. P1 | NS | NS | NS | NS | NS | NS | NS |
| D1P1 vs. P3 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | NS |
| D1P1 vs. CRC P1 | NS | NS | NS | NS | NS | NS | NS |
| P1 vs. P3 | <0.01 | <0.01 | <0.001 | <0.001 | <0.001 | <0.001 | NS |
| P1 vs. CRC P1 | NS | <0.01 | NS | <0.01 | NS | NS | NS |
| P3 vs. CRC P3 | <0.01 | <0.01 | <0.001 | <0.001 | <0.001 | <0.001 | <0.01 |
| CRC P1 vs. CRC P3 | NS | <0.01 | NS | <0.001 | NS | <0.01 | NS |
Definition of abbreviations: CF = cystic fibrosis; CFTRinh-172 = cystic fibrosis transmembrane regulator inhibitor-172; CRC = conditionally reprogrammed cell; D1P1 = 1 day to passage 1; ΔIsc = change in short-circuit current; ddPCR = droplet digital PCR; NS = not significant; P1 = passage 1; P3 = passage 3.
Biological n = 3–4 for the 100% condition; biological n = 6–7 for all other conditions; n = 2–3 for each donor. Two-way ANOVA with Bonferroni’s post hoc test.
Table 2.
P Values for Percent Non–Cystic Fibrosis Cells Obtained from ddPCR Analysis for Five Candidate Lung Progenitor Populations
| Population |
P Values |
|||||
|---|---|---|---|---|---|---|
| 0% vs. 1% Non-CF Cells Seeded |
0% vs. 5% Non-CF Cells Seeded |
0% vs. 10% Non-CF Cells Seeded |
||||
| CFTRinh-172 ΔIsc | Relative Cell No. | CFTRinh-172 ΔIsc | Relative Cell No. | CFTRinh-172 ΔIsc | Relative Cell No. | |
| D1P1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| P1 | <0.001 | <0.01 | <0.001 | <0.001 | <0.001 | <0.001 |
| P3 | NS | NS | <0.05 | NS | <0.001 | <0.001 |
| CRC P1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| CRC P3 | <0.05 | <0.01 | <0.001 | <0.001 | <0.001 | <0.001 |
Biological n = 3–4 for the 100% condition; biological n = 6–7 for all other conditions; n = 2–3 for each donor. Two-way ANOVA with Bonferroni’s post hoc test.
Using this assay, we also found that the murine NIH3T3J2 feeder cells used in the CRC method can be replaced with human MRC5 feeder cells, which may be useful to avoid xenogenic reagents and potential immunogenicity. Cells expanded with MRC5 fibroblasts generated a similar number of non-CF cells in chimeric ALI cultures as cells expanded with NIH3T3J2 cells at all doses (Figure E5A and Table E9). Functional reconstitution of CFTR activity by MRC5-expanded HBECs was slightly lower, only reaching significance at the 10% dose (Figure E5B and Table E10). These findings were corroborated by CFE data (Figure E5C and Table E9). Thus, the competitive repopulation assay described here is useful for comparing proliferative, competitive, and functional properties of candidate progenitor populations.
CFTR Ion Transport Is a Nonlinear Function of the Proportion of Non-CF Cells
CFTR activity, measured as the short-circuit current response to CFTRinh-172, was examined as a function of the percent of non-CF cells between Days 24–32, the days on which Ussing chamber studies were performed (Figure 6). For each of the five HBEC populations studied, a positive dose–response between CFTR function and the proportion of non-CF cells was seen. However, the relationship did not appear to be linear. An extra sum-of-squares F test was performed to compare the goodness of fit between linear and one-phase decay models. For each population, a one-phase decay model was a significantly better fit (P < 0.0001). The proportion of non-CF cells required to achieve 50% CFTR function (i.e., half-life in the one-phase decay equation) was calculated (Figure 6F). These values ranged from 7.1% to 15.2%. Thus, a nonlinear relationship exists between CFTR function and the proportion of non-CF cells present.
Figure 6.
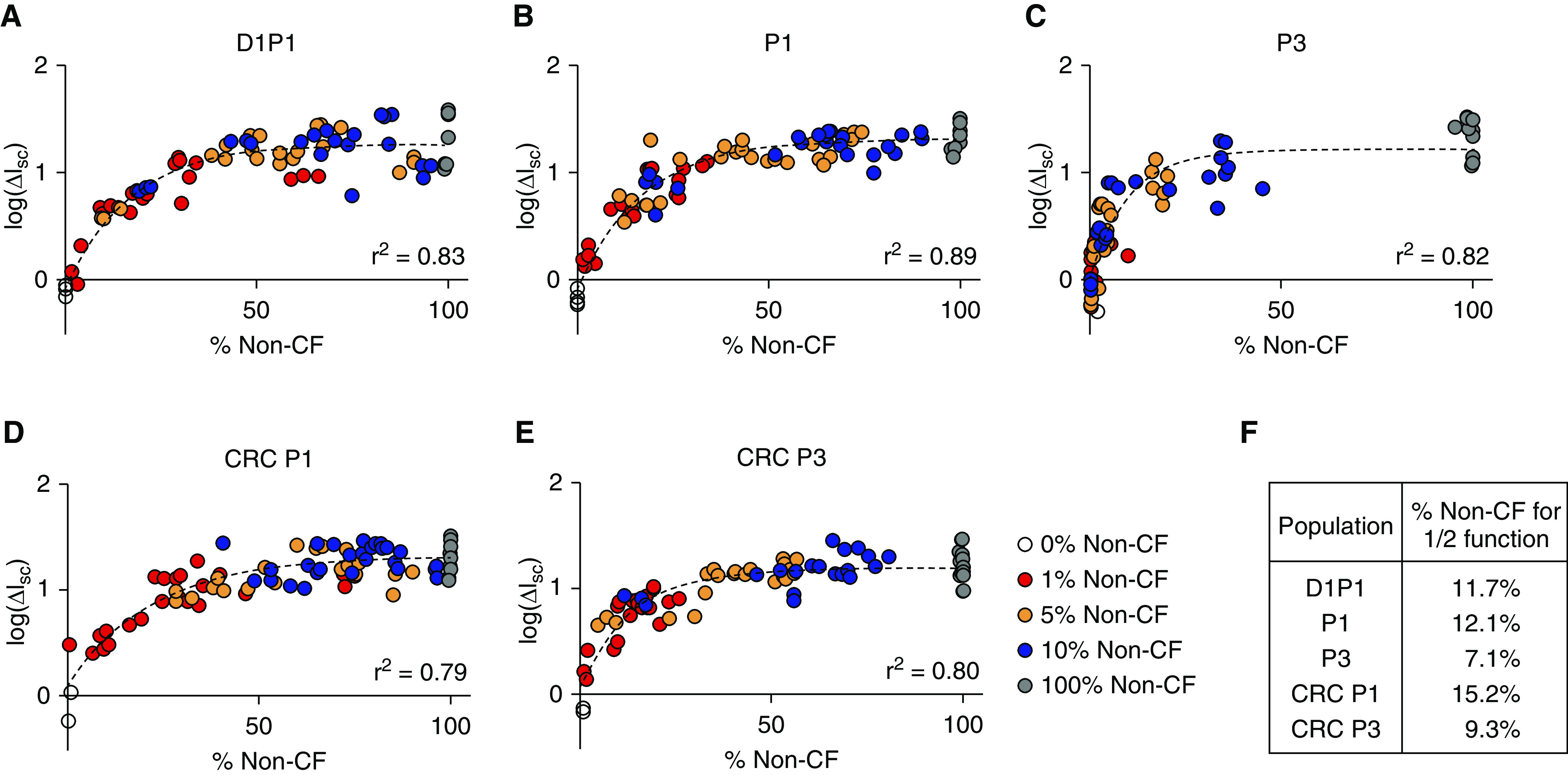
CFTR ion transport function exceeds the proportion of non-CF cells seeded. The log of the CFTRinh-172 short-circuit current response as a function of the percent non-CF (A) D1P1, (B) P1, (C) P3, (D) CRC P1, and (E) CRC P3 cells between Days 24–32 (the time of Ussing studies). Biological n = 3–4 donors for the 100% condition; n = 6–7 for all other conditions; two to three replicates per donor. An extra sum-of-squares F test was performed for each population to compare the goodness of fit between linear and one-phase decay models. In each case, a one-phase decay model was a better fit for the data (see Table E10). (F) The percentage of non-CF cells required to achieve 50% function was calculated by constraining one-phase decay models to plateau at the mean 100% non-CF CFTRinh-172 response.
CRC Culture Produces More FOXI1+ Ionocytes than Conventional Culture Methods
The nonlinear relationship described previously here supports the notion that functional contribution may vary between distinct cell types. Recent single-cell RNA sequencing studies identify the CFTR-high ionocyte, a rare cell type characterized by expression of the FOXI1 transcription factor, as a potential workhorse of CFTR ion transport (42, 43). To look for CFTR-high cells in our model, we probed conventionally grown P1 and CRC P1 ALI cultures seeded with equivalent cell numbers for CFTR and FOXI1 protein expression by whole-mount immunostaining (Figure 7). All CFTR-high cells had a corresponding FOXI1+ nucleus, indicating that these cells are ionocytes. Occasionally, a FOXI1+ nucleus could be seen with no visible CFTR expression, but these tended to be closer to the basolateral surface and likely represent ionocytes in the process of differentiating. On average, we found that CRC P1 cultures had four times as many FOXI1+ nuclei than conventionally grown P1 cultures (Figure 7E; P < 0.001). From this observation, we concluded that ionocyte abundance is dependent on in vitro expansion methods.
Figure 7.
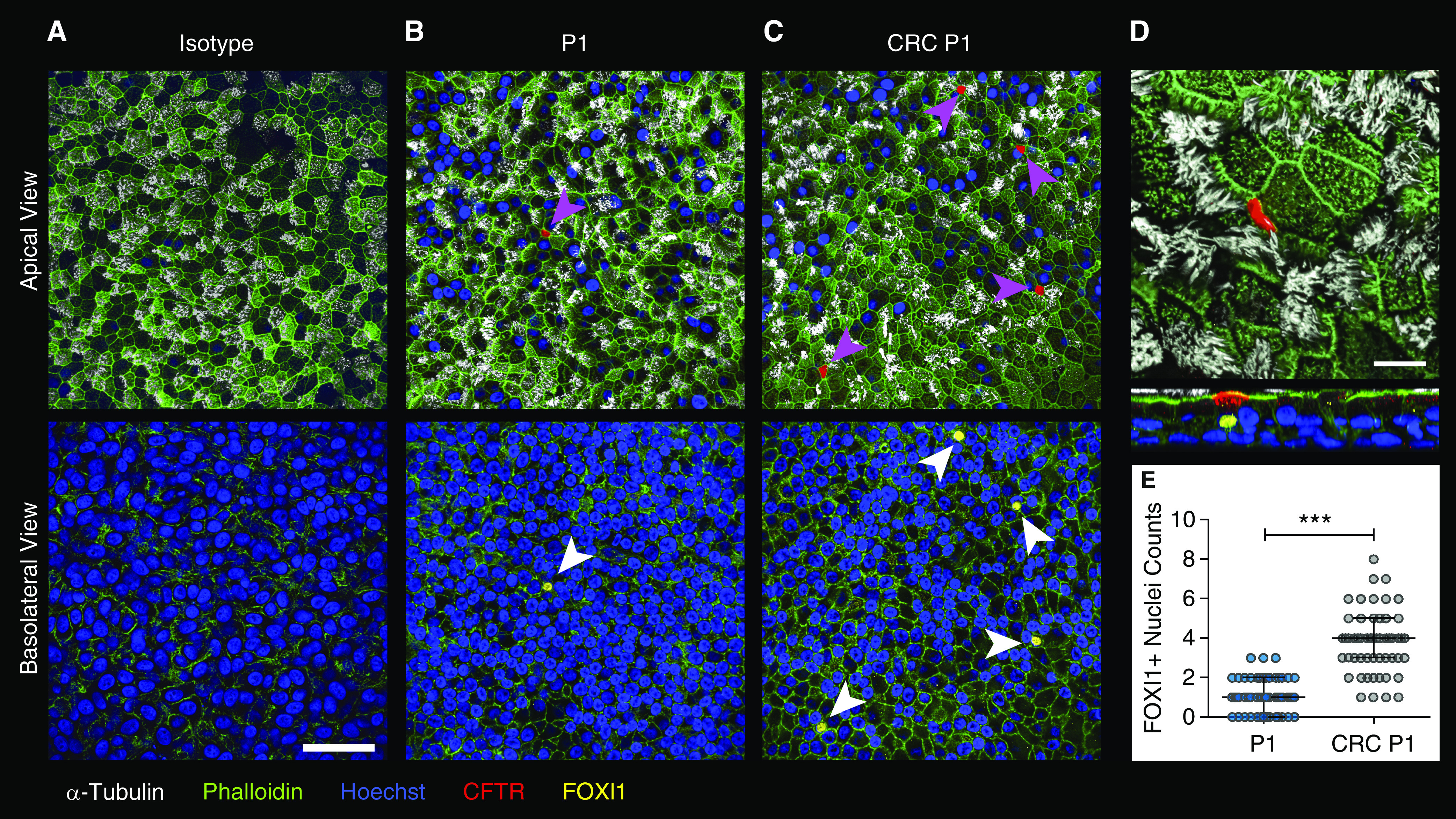
CRCs generate greater numbers of ionocytes. (A–C) Representative apical and basolateral views of non-CF P1 (A and B) and CRC P1 (C) HBEC ALI cultures. (A) Isotype control for both forkhead box I1 (FOXI1) and CFTR antibodies, (B) P1, and (C) CRC P1 stained for α-tubulin (white, ciliated cells); green, phalloidin (F actin); blue, Hoescht; red, CFTR; and yellow, FOXI1. Magenta arrowheads indicate CFTR-high cells. White arrowheads indicate FOXI1+ nuclei. Scale bar, 50 μm. (D) Apical and X–Z views of a CFTR-high expressing FOXI1+ ionocyte. Scale bar, 10 μm. (E) Quantitation of FOXI1+ nuclei in P1 and CRC P1 ALI cultures. Biological n = 3 donors; two replicates per donor. Nine representative 212 × 212-μm fields were quantified per ALI culture. ***P < 0.001. Data are presented as median and interquartile range.
Discussion
Cell therapy is a potential mutation-agnostic approach for treating CF. However, an optimal progenitor population must be identified before translation to the bedside. In this study, we evaluated NGFR expression in HBECs cultured by conventional and CRC methods for varying lengths of time. We also analyzed CFE, competitive growth, and restoration of CFTR ion transport as complimentary measures of stemness, hypothesizing that airway epithelial cells with the greatest stemness are most likely to be effective for cell therapy.
In prior studies, NGFR expression correlated with greater CFE of mouse basal cells. Likewise, human NGFR+/integrin subunit alpha 6 (ITGA6+) basal cells demonstrated greater CFE than NGFR−/ITGA6− populations in the absence of stromal and columnar epithelial cells (28). NGFR is also expressed at high levels in a mouse squamous lung cancer model, indicating that NGFR may be a marker of cells with high proliferative capacity (44). In the current studies, the percentage of NGFR+ cells diminished at later passages in conventionally grown cells but was maintained in cells grown by the CRC method, correlating loosely with our CFE data. When D1P1 HBECs were sorted based on NGFR expression, the NGFR− cells retained minimal clonal growth capacity and performed poorly in the competitive repopulation assay, recovering very little CFTR function. However, the CRC method rescued NGFR expression and restored the ability of NGFR− cells to effectively compete in chimeric cultures. The ability to rescue stemness would be highly beneficial to the development of cell therapy.
Effective cell therapy using endogenous airway epithelial progenitors must address the challenge of low initial cell numbers. Typically, a limited number of epithelial cells are obtained in bronchial lavage (∼5.5 × 103 [45]), induced sputum (<2 × 103 [46]), or by endobronchial biopsy (∼2 × 106 [47]). Furthermore, lung engraftment is inefficient (45, 48–52), with engraftment levels approaching 20% only after repeated administration of cells (53) or by using a cancer cell line (54). Thus, an effective cell therapy will likely require large-scale expansion of meager patient-derived samples. We found that the CRC method maintains high CFE and differentiation potential for multiple passages. These results are consistent with previous work demonstrating that airway epithelial cells grown using the CRC method are highly proliferative and capable of many population doublings (26, 33, 55). The potential utility of this technique to produce therapeutic cells is corroborated by the studies of Butler and colleagues (47), who used it to repopulate a tissue-engineered airway.
The work presented here also highlights an important challenge for cell therapy of CF: extended in vitro expansion of airway epithelial cells often results in decreased ion transport function (Figure 4C and References 33, 46, and 56). Modifications of the CRC method have been reported to slow the decline in CFTR activity, but forskolin-stimulated CFTR currents still decrease with time in culture (57). One study used a feeder-free expansion protocol and reported no decline in CFTR modulator effects in ALI cultures created at 15 and 30 population doublings, but amiloride-sensitive and UTP-stimulated currents declined and the magnitude of forskolin-stimulated current in corrected CF cultures was quite low (58). In our studies, CRC expansion generated cells that restored ion transport more effectively than conventional culture, but the average short-circuit response to CFTRinh-172 still decreased by P3. Studies to better understand the mechanism of functional CFTR loss in highly expanded HBECs and further optimization to prevent functional CFTR loss are warranted.
In the competitive repopulation assay, we found that non-CF donor cells often dramatically outcompeted CF donor cells. It was not uncommon for 10% seeded non-CF cells to comprise 75% of the mature ALI culture 24–32 days later. A possible explanation may be an innate difference in the proliferative capacity of CF and non-CF donor cells. Several studies have suggested differences in proliferation, wound repair, and lung epithelial development, depending on CFTR status (59–62). However, this idea has been countered through extensive comparative studies by Hayes and colleagues (63). Alternatively, the progenitor populations obtained from CF lungs may have undergone more population doublings in response to chronic infection, inflammation, and damage, providing the isolated non-CF cells a proliferative advantage. We note that we used the UNCCF7T cell line for many of the competitive repopulation studies, but we observed no differences between the cell line and the parent primary CF cells. Future competitive repopulation studies using genetically corrected and noncorrected CF cells from the same donor will be informative.
Mixing of CF and non-CF cells in ALI cultures has been performed previously (64, 65). A study performed by Li and colleagues (65) used flow cytometry to assess chimerism after mixing α6β4+ GFP-labeled distal lung progenitors from a non-CF donor with CF bronchial cells. They did not observe a competitive growth advantage of non-CF cells using these methods, with 1% non-CF cells at initial seeding resulting in 1% non-CF cells 2 weeks later. There are several possible explanations for this discrepancy. First, the study by Li and colleagues mixed distal lung progenitors with tracheobronchial progenitors, whereas our study used only HBECs. Second, we used a much lower total seeding density, providing the donor and host cells greater opportunity to compete for attachment and proliferation before becoming confluent. Third, their analysis depended on the longevity of GFP expression (i.e., the absence of vector silencing), whereas, to our knowledge, our study is the first to use ddPCR to rigorously quantify the proportion of CF and non-CF cells after differentiation into a mature epithelium.
We sought to determine the proportion of cells needed to restore CFTR function. A prior study demonstrated that 25% CFTR-corrected cells restored normal mucociliary transport in the CF HBEC ALI model (66). An important earlier gene therapy study found that cultures coseeded with only 6–10% CFTR-corrected cells generated ion transport properties similar to cultures comprising 100% corrected cells (64). In our competitive repopulation assay, CFTR activity was significantly increased from a null baseline when the proportion of non-CF cells in the chimeric ALI culture reached or exceeded 15% (i.e., after a 1% initial seeding density of CRC P3 cells). Seeding cocultures at 5–10% non-CF cells led to an expansion to ∼40–80% non-CF cells at the time of Ussing analysis, resulting in chimeric cultures functionally equivalent to those comprising 100% non-CF cells (Figure 6 and Table E8). The nonlinear relationship between CFTR function and the proportion of non-CF cells that we observed in this study may indicate a lower bar for CF cell therapy. In other words, even moderate levels of cell replacement may produce clinically significant CFTR correction.
The studies cited previously here (64, 66) raise the possibility that CFTR-high cells and cell–cell communication within the epithelial sheet might facilitate overall ion transport, a notion supported by the nonlinear correlation between CFTR ion transport and the proportion of non-CF cells. Recent studies have identified ionocytes, rare epithelial cells enriched with CFTR and marked by the transcription factor FOXI1 in the airway epithelium (42, 43). We found protein-level evidence of FOXI1+ CFTR-high cells in P1 and CRC P1 ALI cultures. Interestingly, FOXI1+ nuclei were fourfold more common in CRC P1 cultures, although the difference in CFTR function between P1 and CRC P1 cultures was minimal. These data indicate that differentiation patterns can be dependent on in vitro expansion methods and that ionocytes may not be the major cell type contributing to CFTR function. Furthermore, studies are needed to define the full repertoire of CFTR-expressing cells and the relative contribution of each cell type to ion transport.
Overall, the competitive repopulation assay described here assesses the proliferative, differentiation, and functional capacity of candidate cell populations, as well as morphological properties of the resulting chimeric cultures. The competitive repopulation assay also proved useful for assessing alternative feeder cells. We observed comparable competitive repopulation capability and ion transport when using human MRC5 fibroblasts versus mouse NIH3T3J2 fibroblasts, consistent with prior reports (26). This assay is therefore a useful method to evaluate candidate progenitor populations for possible use in cell therapy.
An important limitation of this study is that stem cell capacity in vitro may not translate in vivo because of the absence of airway parenchymal matrix and other cellular components. This may be particularly important in small airways, where there is theoretically a greater opportunity for epithelial-to-mesenchymal cell communication because of greater proximity. Future work will be necessary to ascertain how cell populations engraft in both in vitro and in vivo models. Although it appears that low doses of CRC-expanded non-CF cells are capable of outcompeting CF cells when seeded on a collagen-coated membrane in vitro, equivalent capacity has yet to be demonstrated in vivo and is a future goal. Alternative sources of stem and progenitor cells, such as glandular myoepithelial cells (67, 68) or iPSC-derived airway epithelial cells (20), may also be considered in future studies.
In summary, NGFR is highly expressed in early passage conventionally grown cells but is lost in culture over time. In contrast, NGFR expression is maintained and rescued by the CRC culture method. The competitive repopulation assay we employed is a useful tool to compare the therapeutic potential of candidate airway progenitor populations and to assess future advances in culture conditions. For cell therapy of CF to be realized as a potential one-time, mutation-agnostic cure, methods must be developed to enable the harvest of cell numbers sufficient for genetic correction and growth in a manner consistent with use in humans. In vivo models to test the engraftment potential of candidate cell populations and safe host preconditioning regimens will be required before translation. Our studies provide useful early information toward this ambitious goal.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Dr. Scott Magness and Carlton Anderson (Center for Gastrointestinal Biology and Disease Advanced Analytics Core at UNC Chapel Hill) for training, expertise, and use of flow cytometry equipment, and the Marsico Lung Institute Tissue Procurement and Cell Culture Core for providing cells and media. NIH3T3J2 cells and ongoing discussion on the conditionally reprogrammed cell method were kindly provided by Drs. Richard Schlegel and Xuefeng Liu (Georgetown University).
Footnotes
Supported by Cystic Fibrosis Foundation grants RANDEL15XX0, RANDEL17XX0, and BOUCHE15R0, and U.S. National Institutes of Health grant P30DK065988.
Author Contributions: R.E.L., S.M.M., and S.H.R. conceived the project and designed the experiments. R.E.L. and S.M.M. performed colony-forming efficiency and competitive repopulation assays and drafted the manuscript. T.M.M. and R.T. performed the whole-mount immunostaining, imaging, and quantitation. C.A.L. conducted the flow cytometry, sorting, and data analysis. Z.H.B. generated the growth curves. R.E.L. composed all of the illustrations and figures. H.D. performed all of the statistical analyses. C.A.L. and S.H.R. performed the significant manuscript editing. All of the authors read, edited, and approved the final version of the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0384OC on May 21, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliver KE, Han ST, Sorscher EJ, Cutting GR. Transformative therapies for rare CFTR missense alleles. Curr Opin Pharmacol. 2017;34:76–82. doi: 10.1016/j.coph.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cholon DM, Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros. 2018;17:S52–S60. doi: 10.1016/j.jcf.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boon M, Jorissen M, Jaspers M, Augustijns P, Vermeulen FL, Proesmans M, et al. The influence of nebulized drugs on nasal ciliary activity. J Aerosol Med Pulm Drug Deliv. 2016;29:378–385. doi: 10.1089/jamp.2015.1229. [DOI] [PubMed] [Google Scholar]
- 5.Bos AC, Passé KM, Mouton JW, Janssens HM, Tiddens HA. The fate of inhaled antibiotics after deposition in cystic fibrosis: how to get drug to the bug? J Cyst Fibros. 2017;16:13–23. doi: 10.1016/j.jcf.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Warnock L, Gates A. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database Syst Rev. 2015;(12):CD001401. doi: 10.1002/14651858.CD001401.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgener EB, Moss RB. Cystic fibrosis transmembrane conductance regulator modulators: precision medicine in cystic fibrosis. Curr Opin Pediatr. 2018;30:372–377. doi: 10.1097/MOP.0000000000000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27:424–433. doi: 10.1091/mbc.E14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. VX17-445-102 Study Group. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther. 2015;145:19–34. doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Alton EWFW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. UK Cystic Fibrosis Gene Therapy Consortium. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moss RB, Milla C, Colombo J, Accurso F, Zeitlin PL, Clancy JP, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18:726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 13.Sondhi D, Stiles KM, De BP, Crystal RG. Genetic modification of the lung directed toward treatment of human disease. Hum Gene Ther. 2017;28:3–84. doi: 10.1089/hum.2016.152. [DOI] [PubMed] [Google Scholar]
- 14.Kim N, Duncan GA, Hanes J, Suk JS. Barriers to inhaled gene therapy of obstructive lung diseases: a review. J Control Release. 2016;240:465–488. doi: 10.1016/j.jconrel.2016.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sack BK, Herzog RW. Evading the immune response upon in vivo gene therapy with viral vectors. Curr Opin Mol Ther. 2009;11:493–503. [PMC free article] [PubMed] [Google Scholar]
- 16.Ilic D, Ogilvie C. Concise review: human embryonic stem cells—what have we done? What are we doing? Where are we going? Stem Cells. 2017;35:17–25. doi: 10.1002/stem.2450. [DOI] [PubMed] [Google Scholar]
- 17.Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, et al. Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res. 2013;112:523–533. doi: 10.1161/CIRCRESAHA.111.256149. [DOI] [PubMed] [Google Scholar]
- 18.Gotoh S, Ito I, Nagasaki T, Yamamoto Y, Konishi S, Korogi Y, et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014;3:394–403. doi: 10.1016/j.stemcr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konishi S, Gotoh S, Tateishi K, Yamamoto Y, Korogi Y, Nagasaki T, et al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 2016;6:18–25. doi: 10.1016/j.stemcr.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berical A, Lee RE, Randell SH, Hawkins F. Challenges facing airway epithelial cell–based therapy for cystic fibrosis. Front Pharmacol. 2019;10:74. doi: 10.3389/fphar.2019.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell. 2017;20:844–857.e6. doi: 10.1016/j.stem.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551:327–332. doi: 10.1038/nature24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 24.Randell SH, Fulcher ML, O’Neal W, Olsen JC. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol. 2011;742:285–310. doi: 10.1007/978-1-61779-120-8_18. [DOI] [PubMed] [Google Scholar]
- 25.Ma Q, Ma Y, Dai X, Ren T, Fu Y, Liu W, et al. Regeneration of functional alveoli by adult human SOX9+ airway basal cell transplantation. Protein Cell. 2018;9:267–282. doi: 10.1007/s13238-018-0506-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynolds SD, Rios C, Wesolowska-Andersen A, Zhuang Y, Pinter M, Happoldt C, et al. Airway progenitor clone formation is enhanced by Y-27632–dependent changes in the transcriptome. Am J Respir Cell Mol Biol. 2016;55:323–336. doi: 10.1165/rcmb.2015-0274MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12:439–451. doi: 10.1038/nprot.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3:545–556. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson JK, Rulands S, Wilkinson AC, Wuidart A, Ousset M, Van Keymeulen A, et al. Clonal dynamics reveal two distinct populations of basal cells in slow-turnover airway epithelium. Cell Rep. 2015;12:90–101. doi: 10.1016/j.celrep.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller SM, Lee RE, Randell SH. Assessing airway epithelial progenitors for CF cell therapy [abstract] Pediatr Pulmonol. 2017;52:S288–S289. [Google Scholar]
- 32.Fulcher ML, Randell SH. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol. 2013;945:109–121. doi: 10.1007/978-1-62703-125-7_8. [DOI] [PubMed] [Google Scholar]
- 33.Gentzsch M, Boyles SE, Cheluvaraju C, Chaudhry IG, Quinney NL, Cho C, et al. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol. 2017;56:568–574. doi: 10.1165/rcmb.2016-0276MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci USA. 2012;109:20035–20040. doi: 10.1073/pnas.1213241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gentzsch M, Ren HY, Houck SA, Quinney NL, Cholon DM, Sopha P, et al. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol. 2016;311:L550–L559. doi: 10.1152/ajplung.00186.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, et al. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol. 2009;296:L82–L91. doi: 10.1152/ajplung.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh A, Coakley RC, Mascenik T, Rowell TR, Davis ES, Rogers K, et al. Chronic e-cigarette exposure alters the human bronchial epithelial proteome. Am J Respir Crit Care Med. 2018;198:67–76. doi: 10.1164/rccm.201710-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoch KG, Lori A, Burns KA, Eldred T, Olsen JC, Randell SH. A subset of mouse tracheal epithelial basal cells generates large colonies in vitro. Am J Physiol Lung Cell Mol Physiol. 2004;286:L631–L642. doi: 10.1152/ajplung.00112.2003. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Matsumoto K, Brockway BL, Rackley CR, Liang J, Lee JH, et al. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells. 2012;30:1948–1960. doi: 10.1002/stem.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McQualter JL, Bertoncello I. Clonal culture of adult mouse lung epithelial stem/progenitor cells. Methods Mol Biol. 2015;1235:231–241. doi: 10.1007/978-1-4939-1785-3_17. [DOI] [PubMed] [Google Scholar]
- 42.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560:377–381. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604. doi: 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pollock K, Albares L, Wendt C, Hubel A. Isolation of fibroblasts and epithelial cells in bronchoalveolar lavage (BAL) Exp Lung Res. 2013;39:146–154. doi: 10.3109/01902148.2013.781720. [DOI] [PubMed] [Google Scholar]
- 46.Mou H, Vinarsky V, Tata PR, Brazauskas K, Choi SH, Crooke AK, et al. Dual SMAD signaling inhibition enables long-term expansion of diverse epithelial basal cells. Cell Stem Cell. 2016;19:217–231. doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butler CR, Hynds RE, Gowers KH, Lee DdoH, Brown JM, Crowley C, et al. Rapid expansion of human epithelial stem cells suitable for airway tissue engineering. Am J Respir Crit Care Med. 2016;194:156–168. doi: 10.1164/rccm.201507-1414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rejman J, Colombo C, Conese M. Engraftment of bone marrow-derived stem cells to the lung in a model of acute respiratory infection by Pseudomonas aeruginosa. Mol Ther. 2009;17:1257–1265. doi: 10.1038/mt.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong AP, Keating A, Lu WY, Duchesneau P, Wang X, Sacher A, et al. Identification of a bone marrow–derived epithelial-like population capable of repopulating injured mouse airway epithelium. J Clin Invest. 2009;119:336–348. doi: 10.1172/JCI36882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leblond AL, Naud P, Forest V, Gourden C, Sagan C, Romefort B, et al. Developing cell therapy techniques for respiratory disease: intratracheal delivery of genetically engineered stem cells in a murine model of airway injury. Hum Gene Ther. 2009;20:1329–1343. doi: 10.1089/hum.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu TJ, Tzeng YK, Chang WW, Cheng CA, Kuo Y, Chien CH, et al. Tracking the engraftment and regenerative capabilities of transplanted lung stem cells using fluorescent nanodiamonds. Nat Nanotechnol. 2013;8:682–689. doi: 10.1038/nnano.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosen C, Shezen E, Aronovich A, Klionsky YZ, Yaakov Y, Assayag M, et al. Preconditioning allows engraftment of mouse and human embryonic lung cells, enabling lung repair in mice. Nat Med. 2015;21:869–879. doi: 10.1038/nm.3889. [DOI] [PubMed] [Google Scholar]
- 53.Duchesneau P, Wong AP, Waddell TK. Optimization of targeted cell replacement therapy: a new approach for lung disease. Mol Ther. 2010;18:1830–1836. doi: 10.1038/mt.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gui L, Qian H, Rocco KA, Grecu L, Niklason LE. Efficient intratracheal delivery of airway epithelial cells in mice and pigs. Am J Physiol Lung Cell Mol Physiol. 2015;308:L221–L228. doi: 10.1152/ajplung.00147.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horani A, Nath A, Wasserman MG, Huang T, Brody SL. Rho-associated protein kinase inhibition enhances airway epithelial basal-cell proliferation and lentivirus transduction. Am J Respir Cell Mol Biol. 2013;49:341–347. doi: 10.1165/rcmb.2013-0046TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinovich KM, Iosifidis T, Buckley AG, Looi K, Ling KM, Sutanto EN, et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci Rep. 2017;7:17971. doi: 10.1038/s41598-017-17952-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peters-Hall JR, Coquelin ML, Torres MJ, LaRanger R, Alabi BR, Sho S, et al. Long-term culture and cloning of primary human bronchial basal cells that maintain multipotent differentiation capacity and CFTR channel function. Am J Physiol Lung Cell Mol Physiol. 2018;315:L313–L327. doi: 10.1152/ajplung.00355.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang C, Lee HJ, Shrivastava A, Wang R, McQuiston TJ, Challberg SS, et al. Long-term in vitro expansion of epithelial stem cells enabled by pharmacological inhibition of PAK1-ROCK-myosin II and TGF-β signaling. Cell Rep. 2018;25:598–610.e5. doi: 10.1016/j.celrep.2018.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trinh NT, Bardou O, Privé A, Maillé E, Adam D, Lingée S, et al. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur Respir J. 2012;40:1390–1400. doi: 10.1183/09031936.00221711. [DOI] [PubMed] [Google Scholar]
- 60.Meyerholz DK, Stoltz DA, Gansemer ND, Ernst SE, Cook DP, Strub MD, et al. Lack of cystic fibrosis transmembrane conductance regulator disrupts fetal airway development in pigs. Lab Invest. 2018;98:825–838. doi: 10.1038/s41374-018-0026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larson JE, Cohen JC. Improvement of pulmonary hypoplasia associated with congenital diaphragmatic hernia by in utero CFTR gene therapy. Am J Physiol Lung Cell Mol Physiol. 2006;291:L4–L10. doi: 10.1152/ajplung.00372.2005. [DOI] [PubMed] [Google Scholar]
- 62.Larson JE, Delcarpio JB, Farberman MM, Morrow SL, Cohen JC. CFTR modulates lung secretory cell proliferation and differentiation. Am J Physiol Lung Cell Mol Physiol. 2000;279:L333–L341. doi: 10.1152/ajplung.2000.279.2.L333. [DOI] [PubMed] [Google Scholar]
- 63.Hayes D, Jr, Kopp BT, Hill CL, Lallier SW, Schwartz CM, Tadesse M, et al. Cell therapy for cystic fibrosis lung disease: regenerative basal cell amplification. Stem Cells Transl Med. 2019;8:225–235. doi: 10.1002/sctm.18-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 65.Li X, Rossen N, Sinn PL, Hornick AL, Steines BR, Karp PH, et al. Integrin α6β4 identifies human distal lung epithelial progenitor cells with potential as a cell-based therapy for cystic fibrosis lung disease. PLoS One. 2013;8:e83624. doi: 10.1371/journal.pone.0083624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang L, Button B, Gabriel SE, Burkett S, Yan Y, Skiadopoulos MH, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. doi: 10.1371/journal.pbio.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lynch TJ, Anderson PJ, Rotti PG, Tyler SR, Crooke AK, Choi SH, et al. Submucosal gland myoepithelial cells are reserve stem cells that can regenerate mouse tracheal epithelium. Cell Stem Cell. 2018;22:653–667, e5. doi: 10.1016/j.stem.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tata A, Kobayashi Y, Chow RD, Tran J, Desai A, Massri AJ, et al. Myoepithelial cells of submucosal glands can function as reserve stem cells to regenerate airways after injury. Cell Stem Cell. 2018;22:668–683, e6. doi: 10.1016/j.stem.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.