

Abstract
The expanding knowledge on the hepatitis B virus (HBV) life cycle has improved the understanding of the human disease caused by this virus and led to the development of better treatments. It is important for the clinician taking care of patients with chronic hepatitis B to have a basic understanding of the molecular events involved in the HBV life cycle, so that they can better appreciate the natural history and atypical presenta- tions of the disease and can develop individual management plans based on readily available virologic tests. This article serves to introduce the clinically relevant aspects of the HBV life cycle as they pertain to patient management.
Keywords: Hepatitis B virus, Polymerase, Antiviral Therapy, cccDNA
VIROLOGY
The Virus
The human HBV is a member of the Hepadnaviridae family.1 Humans and higher-order primates are the only known hosts for human HBV. Other HBVs related to the human virus specifically infect hosts such as the ground squirrels, woodchucks, Peking ducks, and herons and serve as models of disease caused by human HBV.1
As visualized by electron microscopy, serum from patients with acute and chronic HBV infection exhibit 3 types of particles.2 The largest particle, a 42-nm spherical structure with a lipid bilayer, is referred to as the Dane particle and represents the intact virion. The outer lipid bilayer derived from host hepatocytes is studded with the hepatitis B surface antigen (HBsAg) and surrounds an inner nucleocapsid with an icosahedral structure composed of hepatitis B core antigen (HBcAg). The HBV genome and the viral polymerase reside within the nucleo- capsid. The other 2 particles visualized by electron microscopy appear either as small spherical particles or as filaments approximately 20 to 22 nm in size. These particles are devoid of any HBV genome and are composed of just host lipids and HBsAg; therefore, these particles are noninfectious.3 The subvirion particles are usually present in significant excess than the Dane particles, but why they are produced in such great numbers is unknown. It has been speculated that these noninfectious particles and filaments may serve as a decoy for the immune system and aid in the establishment of chronic infection. All 3 particles have a common antigen on their surface termed the HBsAg. Detection of HBsAg is the basis of the serologic assay for the diagnosis of chronic hepatitis B.
Genomic Organization
HBV is one of the smallest DNA viruses to infect humans. The HBV genome is a partially double-stranded circular DNA of approximately 3.2 kb in length.1,4 A unique feature of the genome is that the 2 DNA strands are asymmetric.4 The minus strand is genome length, whereas the complimentary positive strand is of variable length. There are 4 open reading frames (core [C], polymerase [P], surface [S], and X) that partially overlap because of the compact nature of the genome (Fig. 1). Four viral messenger RNAs (mRNAs) are generated from the viral genome.1,4 The C and S genes have a single open reading frame with multiple in-frame start codons that give rise to related but functionally different gene prod- ucts. The mRNAs are designated by their lengths as a 3.5-kb pregenomic mRNA, a 2.4-kb large surface mRNA, a 2.1-kb middle and small surface mRNA, and a 0.7-kb X mRNA.1,4 The 3.5-kb length mRNA actually exists as 2 subspecies: a slightly longer precore-core 3.5-kb mRNA and a shorter pregenomic 3.5-kb mRNA. The precore-core mRNA yields the precore –core protein, which undergoes posttranslational modification to give rise to the secretory hepatitis B e antigen (HBeAg) protein, whereas the pregenomic mRNA gives rise to the core and polymerase proteins and also serves as the template for replication of the virus. The biologic function of the HBeAg remains obscure. HBeAg is not required for viral replication but seems to be necessary for the establishment of chronic infection by acting as a tolerogen.5 Clinically, detection of HBeAg corre- lates with high-level viremia.6 Reduction in HBeAg level either spontaneously or during treatment is an important milestone in the natural history of the disease because it serves to indicate a transition from a high to a low viral replicative state.7 The S gene gives rise to 2 mRNA transcripts: a 2.4-kb large surface mRNA and a 2.1-kb middle and small surface mRNA. These 2 transcripts yield the 3 surface antigen proteins: large, middle, and small.1,4 All the three HBsAgs share the same carboxy-terminal region. The 0.7-kb X mRNA translates to yield the X protein. The function of the X protein (hepatitis B X antigen) remains incom- pletely understood, but emerging evidence seems to support a role in productive HBV replication, transcriptional activation, and DNA repair.8,9
Fig. 1.
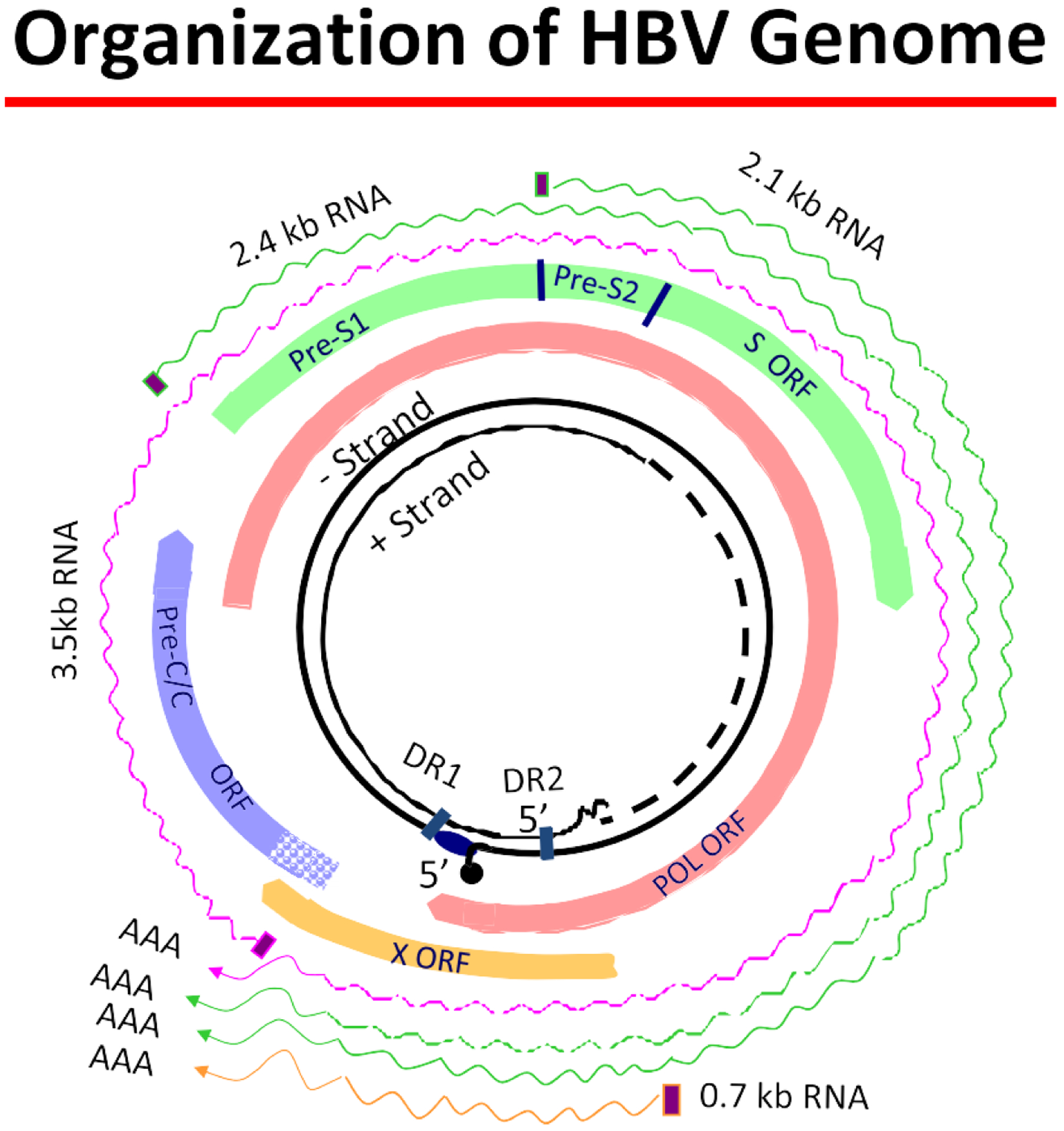
The viral genome is represented by the inner, black circles. It is a relaxed circular partially double-stranded DNA. The DNA’s circularity is maintained by 5°-cohesive ends containing 2 11-nucleotide direct repeats that are important for viral replication. The 2 strands are asymmetric. The minus strand is genome length and is covalently linked at its 5°end to the terminal protein domain of polymerase. The positive strand is of variable length and has a capped oligoribonucleotide on its 5° end. The genome has 4 open reading frames, which overlap due to the compact nature of the genome. These are all encoded by the minus strand and consist of the polymerase, precore/core, pre-S1/pre-S2, s/pre-s, and X. Finally, the outer wavy lines represent 4 viral messenger RNA (mRNA) species. The 3.5-kb pregenomic mRNA is genome length and the other 3 are subgenomic length. All terminate in a poly A tail.
Viral Life Cycle
The first step of initiating a productive infection involves the attachment of the mature Dane particle to the outer hepatocyte membrane through an unidentified host receptor. Evidence suggests that the large HBsAg plays a role in the attachment process.10,11 After attachment, the Dane particle is internalized into the cytoplasm and undergoes a series of transformations involving disassembly of the lipid bilayer and dissolution of the nucleocapsid.1,4 The relaxed circular form of the viral DNA is delivered to the nucleus where host and viral polymerases repair the partially double-stranded DNA to form the covalently closed circular DNA (cccDNA). The conversion of the relaxed circular DNA to a cccDNA involves the extension of the posi- tive strand to full length, removal of the relaxed circular DNA’s terminal modifications, and covalent ligation of the DNA ends.1,4,12,13 The cccDNA serves as the template for transcription of all the viral mRNAs.
The HBV mRNAs are then transported into the cytoplasm where translation of the viral proteins, self-assembly of core protein molecules into nucleocapsids, and progeny genome replication occurs.1,4 Replication requires encapsidation of the pre- genomic RNA by the core particle. This process is initiated by the coordinated binding of the viral polymerase to a secondary stem-loop structure, termed epsilon, on the pregenomic 3.5-kb mRNA, which triggers encapsidation by the core protein to form the viral nucleocapsid.14
After binding of the HBV polymerase to epsilon on the pregenomic mRNA, the poly- merase then serves as a protein primer to initiate negative-strand DNA synthesis. Elongation of a newly synthesized negative DNA strand subsequently proceeds with the pregenomic 3.5-kb mRNA serving as the template. After completion of the negative-strand DNA synthesis, the 3.5-kb mRNA template is degraded by the viral ribonuclease (RNase) H domain of the polymerase. Thereafter, the partially duplex HBV genome is synthesized using the newly synthesized negative strand as the template. The positive-strand synthesis is rarely complete, and thus the variable length of the positive strand in the HBV virion. Finally, the partially duplex HBV genome circularizes to assume the partially double-stranded, relaxed circular conformation within the nucleocapsid.1,4 Although HBV replicates via an RNA intermediate step, integration of the HBV DNA into the host DNA is not a requirement for replication as opposed to human immunodeficiency virus (HIV) in which the integration is essential for the life cycle.
Once the viral genome synthesis is completed, the immature viral nucleocapsid has 2 fates: it can acquire a lipid membrane with the HBsAg proteins in the endoplasmic reticulum to form mature Dane particles that are secreted from the hepatocyte or it can be transported back to the nucleus to replenish the pool of cccDNA.
The cccDNA
The cccDNA is central to the durability of HBV infection. It resides in the nucleus of infected hepatocytes as a stable, nonintegrated episomal DNA.15 The cccDNA assumes a supercoiled conformation that is reminiscent of a chromosomal structure complete with histone binding.16,17 The number of cccDNA molecules in each infected hepatocyte nucleus varies, ranging from 1 to 50 per cell nuclei.16,17 Unlike the hepa- tocyte chromosome, the cccDNA is not replicated by the host DNA replication machinery. Rather, maintenance of cccDNA is through an intracellular recycling pathway whereby newly synthesized but immature nucleocapsids are recycled from the cytoplasm to the nucleus to replenish the cccDNA level.18 Factors that regulate the number of cccDNA molecules in each nucleus remain incompletely understood. Immunologic, virologic, and epigenetic factors, including transcription factors, have been suggested to regulate cccDNA levels.15,19
cccDNA seems to be very stable within the hepatocyte and has been shown to persist after antiviral therapy and even after clearance of HBsAg in both animal and human studies.20–25 Because cccDNA serves as the template for the viral mRNAs, its stability in hepatocytes means that it can be a constant source of future viral progeny. Thus, cccDNA plays a significant role in reactivation of disease after stop- ping of antiviral treatment, following withdrawal of immunosuppression, in de novo hepatitis B after transplantation of anti-hepatitis B core grafts, and in the development of drug resistance. A particular concern over the use of oral nucleoside or nucleotide analogues is the archiving of resistant mutations in cccDNA, which may contribute to the development of multidrug-resistant viruses. Persistence of cccDNA is a major challenge in successfully eradicating HBV. Two leading mechanisms seem to be important for elimination of cccDNA. The first mechanism is based on a cytolytic mechanism by which infected hepatocytes are killed and replaced by regenerating noninfected hepatocytes.26,27 The second mechanism is based on a noncytolytic mechanism, whereby antiviral cytokines downregulate HBV gene expression and eliminate HBV from hepatocytes without cell death.28,29 Evidence exists for both mechanisms.
Given the central role of cccDNA in HBV replication, it is an attractive target for ther- apeutic intervention. Current treatment strategies have had little effect on cccDNA levels. In particular, nucleoside or nucleotide analogue inhibitors of the HBV poly- merase do not have a direct effect on cccDNA. Rather, they indirectly affect cccDNA levels through the abrogation of the intracellular recycling pathway. Several studies have examined the change in cccDNA levels during therapy with currently available nucleoside or nucleotide analogues used as monotherapy and in combination with peginterferon (Fig. 2).21,22,24,30,31 In all cases, with the exception of peginterferon plus adefovir dipivoxil,30 therapy for 1 year was associated with only modest reduction in cccDNA levels and none resulted in elimination of cccDNA. In contrast, serum HBV- DNA levels decreased by 4.7 to 5.5 log10 and cytoplasmic HBV-DNA levels in the hepatocytes decreased by about 2 log10. Based on currently available treatment data, mathematical modeling has projected that a therapeutic duration of 12 to 14 years would be necessary to achieve eradication of cccDNA from the liver. Recent data identifying cellular factors involved in the regulation of cccDNA may lead to the development of new antiviral therapies.32,33
Fig. 2.
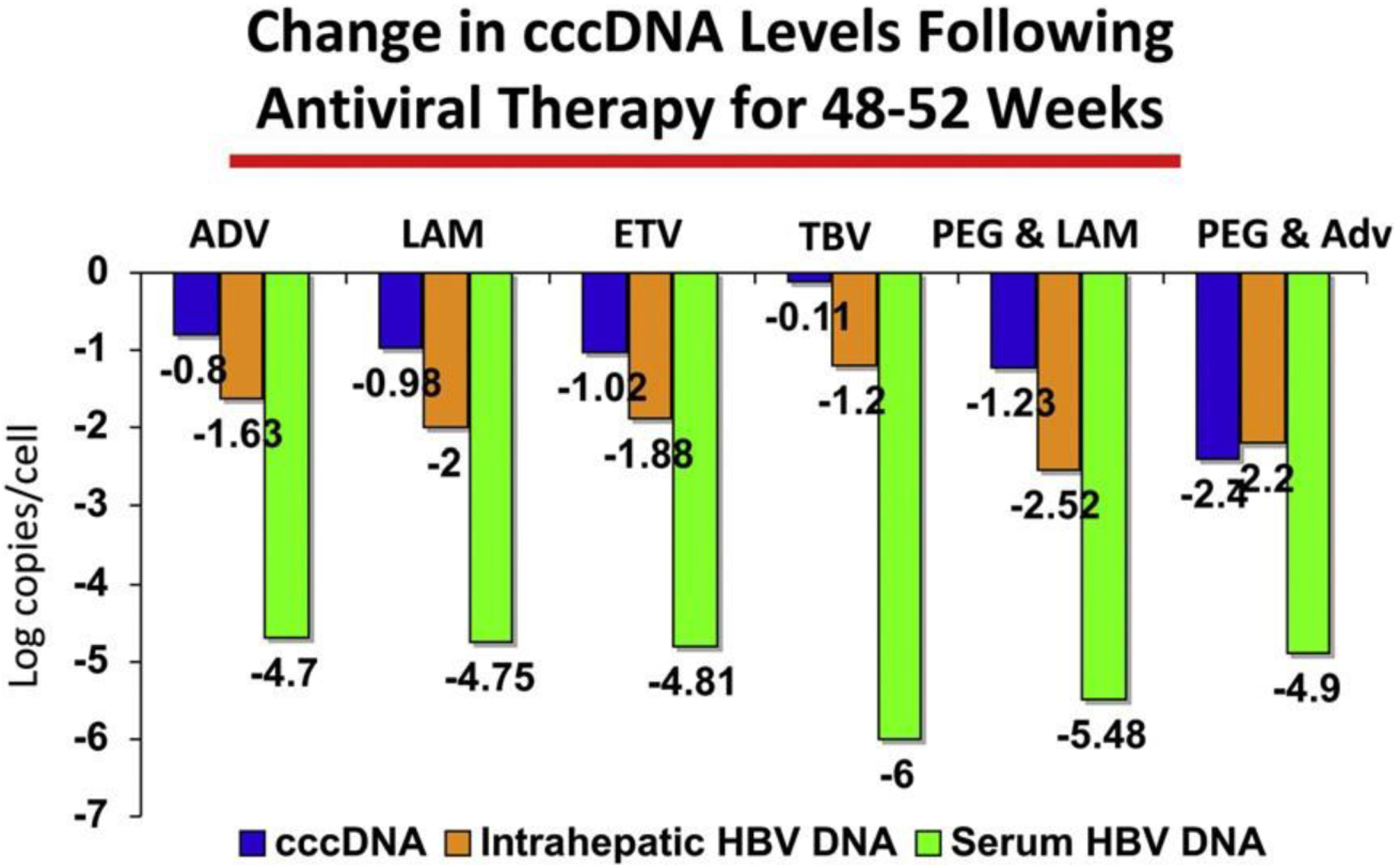
Comparison of change in cccDNA, intrahepatic, and serum HBV-DNA levels after anti- viral therapy for 48 to 52 weeks.
The Polymerase
The HBV polymerase has a central role in the synthesis of new virions. Knowledge of the structure-function relationship of the HBV polymerase has allowed a better understanding of how current nucleoside or nucleotide therapy works and conversely why it fails.34 The HBV polymerase is a multifunctional protein that is organized into 4 distinct domains beginning with a terminal protein domain at the amino-terminus region followed by a spacer region domain, a reverse transcriptase domain, and ending with an RNase H domain at the carboxy-terminus region. The terminal protein and spacer domains are unique to the HBV polymerase, with no phylogenetically related structures present on other viral polymerases.35 The terminal protein domain is absolutely essential for replication of the HBV genome.36,37 Similar to the replication schemes of other viruses, HBV uses a protein instead of the classic RNA or DNA termini as the primer for initiating DNA synthesis. Specifically, a tyrosine residue within the terminal protein domain acts as a primer and covalently links to the first nucleotide of the nascent negative-strand DNA.38,39 In contrast, the function of the spacer region is unknown and is dispensable without affecting polymerase func- tion. Unlike the terminal protein and spacer domains, the reverse transcriptase and RNase H domains share sequence motifs with other viral polymerases.35 The reverse transcriptase domain can be subdivided into 7 subdomains designated A to G.35 The subdomain C encompasses the DNA polymerization active site of the HBV polymerase and is the site of action for oral nucleoside or nucleotide analogues. The RNase H domain degrades the pregenomic mRNA template after negative- strand synthesis.
The crystal structure of the HBV polymerase has not been resolved. However, based on sequence homology with the HIV-1 reverse transcriptase and on the similar pattern of resistant mutations that occur with antiviral compounds that have both HBV and HIV activity, several predictions about the HBV polymerase can be made. Like other viral polymerases, HBV polymerase also has a right-handed structure with fingers, palm, and thumb regions.35 The palm region serves as the catalytically active site of the reverse transcriptase domain, where incoming nucleotides are attached to the elongating DNA of a new HBV genome. Structurally, the subdomains A, C, and perhaps D participate directly in nucleotide binding and catalysis.35 Subdomains B and E are involved with precise positioning of the primer-template complex relative to the incoming nucleotide in the active site and are components of the fingers and thumb regions.35
Nucleoside or nucleotide analogues inhibit HBV-DNA synthesis by acting as DNA chain terminators. Structurally, these analogues lack the 3°-hydroxyl group that permits covalent bonding to an incoming nucleotide on the elongating DNA chain. Therefore, after incorporation of the nucleoside or nucleotide analogue into the nascent DNA chain, subsequent nucleotides are unable to covalently bond with the analogue resulting in termination of replication. These analogues are potent inhibitors of HBV replication. However, a major challenge of nucleoside or nucleotide analogue therapy is the development of resistance mutations. Structure-function modeling helps to understand the mechanisms of resistance. In the case of lamivudine, structure-function modeling suggests that HBV polymerase–resistant mutations alter the spatial conformation of the nucleotide-binding pocket by steric hindrance, thus limiting lamivudine access to the polymerase.40,41 These changes also affect the natural substrate but to a lesser degree. An alternate model suggests that lamivudine may bind to the resistant polymerase but in a manner that is strained relative to the configuration of the wild-type polymerase.41 In this strained configuration, the position of lamivudine on the replication complex is not suitable for efficient catalysis.
OTHER THERAPEUTIC TARGETS
The widespread problem of drug resistance associated with nucleoside and nucleo- tide analogues targeting the HBV polymerase has led investigators to explore other targets for drug development. The identification of other targets for antiviral drug development stems from the knowledge gained through the understanding of the basic molecular biology of the HBV life cycle (Fig. 3). Stepwise advancements have been made in understanding the virion attachment to the hepatocyte, the assembly of nucleocapsids, and the mature virus and in the science of antisense and short inter- fering RNAs (siRNAs).
Fig. 3.
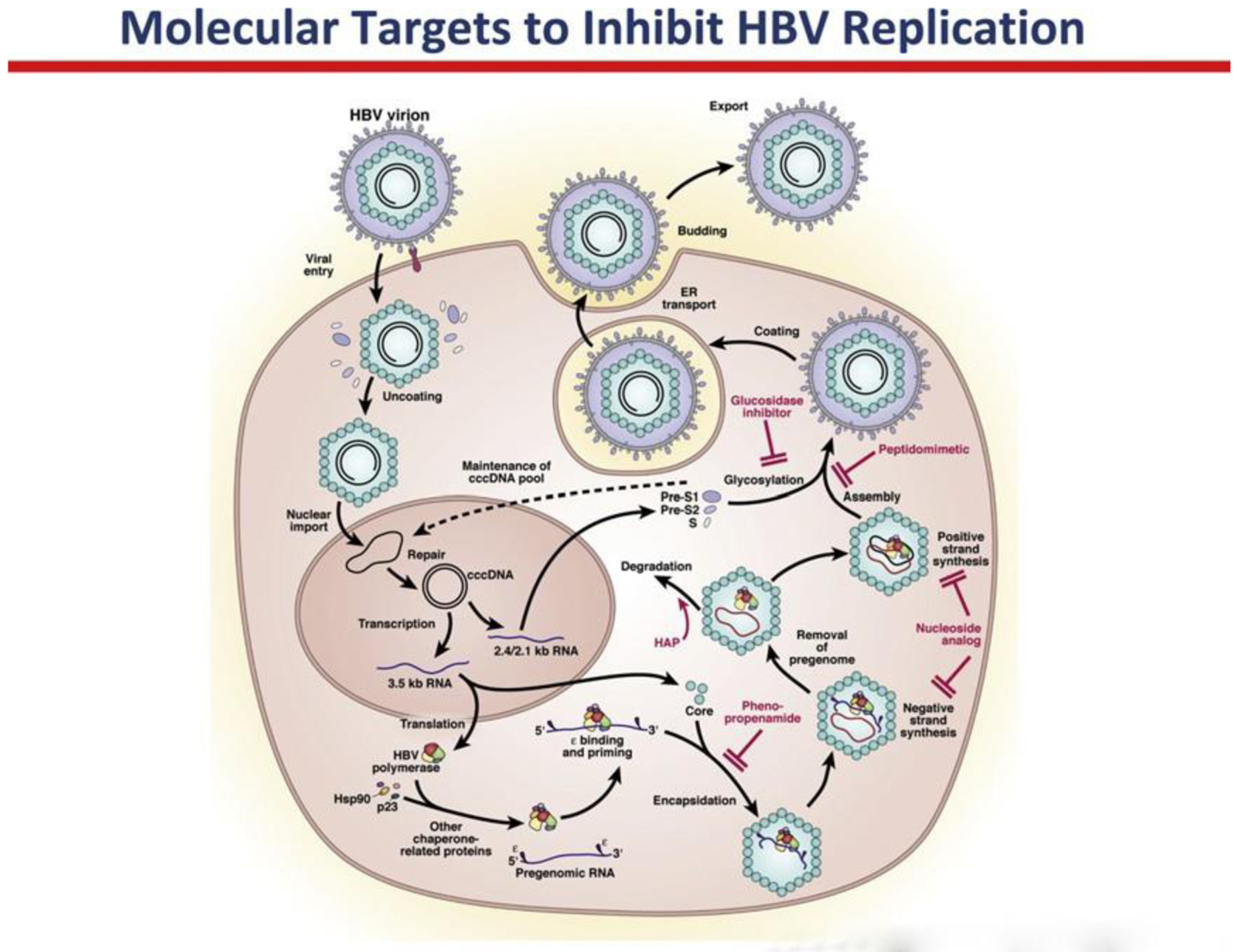
The small compact genome and the requirement of host cellular enzymes for many stages of the HBV life cycle have been an impediment to antiviral drug development. However, a better understanding of the HBV life cycle has identified several molecular targets and compounds that can disrupt new virion formation. These include viral entry inhibitors, encapsidation inhibitors, and agents that prevent viral assembly and coating. Heteroaryldihydropyrimidines (HAPs) bind to the core particles and lead to their degrada- tion. Another group of compounds that inhibit the encapsidation step are phenylpropena- mides. These compounds directly inhibit formation of the nucleocapsid. Antisense RNA and short interfering RNA (siRNA) have been shown to inhibit viral assembly in vitro and in animal models. Glucosidase inhibitors block the glycosylation of surface proteins during the viral assembly step. Recently, pre-S1 derived lipopeptides were shown to be potent inhibitors of HBV entry and may be useful after liver transplantation and perhaps in chronic hepatitis B. (From Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resis- tance in chronic hepatitis B. Gastroenterology 2007;132:1574–85; with permission.)
Inhibitors of Nucleocapsid Assembly
The HBcAg serves as the structural unit in the assembly of the viral nucleocapsid. As previously discussed, initiation of nucleocapsid formation occurs with the interaction of the HBV polymerase with epsilon on the pregenomic 3.5-kb mRNA, signaling the recruitment of HBcAg. Subsequently, HBcAg units aggregate on the initial complex and through a process of self-assembly lead to nucleocapsid formation. Heteroaryldi- hydropyrimidines (HAPs) are a class of compounds that have been shown to interfere with the HBcAg self-assembly into nucleocapsids and therefore inhibit HBV replication in a transgenic mouse model.42 The precise molecular mechanism by which HAPs disrupt nucleocapsid formation is unclear. Some studies suggest that HAPs accel- erate nucleocapsid formation but in a disordered fashion.43–45 The misassembled nucleocapsid is subsequently recognized by the host’s unfolded-protein response and is subjected to degradation by cellular proteosomes.43,44 This approach of inhib- iting HBV replication represents a promising direction for drug development.
Another class of compounds with nucleocapsid-assembly inhibitory activity are the phenylpropenamides. One derivative, AT-61, was found to be a potent inhibitor of HBV replication in tissue culture models.46,47 AT-61 was equally effective at inhibiting both the formation of intracellular immature core particles and the release of virions from hepatocytes. It seems to be active at one of the steps between the synthesis of viral RNA and the packaging of pregenomic mRNA into immature core particles.46,47 A related compound, AT-130, with more potent activity was reported to block HBV replication at the level of pregenomic mRNA packaging into nucleocapsids.48 To date, no human studies with these compounds have been reported.
Inhibitors of Viral Assembly
Glycosylation of the HBsAg is a requirement for viral assembly and proper function of the protein.1 N-glycosylation of proteins is important for proper folding of proteins by mediating interactions between the lectinlike endoplasmic reticulum chaperone proteins, calnexin and calreticulin. However, the N-glycans must first be modified by a-glucosidases to facilitate this interaction. These interactions can be prevented by inhibitors of a-glucosidases, which cause some proteins to be misfolded and retained within the endoplasmic reticulum. Several iminosugar compounds have been shown to have antiviral activity, although their mechanism of action is incom- pletely understood. An iminosugar compound, N-nonyl-deoxynojirimycin, was shown to inhibit woodchuck hepatitis virus in an animal model of infection.49 Evidence sug- gested that the antiviral activity was by a direct consequence of glucosidase inhibition and disruption of virion assembly via a dominant negative effect.49 However, another related compound N-nonyl-deoxygalactojirimycin (N-nonyl-DGJ), an alkyl derivative of galactose, had no effect on glycoprocessing yet retained anti-HBV activity.50 This property of N-nonyl-DGJ suggests that it exerted its antiviral action at a point before nucleocapsid envelopment by host membranes and may prevent the proper encapsi- dation of the HBV pregenomic mRNA.50 As with any agent that targets a host protein, there is the potential for toxicity, and these agents have been inadequately assessed in humans.
Inhibitors of Viral Entry
The development of the HBV-susceptible cell line HepaRG has facilitated systematic investigations of HBV entry and resulted in the discovery of entry inhibitors derived from viral envelope proteins. Several N-terminal myristoylated or steroylated pre-S1 peptides encompassing the amino acid positions 2 to 47 or 2 to 39 have been shown to block virus infectivity with a 50% inhibitory concentrations ranging from 8 to 300 nM.51 The postulated mechanism of action is unknown and may be through interaction and inactivation of a cellular receptor or viral protein. At present, the potential role of viral peptide–derived lipoproteins would be primarily preventative and limited to postexposure prophylaxis, prevention of vertical transmission, or graft reinfection after liver transplant. Whether these agents would have any role in patients with chronic infection is currently unknown.
GENETIC APPROACHES
RNA interference is an evolutionary conserved mechanism that uses short RNAs in association with an effector complex referred to as the RNA-induced silencing complex to regulate gene expression in a sequence-specific manner.52 Two classes of small RNAs mediate this process: microRNAs (miRNAs) and siRNAs.52 HBV is a promising target for an RNA interference approach because its compact genome lacks significant redundancy. Several approaches have been aimed at using in vitro and animal models with varying results. siRNA sequences targeting most of the subgenomic viral mRNAs have been tested and resulted in w60% to 80% reduction in protein expression from the targeted gene.52 Short hairpin RNAs have also been used and possess the advantage of being expressed from plasmids or viral vectors. Results are similar to those obtained with siRNAs. A promising approach may be the use of trimeric miRNA shuttle cassettes, which allows targeting of multiple sequences from a single transcript.
At present, this technology is still nascent with many technical challenges that need to be resolved before it can find widespread human application. The following chal- lenges are common to this therapeutic approach for a multitude of diseases: how to effectively deliver the siRNA or miRNA, how to achieve long-term gene silencing, how to deliver the molecule to the target site of action, and how to deal with the poten- tial for toxicity.
SUMMARY
A comprehensive, detailed understanding of the HBV life cycle and the host-virus interactions has led to the development of successful oral therapy for chronic hepatitis B and the identification of several promising new targets. The development of agents directed toward these targets may lead to new therapies for patients with chronic hepatitis B. With the improved knowledge gained from studying the molecular biology of HBV, novel approaches to inhibition of viral replication are being explored, such as viral entry inhibitors, nucleocapsid inhibitors, and inhibitors of viral assembly. However, the ultimate goal of therapy is to identify strategies to eliminate cccDNA from infected hepatocytes, which would have the potential to be curative and to prevent transmission and could reduce an infected individual’s risk for hepatocellular carcinoma. The future seems optimistic for addressing these goals.
Financial support:
This research was supported by the Intramural Research Program of the National Institutes of Health, Nutrition, National Institute of Diabetes and Digestive and Kidney Diseases.
REFERENCES
- 1.Seeger C, Zoulim F, Mason WS. Hepadnaviridae In: Knipe DM, Roizman B, Howley PM, et al. , editors. Fields virology. 5th edition Philadelphia: Lippincott- Raven Publishers; 2007. [Google Scholar]
- 2.Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoe- lectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974;75:489–502. [PMC free article] [PubMed] [Google Scholar]
- 3.Gavilanes F, Gonzalez-Ros JM, Peterson DL. Structure of hepatitis B surface antigen. Characterization of the lipid components and their association with the viral proteins. J Biol Chem 1982;257:7770–7. [PubMed] [Google Scholar]
- 4.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000;64: 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepa- titis B virus infection. Hepatology 2003;38:1075–86. [DOI] [PubMed] [Google Scholar]
- 6.Perrillo R, Campbell C, Wellinghoff W, et al. The relationship of hepatitis B e antigen, DNA polymerase activity, and titer of hepatitis B surface antigen with ongoing liver injury in chronic hepatitis B virus infection. Am J Gastroenterol 1982;77:445–9. [PubMed] [Google Scholar]
- 7.Hoofnagle JH, Dusheiko GM, Seeff LB, et al. Seroconversion from hepatitis B e antigen to antibody in chronic type B hepatitis. Ann Intern Med 1981;94: 744–8. [DOI] [PubMed] [Google Scholar]
- 8.Cross JC, Wen P, Rutter WJ. Transactivation by hepatitis B virus X protein is promiscuous and dependent on mitogen-activated cellular serine/threonine kinases. Proc Natl Acad Sci U S A 1993;90:8078–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol 2004;78:12725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glebe D, Urban S, Knoop EV, et al. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005;129:234–45. [DOI] [PubMed] [Google Scholar]
- 11.Klingmuller U, Schaller H. Hepadnavirus infection requires interaction between the viral pre-S domain and a specific hepatocellular receptor. J Virol 1993;67:7414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kock J, Schlicht HJ. Analysis of the earliest steps of hepadnavirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J Virol 1993;67:4867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sohn JA, Litwin S, Seeger C. Mechanism for CCC DNA synthesis in hepadnavi- ruses. PLoS One 2009;4:e8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollack JR, Ganem D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J Virol 1993;67:3254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoulim F. New insight on hepatitis B virus persistence from the study of intrahe- patic viral cccDNA. J Hepatol 2005;42:302–8. [DOI] [PubMed] [Google Scholar]
- 16.Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986;47:451–60. [DOI] [PubMed] [Google Scholar]
- 17.Newbold JE, Xin H, Tencza M, et al. The covalently closed duplex form of the hep- adnavirus genome exists in situ as a heterogeneous population of viral minichro- mosomes. J Virol 1995;69:3350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu TT, Coates L, Aldrich CE, et al. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 1990;175:255–61. [DOI] [PubMed] [Google Scholar]
- 19.Levrero M, Pollicino T, Petersen J, et al. Control of cccDNA function in hepatitis B virus infection. J Hepatol 2009;51:581–92. [DOI] [PubMed] [Google Scholar]
- 20.Mason WS, Cullen J, Moraleda G, et al. Lamivudine therapy of WHV-infected woodchucks. Virology 1998;245:18–32. [DOI] [PubMed] [Google Scholar]
- 21.Yuen MF, Wong DK, Sum SS, et al. Effect of lamivudine therapy on the serum covalently closed-circular (ccc) DNA of chronic hepatitis B infection. Am J Gas- troenterol 2005;100:1099–103. [DOI] [PubMed] [Google Scholar]
- 22.Wong DK, Yuen MF, Ngai VW, et al. One-year entecavir or lamivudine therapy results in reduction of hepatitis B virus intrahepatic covalently closed circular DNA levels. Antivir Ther 2006;11:909–16. [PubMed] [Google Scholar]
- 23.Bourne EJ, Dienstag JL, Lopez VA, et al. Quantitative analysis of HBV cccDNA from clinical specimens: correlation with clinical and virological response during antiviral therapy. J Viral Hepat 2007;14:55–63. [DOI] [PubMed] [Google Scholar]
- 24.Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004;126:1750–8. [DOI] [PubMed] [Google Scholar]
- 25.Moraleda G, Saputelli J, Aldrich CE, et al. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepa- titis virus. J Virol 1997;71:9392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fourel I, Cullen JM, Saputelli J, et al. Evidence that hepatocyte turnover is required for rapid clearance of duck hepatitis B virus during antiviral therapy of chronically infected ducks. J Virol 1994;68:8321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo JT, Zhou H, Liu C, et al. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections. J Virol 2000;74:1495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thimme R, Wieland S, Steiger C, et al. CD8(1) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 2003;77:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guidotti LG, Rochford R, Chung J, et al. Viral clearance without destruction of in- fected cells during acute HBV infection. Science 1999;284:825–9. [DOI] [PubMed] [Google Scholar]
- 30.Wursthorn K, Lutgehetmann M, Dandri M, et al. Peginterferon alpha-2b plus ade- fovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 2006;44:675–84. [DOI] [PubMed] [Google Scholar]
- 31.Sung JJ, Wong ML, Bowden S, et al. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology 2005;128:1890–7. [DOI] [PubMed] [Google Scholar]
- 32.Belloni L, Pollicino T, De Nicola F, et al. Nuclear HBx binds the HBV minichromo- some and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A 2009;106:19975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turin F, Borel C, Benchaib M, et al. n-Butyrate, a cell cycle blocker, inhibits early amplification of duck hepatitis B virus covalently closed circular DNA after in vitro infection of duck hepatocytes. J Virol 1996;70:2691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology 2007;132:1574–85. [DOI] [PubMed] [Google Scholar]
- 35.Bartholomeusz A, Tehan BG, Chalmers DK. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir Ther 2004;9:149–60. [PubMed] [Google Scholar]
- 36.Bartenschlager R, Junker-Niepmann M, Schaller H. The P gene product of hepa- titis B virus is required as a structural component for genomic RNA encapsida- tion. J Virol 1990;64:5324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartenschlager R, Schaller H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J 1992;11: 3413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weber M, Bronsema V, Bartos H, et al. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J Virol 1994;68:2994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zoulim F, Seeger C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol 1994;68:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investi- gation Group. Hepatology 1998;27:1670–7. [DOI] [PubMed] [Google Scholar]
- 41.Das K, Xiong X, Yang H, et al. Molecular modeling and biochemical characteriza- tion reveal the mechanism of hepatitis B virus polymerase resistance to lamivu- dine (3TC) and emtricitabine (FTC). J Virol 2001;75:4771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deres K, Schroder CH, Paessens A, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003;299:893–6. [DOI] [PubMed] [Google Scholar]
- 43.Bourne CR, Finn MG, Zlotnick A. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J Virol 2006;80:11055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stray SJ, Bourne CR, Punna S, et al. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 2005; 102:8138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stray SJ, Zlotnick A. BAY 41–4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit 2006;19:542–8. [DOI] [PubMed] [Google Scholar]
- 46.King RW, Ladner SK, Miller TJ, et al. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta- L-2’,3’-dideoxy-3’-thiacytidine. Antimicrob Agents Chemother 1998;42:3179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaney WE 4th, Edwards R, Colledge D, et al. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob Agents Chemother 2002;46:3057–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feld JJ, Colledge D, Sozzi V, et al. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res 2007; 76:168–77. [DOI] [PubMed] [Google Scholar]
- 49.Block TM, Lu X, Mehta AS, et al. Treatment of chronic hepadnavirus infection in a woodchuck animal model with an inhibitor of protein folding and trafficking. Nat Med 1998;4:610–4. [DOI] [PubMed] [Google Scholar]
- 50.Mehta A, Carrouee S, Conyers B, et al. Inhibition of hepatitis B virus DNA replica- tion by imino sugars without the inhibition of the DNA polymerase: therapeutic implications. Hepatology 2001;33:1488–95. [DOI] [PubMed] [Google Scholar]
- 51.Petersen J, Dandri M, Mier W, et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol 2008; 26:335–41. [DOI] [PubMed] [Google Scholar]
- 52.Wilson R, Purcell D, Netter HJ, et al. Does RNA interference provide new hope for control of chronic hepatitis B infection? Antivir Ther 2009;14:879–89. [DOI] [PubMed] [Google Scholar]