

Abstract
Purpose:
Angiosarcomas (AS) are soft tissue sarcomas with endothelial differentiation and vasoformative capacity. Most AS show strong constitutive expression of the endothelial adhesion receptor CD31/PECAM-1 pointing to an important role of this molecule. However, the biological function of CD31 in AS is unknown.
Experimental design:
The expression levels of CD31 in AS cells and its effects on cell viability, colony formation and chemoresistance was evaluated in human AS clinical samples and in cell lines through isolation of CD31high and CD31low cell subsets. The redox-regulatory CD31 function linked to YAP signaling was determined using a CD31 blocking antibody and siRNA approach and was further validated in CD31-knockout endothelial cells.
Results:
We found that most AS contain a small CD31low cell population. CD31low cells had lost part of their endothelial properties, were more tumorigenic and chemoresistant than CD31high cells due to more efficient reactive oxygen species (ROS) detoxification. Active downregulation of CD31 resulted in loss of endothelial tube formation, nuclear accumulation of YAP, and YAP-dependent induction of antioxidative enzymes. Addition of pazopanib, a known enhancer of proteasomal YAP degradation re-sensitized CD31low cells for doxorubicin resulting in growth suppression and induction of apoptosis.
Conclusions:
Human AS contain a small aggressive CD31low population that have lost part of their endothelial differentiation programs and are more resistant against oxidative stress and DNA damage due to intensified YAP signaling. Our finding that the addition of YAP inhibitors can re-sensitize CD31low cells towards doxorubicin may aid in the rational development of novel combination therapies to treat AS.
Keywords: Angiosarcomas, CD31/PECAM-1, oxidative stress, pazopanib, YAP
INTRODUCTION
Angiosarcomas (AS) are rare vasoformative sarcomas arising in the skin, soft tissues and various other locations including breast, liver, spleen and bone (1). Patients with long standing lymphedema (so-called Stewart-Treves-syndrome) (2, 3) or with a history of irradiation (4), e.g. for breast cancer, are at increased risk. The median overall survival rates for AS drop from 51 months in localized to only 12 months in metastatic tumors (5–7). Current therapeutic approaches include surgical excision for localized tumors and systemic therapy using agents such as doxorubicin or paclitaxel (8). However, most AS soon become resistant to chemotherapy and no treatment has so far been shown to prolong overall survival (7, 8). Clinical trials using anti-angiogenic agents such as bevacizumab or multikinase inhibitors targeting angiogenesis pathways such as sorafenib, imatinib or sunitinib were also mostly disappointing (9, 10).
The pathohistological diagnosis of AS rests on the morphological and immunohistochemical identification of endothelial differentiation using antibodies against e.g. CD34, CD31, D2–40/podoplanin, and ERG. For largely unknown reasons, platelet-endothelial cell adhesion molecule 1 (PECAM1/CD31) is a very robust marker of endothelial differentiation in AS and is usually expressed even in rare variants (11–13), pointing to an important role of PECAM1/CD31 in the biology of these tumors, whereas other endothelial markers, such as CD34, are frequently lost (11–13). CD31 expression is normally restricted to endothelial cells, circulating platelets, monocytes, and circulating T cell subsets. PECAM1/CD31 is an adhesion receptor that has several important functions including immune cell adhesion and transendothelial migration, coagulation, angiogenesis and integrin activation (14, 15). CD31 has been shown to play an important role in the regulation of apoptosis as well as cell adhesion-mediated cell growth and in the maintenance of vascular permeability and integrity (16, 17). It was recently demonstrated that genetic depletion of CD31 in endothelial cells resulted in reduced VE-cadherin expression, increased survivin expression, and nuclear localization of Yes-associated protein (YAP), an important mediator of the Hippo pathway. This led to increased proliferation and decreased apoptosis through transcriptional activation of genes required to promote cell growth and inhibit apoptosis (17).
In order to study the biological function of CD31 in AS, we analyzed the expression of CD31 in human AS tissue samples and in AS-derived cell lines. We here show that suppression of CD31 protects AS from oxidative DNA injury and ROS production. Mechanistically, this redox-protection is achieved through nuclear accumulation of YAP. Our data suggest activation of the Hippo pathway as a general mechanism in malignant endothelial tumors with potential relevance for the development of more effective treatments for these aggressive neoplasms.
MATERIAL AND METHODS
Human tissue microarray and immunohistochemical analysis.
All AS samples were taken from a previously published cohort (46). Immunohistochemical staining of CD31 was performed on 3 μm paraffin sections using standard procedures on a Dako Omnis platform (CD31 (JC70A), ready-to-use, Dako, Danmark). All experiments with human AS samples were obtained with full patient consent and carried out in compliance with the ethical regulations approved by the Ethics Committee of the University Medical Center Göttingen (Gö 26/8/15) and of the University Medical Center Mannheim (Ma 2012–289N-MA).
Cells and cell culture.
The following primary human endothelial cell lines were used: HUVEC and HDMEC cells were purchased from Promocell (Heidelberg, Germany) and passaged for seven times maximum. The angiosarcoma cell lines ASM (provided from Vera Krump-Konvalinkova, University Mainz, Germany) (19), HAMON (provided by Daichi Hoshina) (21), ISO-HASc.1 (subclone of the ISO-HAS cell line generated and provided by James Kirkpatrick, University Mainz, Germany) (20) and the transformed fusion cell line EA.hy926 cell line (DMSZ, Germany). Murine endothelial cells from CD31+/+ and CD31−/− mice were extracted as previously described (28). HUVEC, HDMEC and ASM were cultured in Endothelial cell growth medium, advanced (Provitro, Germany), HAMON cells were cultured in EGM-2 BulletKit endothelial cell medium (Lonza, Cologne, Germany), ISO-HASc.1, EA.hy926 and murine endothelial cells were cultured in DMEM medium supplemented with 10% serum, 1% L-glutamine and 1% penicillin/streptomycin (all from Thermo Fisher Scientific, USA).
Antibodies and reagents.
The following antibodies were used: CD31 antibody clone JC70A from DAKO (used for immunhistochemistry and immunoblot analysis), clone D8V9E from Cell Signaling, Frankfurt, Germany (immunoblot analysis). vWF (clone A0082) and CD44 (clone M7082) were purchased from DAKO. Antibodies against YAP (#14074), p-H2AX (γ-H2AX) (#9718), caspase-3 (#9665), caspase-7 (#12827), PARP (#9542), Beclin-1 (#3495), LC3B (#12741), total GSK3β (#12456) and p-GSK3β (Ser9) (#5558) and p-AKT (Ser473) (#4060), Catalase (#12980) were all purchased from Cell Signaling, mouse monoclonal β-actin (clone AC-74) from Sigma. Antioxidants N-Acetyl-L-cysteine (NAC), Ferrostatin-1 (Fer-1) and vitamine E (trolox); sodium arsenite (AsN), proteasome blocker MG-132 (MG), HSP90 inhibitor 17-AAG, MEK inhibitor U0126 and p38MAPK inhibitor SB203580 were all purchased from Sigma-Aldrich, Taufkirchen, Germany. JNK inhibitor SP600125 and multikinase inhibitor pazopanib were from Selleckchem, Munich, Germany and selective p38α inhibitor JX401 from Tocris, Wiesbaden, Germany.
Protein extracts, western blot analyses and angiogenesis array analysis.
Cells at 60–70% density were treated with indicated conditions and time points. After subsequent washing in PBS, cells were scraped in RIPA lysis buffer. Total protein concentrations were analysed using Biorad protein assay. Cell extracts were immunoblotted as described elsewhere (47). For determination of differential expression of pro- and anti-angiogenic factors, total protein extracts from indicated cell lines (200 μg total) were analyzed using the Human Angiogenesis Antibody Array (R&D Systems). All arrays were analyzed and quantified using Image J software (NIH).
Immunofluorescence of YAP, CD31 and ERG.
Cells were cultivated on glas coverslips to a confluence of 70–80% and after washing fixated in 4% buffered paraformaldehyde (PFA) for 10 minutes at RT. After PBS washing nuclei were permeabilized in 0.3% Triton-X for 10 minutes at RT. After washing unspecific binding sites were blocked with 1% BSA for 30 minutes at RT. After an additional washing cells were subjected to primary YAP antibody (#14074, Cell Signaling) in a 1:100 dilution at room temperature for 1 hour. Cells were then incubated with a fluorescent secondary antibody Alexa488 (1:600) and DAPI (1:2500) (both Thermo Fisher, USA) for 1 hour at room temperature. Coverslips were then upside down embedded in Mowiol and fluorescent signals were scanned on a confocal laserscan microscope with a 60x optical enlargement (Olympus, Germany). Immunofluorescence stainings on formalin-fixed, paraffin-embedded tissues were performed on 3 μm sections. After deparaffinization, rehydration and antigen retrieval in citrate buffer (pH 6.1), the rabbit monoclonal anti-ERG (EP111) and the mouse anti-CD31 monoclonal antibody (PECAM-1) (JC70A) (both Dako, Glostrop, Denmark) were incubated in the Autostainer (Dako, Glostrop, Denmark) according to the recommended protocol. Thereafter, the sections were sequentially incubated for 30 min with a secondary anti-rabbit antibody labeled with Alexa488 (Thermo Fisher, USA) and a secondary anti-mouse antibody labeled with CY3 (Thermo Fisher, USA). Samples were embedded in Mowiol and fluorescent signals were detected using a confocal laser scanning microscope with a 60× optical and 1.5× digital enlargement (Olympus, Germany).
Tube formation assay.
In brief, 100 μl of Matrigel (BD Bioscience) was added to each well of a 96-well plate and allowed to polymerize for a minimum of 30 min at 37°C. Indicated cell lines were re-suspended in endothelial cell medium and seeded in each well at a concentration of 1 × 105 cells/well. Cells were monitored under a light microscope to determine the formation of tube-like structures and representative pictures were taken at indicated time points. HUVEC cells were used as a positive control forming capillary-like structures as early as 4–6 hrs and matured after 24 hrs.
Knockdown experiments using siRNA.
Cells were transfected with 80 nM siRNA against either CD31 (Hs_PECM1_7, cat. nr. SI05098863) or YAP (Hs_YAP1_5, cat. nr. SI0266254) (Qiagen, Hilden, Germany) using HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) according to the manufactureŕs protocol. Briefly, 100 μl transfection medium containing 12 μl HiPerFect, 9.6 μl siRNA / negative control (20 μM) and 88.4 μl RPMI160 medium without FCS was incubated at room temperature for 20 min and added to 3×105 cells in 2.3 ml medium just after seeding. Cells were incubated for 24 hrs and further processed for immunoblot analysis.
Flow cytometric analysis of CD31, ROS species and cell trace experiments.
For determination of CD31 expression, cells were incubated with CD31-PE (monoclonal, 390, Biolegend) for 30 min, washed twice with ice-cold PBS and analyzed and sorted by a BD FACS Aria. For analyses of intracellular cytoplasmic ROS, cells were incubated in FCS-free medium containing 20 μM 2’,7’ –dichlorofluorescin diacetate (DCFDA) for 10 min at 37°C, for assessing lipid peroxidation incubated with 20 μM C11-BODIPY581/591 dye for 30 min and for mitochondrial ROS incubated with 20 μM MitoSOX dye for 30 min (all from Invitrogen), washed twice and analyzed via flow cytometry. For cell tracking indicated cell lines were incubated with CellTrace Violet dye (final concentration 5 μM) for 20 min at 37°C protected from light. After quenching unbound dye, stained cells were combined with unstained cells and left untreated or incubated with increasing doxorubicin doses. After 24 hrs incubation, all cells were washed in PBS and analyzed via flow cytometry using 405 nm laser and 450 nm emission filter. Viable marked cells were quantified and presented as percent labeled cells to untreated controls.
Measurement catalase activity.
CD31high and CD31low cells were tested for catalase activity using the OxiSelectTM Catalase Activity Assay (Cell Biolabs, USA) according to the manufacturer’s protocol. In brief, cells were harvested and sonicated in cold PBS with 1mM EDTA. Samples were centrifuged at 10.000 × g for 15 min at 4°C and supernatant was used for catalase activity measurement. 20 μL of the diluted catalase standards or unknown samples were incubated exactly for 1 min with 50 μL hydrogen peroxide working solution (12 mM). Then the reaction was stopped with 50 μL catalase quencher. 5 μL of each reaction were mixed with 250 μL of the chromogenic working solution. After 40 min incubation absorbance was measured at 520 nm. Data are presented as relative values.
RNA extraction and Nanostring analysis.
For RNA isolation cells were harvested by trypsination and pelleted by centrifugation at 800g. The dry pellet was lysed in 1ml Trizol (Invitrogen) and processed according to the manufacturer’s protocol. 50 ng of total RNA was used to measure gene expression. Expression profiles were established using a nCounter custom made Panel for oxidative genes and the nCounter Digital Analyzer (nanoString Technologies, Seattle, WA, USA) according to the manufacturer’s protocol.
Viral vector production and transduction.
Unsorted wild-type ASM cells with a previous determined high (48%) CD31low fraction was used. Non-targeting scrambed shRNA (plasmid #1864) was obtained from Addgene. Lentiviral particles were produced in HEK293T cells with plasmids pCMV-Δ-R8.91 (containing gag, pol, and rev genes) and pMD.G (VSV-G-expressing plasmid) following standard protocols. After transduction, cells were kept for 72 hrs under 2 μg/ml puromycin (InvivoGen) resulting in more than 70% dead cells. Surviving clones (initial 27) were picked, expanded and 6 representative clones were immunblotted for CD31.
Cell viability assay and colony formation analysis.
For cell viability analysis equal numbers of cells were seeded into 96-well flat-bottom plates and incubated for indicated time points. Cell viability was determined by adding MTS (Promega, Mannheim, Germany) to each well for 2 hrs at 37°C. For colony formation, cells were diluted in medium and plated in methylcellulose (from R&D) at 1×10³ cells/30 mm plates. For treatment experiments, cells were incubated for 24 hrs with indicated compounds. After treatment cells were trypsinized, pelleted and seeded in methylcellulose as outlined above. After incubation for at least 7–16 days at 37°C in 5% humified CO2, formed compact colonies were counted using inverted microscopy.
In vivo tumorigenic studies using chick chorioallantoic membrane (CAM)-Assay.
CAM assay was used to determine in vivo tumorigenicity as described elsewhere (22). In brief, equal cell numbers (2.5 × 106/egg) of indicated ASM sublines were mixed with matrigel (BD Matrigel) and implanted on the CAM on embryonic day 10 (E10) chicken embryos. On day 10 (E20) after implantation, formed tumors were explanted, weighed, and photographed.
Statistical analysis.
All experiments were performed at least in triplicates unless specified otherwise. Differences between treatment groups were either evaluated using unpaired t-test, one- or two-way ANOVA followed by Bonferroni post-hoc analysis, as indicated. Statistical analyses were performed using GraphPad Prism Software (GraphPad Software Inc.)
RESULTS
Most human angiosarcomas and angiosarcoma-derived tumor cell lines contain a small population of CD31low cells.
CD31 expression was assessed in 17 full section paraffin samples of human angiosarcomas. Of these, the majority (n=11; 65%) showed a small population of CD31low tumor cells (Fig.1A and Supplemental Fig. 1A).
Figure 1. Heterogenous expression of CD31 in human samples in situ and patient-derived angiosarcoma cells in vitro.
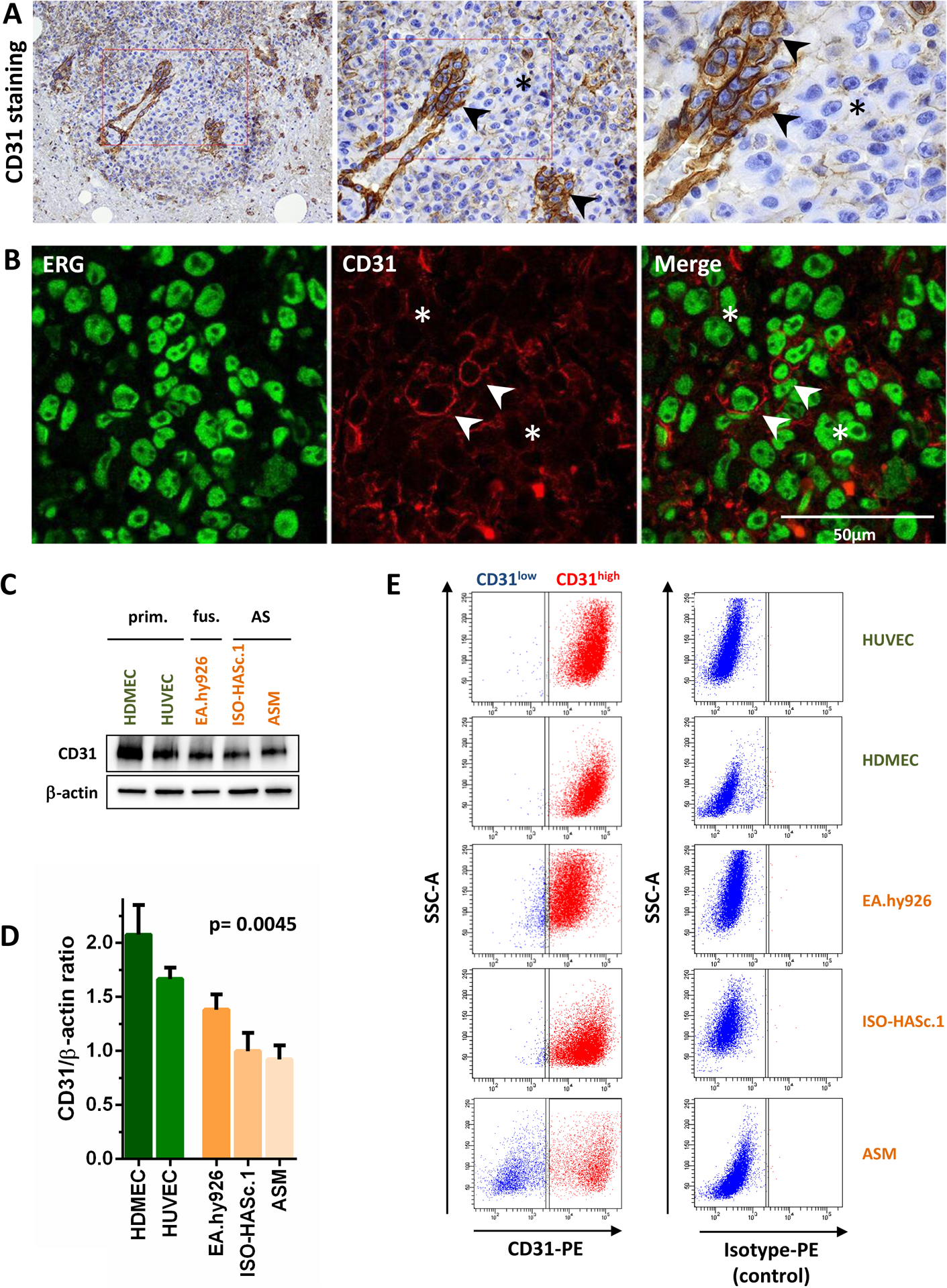
(A) CD31 immunohistochemical staining in an epitheloid human angiosarcoma showing very low expression in this tumor area (*), while neoplastic endothelial cells forming a vascular structure are strongly positive (arrow heads). Other regions of the tumor showed strong expression of CD31. See also Supplemental Fig. 1A for more CD31 stainings. (B) Immunofluorescence double staining of the same case showing multiple ERG positive tumor cell nuclei (green). Few cells show membranous co-expression of CD31 (CD31high, red staining), while most cells in this part of the tumor were CD31low. Arrow heads highlight regions with CD31/ERG co-localization, while (*) marks ERG positive regions with low CD31 expression. (C) Immunoblot analysis and (D) densitometric quantification of CD31 expression in normal primary endothelial cells (HUVEC and HDMEC), the hybrid cell line EA.hy926 (HUVEC fused with lung adenocarcinoma cells), and angiosarcoma (AS) cell lines ISO-HASc.1, HAMON and ASM, showing lower CD31 levels in neoplastic endothelial cells (n=3). (E) FACS analysis using a CD31-PE or isotype-PE (control) antibody was performed in all indicated cell lines. While non-neoplastic primary endothelial cells (HUVEC and HDMEC) presented homogenous CD31 expression, the EA.hy926 cell line as well as the angiosarcoma cell lines ISO-HASc.1 and ASM showed a substantial proportion of CD31low cells. See also Supplementary Figure 1B for a representative CD31 FACS analysis of the HAMON angiosarcoma cell line and Supplementary Figure 1C for statistical analysis of all cell lines.
To analyze CD31 expression in healthy and malignant endothelial cell lines, we used primary endothelial cells derived from human umbilical cord (HUVEC) and dermal endothelial cells (HDMEC), a hybrid immortalized cell line (EA.hy926) in which normal HUVEC cells had been fused with the lung adenocarcinoma cell line A549 (18), and the human AS cell lines ASM, ISO-HASc.1 (a cell line subcloned from the original ISO-HAS cell line) (19, 20) and the newly established HAMON cell line (21). Western blotting showed strong CD31 expression in all cell lines, but higher levels in normal endothelial cells compared to the hybrid cells and AS cell lines (Fig. 1B and C). To further investigate whether CD31 heterogeneity was also present in these cell lines, we performed fluorescence-activated cell sorting (FACS) using a primary phycoerythrin (PE)-labeled CD31 antibody and isotype-specific control antibodies. In the non-neoplastic HUVEC and HDMEC cells, 99.6% of the cells showed high levels of CD31. In contrast, analysis of the tumor cell lines showed a small but consistent CD31low population (ranging from 1.7% in EA.hy926 to 47.8% in ASM), which was reproducible in repetitive tests independent of the passage number or cultivation method although the population size was variable (Fig. 1D and Supplemental Fig. 1B).
We conclude that both human AS tissue samples as well as cell lines contain variable numbers of CD31low tumor cells.
Separation of CD31high and CD31low endothelial cell fractions in angiosarcoma cell lines.
Because ASM cells contained the highest fraction of CD31low cells, we mainly focused on this cell line for further experiments. In order to establish stable sublines of CD31 high vs. low cells, we transduced ASM cells with the empty lentiviral vector pLKO.1 puro under puromycin selection, and subsequently picked resistant single cell colonies (Fig. 2A). Six representative clones were expanded and subjected to immunblot analysis for CD31. For following experiments, ASM subclones #3 (termed CD31low hereafter) and #6 (termed CD31high hereafter) were used. Both subfractions preserved their initial CD31high vs CD31low phenotype even after multiple passages, while the expression level of von-Willebrand-factor (vWF) was similar, suggesting that both cell fractions were endothelial in nature in spite of different CD31 levels (Fig. 2B).
Figure 2. CD31 expression in ASM angiosarcoma cells separates vasoformative ‘organotypic’ CD31high cells from the more aggressive CD31low cells with spindle cell morphology.
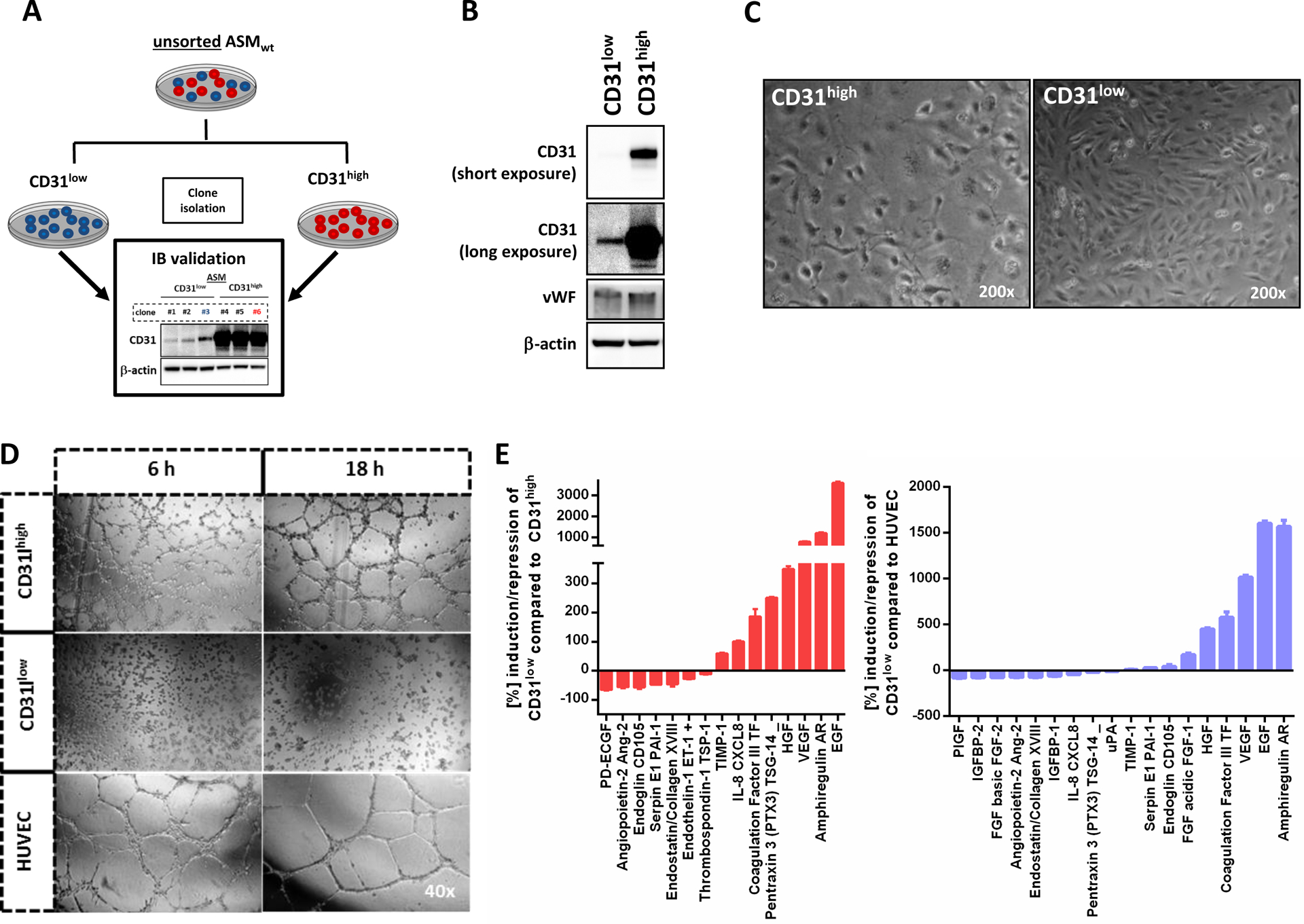
(A) Schematic summary of the experimental protocol used to isolate CD31high and CD31low fractions from the ASM cell line. Unsorted ASMwt (wild-type) cells were stably transduced with the empty pLK0.1 vector under puromycin selection. Subsequently, surviving single clones with picked and immunoblotted (IB validation) for CD31 and classified as CD31high or low CD31low cells. Repetitive CD31-FACS analysis from CD31high (clone #3) and CD31low (clone #6) and Western blot demonstrated stable levels of CD31 up to 44 passages (data not shown). (B) Western blot analysis revealed only differential expression of CD31, but not of von-Willebrand factor (vWF), suggesting that both populations were endothelial cells. Longer exposure was needed to detect the presence of CD31 in CD31low cells. (C) Microscopic images of CD31high and CD31low cells showing a polygonal cell shape in non-confluent CD31high cells and cobblestone pattern upon confluence. In contrast, CD31low cells had a more fibroblastic, spindle cell morphology. (D) HUVEC formed organized networks of tubular endothelial structures which matured after 6 hours and even presented signs of degradation after 18 hours. In CD31high cells, pre-tubular structures could be detected after 6 hours and mature tubes after 18 hours. In strong contrast, CD31low cells did not show tubular structures at any time point, even after extended observation for up to 24 hours (data not shown). (E) In a screening of 55 angiogenesis-associated proteins, 18 factors with both pro- and antiogenic properties were differentially expressed in CD31low compared to CD31high cells and HUVEC (n=2).
CD31 is essential for vasoformation of malignant endothelial cells.
Next, we sought to characterize the ASM CD31low and CD3high sublines. Under normal culture conditions containing 20% serum and low cell density, CD31high cells appeared oval-shaped and built a cobblestone pattern as monolayer, similar to primary HUVEC and HDMEC cells. In strong contrast, CD31low cells were more spindly, elongated and flattened, resembling fibroblasts (Fig. 2C).
To study their capacity to form vascular structures, we performed tube formation assays and included HUVEC as a positive control. As expected, HUVEC formed organized networks of vascular structures after 6 hours that matured after 18 hours. In CD31high cells, pre-tubular structures could be detected after 6 hours and mature tubes after 18 hours. In strong contrast, CD31low cells did not show tubular structures at any time point, even after extended observation for up to 24 hours (Fig. 2D).
To further characterize CD31high and CD31low cells at the molecular level, we measured 55 angiogenesis-related proteins in whole cell lysates of both ASM sublines. Compared to CD31high ASM cells and HUVEC, many angiogenic factors such as angiopoietin-2, PDECGF (thymidine phosphorylase) and endothelin-1 were strongly suppressed in CD31low cells, whereas established angiogenic inhibitors such as TIMP-1 were upregulated. Surprisingly, several other potent factors involved in angiogenesis such as HGF, amphiregulin AR and VEGF were strongly activated along with suppression of the potent angiogenic inhibitor thrombospondin-1, suggesting a complex molecular counterregulation to compensate for the loss of CD31 function (Fig. 2E).
CD31low cells are more resistant to serum starvation and have increased protumorigenic properties in vitro and in vivo.
In order to further study the biological differences between CD31high and CD31low cells, we compared their cell growth under normal culture conditions and under serum-reduced starvation. CD31low cells showed a significantly higher proliferation rate than CD31high cells under normal culture conditions (Fig. 3A) and higher cell survival under serum deprivation (Fig. 3B). Moreover, CD31low cells survived up to 16 days in semi-solid methylcellulose media and were even able to form conglomerates of two-dimensional colonies. In strong contrast, CD31high cells were mainly dispersed as single cells after 7 days that did not survive up to 16 days under these conditions (Fig. 3C). Similar results could be also observed in normal HUVEC cells (data not shown). According to the initial report and own previous observations, ASM and ISO-HASc.1 cells failed to establish tumors when xenografted into immunodeficent mice. One reason could be lack of an established microenvironment needed for tumor formation and propagation (19). Therefore, we utilized the chicken chorioallantoic membrane (CAM) xenograft assay to analyze in vivo tumorigenicity (22). The CAM is a highly vascularized extraembryonic membrane providing optimal delivery of growth supplements, notably with an immature immune system. Indeed, under these conditions, both sublines formed detectable tumors 10 days after implantation. In line with our in vitro results, inoculated CD31low cells formed significantly larger and heavier tumors than their CD31high counterparts (Fig. 3D).
Figure 3. CD31low cells are more resistant to serum starvation and have increased protumorigenic properties.
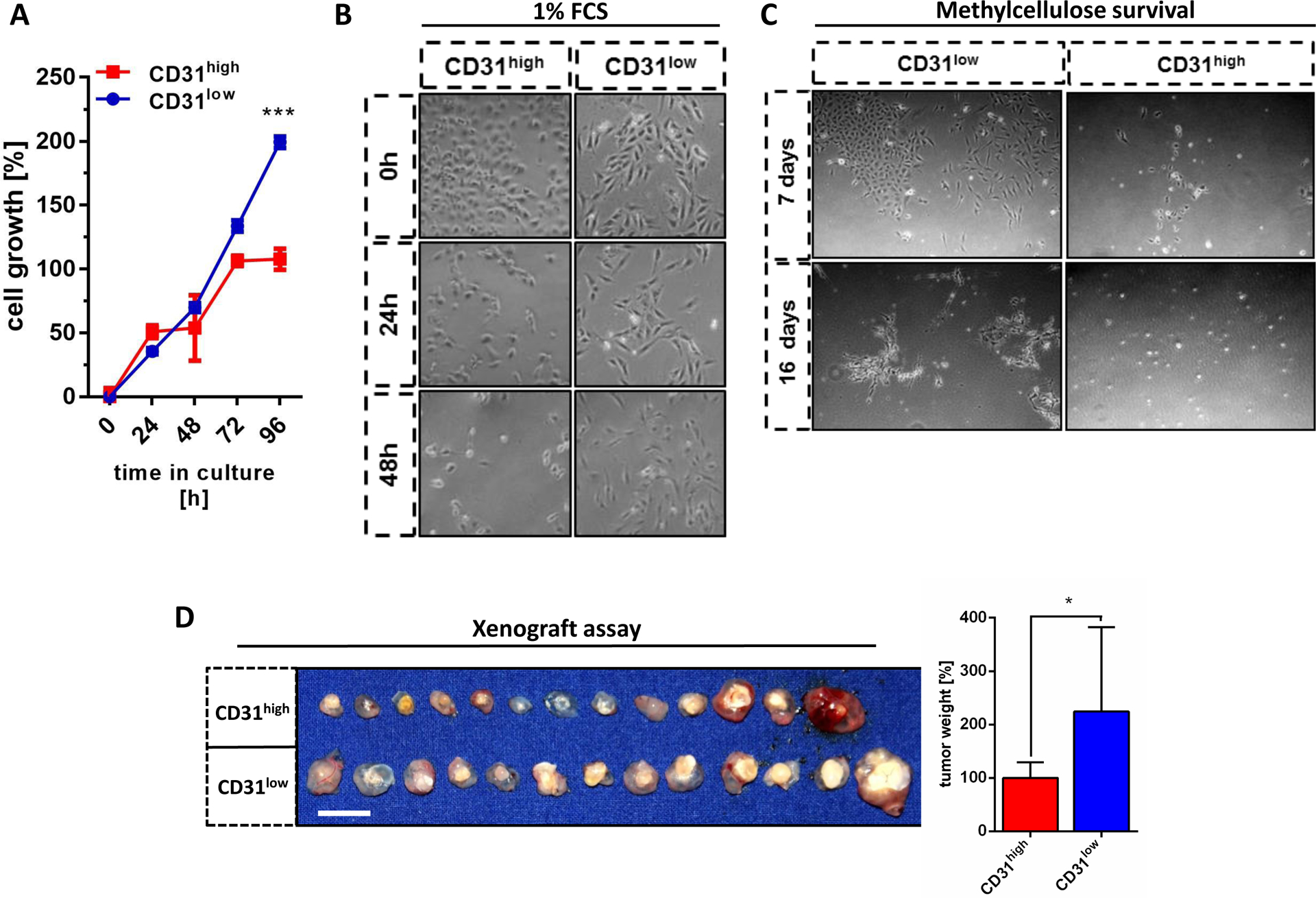
(A) CD31low cells showed higher proliferation rates than CD31high cells under normal culture conditions (n=3) and (B) higher cell survival under serum deprivation (1% FCS). (C) CD31low cells formed stable colonies for up to 16 days in methylcellulose, while CD31high cells were dispersed as single cells at day 7 that did not survive up to 16 days. (D) In a chorio-allantoic membrane (CAM) xenograft assay, 3×106 cells/egg from each cell line were implanted in matrigel and incubated for ten days. CD31low cells formed significantly larger and heavier tumors than their CD31high counterparts after 10 days (n=13). Scale bar: 1 cm. All data are mean ± SEM and were analyzed using two-way ANOVA followed by Bonferroni’s multiple comparisons test (A) or an unpaired t-test (D) (*p < 0.05; ***p < 0.001).
Taken together, these results indicate that CD31low cells represent a highly proliferative, stress-resistant and tumorigenic subpopulation that outcompetes vasculogenic CD31high cells.
CD31low cells are more resistant against doxorubicin.
Anthracycline-based chemotherapy is the backbone of current AS therapy by improving local disease control, but does not result in any survival advantage (8, 23). We treated both CD31 sublines with increasing concentrations of doxorubicin for 24 hours and measured cell survival using MTS assay. At concentrations >500 nM, CD31low cells survived significantly better than CD31high cells with only a 20–30% decrease in cell viability at 10 μM doxorubicin (peak plasma concentration achieved in patients ranging between 5 and 15 μM) (24) (Fig. 4A). In accordance with this observation, western blot analysis showed increasing levels of cleaved PARP, and effector caspases-3 and −7 as indicators of apoptosis only in CD31high, but not in CD31low cells (Fig. 4B and Supplemental Fig. 2A). Since suppressed CD31 levels propagated chemo-resistance, we next asked if doxorubicin treatment results in selection of CD31low cells. We therefore used unsorted wild-type ASM cells with a predominant CD31high (66.6%) and a smaller CD31low (2.7%) subpopulation. Indeed, 1 μM doxorubicin efficiently killed the majority of cells after 24 hours. However, the residual cells that fully recovered after 12 days had a fibroblast-like morphology (Supplemental Fig. 2B) and had lost their vasculogenic capability (Supplemental Fig. 2C). In agreement with this phenotype, western blot analysis demonstrated low CD31 protein levels in doxorubicin-suriving cells and flow cytometric analysis clearly revealed a shift towards CD31low cells as the predominant subpopulation (Fig. 4C and D). To further elucidate this selection for the CD31low phenotype under chemotherapy more precisely, we first labeled CD31high cells with CellTrace Violet dye, mixed them at a 1:1 ratio with unstained CD31low counterparts and challenged the mixture with increasing doxorubicin concentrations (Fig. 4E). After 24 hours treatment, flow cytometric tracking of the cell trace dye showed a dose-dependent depletion of the CD31high population. In strong contrast, the reverse experiment with CD31low cells labeled and mixed with equal numbers of unlabeled CD31high cells clearly showed that doxorubicin treatment left the population size of CD31low cells nearly unaffected (Fig. 4F and G).
Figure 4. CD31low cells are resistant against doxorubicin-induced apoptosis.
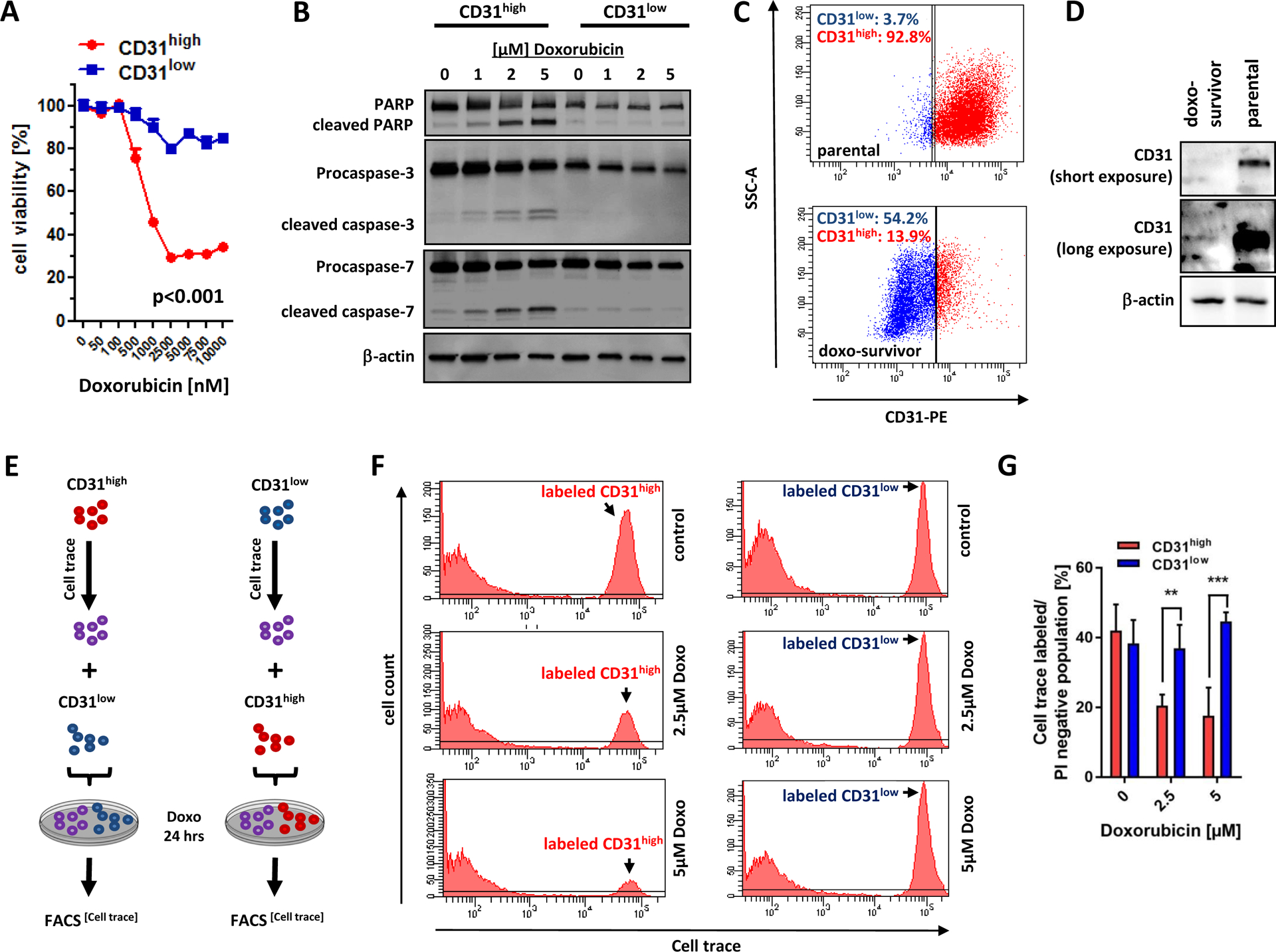
(A) Both sublines were treated with increasing concentrations of doxorubicin for 24 hours and cell survival was determined using MTS assay. At concentrations >500nM, CD31low cells survived significantly better than CD31high cells (p<0.001). (B) Immunoblot analysis of CD31low and CD31high cells treated with increasing concentrations of doxorubicin showed activation of apoptotic enzymes (cleaved PARP, cleaved caspase-3 and −7) only in CD31high but not in CD31low cells. See also Supplementary Figure 2A for densitometric quantification. (C) Parental CD31 heterogeneous ASM cells with a high CD31 subpopulation (CD31high:92.8% vs CD31low: 3.7%) were treated with 1 μM doxorubicin (respective isotype controls are presented in Supplementary Figure 2B). After 24 hours treatment, medium was changed to normal. After seven days the surviving ASM cells (termed doxo-survivors) were analyzed for CD31 expression via flow cytometry and (D) Western Blot. See also Supplementary Figure 2B and C for morphology and functional features of parental and doxo-surviving ASM cells. (E) Schematic description of cell trace experiments. CD31high (or CD31low cells, respectively) were labeled with the tracer dye CellTrace Violet and then mixed with equal numbers of CD31low cells (or CD31high cells, respectively) and then incubated with increasing doses of doxorubicin for 24 hours. (F and G) FACS analysis of these mixed labeled cell populations revealed preferential depletion of CD31high cells, while the size of the labeled CD31low population remained stable. All data are mean ± SEM and were analyzed using two-way ANOVA followed by Bonferroni’s multiple comparisons test (*p < 0.05;**p < 0.01; ***p < 0.001).
In conclusion, these results indicate that treatment with doxorubicin depletes CD31high cells and selects for CD31low cells rather than leading to downregulation of CD31 in CD31high cells.
CD31 critically impacts redox homeostasis in AS
In order to get a deeper insight into the molecular effects of CD31, we first analyzed the steady-state expression level of various specific cell death markers in both CD31 cell lines. No differences in basal activation of apoptosis (caspase-3) and autophagy (beclin-1 and LC3) could be observed. Interestingly, CD31high cells had markedly higher levels of phosphorylated histone variant H2AX (γ-H2AX), a reliable marker of double-strand DNA breaks, than CD31low cells (Supplemental Fig. 3A). In a parallel experiment, we performed a focused pharmaceutical screen for compounds that could restore the diminished colony formation capacity of CD31high cells. Surprisingly, potent antioxidants, such as N-acetyl-L-cysteine (NAC) that replenishes intracellular levels of the antioxidant glutathione (GSH) or the water-soluble vitamin E analogue trolox and ferrostatin-1 (Fer-1), both known lipid-ROS scavengers, were capable to significantly increase the colony formation capacity in semi-solid methylcellulose (Supplemental Fig. 3B and Supplemental Fig. 3C). Vice versa, treatment with the potent ROS inducer sodium arsenite (AsN) dramatically decreased the colony formation of CD31low cells (Supplemental Fig. 3D). These experiments hinted towards marked differences in the redox status between both CD31 subpopulations. In order to test this assumption, we evaluated the endogenous levels of cytoplasmic reactive oxygen species (ROS) via dihydrodichlorofluorescein diacetate (DCF), lipid peroxidation levels via C11 undecanoic acid (C11-BODIPY©), and mitochondrial superoxide level using MitoSox™ dyes. All three ROS indicators conclusively revealed significantly lower levels of cytoplasmic, lipid and mitochondrial-derived ROS entities in CD31low compared to CD31high cells (Fig. 5A). Furthermore, exogenous addition of the ROS inducer H2O2 (500 μM over 20 min) resulted in a significant ROS release in CD31high but not in CD31low cells (Fig. 5B). In a similar manner, serum reduction potently evoked starvation induced oxidative DNA damage only in CD31high cells (Fig. 5C). These results suggest that CD31low cells have a much greater ROS-scavenging capacity than CD31high cells.
Figure 5. CD31low cells maintain lower oxidative stress levels through stabilization of YAP.
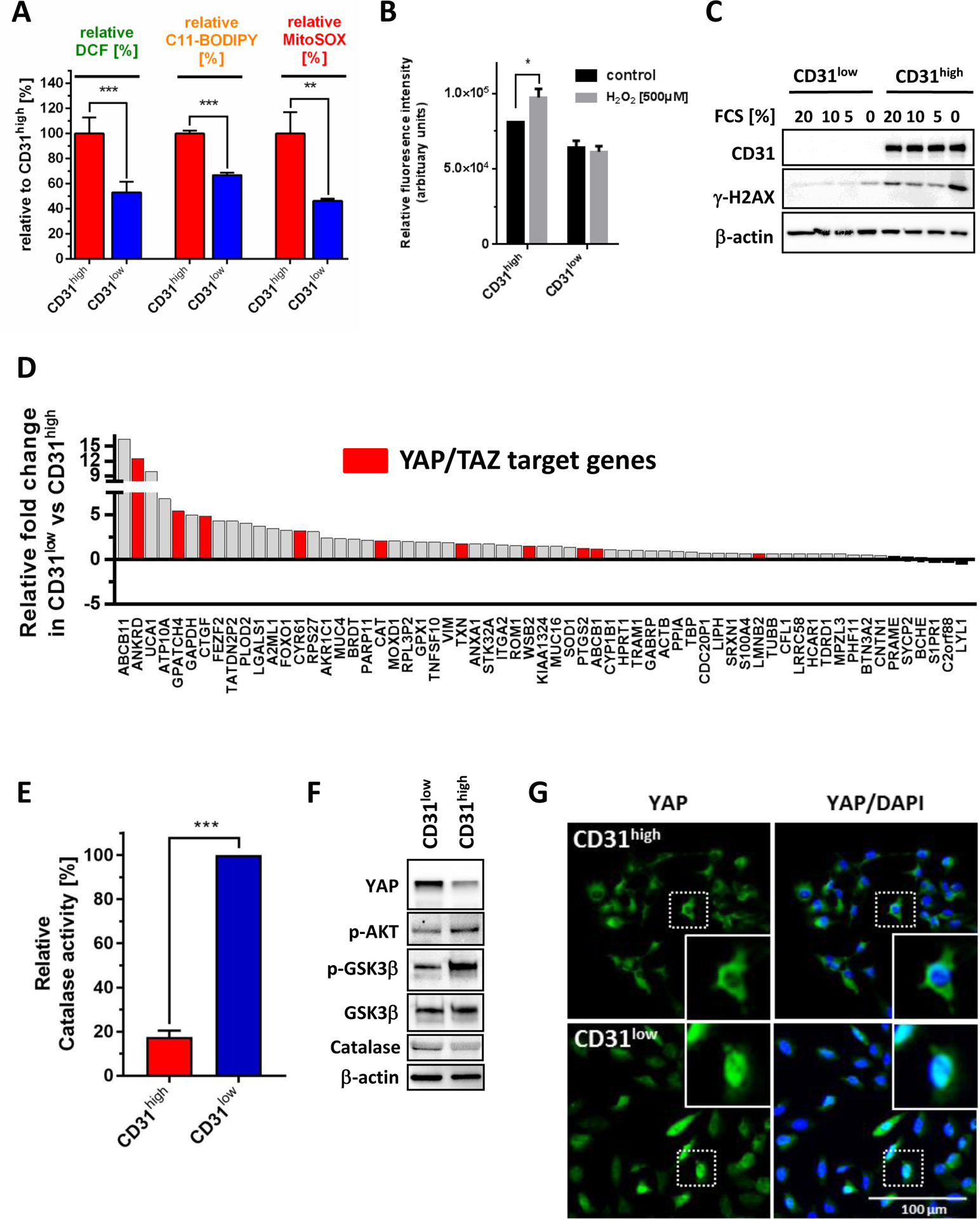
(A) Endogenous cytoplasmic levels of reactive oxygen species (ROS) were measured by dihydrodichlorofluorescein diacetate (DCF, n=6), lipid-ROS by C11-BODIPY© (n=3), and mitochondrial ROS using MitoSox™ (n=3) indicator fluorescence. All three measurements revealed highly significantly lower ROS levels in CD31low compared to CD31high cells. (B) Stimulation with 500 μM H2O2 over 20 min induced ROS only in CD31high but not in CD31low cells. (C) Serum starvation with reduced serum concentrations for 24 hours was used as an inductor of oxidative stress. Immunoblot analysis of γ-H2AX showed significantly more DNA damage at serum baseline levels and in starved CD31high compared to CD31low cells. (D) Differential gene expression analysis based on NanoString nCounter data was performed in CD31low and CD31high cells. Data are presented as relative expression fold-change in CD31low compared to CD31high cells. Known YAP/TAZ target genes are highlighted in red. See also Supplemental Table 1 for details on identified YAP/TAZ target genes. (E) Using an OxiSelect™ Catalase activity assay, CD31low cells showed an 80% greater steady-state enzymatic catalase activity compared to CD31high cells (n=2). (F) Western blot analysis revealed in CD31low cells suppressed levels of phosphorylated Akt (Ser473) and its downstream kinase GSK3β (Ser9) in conjunction with increased expression of YAP and catalase compared to CD31high cells. (G) Immunoflurescence microscopy demonstrated nuclear YAP translocation in CD31low cells, while in CD31high cells YAP was predominantly located in the cytoplasm. All data are mean ± SEM and were analyzed using either unpaired t-test (A, E) or two-way ANOVA (B) (*p < 0.05; ***p < 0.001).
CD31low cells maintain lower intracellular oxidative stress levels through stabilization of YAP.
We next performed a focused gene expression analysis using a custom-designed NanoString array. This technique quantifies specific RNA molecules by dual probe hybridization and allows direct measurement and comparison of absolute transcript levels. Compared to CD31high cells, CD31low cells showed overexpression of 10 previously described YAP/TAZ target genes (Fig. 5D and Supplemental Table 1). As a transcriptional co-activator, YAP binds to promoters of multiple target genes including the antioxidative enzyme catalase (CAT) (25) (Fig. 5D and Supplemental Table 1). CD31low cells showed an 80% greater steady-state enzymatic catalase activity compared to CD31high cells (Fig. 5E). It was previously demonstrated that Akt activation propagates higher YAP phosphorylation causing its enhanced proteasomal degradation (26). In agreement with these findings, CD31high cells showed increased phosphorylation levels of Akt (Ser473) and its downstream target kinase GSK3β (Ser9) (27) (Fig. 5F). Moreover, CD31low cells not only contained markedly higher YAP protein levels, but also showed higher nuclear YAP accumulation compared to CD31high cells (Fig. 5G).
We confirmed the redox-regulating role of CD31 by incubating CD31high cells with a selective antagonizing CD31 antibody (clone JC/70A) that not only effectively blocked tube formation (Fig. 6A), but also resulted in an increased YAP expression along with reduced steady-state ROS and less oxidative DNA damage as assessed by γ-H2AX (Fig. 6B and C). In a similar fashion, genetic CD31 depletion in CD31high cells using an siRNA approach resulted in YAP accumulation and suppressed oxidative stress and DNA damage levels (Fig. 6D and E). Moreover, siRNA-mediated YAP suppression in CD31low cells consequently induced ROS and DNA damage (Fig. 6F and G). To confirm the molecular link between CD31 expression, endothelial cell morphology, YAP signaling and ROS detoxification as a general mechanism, a number of control experiments were performed. First, we demonstrated increased YAP and reduced ROS as well as γ-H2AX levels in the previously established doxorubicin-surviving ASM cells compared to their parental cells (Supplemental Fig. 4A and B). Second, ISO-HASc.1 cells were sorted for the CD31high and CD31low population. Again, ISO-HASc.1 CD31low cells grew better in semi-solid methylcellulose, had more YAP and lower ROS and less DNA damage compared to CD31high cells (Supplemental Fig. 4C–H). Finally, we compared previously established murine endothelial cells isolated from wildtype (CD31+/+) or CD31 knockout (CD31−/−) mice (28). Morphological analysis showed that cells from CD31+/+ animals preferentially grew in cobblestone patterns and formed small colonies, whereas cells from CD31−/− animals had a more elongated, fibroblastic morphology (Supplemental Fig. 4I). The CD31−/− cells had lower Akt activation and higher YAP expression than their wildtype counterparts. Basal ROS and DNA damage levels were significantly lower in CD31−/− cells compared to their wild-type controls (Supplemental Fig. 4J and K). In summary, the aggressive and redox-protective phenotype of CD31-depleted AS can be attributed to YAP activation and subsequent induction of YAP-regulated anti-oxidative target genes.
Figure 6. Validation of CD31 as a YAP-dependent regulator of redox functions in angiosarcoma cells.
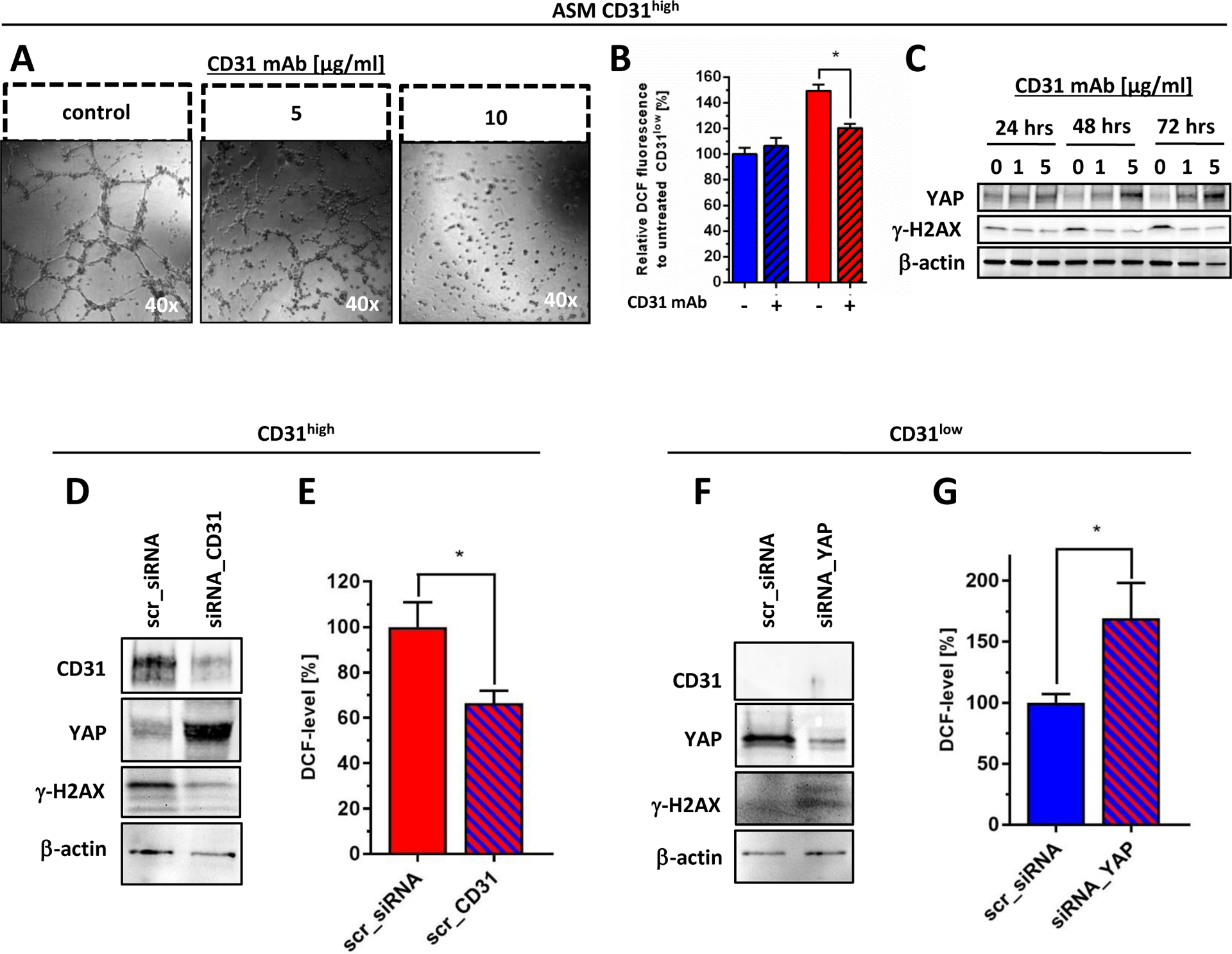
(A) CD31high cells lost their tube forming capacity after treatment with an anti-CD31 blocking monoclonal antibody (CD31 mAb) for 18 hours in a concentration-dependent fashion. (B) Treatment with CD31 mAb for 24 hours effectively reduced basal oxidative stress levels (indicated by flow cytometric DCF staining) selectively in CD31high cells, while similar treatment did not alter ROS level in CD31low cells. (C) Immunoblot anaylsis demonstrated that CD31 mAb treatment in CD31high cells under 1% serum-starvation induced YAP expression that was paralleled by decreased levels of γ-H2AX, indicating attenuated basal oxidative DNA damage. Medium with or without CD31 mAb was replaced every 24 hours. (D) CD31high cells were treated either with selective siRNA against CD31 (siRNA_CD31) or scrambled sequence (scr_siRNA). Immunoblot analysis was performed 48 hours after transfection showing selective knockdown of CD31, together with a marked increase of YAP and suppression of oxidative DNA, indicated by γ-H2AX. (E) Flow cytometric quantification of DCF fluorescence at the same time point showed a significant increase of endogenous oxidative stress levels in YAP-depleted CD31low cells compared to scrambled transfected controls (n=3). (F) Selective knockdown of YAP (siRNA_YAP) in CD31low cells resulted in a marked induction of γ-H2AX compared to scrambled transfected controls (scr_siRNA), 48 hours after transfection. (G) Flow cytometric quantification of DCF fluorescence at the same time point showed a significant increase of endogenous oxidative stress levels in YAP-depleted CD31low cells compared to scrambled transfected controls (n=3). All data are mean ± SEM and were analyzed using either two-way ANOVA (B) or unpaired t-test (E, G) (*p < 0.05).
The multikinase inhibitor pazopanib selectively re-sensitizes the anthracycline-resistant CD31low population to doxorubicin.
In a recent study, the FDA-approved multikinase inhibitor pazopanib was described to enhance proteasomal YAP degradation in cancer cells (29). We therefore analyzed the effects of pazopanib on the doxorubicin-resistant CD31low cell fraction in AS. Increasing doses of pazopanib over 24 hours decreased YAP protein and induced γ-H2AX levels in a concentration-dependent fashion (Fig. 7A). Moreover, pazopanib dramatically reduced catalase activity (Fig. 7B) resulting in a marked induction of total ROS levels (Fig. 7C). In contrast, the same treatment had no significant effect on catalase activity and ROS or γ-H2AX levels in CD31high cells (Fig. 7D–F). Importantly, combined treatment of CD31low cells with pazopanib plus doxorubicin markedly suppressed cell viability and colony formation by inducing apoptosis (Fig. 7 G–I). In conclusion, our results indicate that pazopanib helps to deteriorate the redox-status preferentially in “YAP-addicted” CD31low cells and thereby resensitizes them for chemotherapy (Fig. 7J).
Figure 7. The multikinase inhibitor Pazopanib selectively re-sensitizes the CD31low cell population to doxorubicin.
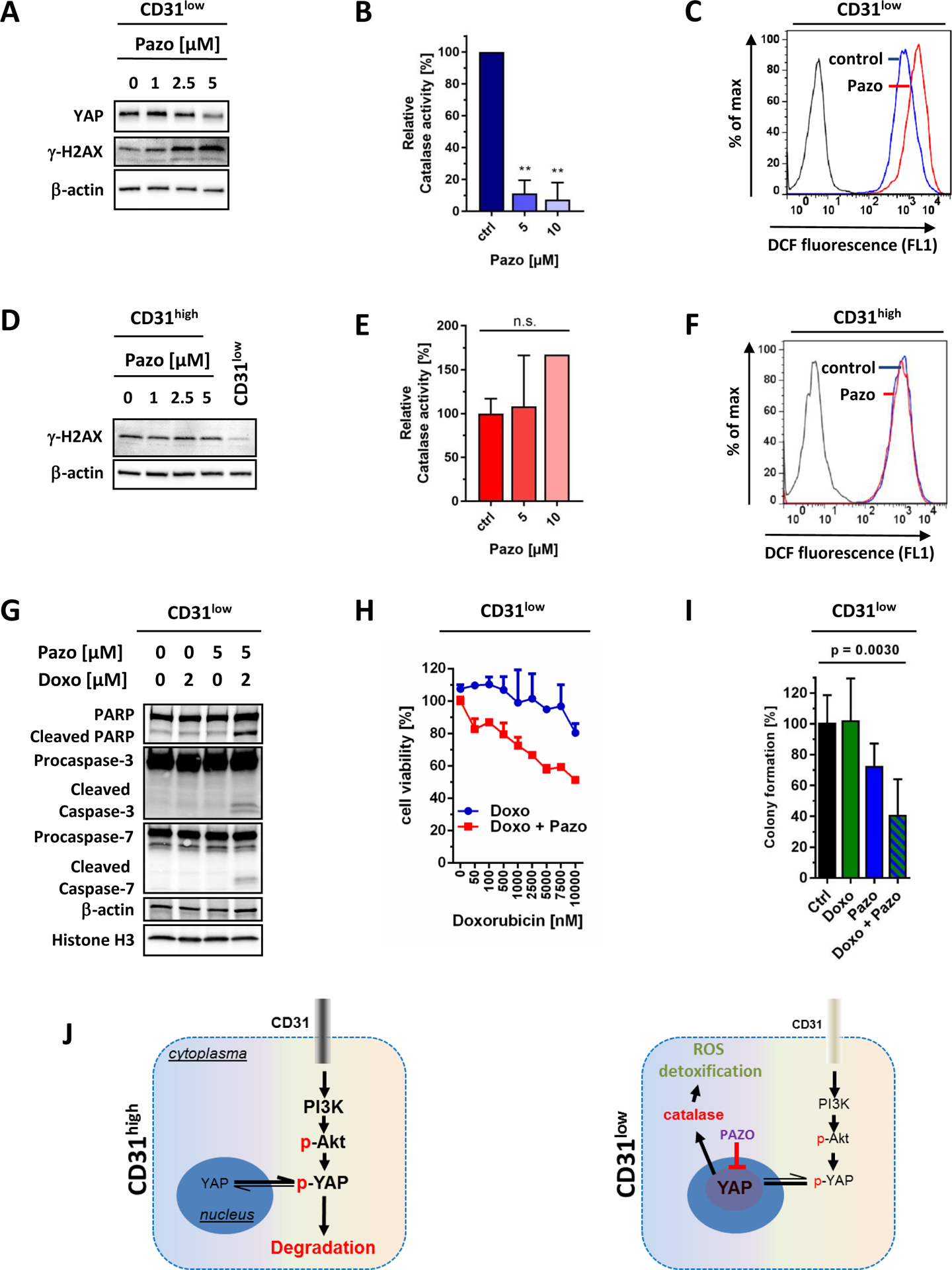
Treatment of CD31low cells with increasing doses of pazopanib (pazo) for 24 hours (A) decreased YAP levels that paralleled with increased oxidative stress-induced DNA damage, indicated by γ-H2AX. (B) Moreover, pazo markedly decreased the activity of the YAP target gene and ROS detoxifier catalase resulting in (C) increased oxidative stress, indicated by a shift of DCF fluorescence. (D) In contrast, the same treatment in CD31high cells had no measurable effect on γ-H2AX, (E) catalase activity, or (F) ROS levels. (G) Combination of pazo and doxorubicin (doxo) in CD31low cells efficiently induced apoptosis, indicated by increased levels of cleaved PARP, caspase-3 and −7, (H) inhibited cancer viability (indicated by MTS assay, n=3), and (I) suppressed colony formation in semi-solid methylcellulose medium (ctrl: n=6; doxo: n=3, doxo+ pazopanib: n=3). All data are mean ± SEM and were analyzed using either unpaired t-test (B, E) or one-way ANOVA (I) (**p < 0.01). (J) Model of CD31 action in endothelial malignancies. High levels of CD31 (CD31high) lead to Akt activation and favor cytoplasmic degradation of YAP. In cells with low CD31 expression (CD31low) and low Akt signaling, YAP is stabilized and translocates into the nucleus, where it functions as a co-transcription factor that increases ROS detoxificating enzymes such as catalase. Pazo decreases YAP protein levels and propagates oxidative stress by blocking anti-oxidative factors.
DISCUSSION
The identification of mechanisms by which malignant neoplasms evade current chemotherapies is a major goal to improve patient outcome. Intratumorous heterogeneity is believed to be one major factor that helps tumors to escape.
We here show for the first time that most if not all tested human AS specimens as well as AS-derived cell lines contain a small population of CD31low endothelial cells that we did not observe in non-neoplastic endothelial cell cultures. This subpopulation has fewer endothelial phenotypic properties, is resistant to starvation-induced oxidative stress, is more tumorigenic, and more resistant to anthracycline-based chemotherapy. It is therefore tempting to speculate that this subpopulation could be responsible for the chemoresistance seen in most AS patients.
One major downstream effect of CD31 seemed to be degradation vs. nuclear accumulation of the putative oncogene and co-transcription factor YAP. YAP and WWTR1 (also called TAZ, for transcriptional co-activator with PDZ-binding motif) are prime mediators of the Hippo pathway, essentially contributing to cell proliferation, stem cell function, tissue regeneration and tumorigenesis. YAP/TAZ are known to function as signaling nexus and integrators of several other prominent signaling pathways such as the Wnt, G protein-coupled receptor (GPCR), epidermal growth factor (EGF), and others (reviewed in (30–32). In addition, it was recently found in cardiomyocytes that YAP in conjunction with the transcription factor FoxO1 positively regulate the transcription of anti-oxidative genes, such as catalase and MnSOD (25). Moreover, YAP overexpression in pancreatic-β-cells conferred apoptosis-resistance via upregulation of the thioredoxin (TXN) antioxidant pathway (33). In good agreement with these data, our gene expression profiling demonstrated that CD31low cells, besides showing marked induction of canonical YAP target genes (ANKRD, CTGF, and CYR61), expressed higher levels of FoxO1, TXN and CAT. This resulted in improved protection from ROS and subsequent DNA damage in CD31low cells (Fig. 5D and Supplemental Table 1).
The finding that deregulation of the Hippo pathway plays an important role in neoplastic endothelial cells is not without precedence: epitheloid hemangioendotheliomas, another important group of malignant vascular tumors, are characterized by a common t(1;3) chromosomal translocation that fuses the Hippo downstream target WWTR1 (also called TAZ, for transcriptional co-activator with PDZ-binding motif), to CAMTA1, a transcription activator in cell cycle regulation (34–36). More recently, a small subset (10%) of hemangioendotheliomas was shown to carry a YAP-TFE3 fusion gene (37). WWTR1/TAZ and YAP are both oncogenes that drive proliferation and tumorigenesis (38, 39). It has recently been estimated that activation of WWTR1 and YAP occurs in about 50% of soft tissue sarcomas including angiosarcomas and is associated with poor prognosis (40).
The nuclear translocation and activity of YAP are determined by post-translational modifications, such as phosphorylation. YAP phosphorylation at its Ser-127 residue causes its cytoplasmic sequestration. Recently, it was shown that Akt can lead to Ser-127 phosphorylation independently from hippokinases LATS1/2 (26, 41). We found that high CD31 expression correlates well with activation of Akt and subsequent sequestration of YAP, while low levels of CD31 and Akt lead to increased nuclear translocation and activation of YAP with transcriptional induction of ROS detoxifying enzymes such as catalase (see scheme in Fig. 7J). Frequent activation of the PIK3CA/AKT pathway in angiosarcomas has been previously noted (42). This advantage in oxidative stress coping was most likely the explanation for the observed resistance of CD31low cells against doxorubicin treatment, while CD31high cells were doxorubicin sensitive. Given the apparent dependence of CD31low cells on YAP signaling, we reasoned that YAP was a good target for reversing chemoresistance in AS. We therefore chose the multi-targeted tyrosine kinase inhibitor pazopanib for two main reasons. First, pazopanib was recently identified as a potent YAP inhibitor causing its cytoplasmic retention and proteasomal degradation in breast cancer cells (29). Second, pazopanib has been approved for the treatment of soft-tissue sarcomas including AS with promising results in ongoing phase 2 trials alone (NCT01462630) or in combination with the endoglin/CD105 blocker TRC0105 (NCT01975519). We showed that pazopanib treatment in vitro at therapeutically relevant doses not only diminished YAP levels, but also suppressed the YAP downstream target catalase resulting in increased ROS levels selectively in CD31low cells. Interestingly, despite higher oxidative stress levels, pazopanib alone had no effect on apoptosis compared to untreated CD31low controls. However, combination with low-dose doxorubicin re-sensitized the cells for apoptosis resulting in a synergistic cytotoxicity and inhibition of colony formation. A recent study (although not in the context of YAP signaling) also observed that pazopanib induced a shift towards a pro-oxidative state in induced pluripotent stem cells (43).
An important question that needs to be addressed in future studies is how population homeostasis is maintained between CD31high and CD31low cells. We found that CD31low cells contained higher levels of the transcription factor Slug and lower VE-cadherin levels than CD31high cells (Supplemental Figure 5). Slug is a key transcriptional repressor of CD31 and VE-cadherin, coordinating the transition of endothelial into mesenchymal fibroblast-like cells (44). Recent studies demonstrated the critical role of Notch signaling that drives Slug induction, independently of TGF-β signaling (45). However, although these findings could explain the differences in CD31 expression and morphology, they cannot answer the question whether transitions between the two populations occur.
In summary, our results show that CD31 plays an important biological role in human AS by regulating endothelial cell function and redox status via the hippo signaling pathway member YAP. Specifically, most if not all human AS contain a small population of tumor cells with a CD31low phenotype and increased YAP levels and improved redox protection. This intrinsic resistance represents a universal mechanism that shields tumor cells against the first contact with cytotoxic agents and provides cells with survival time to acquire longer-lasting resistance mechanisms. The presented data point to an emerging pivotal role of the hippo pathway in malignant endothelial tumors (hemangioendotheliomas and AS) with new clues for the development of more effective treatments of these aggressive neoplasms. The proof-of-concept that combination of pazopanib and doxorubicin could specifically resensitize CD31low cells towards doxorubicin indicates that YAP inhibition together with conventional chemotherapy may be a valid new therapeutic perspective in AS.
Supplementary Material
Translational relevance.
Advanced Angiosarcomas (AS) remain an incurable disease with poor response to chemotherapy. AS cells usually show high constitutive expression of CD31. We here demonstrate that CD31 maintains endothelial cell features in AS and has downstream effects on the hippo pathway by modifying the stability and turnover of YAP. While CD31high AS cells can be effectively killed e.g. through doxorubicin in vitro, we here show that most AS in vivo and in vitro contain an aggressive, clonogenic subpopulation of CD31low cells that relies on enhanced YAP signaling to improve redox status and is doxorubicin resistant. This population can be selectively targeted through a combination of a YAP inhibitor (such as pazopanib) plus doxorubicin. Together, the observed intratumorous heterogeneity could be relevant for the chemoresistance of most AS and for the rational design of novel treatments. The hippo pathway emerges as an important player not only in hemangioendotheliomas, but also in angiosarcomas.
ACKNOWLEDGMENTS
We wish to thank Vera Krump-Konvalinkova (Ludwig-Maximilians-Universität, Munich, Germany) for the ASM cell line, James Kirckpatrick (University Mainz, Germany) for providing the ISO-HAS subclone ISO-HASc.1 and Daichi Hochina (Hokkaido University Graduate School of Medicine) for the HAMON cell line. We furthermore thank Sabrina Becker for flow cytometric analysis, Christina Kiecke for colony formation analysis, Verena Pape for experimental support and Nuno Raimundo (University Medicine Göttingen, Department of Cellular Biochemistry) for providing the MitoSOX dye.
Financial support:
VV was supported by the intramural research grant (Forschungsförderung) of the University Medical Center Göttingen and the Else-Kröner-Fresenius Foundation. XJ was supported by the NCI cancer center core grant P30 CA008748. PS was supported by the Deutsche Krebshilfe (KO.SAR network)
Footnotes
Conflict of interest statement:
The authors declare no conflict of interest whatsoever
REFERENCES
- 1.Weiss SW, Goldblum JR. Malignant vascular tumors In: Weiss SW, Goldblum JR, editors. Enzinger&Weissś Soft tissue tumors: Mosby Elsevier; 2008. p. 703–50. [Google Scholar]
- 2.Stewart NJ, Pritchard DJ, Nascimento AG, Kang YK. Lymphangiosarcoma following mastectomy. Clin Orthop Relat Res. 1995:135–41. [PubMed] [Google Scholar]
- 3.Sordillo EM, Sordillo PP, Hajdu SI, Good RA. Lymphangiosarcoma after filarial infection. J Dermatol Surg Oncol. 1981;7:235–9. [DOI] [PubMed] [Google Scholar]
- 4.Nanus DM, Kelsen D, Clark DG. Radiation-induced angiosarcoma. Cancer. 1987;60:777–9. [DOI] [PubMed] [Google Scholar]
- 5.Fayette J, Martin E, Piperno-Neumann S, Le Cesne A, Robert C, Bonvalot S, et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 cases. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2007;18:2030–6. [DOI] [PubMed] [Google Scholar]
- 6.Lahat G, Dhuka AR, Lahat S, Smith KD, Pollock RE, Hunt KK, et al. Outcome of locally recurrent and metastatic angiosarcoma. Ann Surg Oncol. 2009;16:2502–9. [DOI] [PubMed] [Google Scholar]
- 7.Fury MG, Antonescu CR, Van Zee KJ, Brennan MF, Maki RG. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer journal (Sudbury, Mass). 2005;11:241–7. [DOI] [PubMed] [Google Scholar]
- 8.D’Angelo SP, Munhoz RR, Kuk D, Landa J, Hartley EW, Bonafede M, et al. Outcomes of Systemic Therapy for Patients with Metastatic Angiosarcoma. Oncology. 2015;89:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner MJ, Ravi V, Menter DG, Sood AK. Endothelial cell malignancies: new insights from the laboratory and clinic. npj Precision Oncology. 2017;1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.SP DA, Mahoney MR, Van Tine BA, Adkins DR, Perdekamp MT, Condy MM, et al. Alliance A091103 a phase II study of the angiopoietin 1 and 2 peptibody trebananib for the treatment of angiosarcoma. Cancer Chemother Pharmacol. 2015;75:629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan HC, Edgar MA, Cohen C, Kovach CK, HooKim K, Reid MD. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44–50. [DOI] [PubMed] [Google Scholar]
- 12.Wood A, Mentzel T, van Gorp J, Flucke U, Huschka U, Schneider J, et al. The spectrum of rare morphological variants of cutaneous epithelioid angiosarcoma. Histopathology. 2015;66:856–63. [DOI] [PubMed] [Google Scholar]
- 13.Rao P, Lahat G, Arnold C, Gavino AC, Lahat S, Hornick JL, et al. Angiosarcoma: a tissue microarray study with diagnostic implications. The American Journal of dermatopathology. 2013;35:432–7. [DOI] [PubMed] [Google Scholar]
- 14.Jones KL, Hughan SC, Dopheide SM, Farndale RW, Jackson SP, Jackson DE. Platelet endothelial cell adhesion molecule-1 is a negative regulator of platelet-collagen interactions. Blood. 2001;98:1456–63. [DOI] [PubMed] [Google Scholar]
- 15.Marelli-Berg FM, Clement M, Mauro C, Caligiuri G. An immunologist’s guide to CD31 function in T-cells. J Cell Sci. 2013;126:2343–52. [DOI] [PubMed] [Google Scholar]
- 16.Flynn KM, Michaud M, Canosa S, Madri JA. CD44 regulates vascular endothelial barrier integrity via a PECAM-1 dependent mechanism. Angiogenesis. 2013;16:689–705. [DOI] [PubMed] [Google Scholar]
- 17.Tsuneki M, Madri JA. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J Biol Chem. 2014;289:5357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn K, Pan S, Beningo K, Hupe D. A permanent human cell line (EA.hy926) preserves the characteristics of endothelin converting enzyme from primary human umbilical vein endothelial cells. Life Sci. 1995;56:2331–41. [DOI] [PubMed] [Google Scholar]
- 19.Krump-Konvalinkova V, Bittinger F, Olert J, Brauninger W, Brunner J, Kirkpatrick CJ. Establishment and characterization of an angiosarcoma-derived cell line, AS-M. Endothelium : journal of endothelial cell research. 2003;10:319–28. [DOI] [PubMed] [Google Scholar]
- 20.Krump-Konvalinkova V, Kleideiter E, Friedrich U, Klotz U, Kirkpatrick CJ. Tumorigenic conversion of endothelial cells. Exp Mol Pathol. 2003;75:154–9. [DOI] [PubMed] [Google Scholar]
- 21.Hoshina D, Abe R, Yoshioka N, Saito N, Hata H, Fujita Y, et al. Establishment of a novel experimental model of human angiosarcoma and a VEGF-targeting therapeutic experiment. Journal of dermatological science. 2013;70:116–22. [DOI] [PubMed] [Google Scholar]
- 22.Koch R, Demant M, Aung T, Diering N, Cicholas A, Chapuy B, et al. Populational equilibrium through exosome-mediated Wnt signaling in tumor progression of diffuse large B-cell lymphoma. Blood. 2014;123:2189–98. [DOI] [PubMed] [Google Scholar]
- 23.Budd GT. Management of angiosarcoma. Current oncology reports. 2002;4:515–9. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Zheng H, Tang M, Ryu YC, Wang X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. American journal of physiology Heart and circulatory physiology. 2008;295:H2541–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H, et al. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature communications. 2014;5:3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14–3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23. [DOI] [PubMed] [Google Scholar]
- 27.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. [DOI] [PubMed] [Google Scholar]
- 28.Cheung K, Ma L, Wang G, Coe D, Ferro R, Falasca M, et al. CD31 signals confer immune privilege to the vascular endothelium. Proc Natl Acad Sci U S A. 2015;112:E5815–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C, et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio. 2015;5:542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nature reviews Cancer. 2015;15:73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelleher FC, O’Sullivan H. FOXM1 in sarcoma: role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget. 2016;7:42792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan T, Rafizadeh S, Azizi Z, Lupse B, Gorrepati KD, Awal S, et al. Proproliferative and antiapoptotic action of exogenously introduced YAP in pancreatic beta cells. JCI insight. 2016;1:e86326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Science translational medicine. 2011;3:98ra82. [DOI] [PubMed] [Google Scholar]
- 35.Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes, chromosomes & cancer. 2011;50:644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson T, Zhang L, Hameed M, Rusch V, Travis WD, Antonescu CR. Thoracic epithelioid malignant vascular tumors: a clinicopathologic study of 52 cases with emphasis on pathologic grading and molecular studies of WWTR1-CAMTA1 fusions. Am J Surg Pathol. 2015;39:132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes, chromosomes & cancer. 2013;52:775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong JX. Elucidation of a universal size-control mechanism in Drosophila and ammals. Cell. 2007;130:1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fullenkamp CA, Hall SL, Jaber OI, Pakalniskis BL, Savage EC, Savage JM, et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget. 2016;7:30094–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehsanian R, Brown M, Lu H, Yang XP, Pattatheyil A, Yan B, et al. YAP dysregulation by phosphorylation or DeltaNp63-mediated gene repression promotes proliferation, survival and migration in head and neck cancer subsets. Oncogene. 2010;29:6160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Italiano A, Chen CL, Thomas R, Breen M, Bonnet F, Sevenet N, et al. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: a pattern distinct from other sarcomas with complex genomics. Cancer. 2012;118:5878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choudhury Y, Toh YC, Xing J, Qu Y, Poh J, Huan L, et al. Patient-specific hepatocyte-like cells derived from induced pluripotent stem cells model pazopanib-mediated hepatotoxicity. Sci Rep. 2017;7:41238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manner J, Radlwimmer B, Hohenberger P, Mossinger K, Kuffer S, Sauer C, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, et al. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via downregulation of the alzheimer amyloid precursor protein. J Biol Chem. 2010;285:10678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.