

SUMMARY
Isothermal titration calorimetry is a powerful and widely used method to measure the energetics of macromolecular interactions by recording a thermogram of differential heating power during a titration. However, traditional ITC analysis is limited by stochastic thermogram noise and by the limited information content of a single titration experiment. Here we present a protocol for bias-free thermogram integration based on automated shape analysis of the injection peaks, followed by combination of isotherms from different calorimetric titration experiments into a global analysis, statistical analysis of binding parameters, and graphical presentation of the results, using the integrated public-domain software packages NITPIC/SEDPHAT/GUSSI. The recently developed low-noise thermogram integration approach and global analysis allow for more precise parameter estimates and more reliable quantification of multi-site and multi-component cooperative and competitive interactions. Titration experiments typically take 1–2.5 h each and global analysis usually 10–20 min.
Keywords: isothermal titration calorimetry, protein interactions, cooperativity, global analysis
Editor Summary:
This protocol from Brautigam et al. describes methods for baseline correction and global analysis of Isothermal titration calorimetry data using NITPIC and SEDPHAT. Publication-quality graphs of resulting data can then be created and visualized using GUSSI.
INTRODUCTION
Noncovalent interactions are essential for the functions and the cellular organization of biological macromolecules1. For example, reversible multivalent and cooperative binding of proteins to create dynamic multi-protein complexes is a ubiquitous motif in signal-transduction processes2,3. The study and the optimization of macromolecular interactions are also key to the development of both protein and small-molecule pharmaceuticals4,5. A classical technique of physical biochemistry for studying the energetics of molecular binding events is isothermal titration calorimetry (ITC)6,7. It is widely used in a broad range of applications, including the study of protein and nucleic acid interactions8, supramolecular chemistry9, and lipid membrane research10,11.
For the study of macromolecular binding, ITC is often the method of choice, as it does not require modifications of the molecules such as labeling or surface immobilization. It has the unique virtue of directly measuring the heat of a reaction and thereby affords exquisite sensitivity for changes in solution composition. ITC follows the basic strategy of recording the evolution of heat when titrating a solution containing one macromolecule (the contents of the ITC sample cell) with aliquots of its binding partner (loaded in the ITC syringe) in a series of injections. From the shape of the saturation curve reflected in the titration isotherm of the successive reaction heats, it is possible to deduce the changes in molar enthalpy and in molar Gibbs free energy (or, equivalently, the equilibrium dissociation constants KD or the association constants KA = 1/KD) as well as the molar ratio of the reaction. Thus, a detailed thermodynamic decomposition of driving forces in terms of the thermodynamic parameters ΔG° = RT ln(KD/M), ΔH°, and –TΔS° = ΔG°–ΔH° can be obtained, and, by carrying out experiments at different temperatures, changes in molar isobaric heat capacity ΔCp° can be determined. Furthermore, the combination of titration experiments performed under different conditions in a global analysis allows characterization of the energetics of binding in multi-component systems with more than one binding interface, the study of cooperativity in multi-protein complexes12–15, as well as proton- or ligand-linked interactions16,17.
However, the current standard mode of ITC analysis poses significant limitations. The first relates to the processing of the raw data in ITC, which are called ‘thermograms’. They represent the recorded time course of differential power required to maintain a zero temperature difference between the sample cell and the reference cell while the series of injections is carried out into the sample cell. Thermograms exhibit a series of peaks, each of which corresponds to one injection of reactant, which must be integrated to determine the total heat associated with the stepwise change in solution composition. A critical part of this integration is the assignment of the thermogram baseline18. Unfortunately, the latter cannot be independently measured and is usually subject to adventitious spikes and essentially stochastic fluctuations on short and long timescales. These are particularly limiting in studies of reactions with low enthalpy changes and in studies of high-affinity systems, which require low concentrations for achieving conditions under which significant fractions of both binding partners are still unbound. Particularly, they are exacerbated in new calorimeters with smaller sample volumes that are more sensitive to environmental thermal noise. The algorithms for integration of thermograms that are provided by the instrument manufacturers are not satisfactory and routinely require manual adjustment of the baseline assignment based on visual inspection by the user19–22. Invariably, such procedure introduces bias and even potentially arbitrary offsets if the experimenter uses results of the final isotherm modeling as criterion to readjust the integration. Furthermore, manual adjustment is impossible in a high-throughput setting23,24. Finally, the standard integration does not offer estimates for the measurement error of the integrated reaction heats, although uncertainties may vary considerably between injections.
The second significant limitation is that, in the absence of significant customization, manufacturers’ analysis programs are adequate only for the simplest systems and for the analysis of only one experiment at a time. By contrast, it is now widely recognized that global analysis of multiple titration experiments15,25–31 and ITC analysis in the context of a global multi-method analysis (GMMA)32–36 offer great advantages. Even for comparatively simple systems such as 1:1 binding of two components, the combination of data from multiple experiments, even those comprising only partial binding isotherms, can offer substantial improvement in statistical precision; this possibility goes unused in the standard analysis, resulting in unnecessarily large uncertainties in the final best-fit values of the binding parameters. Further, global analysis of multiple titrations and GMMA are often essential in the study of interactions involving multiple components or multiple binding sites.34 A major hurdle for combination of multiple datasets in standard analysis is the conventional parameterization of the model including an ‘n-value’ subsuming both the reaction’s molar ratio and possible concentration errors, which leads to apparent non-integral reaction molar ratios that are usually inconsistent across multiple (and reverse) titrations. Finally, the standard presentation of results in manufacturers’ programs is very limited, with a static figure output format that can be modified only with advanced expertise and does not lend itself to the presentation of results from global ITC or GMMA analyses.
Development and overview of the protocol
We have recently developed new approaches that address the problems described above: (1) Integration of the raw thermogram data can be carried out by global peak-shape analysis and regularization with truncated singular value decomposition (SVD) in NITPIC18. This method takes advantage of the contrast between the shape similarity of injections and the stochastic nature of baseline fluctuations. It yields high-quality, low-noise isotherms of reaction heats and produces error bars for each data point of the isotherm. Furthermore, it is unbiased, fully automated, and robust, such that it can be applied in a high-throughput modus24. (2) We have established an analysis platform, SEDPHAT15,30, in which the transition from single-set to global ITC analysis and GMMA is seamless and consists in solely adding more datasets of ITC or one of several other biophysical techniques to a graphical user interface. Many published studies have already taken advantage of this platform in analyses of various systems17,37–47. SEDPHAT uses a rational, physically meaningful parameterization of reaction stoichiometry separate from concentration errors or fractions of incompetent macromolecules; therefore, it is capable of globally analyzing pre-programmed cooperative or competitive multi-site and multi-component interactions as well as temperature-, protonation-, or other linked binding models. Further, by default, it exploits the individual error bars of the integrated heats arising from peak-shape analysis and has advanced statistical functions to determine their propagation into the final parameter uncertainties. To facilitate the work with multi-site models, it can generate parameter correlation maps, visualize heat contributions arising from different species formed during the titration, and offers a graphical experimental design tool to optimize the information content of the data given certain experimental constraints30. (3) Finally, we have developed a dedicated program GUSSI to visualize the results of the peak shape and titration isotherm analysis and create publication quality graphs48. It offers new ways of presenting the data that highlight different aspects of the analysis and is particularly useful in presenting global ITC and GMMA analyses.
NITPIC, SEDPHAT, and GUSSI are public-domain software packages that seamlessly interface with each other (Figure 1). Their use requires only knowledge of ITC and familiarity with basic data fitting; no programming or customization for any particular analysis model is needed. In the present protocol, we guide the reader through all steps required for using these tools. As a simple model system that is readily available and easy to reproduce, we describe the determination of the affinity and binding enthalpy corrected for buffer ionization events for the interaction of carbonic anhydrase isozyme II (CAII) with its inhibitor trifluoromethanesulfonamide (TFMSA) through global analysis of titration experiments in buffers of different ionization enthalpies16,17,49,50 (see Box 1). For readers who wish to take a shortcut by solely recapitulating global data analysis, the experimental data used in this protocol are provided in the Supplementary Dataset. To streamline the application solely to ITC data, we have recently created a simplified user-interface of SEDPHAT, termed ITCsy, which the reader may use instead of SEDPHAT; since all ITC-related capabilities of SEDPHAT are also available in ITCsy, we will refer only to SEDPHAT in the following for simplicity.
Figure 1.
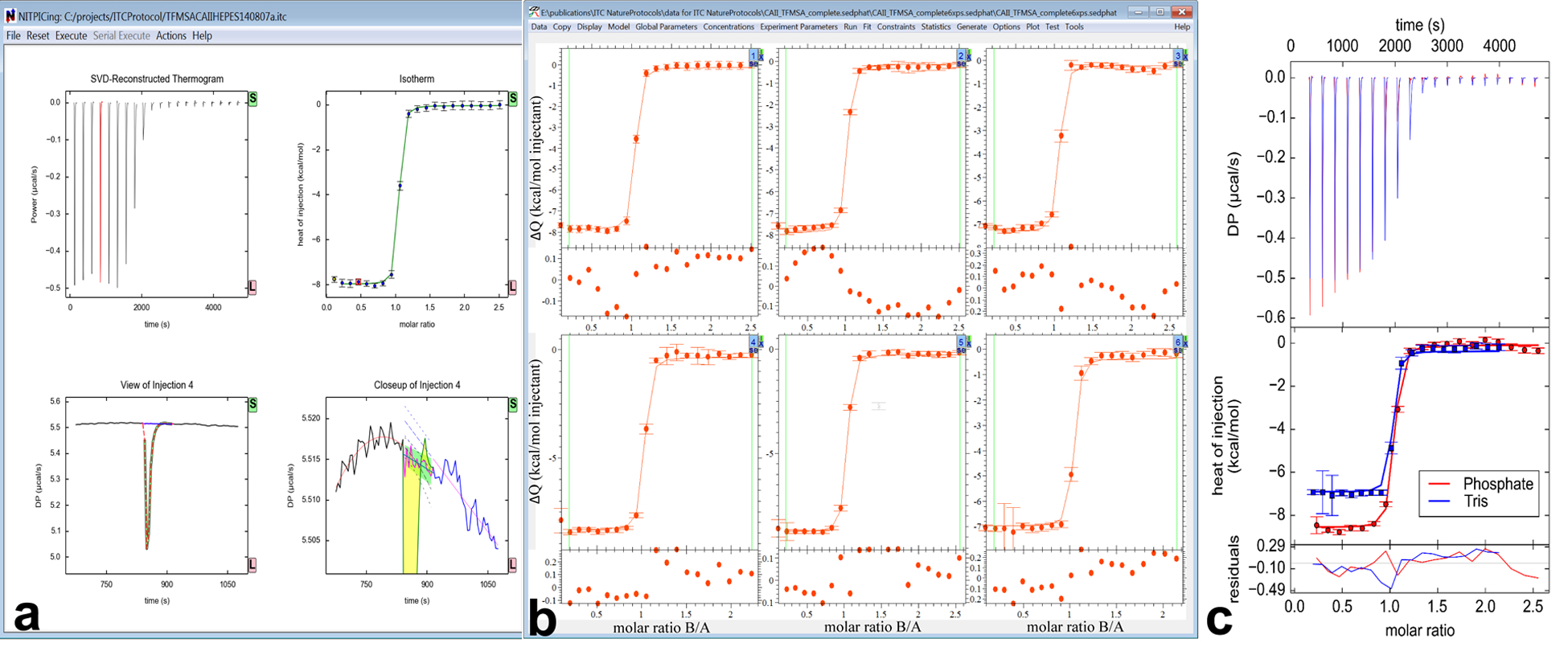
Workflow of global ITC analysis using NITPIC (a), SEDPHAT/ITCsy (b) and GUSSI (c). (a) The NITPIC window is shown after integration of a thermogram (control panel not shown). After a mouse click on one of the injections shown in the reconstructed thermograms (upper left) or isotherm (upper right) plots, a view of the isolated injection appears with raw thermogram data, reconstructed injection shapes and baseline (lower left), and a zoomed-in view of that same injection (lower right) highlighting the integrated area in yellow and the assigned baseline during injection in magenta. The latter plot also shows the extrapolated pre-and post-injection baselines that yield the estimated error of the integral (green). (b) A screenshot of SEDPHAT after importing six isotherms and performing a global analysis. Buttons in the upper right corner of each plot lead to the experimental parameter input (Figure 2). (c) The GUSSI output for two of the ten proton-linkage experiments described in this protocol. The top panel shows the SVD-reconstructed thermograms provided by NITPIC, the middle panel the isotherms, and the bottom panel the residuals. All elements are colored as indicated in the inset legend. DP stands for “Differential Power”.
Box 1 |. Protonation-Linked Binding.
For a simple proton-linked binding system, we consider a protein, P, and a ligand, L, with an ‘intrinsic’ equilibrium association constant KA,int (or, equivalently a dissociation constant KD,int = 21/KA,int) and an ‘intrinsic’ enthalpy change upon binding ΔH°int. The protonation process can occur on the free protein with an equilibrium association constant K(f)p and the corresponding enthalpy change ΔH(f)p, where the subscript ‘p’ indicates protonation and the superscript ‘(f)’ denotes the free protein. Similarly, the protein–ligand complex, PL, can also accept a proton with K(c)p and ΔH°(c)p. Finally, the protonated protein can bind to the ligand with KA,int,p and ΔH°int,p, leading to the thermodynamic binding cycle. See the figure below
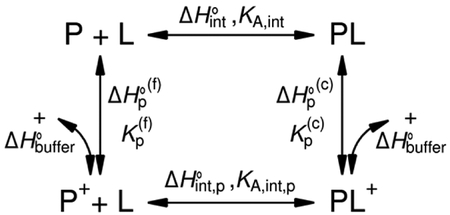
It is obvious that, in the presence of proton linkage, the observed binding constant for protein and ligand, KA,obs, is not equivalent to KA,int; rather, it is altered by the involvement of proton binding. This obscures the characterization of the molecular binding interface. From the reaction scheme above, the relationship between KA,obs and KA,int can be derived as
and the observed enthalpy change, ΔH°obs, is
where ΔnH+ is the change in the fractional proton occupancy of the ionizable group in the complex relative to the free state, ΔH°0 is the ‘intrinsic’ binding enthalpy independent of the buffer, and ΔHbuffer is the contribution from buffer ionization. Experiments in multiple buffers with different ionization enthalpies allow identification of the presence of proton linkage (i.e., a non-zero ΔnH+) and determination of ΔH°0. For the measurement of KA,int and ΔH°int, additional experiments at different pH values are necessary. For a detailed discussion of proton-linkage analysis and global data analysis, see Armstrong & Baker3 and Coussens et al.4
Global analysis of other systems proceeds analogously, and many different two-component and three-component binding schemes involving multiple complexes with various stoichiometries are available in SEDPHAT. After gaining initial familiarity with the approach, workflow, and conventions, advanced functions may be explored beyond the minimal protocol described here. The inclusion of data from other, complementary biophysical techniques in GMMA30,34,35 proceeds along the same steps (replacing NITPIC with other data pre-processing operations). For advanced training, workshops are held at the National Institutes of Health and other places, and questions can be exchanged at SEDPHAT-L@list.nih.gov.
Experimental Design
The data-analysis strategy described in the present protocol will provide statistically improved results even when applied to individual ITC experiments with a single-site system. In this case, the major critical experimental design parameters are the reactant concentrations. Most important is the ratio of the cell concentration to the anticipated dissociation constant (KD) (referred to as the ‘c’-value), which should fall broadly within a range of 1–1,0007. As a target, a value for the cell concentration of 40-fold KD may be chosen51, with a tenfold higher reactant concentration in the syringe such as to achieve near-saturation after the latest injection. The practical execution of the ITC experiment can follow standard experimental protocols as recommended by the instrument manufacturers, and will not be discussed here.
Global ITC analysis can also significantly enhance the accuracy and precision of the results by combining isotherm data from replicate experiments, even including truncated isotherms30,34. However, global analysis is often critical when studying multi-site systems. In this more complex case, the ‘c’-value approach generally ceases to be meaningful for experimental design. Instead, data can be simulated with stochastic noise and reanalyzed to establish whether a given experimental configuration will generate sufficient information. The latter would be indicated by small error bars in the statistical analysis of the parameters of interest, as described below. By pursuing simulations with different predictions, experimental conditions can be identified for a given binding model with hypothesized binding parameters. Even though an initial experiment may lead to better estimates requiring revised experimental design, incomplete or suboptimal titrations can still contribute significantly in the context of a final global fit of all ITC data.
SEDPHAT has graphical functions to facilitate the identification of favorable experimental conditions2: After selection of the interaction model in the Model list, the ‘Generate’ function will create a two- or three-dimensional color temperature plot (for two- or three-component systems, respectively) that depicts differential heats of reaction, or species populations, as a function of loading concentration and orientation of the titration. As described in more detail previously2, dragging a line across concentration regions with significant gradients or suitable features creates simulated titration data, which can be assembled in a global analysis. To further guide the simulation, conditions can be highlighted to create the information with regard to certain parameters, such as affinity, cooperativity, or number of sites.2 The selection of suitable conditions can be adapted to existing constraints in sample concentrations.
Limitations and Comparison with Alternative Methods
For some systems, it may not be possible to arrive at satisfactory results using the approach described here. Trivial potential problems related to any ITC analysis can include a lack of significant reaction enthalpies or ill-defined active concentrations of reactants.
A limitation for ITC analysis in SEDPHAT consists in the finite number of available models. Other published global modeling approaches are more flexible and allow complex models to be created with textual or graphical programming in different environments with different levels of abstraction25–29,31–33. This also includes commercial ITC analysis software such as HypΔH52 and Affinimeter (though the algorithmic standing of the latter is enigmatic, as few technical details are currently published). However, at present, none of these covers integration of thermograms, takes advantage of NITPIC output, or considers estimated errors for the integrated heat of each injection.
More fundamental limitations arise in the analysis of protein complexes where the interacting partners have multiple binding sites. If the different sites are non-equivalent, they can show similar thermodynamic signatures.53 In this case, binding constants can be highly correlated with each other or with binding enthalpies because of insufficient information content of the titration isotherm. A powerful alternative approach capable of analyzing complex interacting systems in a general way is the method of binding polynomials.14,54 Binding polynomials allow a model-free characterization of the binding isotherm but ultimately may require a further step to assign particular binding constants in a binding model.14 A variation of binding polynomials introduces differential equations as models for the titration55, but these fundamentally do not add new capabilities because of the inherently step-wise progression of the titration, which reports only about path-independent states in thermodynamic equilibrium.54
Features unique to SEDPHAT that may overcome insufficient information content of ITC titrations for complex systems are the graphics-based experimental design tool and the ability to seamlessly include orthogonal data from other biophysical techniques into the global fit.
MATERIALS
REAGENTS
CAII from bovine erythrocytes, lyophilized powder (Sigma-Aldrich, cat. no. C2522)
HEPES, buffer grade, ≥99.5% purity (Carl Roth, cat. no. HN78.3)
-
Hydrochloric acid (HCl), 35.0–38.0% (w/w) p.a. (min. 35.0% (w/w)) (Th. Geyer, cat. no. 836.2500)
!CAUTION: Hydrochloric acid is corrosive and irritant. Wear protective gear and perform dilution in a fume hood. Add concentrated acid to water slowly; never add water to concentrated acid.
-
Imidazole, buffer substance, ACS reagent, ≥99% purity (Merck, cat. no. 104716)
!CAUTION: Imidazole is corrosive and toxic. Wear protective gear and avoid inhalation.
MES, ≥99% purity (Carl Roth, cat. no. 4256.4)
Sodium chloride (NaCl), ACS reagent, 99.5% purity (VWR, cat. no. 27810.364)
-
Sodium hydroxide (NaOH), micro granules, p.a. (min. 98.8% purity) (Th. Geyer, cat. no. 1375.1000)
!CAUTION: Sodium hydroxide is corrosive. Wear protective gear.
Sodium phosphate dibasic (Na2HPO4), p.a., ACS reagent, anhydrous, ≥99% purity (Sigma-Aldrich, cat. no. 71640)
Sodium dihydrogen phosphate dihydrate (NaH2PO4 · 2H2O), p.a., ≥99% purity (Carl Roth, cat. no. T879.2)
TFMSA, 95% purity (Sigma-Aldrich, cat. no. 638455)
TRIS, ultra quality, ≥99.9% purity (Carl Roth, cat. no. 5429.3)
EQUIPMENT
High-sensitivity isothermal titration calorimeter, such as available from Malvern Instruments (previously MicroCal) or TA Instruments. The data provided in the Supplementary Dataset and used in this protocol were acquired on an iTC200 from Malvern Instruments. The files contain raw traces of differential power measured during the titration experiments under the different conditions.
Computer able to run Windows programs (any version since XP).
Software: NITPIC version 1.1.7 or higher and GUSSI version 1.1.0 or higher (both http://biophysics.swmed.edu/MBR/software.html); either ITCsy or SEDPHAT version 12.1b or higher (both https://sedfitsedphat.nibib.nih.gov/software/default.aspx).
REAGENT SETUP
CRITICAL Always prepare buffer from ultrapure water with a resistivity >18 MΩ as provided, for instance, by a Millipore filtration system.
0.2 M NaCl Add 1.17 g NaCl to a volumetric flask and bring to a total volume of 100 mL with H2O; mix well to dissolve and store at room temperature (20 °C) for an indefinite period.
-
0.2 M NaOH Dissolve 1 g NaOH in 125 mL H2O and mix it well by stirring and store at room temperature for an indefinite period.
!CAUTION: Sodium hydroxide is corrosive. Wear protective gear.
-
0.2 M HCl Prepare 50 mL of a 1 M stock solution by adding 4.5 mL of ~35% (w/w) HCl to 45.5 mL H2O. Add 4 mL of the 1 M HCl stock solution to 16 mL H2O. Store at room temperature for an indefinite period.
!CAUTION: Hydrochloric acid is corrosive. Wear protective gear and perform dilution in a fume hood. Add concentrated acid to water slowly; never add water to concentrated acid.
HEPES buffer Prepare 50 mL of a 0.1 M stock solution by dissolving 1.19 g HEPES in ~40 mL H2O in a beaker. Adjust the pH to 7.0 by adding a few drops of 0.2 M NaOH while stirring. Transfer the solution to a volumetric flask and bring to 50 mL mark with H2O. Take 25 mL of the 0.1 M HEPES stock solution, add 12.5 mL of 0.2 M NaCl, and add 12.5 mL H2O. Filter the solution using a 0.22-μm filter. Store at +4°C for up to three months.
-
Imidazole buffer Prepare 50 mL of a 0.1 M stock solution by dissolving 340.4 mg imidazole in ~40 mL H2O in a beaker. Adjust the pH to 7.0 by adding a few drops of 0.2 M HCl while stirring. Transfer the solution to a volumetric flask and bring to 50 mL mark with H2O. Take 25 mL of the 0.1 M imidazole stock solution, add 12.5 mL of 0.2 M NaCl, and add 12.5 mL H2O. Filter the solution using a 0.22-μm filter. Store at room temperature for up to three months.
!CAUTION: Imidazole is corrosive and toxic. Wear protective gear and avoid inhalation.
MES buffer Prepare 50 mL of a 0.1 M stock solution by dissolving 976 mg MES in ~40 mL H2O in a beaker. Adjust the pH to 7.0 by adding a few drops of 0.2 M NaOH while stirring. Transfer the solution to a volumetric flask and bring to 50 mL mark with H2O. Take 25 mL of the 0.1 M MES stock solution, add 12.5 mL of 0.2 M NaCl, and add 12.5 mL H2O. Filter the solution using a 0.22-μm filter. Store at +4°C for up to 12 months.
Phosphate buffer Prepare 50 mL of a 0.1 M NaH2PO4 · 2H2O stock solution by adding 780 mg in a volumetric flask and bring to 50–mL mark with H2O while stirring. Similarly, prepare 50 mL of a 0.1 M Na2HPO4 stock solution by adding 709.8 mg in a volumetric flask and bring to 50 mL mark with H2O while stirring. Prepare 50 mL of a 50 mM solution containing 50 mM NaCl by taking 25 mL of the 0.1 M NaH2PO4 · 2H2O stock solution, adding 12.5 mL of 0.2 M NaCl, and adding 12.5 mL H2O. In a separate vessel, take 25 mL of the 0.1 M Na2HPO4 stock solution, add 12.5 mL of 0.2 M NaCl, and add 12.5 mL H2O. Prepare the final phosphate buffer by taking 25 mL of the 50 mM Na2HPO4 containing 50 mM NaCl solution and adding an appropriate volume of 50 mM NaH2PO4 · 2H2O containing 50 mM NaCl solution until pH 7.0 is reached. Filter the solution using a 0.22-μm filter. Store at +4°C for no more than one week.
Tris buffer Prepare 50 mL of a 0.1 M stock solution by dissolving 605.7 mg Tris in ~40 mL H2O in a beaker. Adjust the pH to 7.0 by adding a few drops of 0.2 M HCl while stirring. Transfer the solution to a volumetric flask and bring to 50–mL mark with H2O. Take 25 mL of the 0.1 M Tris stock solution, add 12.5 mL of 0.2 M NaCl, and add 12.5 mL H2O. Filter the solution using a 0.22-μm filter. Store at room temperature for up to six months.
40 μM CAII Dissolve 1.0 mg of lyophilized CAII in 750 μL of each of the above 50 mM buffers containing 50 mM NaCl, pH 7.0. Vortex the solutions gently and centrifuge at 5000 g for 20 min. Determine the final protein concentration in each buffer spectrophotometrically using a molar extinction coefficient of ε280 nm = 55,100 M−1·cm−1; these values should typically be ~40 μM43. Prepare the solution fresh.
400 μM TFMSA Prepare 10 mM stock solution by dissolving 2.00 mg TFMSA in 1.34 mL buffer containing 50 mM NaCl, pH 7.0. Take 40 μL of 10 mM TFMSA and add 960 μL buffer. Prepare the solution fresh.
EQUIPMENT SETUP
Setup of MicroCal iTC200
Recommended and typical experimental settings for an iTC200 are as follows: 20 injections; cell temperature 25°C; reference power 5 μcal/s; initial delay 120 s; stirring speed 1000 rpm or 750 rpm for stirrers with flat or corkscrew (twisted) paddle design, respectively; high feedback mode/gain. Set the injection parameters as follows: injection volume 2 μL; injection duration 4 s; spacing 240 s; filter period 5 s.
Software setup
For data analysis, interprogram communication among NITPIC, SEDPHAT, and GUSSI is path-dependent, and thus is it necessary to set up the computational environment with a specific set of file folders. First, extract the .zip archives of the downloaded software to a temporary location. Create three file folders with the following paths: “C:\sedfit”, “C:\sedfit\NITPIC”, and “C:\sedfit\GUSSI”. Move the extracted file “sedphat.exe” to the C:\sedfit folder. Move the extracted file NITPIC.exe to the C:\sedfit\NITPIC folder and, likewise, the GUSSI.exe file to the C:\sedfit\GUSSI folder. Test each program by double-clicking its respective icon in the Windows Explorer. SEDPHAT will start instantly, but NITPIC and GUSSI may take longer. The latter two programs require acceptance of terms for each actuation.
Advanced users might find it convenient to associate the “.sedphat” and “.xp” file types (i.e., the pre-assembled experiment and analysis configuration files) with sedphat.exe. Such users equipped with Malvern calorimeters may wish to associate the “.itc” file type (raw thermogram files) with NITPIC.exe. However, these file-association steps are not required for the smooth functioning of the protocol presented herein.
When using ITCsy, place it in the same folder as directed for SEDPHAT and, in NITPIC, use the “Set SEDPHAT location” function in the “Actions” menu to select the ITCsy.exe path for downstream data analysis.
PROCEDURE
Data Acquisition
-
1To acquire data for the analysis protocol below, carry out titration experiments according to option A. Alternatively, if using the example dataset, follow option B.
- Carry out titration experiments ● TIMING Approx. 15––25 hr
- Following the manufacturer’s instructions, or steps 1–14 of the protocol by Velazquez-Campoy & Freire20, perform a series of ITC experiments in five different buffers (preferably, each experiment in duplicate). Specifically, inject 400 μM TFMSA from the syringe into 40 μM CAII in the sample cell using as buffer (a) HEPES, (b) imidazole, (c) MES, (d) phosphate, and (e) Tris. The concentration of each of these buffers should be 50 mM in 50 mM NaCl at pH 7.0.
- Download a copy of sample datasets ● TIMING 1 min
- Download the Supplementary Dataset provided with the current protocol. This contains the following files: CAII_TFMSA_HEPES_1, CAII_TFMSA_HEPES_2, CAII_TFMSA_Imidazole_1, CAII_TFMSA_Imidazole_2, CAII_TFMSA_MES_1, CAII_TFMSA_MES_2, CAII_TFMSA_Phosphate_1, CAII_TFMSA_Phosphate_2, CAII_TFMSA_Tris_1, and CAII_TFMSA_Tris_2.
Data Analysis: Thermogram Integration in NITPIC ● TIMING 5–10 min.
-
2Prepare raw thermogram data files. In our experiment, there were ten files, as the titration was carried out twice for five different buffers (HEPES, imidazole, MES, phosphate, and Tris). Data preparation will depend on the instrument used for data acquisition: For Malvern instruments follow option A; for TA instruments, follow option B.
- Malvern instruments data files
- Malvern data files have the extension “.itc” and are ASCII files natively readable in NITPIC. Transfer all ITC titration data to a single folder on the analysis computer.
- TA instruments data files
- Data from TA Instruments calorimeters with extension “.jet” or “.nitc” need to be converted from a proprietary binary format to a NITPIC-readable XML format. This requires loading the data in the program NanoAnalyze, which is freely available from TA Instruments, and re-saving them with a filename ending in “.xml” (note this is not explicitly listed as a file type option), all in the same folder.
-
3
Start NITPIC by double-clicking on its icon. Press the green button on the splash screen to accept the terms of use. The NITPIC window will appear. After selecting a file-opening command from the File menu, select one thermogram file and click on “Execute” to start automated integration. Without requiring user intervention, this process consists of initial shape analysis to determine the injection lengths, extrapolation of experimental pre- and post-injection baselines to determine error estimates for the injection, final shape analysis by SVD, truncation of SVD components to filter rare shape features in individual injections designated to be thermogram noise, reconstruction of the remaining net injections, and integration of the latter18. After a few seconds, a box will appear reporting the conclusion of the calculations (Figure 1a).
▲CRITICAL STEP: ITC titrations may contain contributions from heats of dilution, and it may seem desirable to eliminate them by subtracting the integrated heats from control titrations19,20. This can be accomplished with the “Subtract Control Titration from Current” in the File menu prior to integration. However, by default, data analysis in SEDPHAT will allow for a constant heat offset to be fitted to the isotherm data along with the reaction heats, which makes control titrations in practice often superfluous15.
? TROUBLESHOOTING
-
4
Examine the results. Click on one of the injections in the upper panels. This will cause a close-up display of this injection (Figure 1a). In the lower right panel, verify that the duration of the injection (yellow/green region) is adequate and that the fluctuations of the calculated baseline (magenta) during injection are comparable to the error range of extrapolated pre- and post-injection region (green region). If necessary, adjustments in the overall control parameters can be made (see Troubleshooting table), followed by re-integration. Usually, no further adjustments are needed, as is the case here.
▲CRITICAL STEP: Note that the ‘Isotherm Fitting Parameters’ in the lower right corner are simple estimates solely for initial quality control of integration and to initialize SEDPHAT for data analysis. These values should not be used for any other purposes.
? TROUBLESHOOTING
-
5
From the File menu, execute “Save Everything”. This will generate a file selection box to enter the path and filename of the resulting “.xp” and “.sedphat” files.
-
6
Repeat Steps 3–5 for the remaining datasets, using in Step 5 the function “Insert into Existing SEDPHAT Config.”, selecting the “.sedphat” configuration file in the identically named folder generated in Step 5.
▲CRITICAL STEP: If analyzing a large number of experiments, all integrations can be carried out at once with the “Prepare Files for Serial Integration” function in the File menu, using the “Put results into one SEDPHAT Config.” option.
Data Analysis: Global Analysis in SEDPHAT ● TIMING 20–30 min.
-
7
Start SEDPHAT and load the assembled results from NITPIC using the Data menu function “Read Configuration from File” and selecting the final “.sedphat” configuration from Step 6. This will result in the display of a series of data plots showing the individual isotherms, numbered in the order in which they were loaded (Supplementary Figure 1). The Display function “Show Last Fit Info Again” will write individual experiment filenames and other experiment-specific (‘local’) information across the plot. Immediately after the data are loaded, the user should provide the following information necessary for analysis:
-
8
For all experiments, adjust concentration parameters. These parameters are accessed in the Experimental Parameter window (Figure 2), which is called by clicking on the blue box showing the experimental number in the upper right corner of the experiment plots (Figure 1b). The concentration correction factor (red highlighted field in Figure 2) provides for a multiplicative factor accounting for small concentration errors (similar to the traditional ‘n’-value in single-experiment analysis)15. Click the checkbox marked “corr. factor” under the “syringe conc. [μM]” label and check the radio button “fit local”. In the present experiments, this refers to the TFMSA concentration of this particular experiment, which will be refined independently of other experiments.
▲CRITICAL STEP: If analyzing a single experiment, only the local correction factor of either the syringe or the cell concentration should be refined; not both simultaneously. Because the choice of which to adjust will impact the value of the resulting binding constants, the concentration of the component that is less error-prone should be fixed.
-
9
For all experiments, specify the buffer identity in the Experimental Parameters (blue highlighted fields in Figure 2). This is essential in our experiment because the goal is to explore the proton linkage in the binding of the ligand to the protein. SEDPHAT has an internal table of buffer ionization enthalpies and ionization heat capacities for commonly used buffers. For experiments with HEPES, imidazole, phosphate, and TRIS, click on the respective radio button. For MES, select the “other” radio button and input the proper values for ionization enthalpy and ionization heat capacity (i.e., 3.71 kcal/mol and 3.82 cal/(mol K), respectively56; tabulated ionization enthalpies and heat capacities for different buffers can be found in refs 56 and 57, the latter containing a literature review of published data.
▲CRITICAL STEP: For the buffer ionization model, specifying the buffer pH in the Experimental Parameters is not required, and the entry of the respective field can be left at the default; when changing the entry in the Experimental Parameters, however, all experiments need to specify the same pH.
-
10
Inspect all isotherms and exclude any clearly outlying data points. The latter is achieved by choosing “Exclude Isotherm Data Points” in the “Display” menu. After identifying which dataset contains the data point in question (in the present dataset, this is the replicate of the phosphate-buffered experiments), it is excluded from the analysis by right-clicking near to the data point and dragging a rectangle around it (Figure 3). Data points at the beginning and at end of the isotherm can be excluded by adjusting the green vertical lines, which indicate the data interval to be fitted. Note that the first data point is excluded by default.
-
11
Choose the binding model in the Model menu. For protonation-linked binding analysis, click “A + B ↔ AB Hetero-Association Global Buffer Ionization Enthalpy Analysis”; SEDPHAT offers this model only if at least two experiments are present with differing buffer ionization enthalpies indicated. Generally, when assigning models, the reaction scheme defines the designations “A”, “B”, and “C”, and the orientation of the titration must be selected accordingly in the Experimental Parameters. However, because of the symmetry of macromolecular components in the protonation-linked binding model, this is not required here.
-
12
Save the SEDPHAT configuration. Once all of the datasets have been loaded and all experimental parameters are set, the SEDPHAT configuration should be saved. The configuration, given the suffix “.sedphat”, is a file that points to where all experimental data and parameters can be located on the disk. The best way to save a configuration is as a path-independent group of files. This process is actuated by choosing “Copy All Data And Save As New Config” from the “Data” menu. After choosing a disk location and filename, the program will create a new file folder containing all the data and files needed to retrieve the analysis. This folder is fully self-contained and portable, providing an easy means for researchers to share analytic results.
-
13
Initialize the Global Parameters. The Global Parameters window appears after the user presses “Global Parameters” in SEDPHAT’s main menu. Five global parameters are available (see Box 1): (1,2) Global incompetent fractions as an alternative to concentration correction factors. These are not used here; therefore, values should be set to zero and the checkboxes unchecked. (3) “log10(Ka_app)” is the base-10 logarithm of the equilibrium association constant (the inverse of the equilibrium dissociation constant KD). Enter an initial estimate of 7.0 (corresponding to a KD of 100 nM) and check the respective box to allow refinement of this parameter in the fit. (4) “dHAB_app” is the apparent ΔH of the macromolecular association in kcal/mol. Enter an initial guess of −8.0 and check this parameter for fitting. (5) “nH+” is the number of protons taken up or released upon macromolecular binding. Initialize this at 0.0 and flag for fitting. Press “OK” to accept the inputted parameters. Conclude this step by updating the SEDPHAT configuration (“Update Current Configuration” in the Data menu), accepting the update of experimental parameters as well.
▲CRITICAL STEP: Rational, educated guesses regarding parameter values should be made, allowing rapid convergence of the refinement algorithm.
▲CRITICAL STEP: Simultaneously refining global incompetent fractions and local concentration factors (Step 8) is unsound; either one or the other should be fitted.
-
14
Test the initialized parameters: Click on “Global Run” from the “Run” menu. SEDPHAT draws fit lines and populates residuals plots. At this juncture, some of the fit lines have significant deviations from the data points (Figure 4a); however, the model is close enough for meaningful parameter refinement and likely convergence to the best-fit model.
Figure 2.
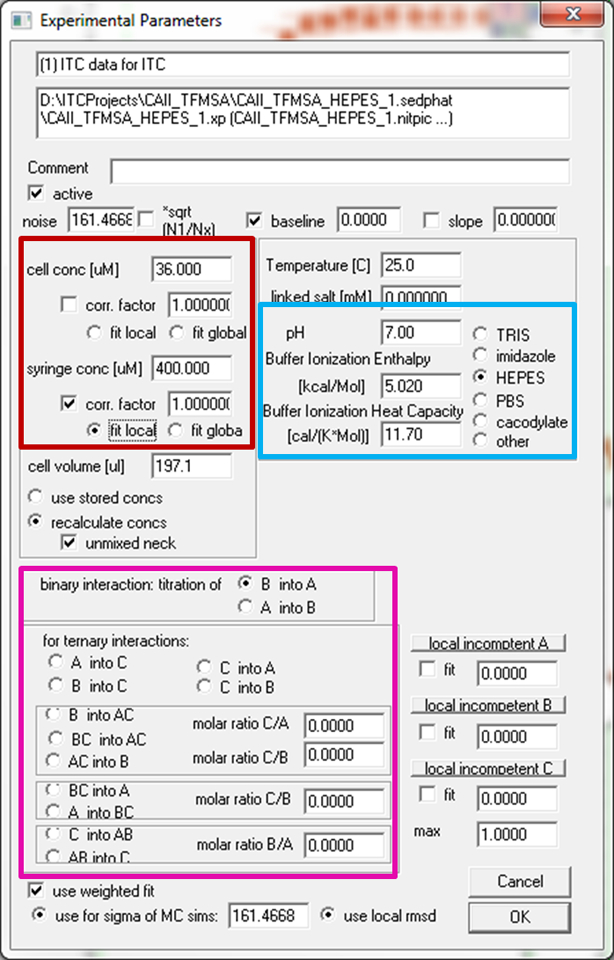
The Experimental Parameter window in SEDPHAT. Highlighted are the fields for specifying the identity of syringe and cell components (magenta) as well as the entries for concentrations (red) and buffer parameters (blue).
Figure 3.
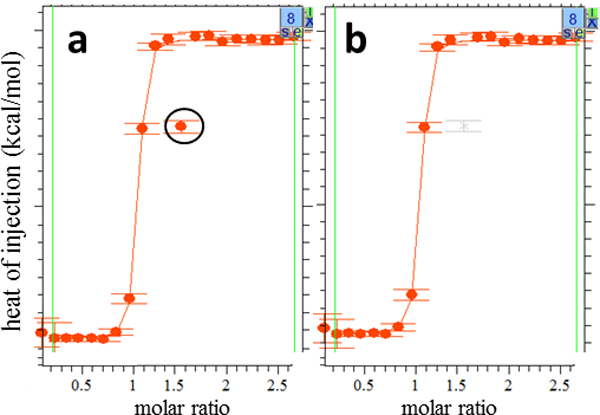
Removing an outlying data point in SEDPHAT. Close-ups of the SEDPHAT global analysis window for experiment 8 is shown (a) before outlier removal, with the outlier data point circled for clarity, and (b) after outlier removal with the “Exclude Isotherm Data Points” function; SEDPHAT shows excluded points in grey. Repeat execution of the same function allows re-inserting points into the pool of points to be fitted.
Figure 4.
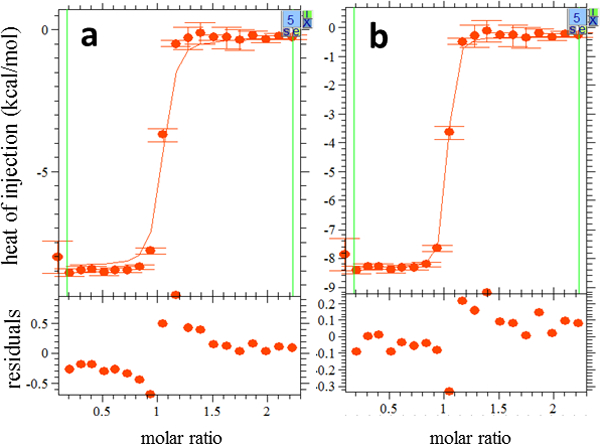
An experiment before and after fitting. The format is the same as Fig. 3. (a) The fit line after a “Global Run” was performed (Step 14). There is a clear mismatch between this line and the experimental data, and the residuals are large. (b) The fit line after final refinement. There is a much closer correspondence between the data and the fit, and the residuals are smaller and clustered around 0.
? TROUBLESHOOTING
-
15
Start a fitting session by selecting “Global Fit” from the “Fit” menu. After computation has concluded, ensure fit convergence by alternating non-linear least-squares minimization algorithms between “Simplex” and “Marquardt–Levenberg” in the Fitting Options. Carry this out until no changes in the global χ2 of the fit occur, which is shown in the second line of fit information in the upper left corner. Then save the fit with the “Update Current Configuration” function from within the Data menu.
-
16
Examine the quality of the fit. A screenshot of the SEDPHAT window after final parameter convergence is shown in Supplementary Figure 2. SEDPHAT will have drawn fit lines corresponding to the model with the fitted parameters applied. In a high-quality fit, these lines will correlate closely with the data points, usually within their error bars (Figure 4b). This correspondence can be easily visualized using the residuals plots below the data plots for each experiment (Figures 3 and 4); these represent the difference between data and fit and should be randomly distributed around zero. Excessively large deviations or strongly systematic residuals indicate a problem with fitted parameters and can even signal that the chosen model is not appropriate for the data at hand. Other measures of fit quality displayed by SEDPHAT are the weighted root-mean-square deviations between the data and the fit, calculated locally for each experiment, and the global weighted reduced χ2. No mathematically or physically rigorous recommendations for the values of these measures can be offered because they are dependent on the nature of the data and the level of noise. However, these values may be used for comparison of different experiments performed under similar conditions.
In this step, also examine if the returned parameter values are physically meaningful, that is, if they fall into a reasonable range. Especially the concentration correction parameters (or incompetent fraction parameters when used) can reveal fundamental errors in the model or in the experiment. In the present case, the concentration correction factors deviate from 1.0 in the range from −2% to + 5%, which is considered reasonable within the experimental precision of determining TFMSA concentrations.
? TROUBLESHOOTING
-
17
Determine the confidence intervals for all relevant parameters. The most reliable and effective method for ITC analysis is the “Automatic confidence interval search w projection method” from the Statistics menu in SEDPHAT, which carries out a search for the contour of the error surface prescribed by F-statistics58. After invoking this function, a series of messages and input boxes will appear with prompts to select the parameter to be searched, set the confidence level, and the limits and step-size of the search. First, execute the error analysis for the log10(Ka_app) parameter, using a confidence level of 0.683 (which would correspond to one standard deviation in the case of a Gaussian error distribution), a maximum of 9.5, minimum of 5.5, and step size of 10–2, allowing Simplex and Marquardt–Levenberg optimization alternating at each step until converged. Record the reported result, and carry out this step equivalently for dHAB_app and nH+ (Table 2). Review the summary of thermodynamic parameters with the function “Display Thermodynamic Information” in the Display menu of SEDPHAT.
TABLE 2 |.
Final best-fit parameter values extracted from the ten-experiment proton linkage study of CAII/TFMSA interaction
| Parameter | Best-fit value | 68.3% confidence interval |
|---|---|---|
| log10(Ka_app) | 7.52a | [7.38, 7.69]a |
| dHAB_app | −8.7 | [−8.9, −8.5] |
| nH+ | 0.19 | [0.16, 0.22] |
In terms of dissociation constant (KD), these values correspond to a best-fit value of 30 nM and a 68.3% confidence interval of (21–42) nM.
? TROUBLESHOOTING
Data Presentation in GUSSI ● TIMING 1–5 min.
-
18
Under the Plot menu in SEDPHAT, choose “GUSSI data, fit residuals”. SEDPHAT will ask which experiment to plot, whether the accompanying thermogram should be included, and what other experiments to be included in the same figure. For illustration, select two experiments with the widest-varying buffer ionization enthalpies: phosphate and TRIS. GUSSI will be spawned automatically.
-
19
Change graph parameters in GUSSI. The default mode of GUSSI for ITC displays three stacked plots (Figure 1c): the thermogram(s) (top), the isotherm(s) with fit line (center), and the residual plot(s) (bottom). The appearance of the plot can be changed with the controls on the right-hand side. When multiple plots are displayed, changes are made only to the “active” plot, and switching active plots is accomplished by left-clicking on the desired plot line. Virtually everything about the plot can be changed, including line colors and thicknesses; marker size and appearance; error bar existence and appearance; axis labels and tick length/direction; existence of the residual plot; existence of the thermogram plots; and existence of and text in a legend. To achieve the display depicted in Figure 1c, change the color of the first inputted line from the default purple to red and add a legend from the Legend menu. Several display modes particular to ITC data are available.
? TROUBLESHOOTING
-
20
Save the GUSSI state and the figure. Saving a GUSSI state file offers a convenient method to save and recreate everything about the plot. Save the figure in one of the output formats: PNG, EPS, or LZW-compressed TIFF at desired resolution and color space.
TIMING
Steps 1: Each titration may last 2.5––3 h. However, sample data to recapitulate the data-analysis part of the protocol are available for download. NITPIC integration of thermograms in Steps 2–6 will take 5–10 min; SEDPHAT analysis in Steps 7–17 20–30 min; and GUSSI plots in Steps 18–19 1–5 min.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1 |.
Troubleshooting table.
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 4 | NITPIC cuts off injections before return to baseline | Slow reaction kinetics when saturation is approached | In “Injection & Baseline Parameters”, toggle radio-button “User” in the Minimum Injection Time field, and set to a visually estimated injection length (%-value relative to total time between injections) |
| 4 | NITPIC includes too much of each injection in the integrations | Low signal-to-noise ratio | In “Injection & Baseline Parameters” increase “Target” and “Max” values (usually tenfold) in the entry boxes for “Cut-off differentials for inj. end” |
| 4 | Calculated thermogram baseline is too smooth or warning message that NITPIC uses too many shape components | Failure in automated shape truncation | In the “SVD Parameters” reduce the maximum # of SVD components |
| 14 | The displayed lines for the model as initialized are far away from the data points | Poor parameter initialization | There is no general mathematical solution for parameter initialization. Revise estimates manually and re-run. Reasonable values for dHAB_app may be obtained by entering the heats of the initial injections. A broad search for log10(Ka_app) by trial and error is often successful. If the inflection points do not match the data, inspect the concentration correction factor. |
| 15 | The resulting parameter values are physically inappropriate | The fit has not converged, too many parameters are optimized simultaneously, some of the parameters might be correlated, or the binding model is inappropriate. | When using incompetent fractions and concentration correction factors, ensure that both are not simultaneously refining. Try a multi-stage approach, first fixing a subset of parameters (for example, fix the concentration correction at 0) while other parameters optimize, then start optimizing them all in a second stage. Use a different model (see below). |
| 16 | The resulting fit does not resemble the data | An incorrect model was used. | Use a different model while incorporating the knowledge obtained from other experiments performed with orthogonal methods if possible. It can be useful to establish whether a data subset can be fit by de-activating individual experiments from consideration (using the small ‘i’ button in the upper right corner of the experiment plot). |
| 16 | An individual experiment is not fit well | Either an incorrect model was used, or an experimental error occurred. | A single aberrant fit can be caused by an erroneous experiment or incorrectly entered experimental parameters. Double-check parameters or repeat the experiment. However, the data may be correct and the conditions may make this particular experiment more informative regarding the binding model, highlighting shortcomings of the model. Considering the unique experimental conditions may lead to the identification of processes that occur in the binding reaction but might have gone unnoticed and are not accounted for. |
| 17 | Confidence interval is too broad or one of the intervals was not determined | Information content of the data is limited, possibly due to shallow or partial transition or low signal/noise ratio, or the model has too many or correlated parameters. | Examine the entire projection of the error surface using the “Generate 1-dim Error Surface Projection” function, and parameter correlations in the “Generate 2-dim Error Surface Projection” function of the Statistics menu. Add more informative data. Use the “Generate” function to simulate experimental data conditions in a search for better experimental conditions |
| 18 | GUSSI does not start up | Insufficient wait, un-answered disclaimer prompt, or location of gussi.exe | Make sure the location of gussi.exe is in a GUSSI subfolder relative to the path of sedphat.exe or itcsy.exe. The disclaimer may be hidden behind other open windows. On some computers, Python libraries will take several seconds to load. |
| 19 | Graph is too wide in GUSSI | Occurs stochastically on some computer systems | Press “Update”. |
ANTICIPATED RESULTS
NITPIC provides unbiased thermogram integration for ITC data with automatic recognition and exclusion of spikes and other adventitious baseline behavior and with error estimates for each individual data point for appropriate weighting in subsequent data fitting. SEDPHAT analysis of these data results in physically meaningful and statistically optimal parameters describing the experimental data well. GUSSI produces well-composed and informative high-quality graphs for global ITC results. This workflow can be carried out in analogous fashion with other systems using other binding models.
In the example presented in the above protocol, repeat titrations using buffers having different ionization enthalpies allowed determination of the number of protons taken up in a protein–ligand interaction (Table 2). In the case at hand, binding of TFMSA to CAII is accompanied by an increase in the degree of protonation of 0.19 (0.16–0.22), meaning that the protein–ligand complex carries, on average, about 0.2 protons more than free TFMSA and free CAII together. This parameter cannot be derived from a single ITC experiment but, with the aid of global analysis of multiple titrations, becomes accessible through linkage equations relating the ‘intrinsic’ protein–ligand interaction to a protonation event (see Box 1). Similarly, global analysis of a set of titrations performed at various temperatures yields, further to ΔG°, ΔH°, and –TΔS°, the change in molar isobaric heat capacity, ΔCp°. An example of such an analysis is shown in Figure 5 for a detergent–cyclodextrin interaction. Finally, global analysis of titrations varying in temperature, buffer, and pH can additionally reveal the intrinsic binding constants, which provide additional insights into the thermodynamics underlying these interactions (see Box 1).16,17,31
Figure 5.
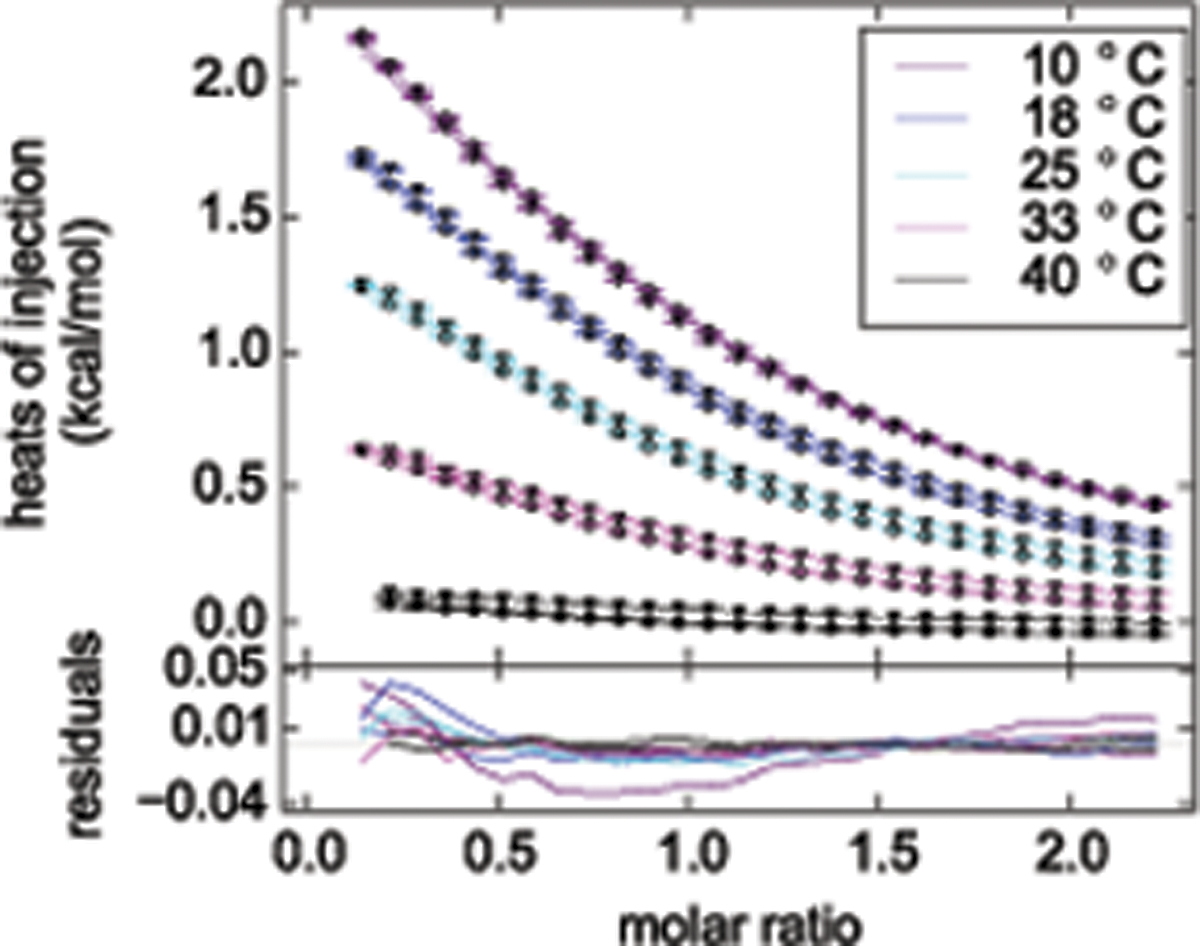
A GUSSI plot of a ten-titration global analysis of randomly methylated β-cyclodextrin (mβCD) binding to the nonionic detergent n-octyl-β-D-maltopyranoside (OM), acquired at different temperatures to determine ΔCp°. Knowledge of the stability and stoichiometry of inclusion complexes is of practical importance, for instance, for optimizing detergent complexation in the process of membrane-protein reconstitution59,60 and thermodynamic parameters such as ΔCp° provide additional insights into the thermodynamics underlying these interactions8. Specifically, we performed titrations of OM with mβCD (circles) as well as of mβCD with OM (triangles) at five different temperatures on a VP-ITC instrument (Malvern Instruments). Raw thermograms were processed using the serial integration function in NITPIC and directly saved as a SEDPHAT configuration. Global data analysis was accomplished with the “A + B ↔ AB Hetero-Association Global Temperature Variation Analysis” model, which SEDPHAT offers only if multiple datasets acquired at different temperatures have been loaded. The excellent agreement over a broad temperature range for both ‘forward’ and ‘reverse’ titrations lends credence to the simple 1:1 binding model assumed. The negative best-fit value of ΔCp° = −127 cal/(mol K) is a signature of hydrophobic interactions, and the 68.3% confidence interval ranging from −136 cal/(mol K) to −120 cal/(mol K) attests to the high precision afforded by global analysis.
Global analysis is particularly useful and often essential in the study of multi-site interactions. Figure 6 illustrates this point with an example taken from the global analysis of interactions among three adaptor proteins involved in signal transduction after T-cell activation. The existence of cooperativity between binding sites can be directly related to structural features of protein complexes and their potential function in signaling pathways. ITC offers a unique opportunity to detect cooperativity in both ΔΔG° and ΔΔH°, with both exhibiting different signatures in the shape of the titrations12, and global ITC analysis is a powerful tool to extract these parameters. For three-component interactions, for example, a number of permutations are possible of which components or component mixtures are in the syringe and the cell, and these can be analyzed seamlessly side-by-side using a variation of the above protocol after selecting a suitable interaction model in SEDPHAT while carefully specifying the identity of each titration in the experimental parameter box (Figure 2). In some cases, the analysis can be made more stringent through embedding additional constraints that arise from prior structural knowledge of the equivalence of sites. To facilitate this improvement, several multi-site binding models in SEDPHAT are phrased alternatively in terms of microscopic or macroscopic sequential binding constants.
Figure 6.
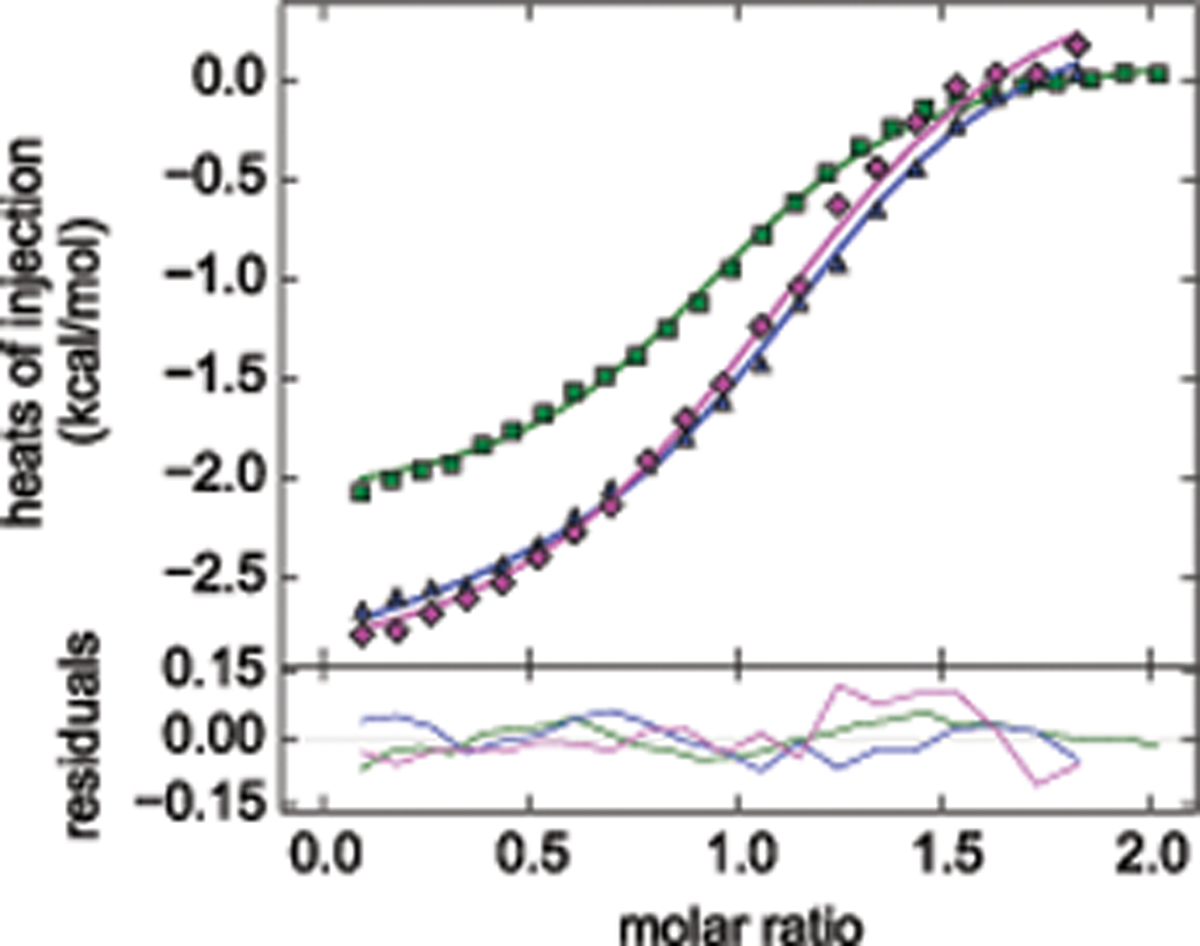
Example of the global ITC analysis of a three-protein interaction analyzed in SEDPHAT and plotted in GUSSI. The experiments examined reversible binding that occurs among the adaptor proteins LAT, Grb2, and Sos1 after T-cell activation: While Sos1 and LAT do not interact and occupy different, single sites on Grb2, multi-phosphorylated LAT is multivalent for Grb2, and Sos1 is bivalent for Grb2, leading to the oligomerization of ternary signaling complexes and initiating signal transduction37. In the experiments shown (reproduced from ref.15), the system is limited to a subset of reactions by using LAT1p, which in its singly phosphorylated form is monovalent for Grb2, and a N-terminal fragment of Sos1 which has only a single site for Grb215. The interaction scheme matches the “A + B +C ↔ AB + C ↔ AC + B ↔ ABC“ variant of the ternary interactions models in SEDPHAT. Plotted are a titration of Grb2 into SoS1NT (green, reduced in scale by a factor ten), a titration of LAT1p into Grb2 (blue) and a titration of LAT1p into the same concentration of Grb2 in an equimolar mixture with Sos1NT (magenta). Global analysis (as described in ref.15) suggests slight cooperativity with ΔΔH° = −4.0 kcal/mol (68% confidence interval from −7.8 to −1.6 kcal/mol) and ΔΔG° = 0.38 kcal/mol (68% confidence interval from 0.11 to 0.65 kcal/mol).
Further extension of the global ITC approach is possible through incorporation of data from complementary biophysical techniques in GMMA. This is particularly powerful for multi-site systems, where global ITC alone may not sufficiently resolve all binding events. GMMA analysis can be accomplished in SEDPHAT with a minor modification of the above protocol, consisting simply of the additional step of loading the complementary datasets, for example, by drag-and-drop, and entering the requisite experimental parameters.30,34 SEDPHAT will then globally refine binding parameters while fitting all datasets, automatically using model functions that match each technique and allowing for experiment-specific additional parameters as needed. Suitable data types include sedimentation equilibrium, sedimentation velocity, surface plasmon resonance, fluorescence quenching and polarization, and various other spectroscopic techniques. An example of a two-site protein interaction studied by GMMA is shown in Figure 7. Thus, familiarity with the protocol above can provide a basis for the study of a variety of different systems of reversibly interacting molecules, using experimental designs and techniques most appropriate to the specific case under investigation.
Figure 7.
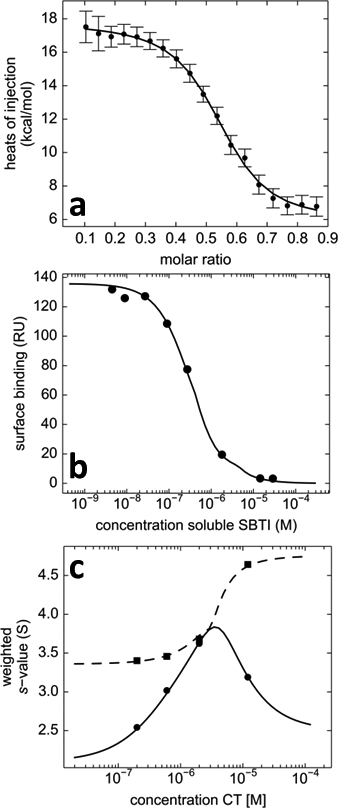
Global multi-method analysis in SEDPHAT of the two-site interaction of α-chymotrypsin (CT) binding to soybean trypsin inhibitor (SBTI), complementing ITC data (a) with surface plasmon resonance (SPR) surface competition isotherm data (b), sedimentation velocity isotherms (c), and fluorescence polarization data (not shown). (a) Normalized heats of reaction measured in calorimetry from the titration of 20 μM CT with aliquots of 84 μM SBTI (symbols); (b) Steady-state SPR biosensor signals from binding of 0.3 μM CT to surface-immobilized SBTI in the presence of different concentrations of soluble SBTI as a competitor (symbols). (c) Weight-average (circles) and reaction boundary (squares) sedimentation coefficients in SV-AUC for 1.8 μM SBTI with different concentrations of CT. Data are taken from the GMMA analysis described in detail in Zhao & Schuck34 consisting of a more comprehensive set of data from 10 experiments, which, in contrast to any single-technique analysis, allowed the thermodynamic binding parameters of both sites to be determined precisely.
Supplementary Material
Supplementary Dataset: Example Data Sets from the duplicat titration experiments of CAII with TFMSA in five different buffers
Supplementary Figure 1: SEDPHAT Initial Screenshot
Supplementary Figure 2: SEDPHAT Final Screenshot
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through International Research Training Group 1830, the Stiftung Rheinland-Pfalz für Innovation, and the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, United States.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests
REFERENCES
- 1.Robinson CV, Sali A & Baumeister W The molecular sociology of the cell. Nature 450, 973–82 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Cebecauer M, Spitaler M, Sergé A & Magee AI Signalling complexes and clusters: functional advantages and methodological hurdles. J. Cell Sci 123, 309–20 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Ladbury JE & Arold ST Noise in cellular signaling pathways: causes and effects. Trends Biochem. Sci 37, 173–178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladbury JE, Klebe G & Freire E Adding calorimetric data to decision making in lead discovery: a hot tip. Nat. Rev. Drug Discov 9, 23–7 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Chaires JB Calorimetry and thermodynamics in drug design. Annu. Rev. Biophys 37, 135–51 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Freire E, Mayorga OL & Straume M Isothermal titration calorimetry. Anal. Chem 62, 950A–959A (1990). [Google Scholar]
- 7.Wiseman T, Williston S, Brandts JF & Lin LN Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem 179, 131–137 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Cooper A Microcalorimetry of protein-protein interactions. Methods Mol. Biol 88, 11–22 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Willerich I & Gröhn F Molecular structure encodes nanoscale assemblies: understanding driving forces in electrostatic self-assembly. J. Am. Chem. Soc 133, 20341–56 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Heerklotz H, Tsamaloukas AD & Keller S Monitoring detergent-mediated solubilization and reconstitution of lipid membranes by isothermal titration calorimetry. Nat. Protoc 4, 686–97 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Tsamaloukas AD, Keller S & Heerklotz H Uptake and release protocol for assessing membrane binding and permeation by way of isothermal titration calorimetry. Nat. Protoc 2, 695–704 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Bains G & Freire E Calorimetric determination of cooperative interactions in high affinity binding processes. Anal. Biochem 192, 203–206 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Brown A Analysis of cooperativity by isothermal titration calorimetry. Int. J. Mol. Sci 10, 3457–77 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freire E, Schön A & Velázquez-Campoy A Isothermal titration calorimetry: general formalism using binding polynomials. Methods Enzym. 455, 127–55 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Houtman JCD et al. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker BM & Murphy KP Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys. J 71, 2049–55 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coussens NP, Schuck P & Zhao H Strategies for assessing proton linkage to bimolecular interactions by global analysis of isothermal titration calorimetry data. J. Chem. Thermodyn 52, 95–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keller S et al. High-precision isothermal titration calorimetry with automated peak shape analysis. Anal. Chem 84, 5066–5073 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce MM, Raman CS & Nall BT Isothermal titration calorimetry of protein-protein interactions. Methods 19, 213–21 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Velázquez-Campoy A & Freire E Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat. Protoc 1, 186–91 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Roselin LS, Lin M-S, Lin P-H, Chang Y & Chen W-Y Recent trends and some applications of isothermal titration calorimetry in biotechnology. Biotechnol. J 5, 85–98 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Velázquez-Campoy A, Ohtaka H, Nezami A, Muzammil S & Freire E Isothermal titration calorimetry. Curr. Protoc. Cell Biol 178, 17.8.1–17.8.24 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Torres FE, Recht MI, Coyle JE, Bruce RH & Williams G Higher Throughput Calorimetry: Opportunities, Approaches and Challenges. Curr. Opin. Struct. Biol 20, 598–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheuermann TH & Brautigam CA High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods (2014). doi: 10.1016/j.ymeth.2014.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman P & Lee JC Functional energetic landscape in the allosteric regulation of muscle pyruvate kinase. 1. Calorimetric study. Biochemistry 48, 9448–55 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishnamoorthy J & Mohanty S Open-ITC: an alternate computational approach to analyze the isothermal titration calorimetry data of complex binding mechanisms. J. Mol. Recognit 24, 1056–66 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Henzl MT Characterization of parvalbumin and polcalcin divalent ion binding by isothermal titration calorimetry. Methods Enzym. 455, 259–97 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Freiburger LA, Auclair K & Mittermaier AK Elucidating protein binding mechanisms by variable-c ITC. Chembiochem 10, 2871–3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schönbeck C, Holm R & Westh P Higher order inclusion complexes and secondary interactions studied by global analysis of calorimetric titrations. Anal. Chem 84, 2305–12 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, Piszczek G & Schuck P SEDPHAT – A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76, 137–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong KM & Baker BM A comprehensive calorimetric investigation of an entropically driven T cell receptor-peptide/major histocompatibility complex interaction. Biophys. J 93, 597–609 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freiburger LA et al. Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nat. Struct. Mol. Biol 18, 288–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herman P & Lee JC Functional energetic landscape in the allosteric regulation of muscle pyruvate kinase. 2. Fluorescence study. Biochemistry 48, 9456–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H & Schuck P Global multi-method analysis of affinities and cooperativity in complex systems of macromolecular interactions. Anal. Chem 84, 9513–9519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H & Schuck P Combining biophysical methods for the analysis of protein complex stoichiometry and affinity in SEDPHAT. Acta Crystallogr D Biol Crystallogr D71, 3–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freiburger L, Auclair K & Mittermaier A Global ITC fitting methods in studies of protein allostery. Methods 76, 149–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houtman JCD et al. Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol 13, 798–805 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Wynn RM, Li J, Brautigam CA, Chuang JL & Chuang DT Structural and biochemical characterization of human mitochondrial branched-chain α-ketoacid dehydrogenase phosphatase. J. Biol. Chem 287, 9178–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duff MR, Grubbs J, Serpersu E & Howell EE Weak Interactions between Folate and Osmolytes in Solution. Biochemistry 51, 2309–18 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Huang L, Serganov A & Patel DJ Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol. Cell 40, 774–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gustchina E et al. Complexes of Neutralizing and Non-Neutralizing Affinity Matured Fabs with a Mimetic of the Internal Trimeric Coiled-Coil of HIV-1 gp41. PLoS One 8, e78187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dellarole M, Sánchez IE & de Prat Gay G Thermodynamics of cooperative DNA recognition at a replication origin and transcription regulatory site. Biochemistry 49, 10277–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krainer G, Broecker J, Vargas C, Fanghänel J & Keller S Quantifying High-Affinity Binding of Hydrophobic Ligands by Isothermal Titration Calorimetry. Anal. Chem 84, 10715–10722 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Moncrieffe MC, Grossmann JG & Gay NJ Assembly of oligomeric death domain complexes during Toll receptor signaling. J Biol Chem 283, 33447–33454 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muscroft-Taylor AC, Soares da Costa TP & Gerrard JA New insights into the mechanism of dihydrodipicolinate synthase using isothermal titration calorimetry. Biochimie 92, 254–62 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Pellizzaro ML et al. Conformer-independent ureidoimidazole motifs--tools to probe conformational and tautomeric effects on the molecular recognition of triply hydrogen-bonded heterodimers. Chem. Eur. J 17, 14508–17 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Krainer G & Keller S Single-experiment displacement assay for quantifying high-affinity binding by isothermal titration calorimetry. Methods 76, 116–123 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Brautigam CA Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzym. 562, 109–133 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Baranauskienė L & Matulis D Intrinsic thermodynamics of ethoxzolamide inhibitor binding to human carbonic anhydrase XIII. BMC Biophys. 5, 12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matulis D & Todd M in Biocalorimetry 2 Appl. Calorim. Biol. Sci (Ladbury JE & Doyle ME) 107–132 (John Wiley & Sons, 2004). at <ISI:000187971200498> [Google Scholar]
- 51.Broecker J, Vargas C & Keller S Revisiting the optimal c value for isothermal titration calorimetry. Anal. Biochem 418, 307–9 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Gans P, Sabatini A, & Vacca A Simultaneous calculation of equilibrium constants and standard formation enthalpies from calorimetric data for systems with multiple equilibria in solution. J. Solution Chem 37, 467–476 (2008) [Google Scholar]
- 53.Brautigam CA Fitting two- and three-site binding models to isothermal titration calorimetric data. Methods (2014). doi: 10.1016/j.ymeth.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vega S Abian O & Velázquez-Campoy A A unified framework based on the binding polynomial for characterizing biological systems by isothermal titration calorimetry. Methods 76, 99–115 (2015) [DOI] [PubMed] [Google Scholar]
- 55.Herrera I & Winnik M Differential binding models for isothermal titration calorimetry: moving beyond the Wiseman isotherm. J. Phys. Chem. B 117, 8659–8672 (2013) [DOI] [PubMed] [Google Scholar]
- 56.Minetti C, Privalov PL & Remeta DP in Proteins Solut. Interfaces Methods Appl. Biotechnol. Mater. Sci (Ruso JM & Pineiro A) 139–179 (Wiley, 2013). [Google Scholar]
- 57.Goldberg R, Kishore N & Lennen RM Thermodynamic quantities for the ionization reactions of buffers. J. Phys. Chem. Ref. Data 31, 231–370 (1999) [Google Scholar]
- 58.Bevington PR & Robinson DK Data Reduction and Error Analysis for the Physical Sciences. (Mc-Graw-Hill, 1992). [Google Scholar]
- 59.Textor M, Vargas C & Keller S Calorimetric quantification of linked equilibria in cyclodextrin/lipid/detergent mixtures for membrane-protein reconstitution. Methods 76, 183–193 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Textor M & Keller S Calorimetric Quantification of Cyclodextrin-Mediated Detergent Extraction for Membrane-Protein Reconstitution. Methods Enzym. in press, (2015). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Dataset: Example Data Sets from the duplicat titration experiments of CAII with TFMSA in five different buffers
Supplementary Figure 1: SEDPHAT Initial Screenshot
Supplementary Figure 2: SEDPHAT Final Screenshot