

Abstract
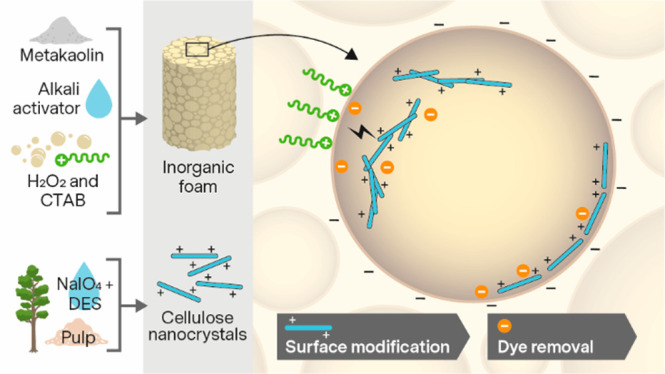
In this work, a surface cationized inorganic–organic hybrid foam was produced from porous geopolymer (GP) and cellulose nanocrystals (CNCs). GPs were synthesized from alkali-activated metakaolin using H2O2 as a blowing agent and hexadecyltrimethylammonium bromide (CTAB) as a surfactant. These highly porous GPs were combined at pH 7.5 with cationic CNCs that had been synthesized from dissolving pulp through periodate oxidation followed by cationization in a deep eutectic solvent. The GP-CNC hybrid foams were employed as reactive filters in the removal of the anionic dye, methyl orange (MO; 5–10 mg/L, pH 7). The effects of a mild acid wash and thermal treatments on the structure, properties, and adsorption capacity of the GPs with CNCs and MO were investigated. The CNCs aligned as films and filaments on the surfaces of the neutralized GPs and the addition of CNCs improved MO removal by up to 84% compared with the reference sample. In addition, CTAB was found to disrupt the attachment of CNCs on the pores and improve adsorption of MO in the GPs with and without CNCs.
Keywords: nanocellulose, DES, micropollutant, wastewater treatment, adsorption, inorganic/organic hybrid
1. Introduction
Inorganic foams composed of ceramic materials,1 metals,2,3 carbon,4 or boron nitride,5 also referred to as cellular solids or cellular solid materials,6 are by definition ensembles of packed cells in a space with solid edges and faces.7 Inorganic foams have been used as lightweight support materials, thermal insulators, in medical applications (scaffolds, implants), electronics, and as absorbents/adsorbents.6,8 Highly porous geopolymer foams are a recent addition to the group of inorganic foams.9 Pores are commonly introduced into the geopolymers by the incorporation of a blowing agent (e.g., H2O2 or metallic Al powder) during synthesis,10,11 but other methods exist, such as mechanical foaming,12 sacrificial templating,13 freeze-casting,14 additive manufacturing,15 and emulsion templating.16
Geopolymers, also known as low-calcium alkali-activated materials, are amorphous to semicrystalline materials synthesized by reacting aluminosilicate precursors (e.g., metakaolin, fly ash) with concentrated aqueous alkaline solutions (e.g., sodium hydroxide and/or sodium silicate).17,18 As a result, a negatively charged three-dimensional and interconnected aluminosilicate (Si–O–Al) framework is formed. The negative charge (originating from four-coordinated Al) is balanced by cations (e.g., Na+ or K+) that are present in the framework cavities. Geopolymers have been extensively studied as replacements for Portland cement,19 but, due to the high intrinsic cation exchange capacity, they have also been used (typically in powdered form) as adsorbents for the removal of cationic dyes (e.g., methylene blue) and heavy metals (e.g., Cu2+, Ni2+, Pb2+).20,21 The introduction of large open pores (μm scale) to the geopolymer structure widens their prospects in adsorption applications9 because of the increased water permeability and available surface area. Consequently, these porous spheres22,23 and monoliths24,25 can be used instead of powders as adsorption media, which simplifies the separation of adsorbent and regeneration.
The pollution of the aquatic environment with emerging contaminants (ECs), such as pharmaceuticals, hormones, and dyes, is suspected to have adverse impacts on humans and ecosystems.26,27 Particular attention should be paid to small, polar, anionic, and nonvolatile molecules as these properties increase their mobility in the environment and hinder removal during conventional wastewater treatment processes.28 Adsorption is considered to be a prominent pretreatment method29 in the EC removal because of its simplicity, efficiency, and the possibility of using abundant and low-cost materials, such as agricultural30 or mineral waste (e.g., fly ash)21 as raw materials. However, negatively charged ECs, such as anionic dyes, are often challenging to remove with inorganic adsorbent materials due to the intrinsic anionic surface charge of the framework. Geopolymers, for example, typically have negative ζ-potential at pH 2–12.22,24 However, a fly ash-based geopolymer has been reported to be capable of adsorbing anionic surfactant sodium dodecyl benzenesulfonate (SDBS).31 According to the ζ-potential measurement, the geopolymer particles were positively charged at pH < 7.04 and negatively charged at pH > 7.04, which explained the highest removal efficiency obtained at pH 2 and the sharp decrease in the surfactant removal at higher pH values. However, such acidic conditions are not feasible in larger-scale applications. Thus, effective removal of persistent anionic pollutants at near-neutral pH requires the surface chemistry of a geopolymer foam to be altered.
Cellulose nanoparticles, i.e., cellulose nanofibers (CNFs) and cellulose nanocrystals (CNCs), are biobased materials that are recognized for their potential to act as adsorbents in water treatment applications due to their hydrophilicity, high surface area-to-volume ratio, low toxicity, and the possibility to add a wide variety of functional groups to the cellulose backbone.32 One novel and sustainable approach to cellulose modification is the use of deep eutectic solvents (DESs). DESs can be produced from safe bulk chemicals, and they have been shown to act not only as a reaction medium but also as a reagent in the production of functionalized CNFs and CNCs.33−37 The most prominent challenge related to the use of nanosized cellulose particles alone as adsorbents is the difficulty in separating the exhausted particles from the solution.38 Therefore, cellulose nanoparticles are often used in combination with other materials, such as (nano)clay minerals,39 metal particles,40 and metal–organic frameworks (MOFs).41 These hybrid materials can be removed after treatment, e.g. by an external magnetic field40 or through charge neutralization with other nanoconstituents, such as halloysite nanotubes or kaolin.39
Porous geopolymer foams are a promising substrate for cellulose nanoparticles due to their large surface areas and rigid nature. Cellulose42,43 and natural cellulosic fibers (e.g., flax, bamboo, cotton)44−46 have been used as a reinforcement agent in the geopolymer matrix due to their low densities, affordability, and good mechanical properties. However, the combination of cellulose nanoparticles with geopolymers has scarcely been studied,47 and indeed no information is available on the surface modification of geopolymers with cellulose nanoparticles. One of the biggest obstacles hindering geopolymer surface modification with cellulose nanoparticles is the presence of free alkali in the material after geopolymerization,48,49 which can trigger the degradation of the biopolymer if there is prolonged contact.47 Therefore, extra effort must be made to effectively reduce the alkalinity of the geopolymer. In the case of geopolymer blocks, this has been achieved through excessive washing with water11,22 or acids.42,43
In this work, we report the surface modification of a porous metakaolin geopolymer foam with cationic CNCs produced from wood cellulose through periodate oxidation and a DES-mediated imination reaction. The geopolymer–CNC hybrid foams were then demonstrated as reactive filter media for the removal of the aqueous anionic dye, methyl orange (MO), at pH 7. In addition, the effects of acid and thermal treatments on the structure, properties, and adsorption capacity of geopolymer foams with CNCs and MO were investigated. The chemical and structural characteristics of CNCs were analyzed using energy-filtered transmission electron microscopy (EFTEM), diffuse reflectance infrared Fourier transform spectroscopy (DRIFT), polyelectrolyte titration, and ζ-potential measurement. The geopolymer foams were analyzed using X-ray diffraction (XRD), X-ray fluorescence (XRF), field emission scanning electron microscopy (FESEM), Brunauer–Emmett–Teller and Barrett–Joyner–Halenda methods (BET-BJH), thermogravimetric analysis (TGA), and ζ-potential measurement. The MO removal was quantified using ultraviolet–visible spectroscopy (UV–vis).
2. Materials and Methods
2.1. Raw Materials and Chemicals
All reagents were used as-received, and deionized water was used throughout the experiments. Metakaolin (MetaMax, 53.0 weight-% SiO2, 44.5 weight-% Al2O3; BASF, Germany), sodium silicate solution (extra pure, 7.5–8.5% Na2O, 25.5–28.5% SiO2, molar SiO2/Na2O = 3.5, water content ≈ 64 weight-%; Merck, Germany), NaOH (98.7%; VWR, Czech Republic), H2O2 (30%; VWR, France), and CTAB (hexadecyltrimethylammonium bromide, ≥ 98%; Sigma-Aldrich, India) were used in the production of the geopolymer foams. Acetic acid (100%; Merck, Germany) was diluted to 0.1 M before use in the acid wash of the geopolymer foams.
Wet disintegrated sheets of commercial softwood dissolving pulp (cellulose 96.2%, hemicelluloses 3.5%, total lignin <0.5%, acetone soluble extractives 0.17%; Domsjö Fabriker AB, Sweden) was used as the cellulose raw material. LiCl (≥99%; Sigma-Aldrich, Germany), sodium metaperiodate (≥99.0%; Sigma-Aldrich, India), glycerol (99.5%; VWR, Belgium), aminoguanidine hydrochloride (>98.0%; TCI, Japan), and ethanol (96%; VWR, France) were used in the chemical modification of the cellulose fibers.
Methyl orange (85%; Sigma-Aldrich, U.S.A.) was used in the dye removal experiments. For pH adjustment, 0.1 M/1 M NaOH and 0.1 M HCl (Oy FF-Chemicals Ab, Finland) were used. Commercial 0.5 M buffer solutions were used in the charge density measurements of the cationic CNCs (citric acid/sodium citrate dihydrate pH 3, citric acid/sodium citrate dihydrate pH 5, sodium phosphate monobasic/sodium phosphate dibasic pH 7, sodium phosphate monobasic/sodium phosphate dibasic pH 8, sodium bicarbonate/sodium carbonate pH 9, and sodium bicarbonate/sodium carbonate pH 10; Alfa Aesar, Germany). Anionic sodium polyethylene sulfonate (Pes-Na, 0.001 N; BTG Instruments GmbH, Germany) was employed as the titrant. TiO2 (rutile, ≥ 99.9%; Aldrich, Japan) was used as an internal standard in XRD measurements of geopolymer samples, and uranyl acetate (98%; Polysciences Inc., U.S.A.) was used for negative staining of the CNCs prior to EFTEM imaging.
2.2. Synthesis of Geopolymer Foams
Alkali activator solution was prepared by mixing the sodium silicate solution (100 g) with NaOH pellets (11.8 g) overnight in a closed plastic bottle. CTAB (Figure 1A) (0.1 g dissolved in 1 mL of water) was added to the activator solution and mixed effectively for 10 min. Metakaolin (63.1 g) was gradually added into the activator solution over 3 min using an overhead mixer (IKA Eurostar 60 digital, Germany) at 1200 rpm, after which the mixing was continued for 7 min. Then, H2O2 solution (5.4 mL) was added to the mixture, and the mixing was continued for another 1 min at 1200 rpm. The homogenized mixture was poured into cylindrical poly(methyl methacrylate) (PMMA) plastic molds (inner height of 101 mm, diameter of 44 mm, and volume of 0.15 L), which were placed inside a plastic bag. The samples were cured at 60 °C for 4 h. The geopolymer had the following molar ratios: SiO2/Al2O3 = 3.65, Na2O/SiO2 = 0.27, Na2O/Al2O3 = 1.00, and H2O/Na2O = 14.1. The fresh paste had a water content of 40% (w/w).
Figure 1.
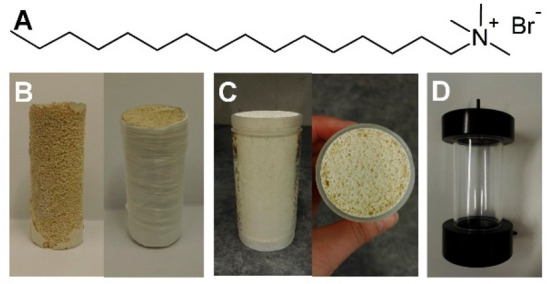
(A) Chemical structure of surfactant CTAB; (B) heat-treated geopolymer foam sample (GP-H) after demolding and before insertion into the column; (C) geopolymer foam (GP) molded directly into the column; and (D) column combined with top and bottom parts containing the liquid inlet and outlet.
Half of the cured geopolymer foams (referred to as GP) were demolded, and the top and bottom parts of the samples were gently smoothened with abrasive paper to reveal the pores. The samples were rinsed with water, placed in Teflon cups and treated hydrothermally at 121 °C and 1 bar for 1 h using an autoclave (Steralis Stèrimax S18–W1, France). The samples (referred to as GP-HT) were rinsed with water, and the excess water was removed with pressurized air. Organic matter was burned away from the samples in a muffle furnace (Nabertherm L5/11/P320, Germany) (heated to 105 °C in 30 min and kept at 105 °C for 4 h, then heated to 525 °C in 2.5 h and kept at 525 °C for 3 h). After the heat treatments, the samples (referred to as GP-H) were fixed into PMMA plastic columns that had the same dimensions as the molds. As the hydrothermal and furnace treatments caused the GP-H samples to shrink slightly, poly(tetrafluoroethylene) (PTFE) tape (Unipak Unitape, Denmark), Parafilm M (Bemis Company Inc., U.S.A.) and silicone paste (Henkel Pattex Healthy Kitchen & Bathroom Sanitary Silicone, Sweden) were used to seal the samples tightly against the column walls (Figure 1B), after which the samples were dried at room temperature overnight. The other half of the cured geopolymer samples (GPs) were kept in the molds, and only the top and bottom parts were smoothened with abrasive paper (Figure 1C).
2.3. Cationization of Cellulose and Disintegration into Nanocrystals
Cationized dialdehyde cellulose (CDAC) was synthesized according to a previously reported34 two-step procedure (Scheme 1). In the first step, cellulose was converted to dialdehyde cellulose (DAC) by selectively oxidizing the vicinal hydroxyl groups to aldehyde groups using sodium metaperiodate. LiCl disrupted the hydrogen bond network of cellulose and improved the oxidation efficiency by exposing more hydroxyl groups to periodate. In the second step, DAC was cationized in an aminoguanidine hydrochloride–glycerol (AhG) DES, which acted as both a reaction medium and a reagent (aminoguanidine hydrochloride).
Scheme 1. Cationization of Oxidized Cellulose Pulp in Aminoguanidine Hydrochloride–Glycerol Deep Eutectic Solvent.

In the synthesis of DAC, 1% (w/w) dissolving pulp (10 g abs.) in deionized water was reacted with sodium metaperiodate (8.2 g) in the presence of LiCl (18 g) for 3 h at 75 °C. Ethanol (500 mL) was added to stop the reaction, after which the mixture was filtered to collect DAC. The DAC fibers were rinsed with 50:50 ethanol:water solution (500 mL), then mixed with ethanol (500 mL) for 15 min, filtered, and finally washed with ethanol (250 mL). The yield of the first reaction was 80%. In the synthesis of CDAC, AhG DES was first prepared by heating and mixing aminoguanidine hydrochloride (52.5 g) and glycerol (87.5 g) in a beaker at 90 °C in an oil bath until a clear liquid was formed. The temperature was adjusted to 80 °C, after which DAC (7 g abs.) was added. The DES-DAC mixture was effectively mixed for 10 min, after which ethanol (250 mL) was added, under stirring, to the mixture. After filtration, the collected CDAC was thoroughly washed with ethanol (2 × 500 mL). Finally, CDAC was mixed with deionized water (500 mL) for 1 h and collected by filtration. The yield of the second reaction was 110%.
Cationic CNCs were produced from 1% (w/w) aqueous CDAC dispersion by mechanical disintegration using a microfluidizer (Microfluidics M-110EH-30, U.S.A.). The CDAC dispersion was passed twice through 400 and 200 μm chambers at a pressure of 1000 bar. The dry matter content of the resulting CNC suspension was determined from oven-dried (105 °C) samples.
2.4. Production of CNC-Geopolymer Foam Hybrids
Geopolymer samples GP and GP-H (directly molded or fixed afterward in columns, Figure 1B and 1C) were washed with 0.1 M acetic acid to remove the excess alkali before combining with CNCs. Acetic acid solution (1 L) was circulated through the column (bottom-up direction, Figure 1D) overnight at 15.5 rpm (8.5 mL/min) using a peristaltic pump (Abimed Gilson Minipuls 3, Germany) with silicone tubing (inner diameter 2 mm). Then, the geopolymer samples (referred to as GP-AA and GP-H-AA) were rinsed until neutrality by pumping deionized water through the column at 48 rpm (0.96 L/h). The 1% (w/w) CNC suspension was diluted with deionized water to 0.1% (w/w) (600 mL), and the pH was adjusted to 7.50 with 1 M NaOH. A sample (1 mL, equal to 1 mg of CNCs) was taken from the CNC suspension for initial charge density determination. Then, the remaining CNC suspension was circulated through the geopolymer GP-AA and GP-H-AA columns in bottom-up direction using a peristaltic pump at 10 rpm (5 mL/min) for 6 days (Figure S1Aof the Supporting Information, SI). A sample (1 mL) was taken from the CNC suspension at regular intervals (1–3 days) for charge density measurement, and the pH of the suspension was recorded. The formed hybrids were referred to as GP-AA-CNC and GP-H-AA-CNC.
2.5. Dye Removal Experiments
Acid-washed geopolymers and heat-treated acid-washed geopolymers with and without CNCs (GP-AA, GP-AA-CNC, GP-H-AA, and GP-H-AA-CNC) were applied as filtration columns for MO removal. The columns were not dried between the acid wash/CNC addition and dye removal experiments because drying agglomerates CNCs. The quantification of MO in the experiments was done using UV–vis spectroscopy (Shimadzu UV-1800, Japan) at 464 nm using quartz cuvettes. The spectrum was recorded at 200–800 nm for each sample to detect any changes in the location of the absorbance maximum. Calibration was performed for the MO concentration range of 0–20 mg/L at pH 7 (Figure S2). Deionized water was used for baseline corrections. An MO solution (5 or 10 mg/L) was prepared by diluting the MO stock solution (1000 mg/L) with deionized water using a volumetric flask (100 mL). The pH of the resulting MO solution was adjusted to 7.00 by adding a few microliters of 0.1 M HCl/NaOH. Using a peristaltic pump at 12 rpm (5 mL/min), the solution was dripped onto the surface of each column, and the filtrate was collected (Figure S1B). The pH, the weight, and the absorbance of the filtrate were recorded. Then, the filtrate solution was passed through the columns again, and the pH, weight, and absorbance were recorded. The procedure was repeated five times in total. The percentage of removed MO from solution was calculated using eq 1:
| 1 |
where C0 (mg/L) is the initial MO concentration, and Ct (mg/L) is the MO concentration after each filtration cycle.
2.6. Material Characterization
For XRF, XRD, TGA, and ζ-potential analyses, powdered geopolymer samples were used. The synthesized geopolymer foams were first crushed by hand and, when needed, mixed overnight with 0.1 M acetic acid. The acid was removed by centrifugation at 7517 rpm for 5 min at 20 °C (Beckman Coulter Avanti J-26 XPI, U.S.A.) and consequently washed with water (500 mL) three times. After each wash, the water was removed by centrifugation. The resulting samples were dried in an oven at 105 °C overnight and stored in a desiccator prior to analysis.
2.6.1. Characterization of Chemical Composition of Geopolymers by XRF
The chemical composition of the geopolymer samples was determined using a 4 kV wavelength dispersive XRF spectrometer (Malvern Panalytical Axios mAX, The Netherlands). The samples (8.25 g) were ground to a fine powder and compressed into tablets before the analysis. The XRF data was interpreted using Omnian software (Malvern Panalytical, The Netherlands).
2.6.2. Characterization of Thermal Decomposition of Geopolymers by TGA
The TGA measurements were performed with a PrepASH 340 ash analyzer (Precisa, Switzerland) under air. Samples (0.5–1.7 g) were heated in a crucible from 25 to 105 °C in 30 min, kept at 105 °C for 4 h, heated to 525 °C in 2.5 h, kept at 525 °C for 3 h, and finally heated to 950 °C in 1 h and kept at 950 °C for 3 h. The loss on ignition at 525 and 950 °C (LOI525 and LOI950, respectively) was determined by comparing the dry weight at 525 and 950 °C with the dry weight at 105 °C, respectively.
2.6.3. Characterization of Geopolymer Structure by XRD
XRD analysis was performed using a Rigaku SmartLab 9 kW diffractometer (Japan) with CuKβ radiation over a 2θ range of 5–130° at a scanning speed of 4° min–1 and a step of 0.02°. Before analysis, samples GP-AA and GP-H-AA underwent additional grinding for 3 min at a speed of 1400 min–1 using a vibratory disc mill (Retsch RS 100, Germany), after which they were sieved to <62 μm. Other samples were ground by hand using a mortar and sieved to <105 μm. Rutile (10% w/w) was added to the samples as an internal standard for the quantification of crystalline phases.
2.6.4. Characterization of Surface Charge of Geopolymers and CNCs by ζ-Potential
Before analysis, the geopolymer samples were ground for 3 min at a speed of 1400 min–1 using a vibratory disc mill (Retsch RS 100, Germany) and sieved to <62 μm. The true densities of the powders were determined using a gas pycnometer (Micrometrics AccuPyc II 1340, U.S.A.), using helium gas as the displacement medium. The particle sizes of the powders were measured by laser diffraction (Beckman Coulter LS 13 320, U.S.A.) from a 0.2% (w/v) water slurry that had been ultrasonicated (37 kHz, pulse mode, 100% power) with overhead mixing for 15 min before measurement. The geopolymer sample (∼0.8 g) was poured into the graduated cylinder containing deionized water (500 mL), after which the suspension was homogenized by agitating the capped cylinder 10 times. The suspension was allowed to settle until a solid concentration of 0.1% (w/w) composed of particles below 10 μm was reached (calculated using Stokes Law). Then, a sample (100 mL) of the supernatant was taken, and the ζ-potential was measured (Malvern Pananalytical Zetasizer Nano Z, U.K.) in the pH range 2–11.5 (adjusted with NaOH/HCl). The 1% (w/w) CNC suspension was diluted with deionized water to 0.1% (w/w) before ζ-potential measurement.
2.6.5. Characterization of Geopolymer Nanometer-Scale Porosity and Specific Surface Area
Specific surface areas and pore volumes of the geopolymer samples were determined from N2 gas adsorption–desorption isotherms using a Micromeritics ASAP 2020 instrument (U.S.A.). The samples were pretreated at 70 °C for 24 h before analysis. The specific surface area was calculated based on the Brunauer–Emmett–Teller (BET) isotherm, and pore-size distributions were calculated from the desorption data using Barrett–Joyner–Halenda (BJH) method.50 Total porosity was calculated using eq 2:
| 2 |
where ρt is the true density (g/cm3) obtained from pulverized (<105 μm) samples using the helium pycnometer (Micrometrics AccuPyc II 1340, U.S.A.), and ρg is the geometric density (g/cm3) determined from dry samples (foam blocks) using an analytical balance and a caliper.
2.6.6. Characterization of Charge Density of CNCs by Polyelectrolyte Titration
The 1% (w/w) CNC suspension was diluted with deionized water to 0.1% (w/w). Buffer stock solutions (0.5 M) in the pH range of 3–10 were diluted to 0.5 mM with deionized water, and the pH was adjusted with 0.1 M NaOH/HCl. The samples for measurement were prepared by mixing 1 mL CNC suspension and 9 mL buffer solution in the sample cell of the particle charge detector (Mütek PCD 03, U.S.A.), after which the mixture was titrated with Pes-Na. CNC samples (1 mL) from the geopolymer-CNC combination experiments were mixed with 9 mL of deionized water, and the mixture was titrated with Pes-Na. The amount of CNC in the solution at each data point was calculated by comparing the Pes-Na consumption with the consumption at t = 0 (charge density of 1 mg of CNCs).
2.6.7. Characterization of Cellulose Structure by DRIFT Spectroscopy
DRIFT spectra were collected with a Bruker Vertex 80v spectrometer (U.S.A.) for the oven- or freeze-dried cellulose samples (pulp, DAC, CDAC). The spectra were recorded in the wavenumber 600–4000 cm–1 using 40 scans with a resolution of 2 cm–1.
2.6.8. Characterization of the Morphology of CNCs by EFTEM
EFTEM samples were prepared by dropping the diluted nanocellulose suspension (0.01% v/v) on a carbon-coated copper grid. Uranyl acetate (2% w/v) was used in the negative staining of the samples, after which the samples were dried at room temperature. Images were captured with a TVIPS TemCam-F416 camera (Germany), using 100 kV as an accelerating voltage (JEOL JEM-2200FS, Japan). The widths and lengths of individual nanocrystals (n = 120) were measured from images with ImageJ/FIJI processing software (National Institutes of Health, U.S.A.). The height and width distribution peaks were evaluated through Gaussian fitting (OriginPro 2018 software; Origin-Lab Corporation, U.S.A.).
2.6.9. Characterization of Geopolymer and CNC–Geopolymer Hybrid Morphologies by FESEM
FESEM samples were prepared by cutting pieces from the top and middle parts of the CNC-geopolymer hybrids (used in MO removal, wet), after which they were frozen in liquid N2 and dried in a freeze-dryer (LaboGene Scanvac CoolSafe, Denmark) overnight. Geopolymer samples that did not contain CNCs were dried at 105 °C prior to freezing and freeze-drying. Images were captured from carbon-coated samples using 5 kV accelerating voltage (Zeiss Ultra Plus, Germany).
3. Results and Discussion
3.1. Characteristics of Geopolymer Foams and the Impact of Thermal Treatments and Acid Wash
The alkali-activation process used in the production of the geopolymer foams included the following main steps: (1) dissolution of aluminosilicate source (in our case metakaolin), (2) speciation and equilibrium of aluminate and silicate, (3) gelation, (4) reorganization, and (5) polymerization and hardening.51 The added H2O2 decomposed into oxygen gas (O2) and water in the alkaline geopolymer paste.52 The interface between O2 gas bubbles and aqueous phase in the fresh geopolymer paste was stabilized (e.g., their coalescence was prevented) with surfactant CTAB. The entrapment of these bubbles in the geopolymer paste resulted in the formation of a highly porous foamy structure during curing. The raw material selected for the geopolymer foam production was metakaolin because of its purity and consistent chemical composition compared with industrial byproducts (e.g., fly ash).53 CTAB was used as a surfactant because, based on the preliminary tests, it was found to produce a highly porous and water permeable structure with homogeneous pore-size distribution when combined with H2O2.
The cured geopolymer foams required further processing before they were brought together with CNCs because cellulose and cellulose nanoparticles are prone to degradation in alkaline conditions47 and geopolymers are known to be highly alkaline due to residual NaOH remaining in the pore water after synthesis.48,49 Consequently, there are four main sample types discussed in this section: Cured geopolymer foams (GP), acid-washed cured geopolymer foams (GP-AA), heat-treated cured geopolymer foams (GP-H), and heat-treated and acid-washed cured geopolymer foams (GP-H-AA). In some procedures, metakaolin geopolymers are ground to a fine powder and washed numerous times with water before use in adsorption experiments.54,55 This method was, however, not applicable in our case because of the highly porous, continuous monolith structure of the fabricated foams. Thus, the foams were washed overnight with dilute acid to guarantee their efficient neutralization. On the basis of the preliminary experiments, 0.1 M acetic acid was chosen (samples referred to as GP-AA and GP-H-AA) as it was less corrosive to the geopolymer than 0.1 M HCl.
Heat treatment was selected as an adjacent strategy for alkalinity reduction. First, GP samples were treated hydrothermally using an autoclave (samples referred to as GP-HT) to ensure that the components had fully reacted and that the amount of unreacted NaOH was as low as possible. Hydrothermal treatment of cured metakaolin geopolymers can also result in the formation of zeolites when continued for 10–18 h at 90–140 °C.56−59 In this case, however, zeolite formation was not expected due to the short duration of the hydrothermal treatment (1 h). The GP-HT samples were then heated to a maximum temperature of 525 °C (annealing, samples referred to as GP-H). This was to reduce the amount of CTAB surfactant (i.e., organic carbon) from the GP samples because it was suspected that CTAB can introduce a cationic charge60 to the GP structure and interfere with the adsorption of cationic CNCs through electrostatic repulsion. After the heat treatment, the GP-H samples were washed with acetic acid to remove any residual NaOH (samples referred to as GP-H-AA).
According to the XRD results (Figure 2), all geopolymer samples had a broad peak centered at around 30° 2θ, which confirmed that the samples remained amorphous61 despite the heat and/or acid treatments, as expected. The peaks originated from rutile that was used as an internal standard (containing anatase impurities). The morphologies of the different geopolymer samples did not differ significantly from each other either (Figure S3). Interconnected pore structures were observed in all samples and the largest pores were approximately 1 mm in diameter. The GP-HT sample was not studied further because it was unlikely that it would greatly differ from the GP sample.
Figure 2.
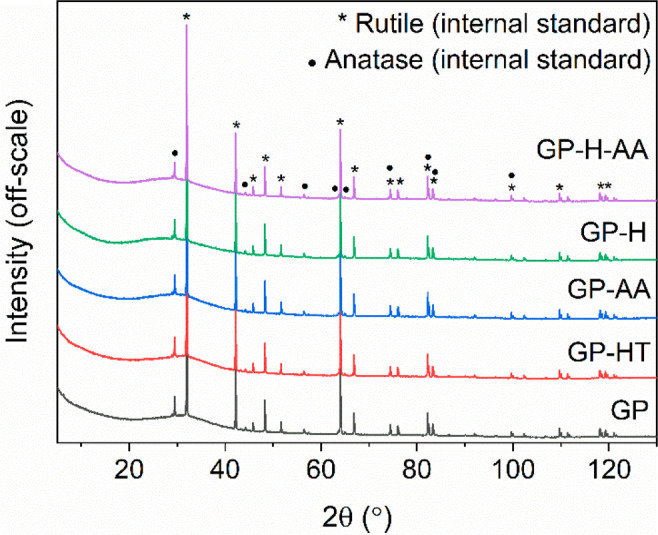
Diffractograms of geopolymer (GP), hydrothermally treated geopolymer (GP-HT), acid-washed geopolymer (GP-AA), heat-treated geopolymer (GP-H), and heat-treated acid-washed geopolymer (GP-H-AA). Samples contained 10% (w/w) of rutile as an internal standard (containing anatase impurity).
XRF results (Table 1) showed that the acid wash reduced the alkaline metal content of the samples because the amount of Na2O was 41% and 35% lower in GP-AA and GP-H-AA samples compared with nonwashed samples GP and GP-H, respectively. The Na/Al molar ratio was close to 1 in the GP and GP-H samples, which indicated that there was little excess sodium (Na) in the geopolymers and that the heat treatment did not reduce the alkaline metal content. However, the amount of bromine (Br) was 54% and 62% lower in samples GP-H and GP-H-AA, respectively, compared with the GP sample, which suggested that heat treatment had decreased the amount of CTAB. LOI525 results (Table 1) supported this conclusion since the weight loss observed for samples GP and GP-AA was considerably higher than for samples GP-H and GP-H-AA. The higher LOI525 result obtained for the GP-H-AA sample compared with GP-H is likely an outcome of the residual acetic acid or sodium acetate in the sample. The low amount of bromine in the GP-AA sample can be explained by the sample preparation method and uncertainty of the analysis since the LOI525 value is in line with the result obtained for the GP sample (Figure S4).
Table 1. Chemical Composition and Loss on Ignition of the Studied Samples.
| composition (wt %) | GP | GP-AA | GP-H | GP-H-AA |
|---|---|---|---|---|
| SiO2 | 48.1 | 52.0 | 48.8 | 51.4 |
| Al2O3 | 22.4 | 24.7 | 23.1 | 24.3 |
| Na2O | 15.2 | 8.9 | 15.0 | 9.7 |
| TiO2 | 0.9 | 1.0 | 0.9 | 1.0 |
| FeO | 0.2 | 0.2 | 0.2 | 0.2 |
| K2O | 0.06 | 0.06 | 0.07 | 0.07 |
| P2O5 | 0.04 | 0.04 | 0.02 | 0.03 |
| CaO | 0.01 | 0.02 | 0.01 | 0.01 |
| MgO | 0.01 | |||
| Cl | 0.03 | 0.01 | 0.02 | 0.02 |
| Br | 0.013 | <0.003a | 0.006 | 0.005 |
| LOI525 | 8.1 | 8.5 | 2.3 | 5.4 |
| LOI950 | 8.3 | 8.9 | 2.6 | 5.8 |
| Na/Al molar ratio | 1.1 | 0.6 | 1.1 | 0.7 |
Detection limit for bromine.
The acetic acid wash increased the surface area and decreased the pore width of both the GP and GP-H samples (Table 2), which indicated that the acetic acid caused slight etching of the samples by removing unreacted NaOH. In addition, the increase in surface area was more pronounced with samples that were not heat-treated, which can possibly be explained by the higher levels of nonreacted NaOH present in the matrix. On the basis of the BJH pore volume determinations, all samples were mostly mesoporous62 (Table 2, Figure S5). The total porosities of samples GP and GP-H were 93.3 and 93.0%, respectively (GP-AA and GP-H-AA were not analyzed because they were fixed in the columns).
Table 2. Surface Areas, Average Pore Widths, And Pore Volumes of Geopolymer Foams Analyzed by BET-BJH.
| GP | GP-AA | GP-H | GP-H-AA | |
|---|---|---|---|---|
| BET surface area (m2/g) | 32.33 ± 0.11 | 47.81 ± 0.11 | 33.14 ± 0.06 | 36.46 ± 0.11 |
| average pore width (nm) | 15.58 | 12.85 | 13.30 | 11.26 |
| macropore volume (cm3/g)a | 0.0007 | 0.0006 | 0.0063 | 0.0009 |
| mesopore volume (cm3/g)a | 0.186 | 0.220 | 0.206 | 0.179 |
| micropore volume (cm3/g)a | 4.5 × 10–6 | 8.4 × 10–4 | 3.7 × 10–4 | 6.6 × 10–4 |
Macropores: d0 > 50 nm, mesopores: 2 nm ≤ d0 ≤ 50 nm, and micropores: d0 < 2 nm
Recently, the pore size distribution of a geopolymer foam produced using a very similar mix design to ours (i.e., direct foaming using CTAB and H2O2, Table S1) was analyzed by X-ray microtomography and BET/BJH.63 The average pore width, surface area, and pore volume determined by BET were 11.7 nm, 31.0 m2/g, and 0.139 cm3/g, respectively. These results correspond well to our results presented in Table 2. However, when the same sample was analyzed by X-ray microtomography, pore size distribution of 10–850 μm was observed, with the average pore width of 0.399 ± 0.221 mm. The reason for the different results is that microtomography can detect pore sizes > ∼ 10 μm, while the BET/BJH method is able to detect pore sizes <300 nm. In the same study63 the porosities of geopolymer foams determined by X-ray microtomography and gas pycnometer were compared. The total and closed porosities determined with X-ray microtomography were 71.8% and 1%, respectively, which was a clear indication of the interconnectivity of the pores. The open (∼total) porosity determined with gas pycnometer (88.8%) was close to values obtained in this study for samples GP and GP-H. The result was higher than that which was acquired using X-ray microtomography, because of the presence of pores <10 μm.
In summary, the different pretreatments were successful in altering the properties of the GPs. Acid wash reduced the alkalinity and increased the surface area of the samples, while annealing reduced the amount of CTAB on the samples. Considering the similarities in the mix design and in the results of the current and previous research, it can be assumed that the geopolymer foams were highly porous and had interconnected pores. The largest pores in our samples were approximately 1 mm in diameter (Figure S3), which indicates that the pore size ranged from ∼10 nm to 1 mm.
3.2. Characteristics of CNCs
According to EFTEM images (Figure 3), the CNCs consisted of rod-like nanoparticles. The CNCs had a width distribution in the range of 5–17 nm, which is comparable to the result obtained for birch pulp (5.7 ± 1.3 nm) under the same conditions.34 The length distribution ranged from 30–170 nm. The average cationic charge density of CNCs was 2.4–3 mequiv/g in the pH range of 3–9 (Figure 4A), which is in line with previously reported results.34 The slight decrease in the charge density in pH 10 is likely attributed to β-alkoxy fragmentation of the dialdehyde cellulose backbone in alkaline conditions and/or deprotonation of the functional groups (guanidinium ions).64,65 The DRIFT spectra (Figure 4B) confirmed the success of the chemical functionalization in both synthesis steps. The spectrum of DAC (the product of step 1) exhibited a band at 1731 cm–1, which is characteristic of an aldehyde carbonyl group. In contrast, the spectrum of CDAC (the product of step 2) had a band at 1680 cm–1 attributed to C=N bond vibration and a shoulder band at ∼1642 cm–1 attributed to N–H bond bending.66,67
Figure 3.
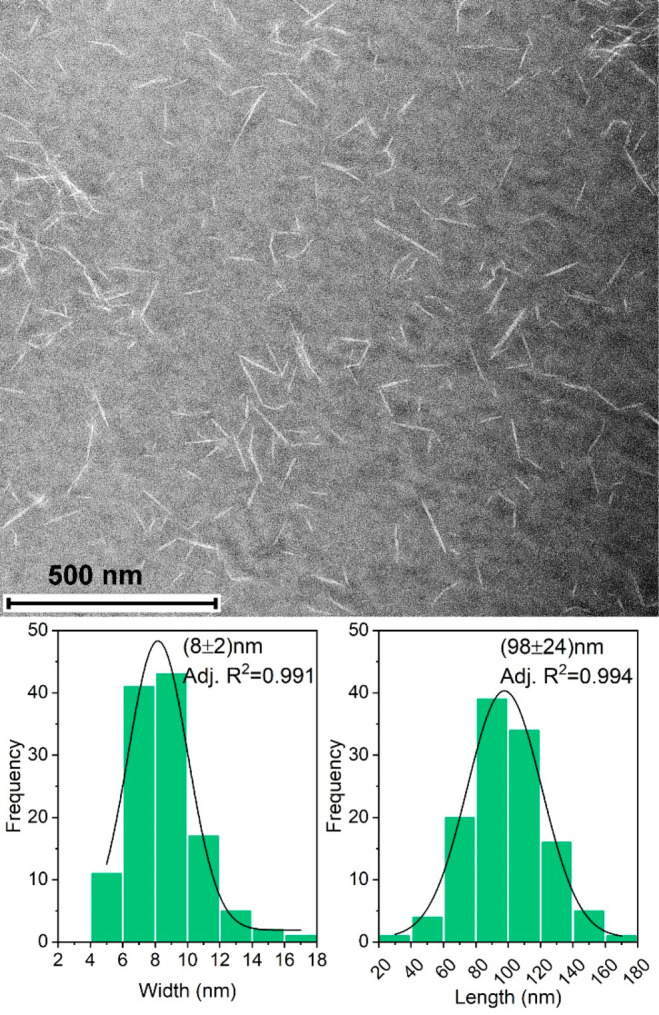
Energy-filtered transmission electron microscope (EFTEM) image of cellulose nanocrystals, and the width and length distributions. Gaussian fitting (n = 120) was used to evaluate the peak centers.
Figure 4.
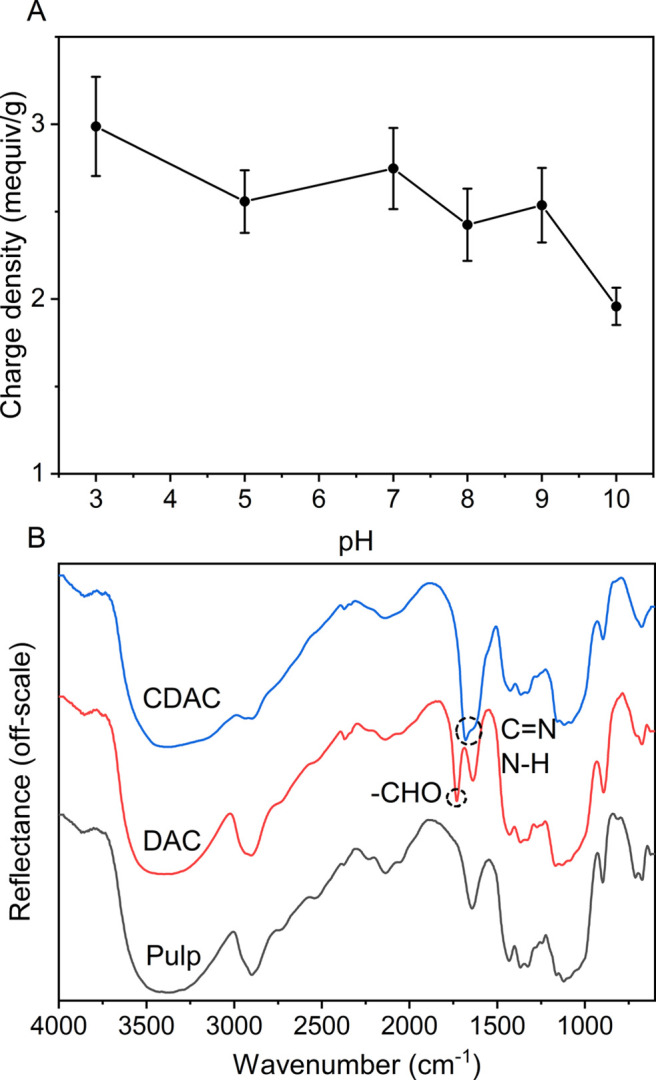
(A) Charge density of cellulose nanocrystals as a function of pH. The error bars represent the sample standard deviation (n = 12). (B) Diffuse reflectance infrared Fourier transform (DRIFT) spectra of cellulose pulp, dialdehyde cellulose (DAC), and cationized dialdehyde cellulose (CDAC).
3.3. Preparation of CNC–Geopolymer Hybrid Foams
Acid-washed samples GP-AA and GP-H-AA were selected for surface modification with cationic CNCs and application testing in dye removal. Especially, it was addressed, if the heat treatments (hydrothermal and annealing) had altered the surface properties of the geopolymer foam, and thus had an impact on the surface modification and dye adsorption compared with the nonheat-treated sample. A good permeability to the dilute CNC suspension was expected as GP and GP-H samples had very high total porosities (93.3% and 93.0%, respectively). The amount of CNC retained onto the geopolymers increased steadily, reaching 11–18 mg/g GP (243–346 mg, corresponds to 40–58% of CNC) during the 6 days of treatment (Figure 5). The average surface coverages of GP-AA-CNC and GP-H-AA-CNC were 2.2% and 3.4%, respectively (the parameters and calculation steps are presented in Table S2). The maximum theoretical surface coverage would have been 7.2%, with the assumption that all CNCs (0.6 g) had adsorbed on the geopolymer. Possible reasons for the observed lower surface coverages are the setup used for the combination (flow instead of sinking), insufficient surface charges on CNCs and geopolymers, and size exclusion. On the basis of the BET/BJH analysis (Table 2), the geopolymers were mainly mesoporous (2–50 nm) with the average pore width of ∼11–13 nm. On the basis of the EFTEM image analysis CNCs had a width distribution of 5–17 nm (Figure 3), which indicated that the surface modification of the smallest pores of the geopolymers was rather unlikely. It can be deduced that CNCs covered the macropores (>50 nm), but because the geopolymer foam was mainly mesoporous, the total surface coverage remained low.
Figure 5.
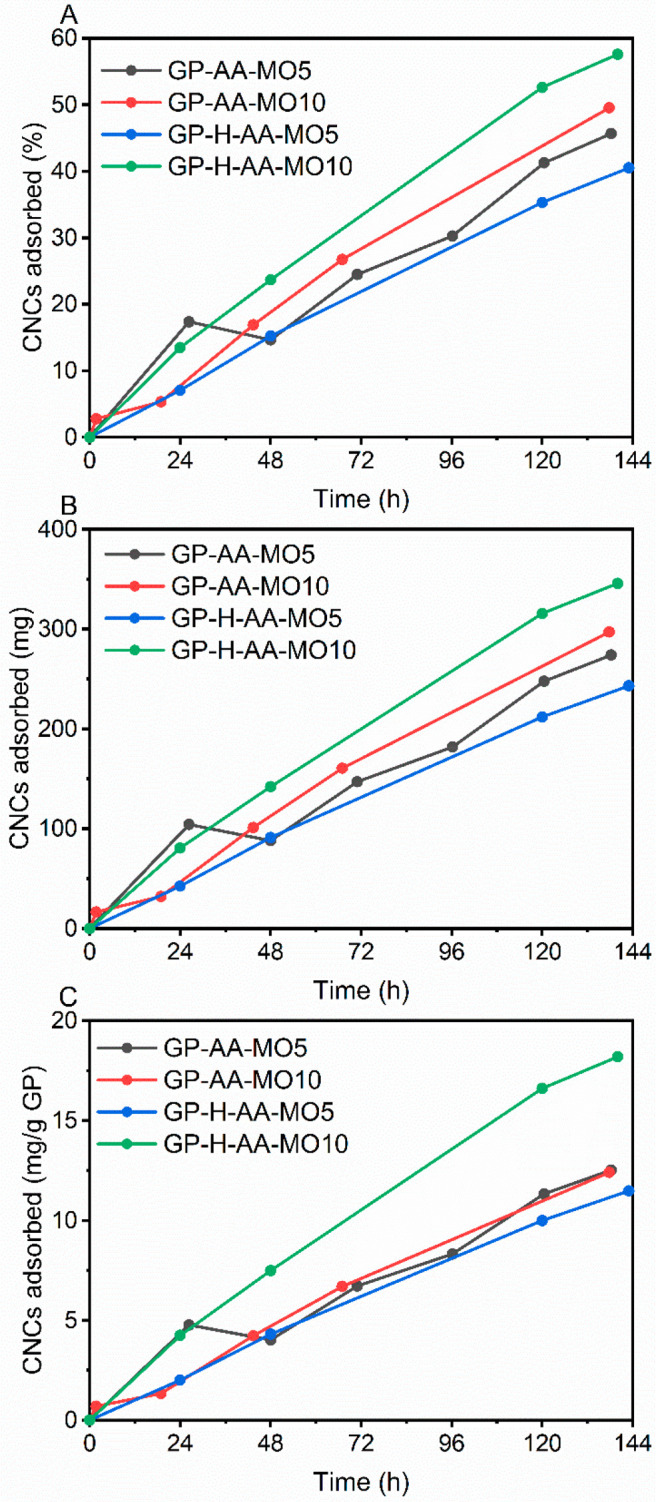
Portion of cellulose nanocrystals (CNCs) adsorbed (A:%; B: mg; C: mg per 1 g of geopolymer) as a function of contact time onto the acid-washed geopolymer (GP-AA) and heat-treated acid-washed geopolymer (GP-H-AA) samples. MO5 and MO10 refer to the methyl orange concentrations in the consecutive dye removal experiments.
The slightly higher surface coverage of GP-H-AA sample compared with GP-AA sample is mainly attributed to the lower BET surface area of GP-H-AA sample (Table 2), because the amount of CNCs retained in the geopolymers was not significantly different (Figure 5). Higher surface coverage values could be achieved by increasing the dry matter content of the CNC suspension, but then again, the risk of blockage of the pores and the cake formation on the surface of the monolith increases, as well as the total costs of the process.
The CNC adsorption and surface coverage results implied that the geopolymer surfaces interacted in a similar manner with CNCs despite the different pretreatments and suggested that the geopolymer possessed an opposite surface charge to that of CNCs. This was confirmed with the ζ-potential result of geopolymers (Figure 6). Both samples had an anionic surface charge in the pH range of 3–11.5 and point of zero charge (PZC) at pH 2.5 (Figure 6). The samples exhibited a similar behavior where the anionic charge increased steeply (i.e., became more anionic) above pH 5 and stabilized at pH 6.9 and 7.7 in the case of GP-AA and GP-H-AA samples, respectively. The results are in line with the results obtained previously for metakaolin geopolymer monoliths,24 where a negative surface charge in pH 2–12 and charge stabilization at pH > 5.6 was observed.
Figure 6.
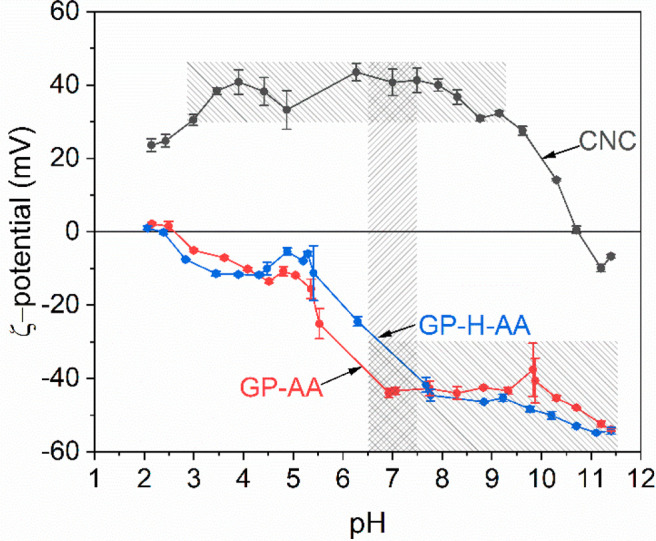
ζ-Potentials of cellulose nanocrystals (CNCs), acid-washed geopolymers (GP-AA) and heat-treated acid washed geopolymers (GP-H-AA) as a function of pH. Each data point represents an average value of three measurements together with the sample standard deviation. The vertical shaded area highlights the pH range during the CNC-GP combination, and the horizontal shaded areas highlight the pH regions where particles form stable colloidal systems.
The ζ-potential of CNCs (Figure 6) was in turn consistent with the charge density result (Figure 4A), indicating that CNCs had a stable cationic charge in pH range 3–9.5 (ζ-potential > +30 mV, shaded horizontal area in Figure 6). The PZC was at pH ≈ 10.5. The pH of the CNC solution decreased from the original value of 7.5 to 6.8–7.0 during the surface modification of the geopolymer, presumably due to the residual acetic acid in the geopolymer. As can be seen from Figure 6 (shaded vertical area), the CNCs and both geopolymer samples exhibited high cationic and anionic surface charges in this region, respectively. The higher initial pH (e.g., 7.6–8) of the CNC solution was not suitable, evident as CNCs precipitated when brought into contact with geopolymers. This may be because small particles were released from the geopolymer foam as a result of the fluid flow and fastening of the caps (containing liquid inlet and outlet, Figure 1D), and these highly charged (ζ-potential −45 mV, Figure 6) particles neutralized the cellulose nanoparticles in the solution in this pH region (the CNC suspension is stable at pH 8, Figure 4A). At pH > 8, the appearance of the CNC solution changed (e.g., bubbling, yellowing) when in contact with geopolymers, which is likely caused by the degradation of dialdehyde cellulose backbone in alkaline conditions.64
Geopolymer surface modification by CNCs was confirmed by FESEM (Figure 7). Individual rod-like CNCs formed a fibrous, assembled nanoscale network in both GP-AA and GP-H-AA samples, as also supported by the CNC surface coverage (adsorption) results (Figure 5). As the FESEM samples were taken from filtration columns that had already been used in dye removal experiments, the CNCs were noted to be fixed on the surfaces and pores of the geopolymer by strong electrostatic forces. On the basis of the FESEM images, the CNCs existed in two different forms on the geopolymer surfaces. First, CNCs had arranged into long filaments that spread across the pores (Figure 7A2 and 7B2). Second, CNCs formed a film (i.e., CNCs lying flat on the surface), evidenced by shrunken and broken CNC layers seen across the samples (Figures 7A3, A4, B3, and B4). On the basis of the FESEM images the films were not uniform and they had a multilayer structure with an approximate thickness of tens to hundreds of nm (the width distribution of individual CNCs was 5–17 nm, Figure 3). Both filament and film formation are typical for CNCs.68,69 It seemed that CTAB, despite being cationic, did not hinder the surface modification of the geopolymers by cationic CNCs. One possible explanation for this is the prolonged contact between CNCs and the geopolymer, which enabled the CNC particles to find the optimum location (i.e., the absence of CTAB) to attach onto.
Figure 7.

Field emission scanning electron microscope (FESEM) images of (A1–A4) acid-washed geopolymer foams modified with cellulose nanocrystals (GP-AA-CNC) and (B1–B4) heat-treated acid-washed geopolymer foams modified with cellulose nanocrystals (GP-H-AA-CNC) with different magnifications. Arrows mark the locations of the CNCs.
The geopolymers were observed to retain 1.9–2.4 g of water per 1 g of geopolymer that originated either from the washing step (GP-AA and GP-H-AA samples) or from the dilute CNC suspension (GP-AA-CNC and GP-H-AA-CNC samples). This can be explained by the high surface area and porosity of the geopolymers (Table 2). Despite being hydrophilic, the addition of CNCs on the geopolymer surface did not improve the water retention capacity, possibly because of their rod-like morphology (Figure 3) and the fact that they were scattered across the geopolymer surfaces, which prevented the hydrogel formation.
3.4. Utilization of CNC Modified Geopolymer Hybrid Foams As Reactive Filter Media for Anionic Dye Removal
Decoration of the large internal surface of the geopolymer with cationic CNCs improved the anionic dye removal efficiency at pH 7 compared with the geopolymer samples without CNCs (Figure 8) in the filtration tests. The initial dye concentration (5 and 10 mg/L) did not have a significant impact on the removal. The increase in the dye removal caused by the addition of CNCs was clearly more pronounced in the heat-treated samples GP-H-AA-CNC (C0 = 5 mg/L: + 35–84%; C0 = 10 mg/L: + 35–58%) than with nonheat-treated samples GP-AA-CNC (C0 = 5 mg/L: + 3–7%; C0 = 10 mg/L: + 12–21%). Surprisingly, the total MO removal was higher with the nonheat-treated samples (with and without CNCs) than with the thermally treated samples (C0 = 5 mg/L: GP-AA 53–68%, GP-AA-CNC 55–70%, GP-H-AA 23–37%, GP-H-AA-CNC 40–50%; C0 = 10 mg/L: GP-AA 45–56%, GP-AA-CNC 54–63%, GP-H-AA 25–37%, GP-H-AA-CNC 36–50%).
Figure 8.
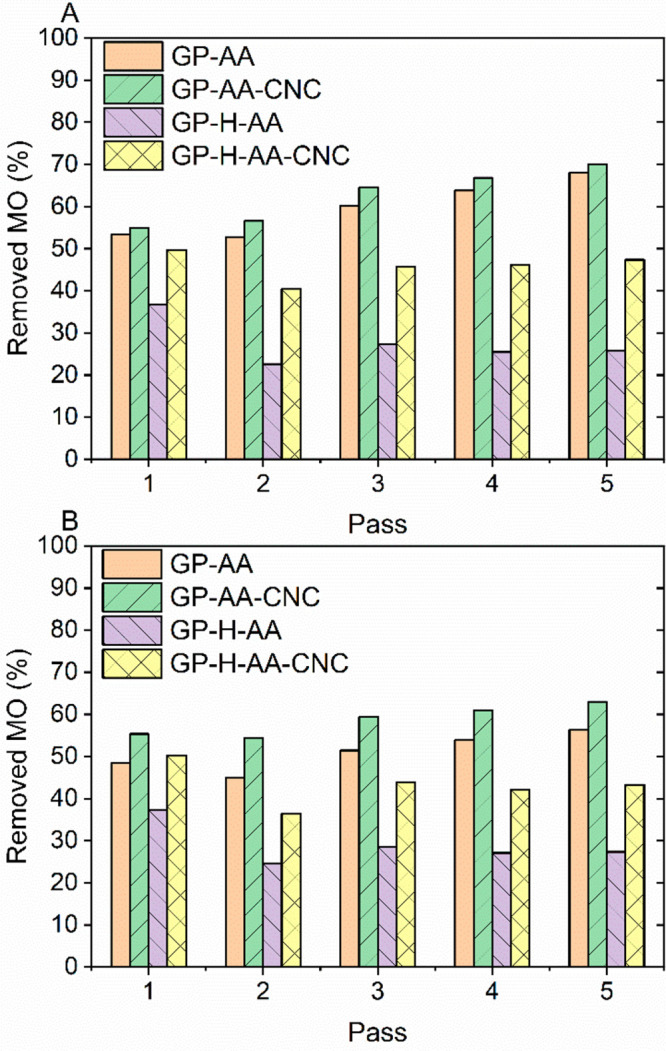
Methyl orange (MO) removal as a function of the number of passes through the acid-washed geopolymer (GP-AA) and heat-treated acid-washed (GP-H-AA) geopolymer columns with and without cellulose nanocrystals (CNCs) at pH 7 using an initial MO concentration of (A) 5 mg/L and (B) 10 mg/L.
The initial hypothesis was that the major constituents responsible for MO adsorption would be CNCs, but, on the basis of these results, it is likely that CTAB also participated in the adsorption. CTAB is a cationic surfactant (Figure 1A) that was added to stabilize the liquid–gas interface during the geopolymer foam synthesis. Considering this, it can be assumed that CTAB was located on the surfaces of the pores across the samples. Thus, the increased dye removal results obtained using the nonheat-treated samples (GP-AA and GP-AA-CNC) can be largely explained by the presence of CTAB as the XRF and LOI results (Table 1) indicated that the nonheat-treated samples contained more CTAB than the heat-treated ones. One possible reason why the addition of CNCs did not significantly improve the dye removal results with the nonheat-treated samples (although being present, see section 3.3) is that the CTAB located on the surfaces of the pores hindered the attachment of CNCs on those surfaces and assisted the aggregation of CNCs instead of a film formation. This aggregation, in turn, lowered the amount of available functional groups in the CNCs for dye adsorption.
In contrast, in the heat-treated samples, CNCs were mainly responsible for the MO adsorption as the amount of CTAB in the geopolymers was reduced. This also explained the low dye removal efficiency of the GP-H-AA samples as they contained no CNCs and only a low amount of CTAB.
In Figure 8, the dye removal efficiency is presented as a function of the passes of the same solution through the filtration column. The trends were different between the two geopolymer samples. For the GP-AA samples (with and without CNCs), the trend followed the anticipated pattern, i.e., the dye removal increased after each pass, indicating that more and more dye was adsorbed onto the filter. With the GP-H-AA samples, however, there was a decrease in the removal efficiency after the first pass, after which it increased slightly and then stabilized. This phenomenon was visible for samples with and without CNCs and in both dye concentrations. A possible explanation for the observed behavior is the ζ-potential of geopolymers (Figure 6). GP-H-AA particles reached stability at pH 7.7, implying that, at pH 7 (the initial pH of the dye solution), the surface charge of the geopolymer was not anionic enough to retain cationic CNCs which had impact on the dye removal. Thus, GP-H-AA filters are expected to perform better at pH > 7.7. For the GP-AA samples, this phenomenon did not occur because the particles had already reached stability at pH 6.9.
One issue that needs further assessment is to investigate how firmly CNCs were attached to the geopolymer surfaces since CNCs were observed in the UV–vis spectra after the filtration cycles (Figure S6 and S7, λ = 200–300 nm). On the basis of the UV–vis spectra intensities, more CNCs were detached from the nonheat-treated geopolymer samples (Figures S6B and S7B) than from heat-treated ones (Figures S6D and S7D), which is a result of the presence of CTAB in the geopolymer structure. However, CNCs were also found in the filtrates of the heat-treated samples. Therefore, future work should focus on improving the attachment of CNCs on the surfaces, e.g., through silane chemistry.70,71 This is especially crucial for the column filtration applications where a continuous flow is applied. Another point to consider in future work is the leaching of the surfactant from the geopolymer as surfactants can have a negative impact on the environment.31 An alternative approach would be to use biobased surfactants (e.g., egg white72).
A compressive strength of ∼0.6 MPa (at 7 d age) has been reported for a geopolymer foam produced using a very similar mix design to ours (Table S1).63 In comparison, sintered ceramic water treatment filters with similar porosities have compressive strengths of approximately 0.9–1.7 MPa.73−75 Considering this background and the fact that our design was almost identical to the reported one, it can be assumed that our filter material has sufficient mechanical properties for the intended application. However, these properties should be studied in detail in the future works. Finally, a more detailed adsorption study (the effect of pH, temperature, time, adsorbent concentration, adsorbate concentration, desorption, and regeneration) should be conducted to gain a full understanding of the potential of these novel hybrid foam materials.
4. Conclusions
The surfaces of the cured geopolymer foams were successfully modified with cationic CNCs after a mild acid-wash treatment. CNCs attached to the geopolymer surfaces through electrostatic interactions and were aligned as continuous films and filaments across the samples. It was found that the addition of CNCs to the geopolymers improved the anionic dye removal by up to 84% in a filtration setup compared with samples without CNCs. In addition, the cationic surfactant CTAB had a crucial role in dye removal: When both CTAB and CNCs were simultaneously present in the geopolymer, CTAB was mostly responsible for the dye adsorption. However, when the amount of CTAB in the geopolymer foam was reduced through heat treatment before CNCs were incorporated on the surface, CNCs significantly increased dye removal. This work demonstrated how the properties of inorganic foams and organic nanomaterials, i.e., rigidity, high porosity and ion exchange capacity of the inorganic foam, and high surface area and functionality of the organic nanoparticles, can be successfully combined to increase the dye removal.
Acknowledgments
Ville Karvonen is acknowledged for his assistance in CNC syntheses and characterizations, Harisankar Sreenivasan for his help with the geopolymer mix design, Kasper Hahtonen for providing the molds and column filtration setup, Jonne Ukkola for his engineering advice, Elisa Wirkkala for her assistance in the geopolymer grinding, Jarno Karvonen for performing the XRF and LS measurements, and Kaisu Ainassaari for performing the BET-BJH measurements. Part of the work was carried out with the support of the Center of Microscopy and Nanotechnology, University of Oulu, Finland.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c05927.
Photographs of the set-ups used in the CNC filtration and MO removal experiments, MO calibration curve and spectra, FESEM images and photographs of the geopolymer samples, TGA curves of the geopolymer samples, incremental and cumulative pore volumes of the geopolymer samples, table comparing the mix designs of geopolymer foams, table of parameters used for the surface coverage calculation, and UV–vis spectra of MO solutions after column filtrations (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
Support from the Advanced Materials Doctoral Program of the University of Oulu Graduate School and the Bionanochemicals project of the Academy of Finland (327810) is acknowledged. P.K. acknowledges financial support from Academy of Finland (grants 322085 and 326291).
The authors declare no competing financial interest.
Supplementary Material
References
- Ge Z.; Chen H.; Ren Y.; Xiao P.; Yang Y.; Zhang T.; Ma Y.; Chen Y. A Universal Method for the Preparation of Dual Network Reduced Graphene Oxide–Ceramic/Metal Foam Materials with Tunable Porosity and Improved Conductivity. Chem. Mater. 2018, 30 (22), 8368–8374. 10.1021/acs.chemmater.8b04081. [DOI] [Google Scholar]
- Ressnig D.; Antonietti M. Ultrafast Syntheses of Silver Foams from Ag 2 NCN: Combustion Synthesis versus Chemical Reduction. Chem. Mater. 2014, 26 (14), 4064–4067. 10.1021/cm501475b. [DOI] [Google Scholar]
- Peng Y.; Liu H.; Li T.; Zhang J. Hybrid Metallic Foam with Superior Elasticity, High Electrical Conductivity, and Pressure Sensitivity. ACS Appl. Mater. Interfaces 2020, 12 (5), 6489–6495. 10.1021/acsami.9b20652. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Ding L.-X.; Liu G.; Chen H.; Wang S.; Wang H. Freestanding, Hydrophilic Nitrogen-Doped Carbon Foams for Highly Compressible All Solid-State Supercapacitors. Adv. Mater. 2016, 28 (28), 5997–6002. 10.1002/adma.201601125. [DOI] [PubMed] [Google Scholar]
- Zeng X.; Ye L.; Yu S.; Sun R.; Xu J.; Wong C.-P. Facile Preparation of Superelastic and Ultralow Dielectric Boron Nitride Nanosheet Aerogels via Freeze-Casting Process. Chem. Mater. 2015, 27 (17), 5849–5855. 10.1021/acs.chemmater.5b00505. [DOI] [Google Scholar]
- Wang X.-B.; Jiang X.-F.; Bando Y. Blowing Route towards Advanced Inorganic Foams. Bull. Chem. Soc. Jpn. 2019, 92 (1), 245–263. 10.1246/bcsj.20180271. [DOI] [Google Scholar]
- Gibson L. J.; Ashby M. F.. Introduction. In Cellular Solids: Structure and Properties; Cambridge Solid State Science Series; Cambridge University Press: Cambridge, 1997; pp 1–14 10.1017/CBO9781139878326.003. [DOI] [Google Scholar]
- Jiang S.; Agarwal S.; Greiner A. Low-Density Open Cellular Sponges as Functional Materials. Angew. Chem., Int. Ed. 2017, 56 (49), 15520–15538. 10.1002/anie.201700684. [DOI] [PubMed] [Google Scholar]
- Novais R. M.; Pullar R. C.; Labrincha J. A. Geopolymer Foams: An Overview of Recent Advancements. Prog. Mater. Sci. 2020, 109, 100621. 10.1016/j.pmatsci.2019.100621. [DOI] [Google Scholar]
- Kränzlein E.; Pöllmann H.; Krcmar W. Metal Powders as Foaming Agents in Fly Ash Based Geopolymer Synthesis and Their Impact on the Structure Depending on the Na /Al Ratio. Cem. Concr. Compos. 2018, 90, 161–168. 10.1016/j.cemconcomp.2018.02.009. [DOI] [Google Scholar]
- Novais R. M.; Buruberri L. H.; Seabra M. P.; Labrincha J. A. Novel Porous Fly-Ash Containing Geopolymer Monoliths for Lead Adsorption from Wastewaters. J. Hazard. Mater. 2016, 318, 631–640. 10.1016/j.jhazmat.2016.07.059. [DOI] [PubMed] [Google Scholar]
- Bai C.; Conte A.; Colombo P. Open-Cell Phosphate-Based Geopolymer Foams by Frothing. Mater. Lett. 2017, 188, 379–382. 10.1016/j.matlet.2016.11.103. [DOI] [Google Scholar]
- Rasouli H. R.; Golestani-Fard F.; Mirhabibi A. R.; Nasab G. M.; Mackenzie K. J. D.; Shahraki M. H. Fabrication and Properties of Microporous Metakaolin-Based Geopolymer Bodies with Polylactic Acid (PLA) Fibers as Pore Generators. Ceram. Int. 2015, 41 (6), 7872–7880. 10.1016/j.ceramint.2015.02.125. [DOI] [Google Scholar]
- Papa E.; Medri V.; Benito P.; Vaccari A.; Bugani S.; Jaroszewicz J.; Swieszkowski W.; Landi E. Synthesis of Porous Hierarchical Geopolymer Monoliths by Ice-Templating. Microporous Mesoporous Mater. 2015, 215, 206–214. 10.1016/j.micromeso.2015.05.043. [DOI] [Google Scholar]
- Franchin G.; Scanferla P.; Zeffiro L.; Elsayed H.; Baliello A.; Giacomello G.; Pasetto M.; Colombo P. Direct Ink Writing of Geopolymeric Inks. J. Eur. Ceram. Soc. 2017, 37 (6), 2481–2489. 10.1016/j.jeurceramsoc.2017.01.030. [DOI] [Google Scholar]
- Medpelli D.; Seo J.-M.; Seo D.-K. Geopolymer with Hierarchically Meso-/Macroporous Structures from Reactive Emulsion Templating. J. Am. Ceram. Soc. 2014, 97 (1), 70–73. 10.1111/jace.12724. [DOI] [Google Scholar]
- Provis J. L.; Van Deventer J. S. J.. 1—Introduction to Geopolymers. In Geopolymers; Provis J. L., van Deventer J. S. J., Eds.; Woodhead Publishing Series in Civil and Structural Engineering; Woodhead Publishing, 2009; pp 1–11. 10.1533/9781845696382.1. [DOI] [Google Scholar]
- Barbosa V. F. F.; MacKenzie K. J. D.; Thaumaturgo C. Synthesis and Characterisation of Materials Based on Inorganic Polymers of Alumina and Silica: Sodium Polysialate Polymers. Int. J. Inorg. Mater. 2000, 2 (4), 309–317. 10.1016/S1466-6049(00)00041-6. [DOI] [Google Scholar]
- Singh N. B.; Middendorf B. Geopolymers as an Alternative to Portland Cement: An Overview. Construction and Building Materials 2020, 237, 117455. 10.1016/j.conbuildmat.2019.117455. [DOI] [Google Scholar]
- Luukkonen T.; Heponiemi A.; Runtti H.; Pesonen J.; Yliniemi J.; Lassi U. Application of Alkali-Activated Materials for Water and Wastewater Treatment: A Review. Rev. Environ. Sci. Bio/Technol. 2019, 18 (2), 271–297. 10.1007/s11157-019-09494-0. [DOI] [Google Scholar]
- Siyal A. A.; Shamsuddin M. R.; Khan M. I.; Rabat N. E.; Zulfiqar M.; Man Z.; Siame J.; Azizli K. A. A Review on Geopolymers as Emerging Materials for the Adsorption of Heavy Metals and Dyes. J. Environ. Manage. 2018, 224, 327–339. 10.1016/j.jenvman.2018.07.046. [DOI] [PubMed] [Google Scholar]
- Novais R. M.; Carvalheiras J.; Tobaldi D. M.; Seabra M. P.; Pullar R. C.; Labrincha J. A. Synthesis of Porous Biomass Fly Ash-Based Geopolymer Spheres for Efficient Removal of Methylene Blue from Wastewaters. J. Cleaner Prod. 2019, 207, 350–362. 10.1016/j.jclepro.2018.09.265. [DOI] [Google Scholar]
- Ge Y.; Cui X.; Liao C.; Li Z. Facile Fabrication of Green Geopolymer/Alginate Hybrid Spheres for Efficient Removal of Cu(II) in Water: Batch and Column Studies. Chem. Eng. J. 2017, 311, 126–134. 10.1016/j.cej.2016.11.079. [DOI] [Google Scholar]
- Novais R. M.; Ascensão G.; Tobaldi D. M.; Seabra M. P.; Labrincha J. A. Biomass Fly Ash Geopolymer Monoliths for Effective Methylene Blue Removal from Wastewaters. J. Cleaner Prod. 2018, 171, 783–794. 10.1016/j.jclepro.2017.10.078. [DOI] [Google Scholar]
- Hertel T.; Novais R. M.; Murillo Alarcón R.; Labrincha J. A.; Pontikes Y. Use of Modified Bauxite Residue-Based Porous Inorganic Polymer Monoliths as Adsorbents of Methylene Blue. J. Cleaner Prod. 2019, 227, 877–889. 10.1016/j.jclepro.2019.04.084. [DOI] [Google Scholar]
- Daughton C. G.; Ternes T. A. Pharmaceuticals and Personal Care Products in the Environment: Agents of Subtle Change?. Environ. Health Perspect. 1999, 107, 907. 10.1289/ehp.99107s6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkaczyk A.; Mitrowska K.; Posyniak A. Synthetic Organic Dyes as Contaminants of the Aquatic Environment and Their Implications for Ecosystems: A Review. Sci. Total Environ. 2020, 717, 137222. 10.1016/j.scitotenv.2020.137222. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Guo W.; Ngo H. H.; Nghiem L. D.; Hai F. I.; Zhang J.; Liang S.; Wang X. C. A Review on the Occurrence of Micropollutants in the Aquatic Environment and Their Fate and Removal during Wastewater Treatment. Sci. Total Environ. 2014, 473–474, 619–641. 10.1016/j.scitotenv.2013.12.065. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Narvaez O. M.; Peralta-Hernandez J. M.; Goonetilleke A.; Bandala E. R. Treatment Technologies for Emerging Contaminants in Water: A Review. Chem. Eng. J. 2017, 323, 361–380. 10.1016/j.cej.2017.04.106. [DOI] [Google Scholar]
- Salleh M. A. M.; Mahmoud D. K.; Karim W. A. W. A.; Idris A. Cationic and Anionic Dye Adsorption by Agricultural Solid Wastes: A Comprehensive Review. Desalination 2011, 280 (1–3), 1–13. 10.1016/j.desal.2011.07.019. [DOI] [Google Scholar]
- Siyal A. A.; Shamsuddin M. R.; Rabat N. E.; Zulfiqar M.; Man Z.; Low A. Fly Ash Based Geopolymer for the Adsorption of Anionic Surfactant from Aqueous Solution. J. Cleaner Prod. 2019, 229, 232–243. 10.1016/j.jclepro.2019.04.384. [DOI] [Google Scholar]
- Köse K.; Mavlan M.; Youngblood J. P.. Applications and Impact of Nanocellulose Based Adsorbents. Cellulose 2020, 27, 2967. 10.1007/s10570-020-03011-1. [DOI] [Google Scholar]
- Selkälä T.; Sirviö J. A.; Lorite G. S.; Liimatainen H. Anionically Stabilized Cellulose Nanofibrils through Succinylation Pretreatment in Urea-Lithium Chloride Deep Eutectic Solvent. ChemSusChem 2016, 9 (21), 3074–3083. 10.1002/cssc.201600903. [DOI] [PubMed] [Google Scholar]
- Li P.; Sirviö J. A.; Asante B.; Liimatainen H. Recyclable Deep Eutectic Solvent for the Production of Cationic Nanocelluloses. Carbohydr. Polym. 2018, 199, 219–227. 10.1016/j.carbpol.2018.07.024. [DOI] [PubMed] [Google Scholar]
- Sirviö J. A.; Ukkola J.; Liimatainen H. Direct Sulfation of Cellulose Fibers Using a Reactive Deep Eutectic Solvent to Produce Highly Charged Cellulose Nanofibers. Cellulose 2019, 26 (4), 2303–2316. 10.1007/s10570-019-02257-8. [DOI] [Google Scholar]
- Sirviö J. A.; Visanko M.; Liimatainen H. Acidic Deep Eutectic Solvents As Hydrolytic Media for Cellulose Nanocrystal Production. Biomacromolecules 2016, 17 (9), 3025–3032. 10.1021/acs.biomac.6b00910. [DOI] [PubMed] [Google Scholar]
- Li P.; Sirviö J. A.; Haapala A.; Liimatainen H. Cellulose Nanofibrils from Nonderivatizing Urea-Based Deep Eutectic Solvent Pretreatments. ACS Appl. Mater. Interfaces 2017, 9 (3), 2846–2855. 10.1021/acsami.6b13625. [DOI] [PubMed] [Google Scholar]
- Selkälä T.; Suopajärvi T.; Sirviö J. A.; Luukkonen T.; Lorite G. S.; Kalliola S.; Sillanpää M.; Liimatainen H. Rapid Uptake of Pharmaceutical Salbutamol from Aqueous Solutions with Anionic Cellulose Nanofibrils: The Importance of PH and Colloidal Stability in the Interaction with Ionizable Pollutants. Chem. Eng. J. 2018, 350, 378–385. 10.1016/j.cej.2018.05.163. [DOI] [Google Scholar]
- Selkälä T.; Suopajärvi T.; Sirviö J. A.; Luukkonen T.; Kinnunen P.; Kling K. I.; Wagner J. B.; Liimatainen H. Efficient Entrapment and Separation of Anionic Pollutants from Aqueous Solutions by Sequential Combination of Cellulose Nanofibrils and Halloysite Nanotubes. Chem. Eng. J. 2019, 374, 1013–1024. 10.1016/j.cej.2019.06.008. [DOI] [Google Scholar]
- Karzar Jeddi M.; Laitinen O.; Liimatainen H. Magnetic Superabsorbents Based on Nanocellulose Aerobeads for Selective Removal of Oils and Organic Solvents. Mater. Des. 2019, 183, 108115. 10.1016/j.matdes.2019.108115. [DOI] [Google Scholar]
- Zhu L.; Zong L.; Wu X.; Li M.; Wang H.; You J.; Li C. Shapeable Fibrous Aerogels of Metal–Organic-Frameworks Templated with Nanocellulose for Rapid and Large-Capacity Adsorption. ACS Nano 2018, 12 (5), 4462–4468. 10.1021/acsnano.8b00566. [DOI] [PubMed] [Google Scholar]
- Szechyńska-Hebda M.; Marczyk J.; Ziejewska C.; Hordyńska N.; Mikuła J.; Hebda M. Optimal Design of PH-Neutral Geopolymer Foams for Their Use in Ecological Plant Cultivation Systems. Materials 2019, 12 (18), 2999. 10.3390/ma12182999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szechyńska-Hebda M.; Marczyk J.; Ziejewska C.; Hordyńska N.; Mikuła J.; Hebda M. Neutral Geopolymer Foams Reinforced with Cellulose Studied with the FT-Raman Spectroscopy. IOP Conf. Ser.: Mater. Sci. Eng. 2019, 706, 012017. 10.1088/1757-899X/706/1/012017. [DOI] [Google Scholar]
- Alzeer M.; MacKenzie K. Synthesis and Mechanical Properties of Novel Composites of Inorganic Polymers (Geopolymers) with Unidirectional Natural Flax Fibres (Phormium Tenax). Appl. Clay Sci. 2013, 75–76, 148–152. 10.1016/j.clay.2013.03.010. [DOI] [Google Scholar]
- Sankar K.; Sá Ribeiro R. A.; Sá Ribeiro M. G.; Kriven W. M. Potassium-Based Geopolymer Composites Reinforced with Chopped Bamboo Fibers. J. Am. Ceram. Soc. 2017, 100 (1), 49–55. 10.1111/jace.14542. [DOI] [Google Scholar]
- Alomayri T.; Shaikh F. U. A.; Low I. M. Characterisation of Cotton Fibre-Reinforced Geopolymer Composites. Composites, Part B 2013, 50, 1–6. 10.1016/j.compositesb.2013.01.013. [DOI] [Google Scholar]
- da Costa Correia V.; Ardanuy M.; Claramunt J.; Savastano H. Assessment of Chemical and Mechanical Behavior of Bamboo Pulp and Nanofibrillated Cellulose Exposed to Alkaline Environments. Cellulose 2019, 26 (17), 9269–9285. 10.1007/s10570-019-02703-7. [DOI] [Google Scholar]
- Lloyd R. R.; Provis J. L.; van Deventer J. S. J. Pore Solution Composition and Alkali Diffusion in Inorganic Polymer Cement. Cem. Concr. Res. 2010, 40 (9), 1386–1392. 10.1016/j.cemconres.2010.04.008. [DOI] [Google Scholar]
- Novais R. M.; Seabra M. P.; Labrincha J. A. Porous Geopolymer Spheres as Novel PH Buffering Materials. J. Cleaner Prod. 2017, 143, 1114–1122. 10.1016/j.jclepro.2016.12.008. [DOI] [Google Scholar]
- Brunauer S.; Emmett P. H.; Teller E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60 (2), 309–319. 10.1021/ja01269a023. [DOI] [Google Scholar]
- Duxson P.; Fernández-Jiménez A.; Provis J. L.; Lukey G. C.; Palomo A.; van Deventer J. S. J. Geopolymer Technology: The Current State of the Art. J. Mater. Sci. 2007, 42 (9), 2917–2933. 10.1007/s10853-006-0637-z. [DOI] [Google Scholar]
- Masi G.; Rickard W. D. A.; Vickers L.; Bignozzi M. C.; van Riessen A. A Comparison between Different Foaming Methods for the Synthesis of Light Weight Geopolymers. Ceram. Int. 2014, 40 (9), 13891–13902. 10.1016/j.ceramint.2014.05.108. [DOI] [Google Scholar]
- MacKenzie K. J. D.28—Innovative Applications of Inorganic Polymers (Geopolymers). In Handbook of Alkali-Activated Cements, Mortars and Concretes; Pacheco-Torgal F., Labrincha J. A., Leonelli C., Palomo A., Chindaprasirt P., Eds.; Woodhead Publishing: Oxford, 2015; pp 777–805 10.1533/9781782422884.5.777. [DOI] [Google Scholar]
- Luukkonen T.; Sarkkinen M.; Kemppainen K.; Rämö J.; Lassi U. Metakaolin Geopolymer Characterization and Application for Ammonium Removal from Model Solutions and Landfill Leachate. Appl. Clay Sci. 2016, 119, 266–276. 10.1016/j.clay.2015.10.027. [DOI] [Google Scholar]
- Luukkonen T.; Věžníková K.; Tolonen E.-T.; Runtti H.; Yliniemi J.; Hu T.; Kemppainen K.; Lassi U. Removal of Ammonium from Municipal Wastewater with Powdered and Granulated Metakaolin Geopolymer. Environ. Technol. 2018, 39 (4), 414–423. 10.1080/09593330.2017.1301572. [DOI] [PubMed] [Google Scholar]
- Zhang J.; He Y.; Wang Y.; Mao J.; Cui X. Synthesis of a Self-Supporting Faujasite Zeolite Membrane Using Geopolymer Gel for Separation of Alcohol/Water Mixture. Mater. Lett. 2014, 116, 167–170. 10.1016/j.matlet.2013.11.008. [DOI] [Google Scholar]
- Tang Q.; Ge Y.; Wang K.; He Y.; Cui X. Preparation of Porous P-Type Zeolite Spheres with Suspension Solidification Method. Mater. Lett. 2015, 161, 558–560. 10.1016/j.matlet.2015.09.062. [DOI] [Google Scholar]
- Tang Q.; He Y.; Wang Y.; Wang K.; Cui X. Study on Synthesis and Characterization of ZSM-20 Zeolites from Metakaolin-Based Geopolymers. Appl. Clay Sci. 2016, 129, 102–107. 10.1016/j.clay.2016.05.011. [DOI] [Google Scholar]
- Wang H.; Yan C.; Li D.; Zhou F.; Liu Y.; Zhou C.; Komarneni S. In Situ Transformation of Geopolymer Gels to Self-Supporting NaX Zeolite Monoliths with Excellent Compressive Strength. Microporous Mesoporous Mater. 2018, 261, 164–169. 10.1016/j.micromeso.2017.11.015. [DOI] [Google Scholar]
- Huang P.; Kazlauciunas A.; Menzel R.; Lin L. Determining the Mechanism and Efficiency of Industrial Dye Adsorption through Facile Structural Control of Organo-Montmorillonite Adsorbents. ACS Appl. Mater. Interfaces 2017, 9 (31), 26383–26391. 10.1021/acsami.7b08406. [DOI] [PubMed] [Google Scholar]
- Provis J. L.; Lukey G. C.; van Deventer J. S. J. Do Geopolymers Actually Contain Nanocrystalline Zeolites? A Reexamination of Existing Results. Chem. Mater. 2005, 17 (12), 3075–3085. 10.1021/cm050230i. [DOI] [Google Scholar]
- Rouquerol J.; Avnir D.; Fairbridge C. W.; Everett D. H.; Haynes J. H.; Pernicone N.; Ramsay J. D. F.; Sing K. S. W.; Unger K. K.. Recommendations for the Characterization of Porous Solids; International Union of Pure and Applied Chemistry, 1994, 66 10.1351/pac199466081739. [DOI] [Google Scholar]
- Luukkonen T.; Yliniemi J.; Sreenivasan H.; Ohenoja K.; Finnilä M.; Franchin G.; Colombo P.. Ag- or Cu-Modified Geopolymer Filters for Water Treatment Manufactured by 3D Printing, Direct Foaming, or Granulation. Scientific Reports 2020, 10 ( (1), ) 10.1038/s41598-020-64228-5. [DOI] [PMC free article] [PubMed]
- Calvini P.; Conio G.; Lorenzoni M.; Pedemonte E. Viscometric Determination of Dialdehyde Content in Periodate Oxycellulose. Part I. Methodology. Cellulose 2004, 11 (1), 99–107. 10.1023/B:CELL.0000014766.31671.8d. [DOI] [Google Scholar]
- Clayden J.; Greeves N.; Warren S.; Wothers P.. Organic Chemistry; Oxford University Press: Oxford, 2001. [Google Scholar]
- Sirviö J. A.; Anttila A.-K.; Pirttilä A. M.; Liimatainen H.; Kilpeläinen I.; Niinimäki J.; Hormi O. Cationic Wood Cellulose Films with High Strength and Bacterial Anti-Adhesive Properties. Cellulose 2014, 21 (5), 3573–3583. 10.1007/s10570-014-0351-y. [DOI] [Google Scholar]
- Zhang Y.; Jiang J.; Chen Y. Synthesis and Antimicrobial Activity of Polymeric Guanidine and Biguanidine Salts. Polymer 1999, 40 (22), 6189–6198. 10.1016/S0032-3861(98)00828-3. [DOI] [Google Scholar]
- Hujaya S. D.; Manninen A.; Kling K.; Wagner J. B.; Vainio S. J.; Liimatainen H. Self-Assembled Nanofibrils from RGD-Functionalized Cellulose Nanocrystals to Improve the Performance of PEI/DNA Polyplexes. J. Colloid Interface Sci. 2019, 553, 71–82. 10.1016/j.jcis.2019.06.001. [DOI] [PubMed] [Google Scholar]
- Tardy B. L.; Ago M.; Guo J.; Borghei M.; Kämäräinen T.; Rojas O. J. Optical Properties of Self-Assembled Cellulose Nanocrystals Films Suspended at Planar-Symmetrical Interfaces. Small 2017, 13 (47), 1702084. 10.1002/smll.201702084. [DOI] [PubMed] [Google Scholar]
- Pacaphol K.; Aht-Ong D. The Influences of Silanes on Interfacial Adhesion and Surface Properties of Nanocellulose Film Coating on Glass and Aluminum Substrates. Surf. Coat. Technol. 2017, 320, 70–81. 10.1016/j.surfcoat.2017.01.111. [DOI] [Google Scholar]
- Laitinen O.; Suopajärvi T.; Österberg M.; Liimatainen H. Hydrophobic, Superabsorbing Aerogels from Choline Chloride-Based Deep Eutectic Solvent Pretreated and Silylated Cellulose Nanofibrils for Selective Oil Removal. ACS Appl. Mater. Interfaces 2017, 9 (29), 25029–25037. 10.1021/acsami.7b06304. [DOI] [PubMed] [Google Scholar]
- Bai C.; Colombo P. High-Porosity Geopolymer Membrane Supports by Peroxide Route with the Addition of Egg White as Surfactant. Ceram. Int. 2017, 43 (2), 2267–2273. 10.1016/j.ceramint.2016.10.205. [DOI] [Google Scholar]
- Salvini V. R.; Luchini B.; Aneziris C. G.; Pandolfelli V. C. Innovation in Ceramic Foam Filters Manufacturing Process. Int. J. Appl. Ceram. Technol. 2019, 16 (1), 378–388. 10.1111/ijac.13062. [DOI] [Google Scholar]
- Shivaraju H. P.; Egumbo H.; Madhusudan P.; Anil Kumar K. M.; Midhun G. Preparation of Affordable and Multifunctional Clay-Based Ceramic Filter Matrix for Treatment of Drinking Water. Environ. Technol. 2019, 40 (13), 1633–1643. 10.1080/09593330.2018.1430853. [DOI] [PubMed] [Google Scholar]
- Masturi S.; Aji M. P.; Sustini E.; Khairurrijal M. A. Permeability, Strength and Filtration Performance for Uncoated and Titania-Coated Clay Wastewater Filters. Am. J. Environ. Sci. 2012, 8 (2), 79–94. 10.3844/ajessp.2012.79.94. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.