

Capsid structures reveal a remarkable structural diversity among small bacterial RNA viruses.
Abstract
The single-stranded RNA (ssRNA) bacteriophages are among the simplest known viruses with small genomes and exceptionally high mutation rates. The number of ssRNA phage isolates has remained very low, but recent metagenomic studies have uncovered an immense variety of distinct uncultured ssRNA phages. The coat proteins (CPs) in these genomes are particularly diverse, with notable variation in length and often no recognizable similarity to previously known viruses. We recombinantly expressed metagenome-derived ssRNA phage CPs to produce virus-like particles and determined the three-dimensional structure of 22 previously uncharacterized ssRNA phage capsids covering nine distinct CP types. The structures revealed substantial deviations from the previously known ssRNA phage CP fold, uncovered an unusual prolate particle shape, and revealed a previously unseen dsRNA binding mode. These data expand our knowledge of the evolution of viral structural proteins and are of relevance for applications such as ssRNA phage–based vaccine design.
INTRODUCTION
The single-stranded RNA (ssRNA) bacteriophages are a group of small bacterial viruses known to infect different Proteobacteria. The ssRNA phages have short 3.5- to 4.5-kb positive-sense RNA genomes that encode only three common proteins: the maturation protein (MP), a minor structural protein that functions to adsorb the phage to the bacterial receptor and deliver the genome into the host cell; the coat protein (CP), the major structural component of the capsid; and the catalytic subunit of an RNA-dependent RNA polymerase (RdRp), necessary for replicating the viral genome (1). The ssRNA phages are believed to represent the oldest extant RNA virus lineage (2), and only their RdRp has identifiable homologs in other viruses, while the MP and CP are unique to the ssRNA phages.
The ssRNA phages have played an important role in studies of genome structure and replication, translational control mechanisms, protein-RNA interactions, and other fundamental problems in biology, and they have been convenient models for investigating virus structure, assembly, and evolution. The ssRNA phage MS2 was among the first viruses with a determined atomic resolution capsid structure (3) and with the entire virion, including the packaged genome in a well-defined conformation, reconstructed at near-atomic resolution (4). The ssRNA phage CPs adopt a fold not found in any other viruses, with an N-terminal β hairpin, a central five-stranded antiparallel β sheet, and two C-terminal α helices, and they are further unusual in that two CP molecules very tightly interact to form dimers with a common hydrophobic core. Consequently, the de facto subunits of ssRNA phage particles are CP dimers, composed of a single 10-stranded β sheet lining the interior of the particle and the α helices and β hairpins exposed to the capsid exterior. The complete virion is built of 89 CP dimers and a single copy of a genome-bound MP in place of another CP dimer. The assembled particle is approximately 28 nm in diameter and, disregarding the slight irregularity introduced by the MP, follows T = 3 quasi-equivalent icosahedral symmetry.
To date, high-resolution structures of eight ssRNA phages infecting Escherichia (3, 5–7), Pseudomonas (8, 9), Caulobacter (10), and Acinetobacter (11) have been determined, which have shown that the CP fold is conserved despite high sequence variability. Thus far, the only notable variation in CP structure has been observed in the Acinetobacter phage AP205, in which the N-terminal strand of the β-hairpin has been translocated to the C terminus; however, the close vicinity of monomer N and C termini in the dimer results in the formation of a two-stranded β structure at a position analogous to the β-hairpin in the other phages. Variations in the CP fold are expected given the exceptionally high mutation rates of RNA viruses, but for reasons not entirely clear, the number and diversity of isolated ssRNA phages have remained very low, which has, in turn, limited continued structural studies on these viruses. There appears to be no reason to presume a genuine scarcity of ssRNA bacteriophages in nature, and the extent to which their structural diversity has been sampled and explored remains largely unknown.
The ever-increasing metagenomic sequencing efforts within the last decade have documented an expanse of highly diverse uncultured microbial and viral life forms in every environment that has been examined. In 2016, two studies uncovered more than 200 novel ssRNA phage sequences in various RNA metagenomes (12, 13), and very recently, thousands of additional ssRNA phage genomes have been found (14, 15), demonstrating that the true ubiquity and diversity of these viruses in nature have been greatly underestimated and underexplored. Still, beyond comparative analyses with experimentally well-characterized species, the metagenomic studies are limited in their capacity for providing deeper insight into the biology of the newly found life forms, and in the case of highly diverged bacteriophage genomes, often provide little or no information about the structure, host bacteria, lysis strategies, and other essential characteristics of these viruses. For the previously studied ssRNA phages, expression of a cloned CP gene results in assembly of virus-like particles (VLPs), which lack the MP and package random bacterial RNA instead of the viral genome, but that are otherwise morphologically indistinguishable from native virions. In context of metagenome data, this gives an opportunity to obtain previously unexplored CP-encoding sequences via chemical gene synthesis and recombinantly express them in bacteria to resurrect phage-like particles in the laboratory. Following this approach, we previously acquired more than 100 novel ssRNA phage CP sequences, which in many cases had no recognizable similarity to the previously known phages or to each other, and we were able to obtain 80 different VLPs of 11 distinct CP types, 8 of which were experimentally characterized (16).
With CP sequences diverged beyond recognition, determination of their three-dimensional structure is instrumental in uncovering distant relationships among the ssRNA phages and can offer valuable clues for better understanding the evolution of these viruses in general. In addition, the ssRNA phage VLPs have found applications as carriers for foreign antigens in vaccine development (17), for which detailed knowledge of the location of CP terminal and loop regions is essential to enable structure-guided design of new medications. To address these questions, we have determined crystal structures of 22 metagenome-derived ssRNA phage VLPs, which have revealed substantial deviations from the previously known CP fold and uncovered previously unseen ssRNA phage particle shapes.
RESULTS AND DISCUSSION
Structure determination
Our laboratory currently holds a collection of more than 80 different metagenome-derived ssRNA bacteriophage VLPs, nearly all of which could be produced in sufficient quantity, homogeneity, and purity for crystallographic studies. We thereby set out to perform crystallization trials of our entire ~80 VLP library and were able to obtain crystals for more than half of the VLPs. Diffraction data to a resolution sufficient for model building (<4 Å) were obtained for 22 VLPs covering nine distinct CP types (table S1). The resolution of the determined VLP structures ranges from 2.6 to 4.0 Å, with an average of 3.4 Å, which in all cases allowed us to build all-atom models. In addition, one of the VLPs in our study, EMS014, had disassembled at a particular crystallization condition, and a crystal of CP dimers had grown instead, which in this case allowed for a further 1.25-Å subunit structure to be solved. The majority of the models could be built for all residues except for the AC, Beihai14, and PQ-465 CPs, which had unstructured termini, and the AVE019, Beihai19, ESE014, ESE020, and ESE058 CPs, in which certain flexible loops were present. X-ray data collection, reduction and refinement statistics, and model quality indicators are presented in table S2.
CP fold
Within our sample set of 22 VLPs, only in 10 cases was the CP structure consistent with the canonical MS2 CP fold with the N-terminal hairpin, five-stranded β sheet, and two C-terminal α helices, while notable deviations are observed for the other CPs (Fig. 1). Only the four central β strands and a single C-terminal α helix are retained in all structures, while the terminal regions, particularly the N termini, are variable between different CP types. Pairwise superposition of all available ssRNA phage CP monomers further illustrates that approximately half of the newly determined structures do not have substantial similarity to those of the previously studied phages (fig. S1). Structures of the nine distinct CP types reveal both unsuspected similarities in distant ssRNA phage lineages and unexpected differences in close relatives.
Fig. 1. Coat protein fold of the novel ssRNA bacteriophages.
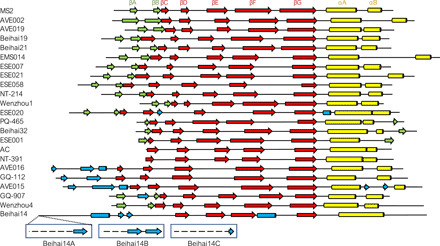
Secondary structure elements are represented as arrows (β strands) and cylinders (α helices). The canonical fold of the bacteriophage MS2 CP with annotated secondary structure elements is included for comparison; for consistency, this nomenclature has been used throughout the text regardless of the actual position of the elements in the particular novel CPs. β strands that constitute the central sheet are shown in red, C-terminal α helices in yellow, β strands that form an N-terminal β-hairpin or an analogous structure in green, and additional secondary structure elements in blue. The different N-terminal structures of Beihai14 quasi-equivalent CP monomers are additionally indicated.
MS2-like CPs
The majority of the currently available metagenomic ssRNA phage CP sequences fall into a broad MS2-like supergroup, and also the greatest percentage of the newly solved VLP structures belong to this similarity cluster. Of the newly characterized MS2-like phages, the Beihai19 and Beihai21 CP structure is similar to that of the previously studied phages of this group, such as MS2 and Qβ; the structures from phages ESE007, ESE058, NT-214, and AVE019 are slightly more diverged, yet still very similar. Although the length of these proteins varies from 123 to 146 residues, the size and position of secondary structure elements remain virtually unchanged, and the structural differences almost entirely arise from differently sized loops and their relative packing. The EMS014 and ESE021 CPs are the most diverged MS2-like CPs in our study and also the longest (156 and 150 residues, respectively), which for the most part can be attributed to markedly extended loops between the two C-terminal α helices. Both CPs are relatively similar with a 27% sequence identity, and both have secondary structure elements in essentially the same positions as in the canonical MS2 fold, but in the ESE021 CP, the αB helices have swapped positions between the two monomers (Fig. 2), which has never been observed in ssRNA phages before. Together with the N-terminal β-hairpins, the extensive interhelix loops in the EMS014 and ESE021 CPs completely wrap the helices and almost entirely isolate them from the outside environment.
Fig. 2. Swapped C-terminal α helices in the ESE021 coat protein.
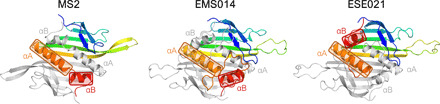
In the MS2 coat protein dimer, the C-terminal α helices αA and αB of one monomer (rainbow-colored) are positioned roughly end to end to each other in a groove formed by the same helices in the other monomer (light gray). The MS2-like EMS014 and ESE021 coat proteins are closely related, but while the fold of the EMS014 CP closely follows that of MS2, in the ESE021 CP, the αB helices are swapped between the two monomers.
Cb5-like CPs
The AVE002 and Wenzhou1 CPs in our study are most closely related to a CP type first described in the previously studied bacteriophage Cb5 (10). Despite undetectable sequence similarity to the Cb5 CP or to each other, the two proteins do not show significant deviations in their structural organization and follow the canonical ssRNA phage CP fold. The AVE002 CP is somewhat longer (140 residues) than that of Cb5 (122 residues), with the difference mostly attributed to an extensive surface-exposed loop between helices αA and αB, but contrary to the MS2-like EMS014 and ESE021 CPs, the AVE002 interhelix loops bend to the opposite direction over the β-hairpins. The Wenzhou1 CP (Fig. 3) spans only 113 residues, but the overall size of the CP dimer is similar to that of the other Cb5-like proteins. The savings in the Wenzhou1 CP arise from a significantly smaller N-terminal β-hairpin and a very short βD strand, which almost reduces the central β sheet to only three strands. The space vacated by the shrunken βD strand is partially occupied by the C terminus, which as a result becomes buried underneath the capsid surface.
Fig. 3. Three-dimensional structure of coat protein dimers.
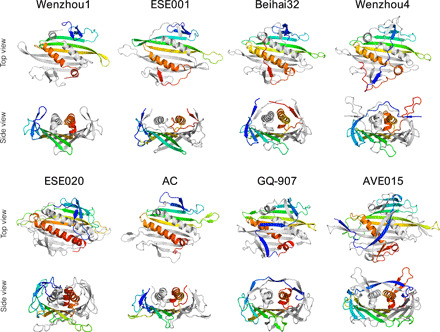
Proteins representing different variations of the CP fold are shown with one of the monomers rainbow-colored blue (N terminus) to red (C terminus) and the other monomer shown in light gray. All CP dimers are shown as seen from outside of the particle (top view) and in a roughly perpendicular view looking lengthwise down the C-terminal α helices (side view).
AP205-like CPs
The remaining group of CPs with previously determined related structures, the AP205-like CPs (11), is represented by phages PQ-465 and ESE001 in our study. The PQ-465 CP shares 30% sequence identity with that of AP205, and there are no noteworthy differences in the three-dimensional structure of the two proteins. The ESE001 CP (Fig. 3) is notably more distinct and was previously recognized as a separate CP type (16) as it could not be reliably aligned to any other CPs. However, the three-dimensional structure of ESE001 VLPs revealed a CP fold with distinctive AP205-like features such as a β-hairpin–like structure formed by the N and C termini, a notable gap between α helices of the two monomers, and stabilizing disulfide bonds between CP subunits. The AP205-like CPs also characteristically have a very short αB helix that lies roughly perpendicular to the αA helix and the β sheet, but in the ESE001 CP, the αB helix is completely eliminated and the protein only has a single C-terminal helix.
Beihai32-like CPs
The remaining VLPs in our study represent six previously uncharacterized ssRNA bacteriophage CP types, and the first of those, putatively named here Beihai32-like CPs, includes proteins from bacteriophages Beihai32 and Wenzhou4, which have lengths of 130 and 146 residues, respectively. Previously, these CPs were included in the MS2-like CP supergroup due to borderline sequence similarity (16), but their three-dimensional structure suggests that these proteins should be recognized as a separate type. Structure superposition–based analysis hints at a remote similarity between the Beihai32 CP and AP205-like CPs (fig. S1), and the N- and C-termini of the Beihai32 CP appear to be arranged similarly to the AP205-like proteins (Fig. 3). The Wenzhou4 CP is closely related to the Beihai32 protein as evidenced both by sequence identity and structure superposition (fig. S2) but does not show notable similarity to the AP205-like proteins. Instead, the Wenzhou4 CP has a rather distinct and unique arrangement of its terminal regions, with the N terminus stretching over the αA helices to the other edge of the dimer and the proline-rich C terminus folding into an unusual Λ-shaped structure that extends some 15 Å above the rest of the protein (Fig. 3). While the C termini lack any secondary structure, the N termini form a pair of β-hairpin–resembling structures at positions analogous to the MS2-like phages.
ESE020
The CP of bacteriophage ESE020 belongs to a distinct cluster of approximately 155-residue-long proteins with no detectable sequence similarity to other CPs, and the ESE020 VLP crystal structure reveals notable differences in the CP architecture (Fig. 3). The ESE020 CP dimer is marked by massively elongated loops connecting b strands E and F (the EF loops), which in the particle extend to the icosahedral threefold symmetry axes where they mediate contacts with neighboring CP molecules. Apparently, because of steric restraints, the EF loops are disordered and not visible around the fivefold axes, and here, analogous contacts are accomplished by the shorter loops between b strands F and G (the FG loops). In other ssRNA phages, interactions around both threefold and fivefold symmetry axes are mediated solely by the FG loops, which often adopt different conformations around each in response to the different spatial environments, but the solution to use a different loop in each case is, so far, unique to the ESE020 CP. Among other distinctive features, the ESE020 CP has a short single-turn α helix between βG and αA, which makes it the only currently known ssRNA phage CP with three C-terminal helices. The N-terminal region of the protein is also different from the other CPs and instead of a β-hairpin contains a small three-stranded β sheet in which the extra strand originates from a curled-up extension of the βD strand.
AC-like CPs
Bacteriophage AC was among the first ssRNA bacteriophage genomes found in RNA metagenome data, and we have determined the three-dimensional structure of AC VLPs, as well as that of another AC-like phage NT-391. The CPs within this group are relatively short (115 and 123 residues for AC and NT-391, respectively) and do not show sequence similarity or clear structural relatedness to any other CP type. For the AC CP, secondary structure-based superposition reveals faint similarity to the MS2-like phages (fig. S1), and from the βC strand onward, the fold of the AC-like CPs closely resembles that of MS2 (Figs. 1 and 3). However, the N-terminal β-hairpins are completely missing in the AC-like phages, which is probably the most minimalistic approach we have observed in the current study; the missing hairpin is, however, partially compensated by a rolled-up corner of the central β sheet at the respective position. Also, contrary to most other ssRNA phages, the C termini of AC-like CPs are positioned relatively far from the N termini, are not involved in any interactions, and extend away from the particle.
GQ-907
The CP of the GQ-907 phage belongs to a small group of distinct sequences previously recognized by us as the ESE017-like CP type (16). There is no detectable sequence similarity between these CPs and other CP types, but by structure superposition, the GQ-907 shows weak resemblance to the Beihai32 CP and to some MS2-like CPs (fig. S1). From the βC strand to the C terminus, the GQ-907 CP fold closely resembles that of MS2 (Fig. 1), but the N termini extend over the α helices in a somewhat similar manner to the Wenzhou4 CP (Fig. 3). The GQ-907 N termini are, however, organized in a distinctive zigzag pattern of three consecutive two-stranded β sheets, with the lateral structures located at the same positions as the N-terminal β-hairpins in MS2-like phages and the central one positioned directly above the α helices.
AVE015-like CPs
The metagenomic data revealed a major previously unknown ssRNA phage lineage with relatively large genomes and distinct CPs approximately 165 residues in length, which we previously designated the AVE015-like CPs (16). We have determined VLP structures of phages AVE015, AVE016, and GQ-112, and while the three CPs do not share more than 20% sequence identity, their three-dimensional structure is similar. The core fold of these proteins still consists of a five-stranded β sheet and two C-terminal α helices, but their N-terminal region is once again completely reorganized (Fig. 3). In a remotely similar manner to the GQ-907 CP, the N termini of AVE015-like CPs intertwine over the αA helices to form a two-stranded β sheet, but a structure analogous to the N-terminal β-hairpin does not exist in these proteins. The AVE015 and AVE016 proteins contain a short α helix in a position roughly analogous to the βB strand, and in all AVE015-like CPs, the N termini complement another short strand to the central β sheet near the FG loop. Other remarkable features of this CP type include notably long loops between β strands E and F and between the C-terminal α helices; the interhelix loop is particularly extensive in the AVE015 CP and forms a prominent surface-exposed β-hairpin.
Beihai14
By far, the most unusual CP in our study is that of the bacteriophage Beihai14, which spans 208 residues and is currently the longest known ssRNA phage CP. The Beihai14 CP is a singleton with no similar sequences found to date, and its relation to other ssRNA phages remains obscure. The familiar CP structure in the Beihai14 protein is reduced to four central β strands and a single C-terminal α helix, but an additional 10-residue α helix has been inserted into the FG loop, which is the first observation in the ssRNA phages (Fig. 4A). The 85 residues preceding the β sheet almost entirely lack defined secondary structure but, apart from the very N termini, adopt a well-defined conformation. Of these, residues 45 to 80 form a giant loop distantly similar to an oversized β-hairpin of the MS2-like phages, which contains a 10-residue α helix that packs alongside the C-terminal αA helix. The 40 N-terminal residues pass under the VLP surface and extend along the intersubunit interface to the fivefold and threefold icosahedral symmetry axes, where they form five-stranded β-barrels or threefold assemblies of β-hairpin–like structures, respectively (Fig. 4B), whereas the very N termini point toward the center of the particle and are not resolved in the structure due to disorder. The N termini in the Beihai14 CP, in a way, resemble the flexible N-terminal arms in several plant ssRNA viruses, where they also mediate intersubunit contacts and are ordered in some but disordered in other CP monomers (18–20). Last, the 20 C-terminal residues of the Beihai14 CP are also unusually arranged and constitute a short surface-exposed loop immediately following the αA helix, after which the backbone threads underneath the N-terminal segment and again resurfaces at the icosahedral quasi-threefold symmetry axis.
Fig. 4. The unusual coat protein of bacteriophage Beihai14.
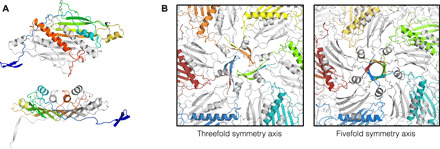
(A) Three-dimensional structure of the Beihai14 CP. The CP dimer is colored and shown in two different orientations as in Fig. 3. (B) Interactions around the threefold and fivefold icosahedral symmetry axes in the assembled particle. The CP monomers with their N-terminal extensions involved in the interactions are shown in color.
Capsid shape and size
With two exceptions, the newly characterized ssRNA phage capsids are of T = 3 quasi-equivalent icosahedral symmetry and range from 28.3 to 32.5 nm in diameter (table S1). The shortest CP in our study, Wenzhou1, forms the smallest VLPs, and the long AVE015-like and Beihai14 CPs are the largest, but overall, the association between the CP length and the size of the assembled particles is not particularly strong (fig. S3). However, the CP length generally correlates with the length of their FG loops that, in turn, determine pore sizes around the icosahedral threefold and fivefold symmetry axes. The size of these pores show great variation, from none at all in Beihai14 VLPs to approximately 25-Å-wide openings around the icosahedral threefold symmetry axes in GQ-907 VLPs; this raises a question of how well the RNA genome is protected inside these particles, as a molecule of ribonuclease would appear to be able to pass through this pore, but apparently, this does not cause issues for the phage in its natural environment. Like the previously studied ssRNA phages, the majority of the novel VLPs are of a roughly spherical shape, and only the AC, AVE015, GQ-112, and Beihai14 capsids are of notably polyhedral appearance (fig. S4).
One of the AC-like CPs in our study, NT-391, is assembled into small ~18.3-nm particles of T = 1 symmetry. A T = 1 particle has over five times smaller volume than a T = 3 capsid, which would leave too little space for packaging the viral genome; hence, the small VLPs can be safely assumed to be artifacts of the recombinant expression system. While uncommon, T = 1 particles have been previously observed in preparations of recombinant AP205 and some mutant MS2 VLPs (21). In our previous studies, we found that NC-443, another CP of the AC-like group with 24% sequence identity to NT-391, forms T = 1 particles as judged by electron microscopy (16); at the same time, the AC CP, which is more similar to the NT-391 CP (35% sequence identity), formed normal T = 3 capsids. It can be noted that both the NT-391 and NC-443, but not AC, CPs make use of intersubunit disulfide bonds, and it cannot be excluded that in the absence of the viral genome, the formation of covalent bonds between CP dimers is somehow involved in triggering assembly of the smaller particles.
Electron microscopy of AVE016 and, to some extent, several other AVE015-like VLPs revealed a mixture of spherical and elongated particles; the elongated particles, however, appeared to be a minority fraction in all cases. While the AVE015 and GQ-112 VLP crystal structures revealed normal T = 3 capsids, unexpectedly, the AVE016 VLP crystals turned out to correspond to prolate particles 28.9 nm in width and 34.6 nm in length. Geometrically, these particles correspond to a fivefold prolate icosahedron of a T = 3, Q = 4 architecture (22) and consist of 210 CP monomers or 105 CP dimers. In a T = 3 particle, the CP molecules are present as three slightly different conformers, whereas the asymmetric unit of the T = 3, Q = 4 AVE016 particle is composed of 21 CP monomers in eight major conformations with their structural differences almost entirely limited to the EF and FG loops (Fig. 5). There have been previous reports of elongated rod-like structures assembled from recombinant ssRNA phage CPs (23, 24), and it cannot be excluded that the prolate AVE016 VLPs likewise are artifacts of the recombinant expression system; however, such homogeneous and well-defined aberrant structures are uncommon and might indicate biological relevance. An AVE016-like phage will have to be isolated and examined in the laboratory to determine the natural virion morphology, but it can be noted that a number of the large tailed double-stranded DNA phages have prolate icosahedral heads (25), and several plant viruses with multipartite genomes form bacilliform particles of variable lengths that package genome segments of different sizes (26). The AVE015-like viruses appear to have large genomes that might approach 5 kb in some cases, and emergence of elongated capsids would be consistent with a requirement for increased genome packaging capacity. It could then be speculated that assembly of the elongated recombinant AVE016 VLPs is triggered by a subpopulation of longer bacterial RNA molecules that do not optimally fit into T = 3 capsids.
Fig. 5. The elongated virus-like particle of bacteriophage AVE016.
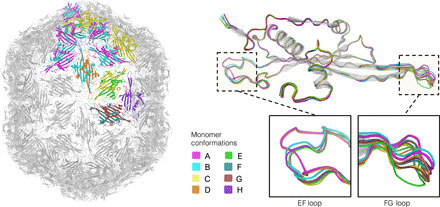
The assembled fivefold-prolate capsid (left) is shown in light gray with the asymmetric unit of the particle, composed of 21 coat protein monomers, shown in color. When backbone atoms of individual CP monomers are superimposed (top right), eight distinct conformations are evident, which mainly differ in positioning of the EF and FG loops (bottom right). The conformers are denoted A to H, with their coloring consistent throughout the figure.
Intersubunit interactions
Contacts between CP dimers within the assembled particles include a wide variety of hydrophobic, stacking, polar, and electrostatic interactions, but the number and type of contacts significantly vary among different VLPs. The solvent-buried surface areas of CP dimers within the capsid differ several fold, with the smallest observed in the AC-like and Beihai32-like VLPs and the largest in capsids formed by the Beihai14 and some of the longer MS2-like CPs (table S1). As with the particle size, there is some, but not a very significant, correlation between the CP length and the surface area they bury in the capsid. Somewhat more unexpectedly, there is also not a very strong relation between subunit interface areas and the previously determined thermal stability of the VLPs (16), suggesting that monomer-monomer interactions, additional RNA-mediated contacts, or other factors besides intersubunit contacts can have a significant influence on the overall particle stability.
In several viruses, capsid-bound metal ions have been shown to bridge negatively charged residues in neighboring subunits and participate in other intersubunit contacts. Bound calcium ions have been previously observed in ssRNA phages PRR1 (9) and Cb5 (10), where they were found to significantly enhance the particle stability. In the newly determined structures, metal ions were observed in the AVE015, AVE019, Beihai19, Beihai32, ESE007, ESE021, and ESE058 VLPs. Except for ESE021, no metal ions were present in the crystallization solution, and the bound ions were modeled as calcium; the ESE021 was crystallized in presence of zinc acetate, and accordingly, zinc ions were modeled in this case. In all cases, the metal ions were located at the quasi-threefold symmetry axes like in the previous Cb5 and PRR1 structures. We also tested the contribution of the metal ions to VLP stability by measuring their thermal denaturation in the presence of EDTA, and with the exception of the AVE015 VLPs where the results were somewhat ambiguous, the particle disintegration temperature of the metal ion–containing particles was reduced by at least 5°C when the chelating agent was added (fig. S5).
A number of ssRNA phages such as Qβ (6), PP7 (8), and AP205 (11) use disulfide bonds between subunits for additional particle stabilization. In all of the known cases, the disulfides are formed between pairs of cysteine residues in CP FG loops, resulting in covalently linked subunits around the icosahedral threefold and fivefold symmetry axes. In our study, disulfide bonds were present in VLPs formed by the AP205-like ESE001 and PQ-465 CPs, the AC-like NT-391 CP, and the MS2-like NT-214 CP. The disulfides are conserved in most of the known AP205-like CPs and, to some extent, also in the AC-like phages but are not found in close relatives of the NT-214 CP, suggesting that the disulfide bonds are fairly easily gained and lost during evolution. The S─S bonds have been shown to considerably increase the overall stability of the particle, but apparently, in nature, this might only be necessary as an adaptation to certain harsh environments and not as a general requirement for phage survival.
RNA binding
The CPs of ssRNA phages MS2 (27), PRR1 (28), Qβ (29), and PP7 (30) bind a specific genomic RNA hairpin at the beginning of the replicase gene which serves to repress translation of the enzyme late in infection. The RNA recognition mode of the PP7 CP is very different from that of the other three phages, and no specific CP-RNA binding has been observed in the distantly related phages Cb5 and AP205, which suggests that the interaction is not universally present in the ssRNA bacteriophages. The interaction appears not to be conserved even within the MS2-like group: For the respective phages in our study, RNA secondary structure predictions did not reveal convincing hairpins around the replicase initiation codon (16), and despite the otherwise significant structural similarity, the residues known to be involved in specific RNA binding were not conserved in these CPs. Sequence-specific RNA interactions are evidently not required when building the virion, and protein-RNA cross-linking studies and asymmetric cryogenic electron microscopy reconstructions of the MS2 and Qβ bacteriophages point to a model in which the capsid assembly is instead nucleated by many surface-exposed RNA hairpins in the folded genome that bind CP with moderate affinity (4, 31, 32). This model is also consistent with packaging of unspecific bacterial RNA into recombinant VLPs, largely achieved through electrostatic interactions between the RNA backbone and positively charged CP residues facing the capsid interior.
In the VLP structures, we generally observed fragmented and uninterpretable RNA density below the capsid surface, with a single notable exception of the Wenzhou1 VLPs, which revealed prominent electron density of double-stranded RNA (dsRNA) at the icosahedral twofold symmetry axes (Fig. 6A). The electron density in this case was interpretable as an A-form RNA double helix, which for simplicity was modeled as 10 A-U base pairs, although due to crystal averaging, no particular RNA bases could be distinguished. The resolution of the Wenzhou1 VLP structure was not sufficiently high for a detailed analysis of the CP-RNA interaction, although the CP side chains involved in binding the RNA backbone could be recognized (Fig. 6B). The structure does not indicate any potential sequence-specific interactions and suggests that the Wenzhou1 CP dimer is adapted for nonspecifically binding a double-stranded region of RNA. The RNA density was observed only under CP “CC” dimers, where both monomers are in the quasi-equivalent C conformation, but not around the fivefold axes, where the they adopt the asymmetric “AB” conformation. The structural basis for this discrimination is not entirely clear as the RNA binding surface in both CP dimer conformations is virtually identical, but superposition of an RNA-bound CC dimer on top of an RNA-free AB dimer inside the particle suggests that spatial constraints around the icosahedral fivefold symmetry axes force the EF loops to be positioned relatively closer to each other, which in turn makes RNA binding to AB dimers unfavorable due to a steric clash between the RNA backbone and the EF loop of a neighboring dimer (Fig. 6C). The observed RNA binding pattern in the Wenzou1 VLPs could be of biological significance and might hint that the native genome contains many dsRNA segments positioned at favorable places for binding to CP CC dimers, which would accomplish a similar function in recognition and packaging of the viral genome as RNA hairpins do in the MS2 and Qβ phages. Wenzhou1 is distantly related to the bacteriophage Cb5, the structure of which revealed intercalated RNA bases between CP subunits with a presumed role in particle stability (10). While no directly analogous interactions were detected in the Wenzhou1 VLPs, it can be envisioned that branched RNA secondary structures could bind to two or more adjacent CP CC dimers in the assembled particle and serve a functionally similar role. The observation of bound RNA in two different Cb5-like phages might suggest that in this group of viruses, RNA generally plays a more important structural role than in the other ssRNA phage lineages. It might also explain the stability issues of many of the Cb5-like VLPs that we have observed, as random bacterial RNA might not always be a sufficiently good substitute for the complex folded genome for holding the particle together.
Fig. 6. dsRNA binding of the Wenzhou1 coat protein.
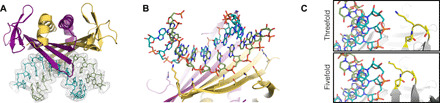
(A) Overview of the protein-RNA interaction. A Wenzhou1 CP dimer is colored yellow-orange and purple as per each monomer and shown bound to a 10–base pair dsRNA fragment colored in teal and pale green as per each strand. An icosahedrally averaged Fo-Fc omit map contoured at 3 σ is presented to illustrate the observed RNA density inside the particle. (B) Detailed view of the protein-RNA interface. The side chains involved in RNA binding are shown in stick representation. (C) Discrimination of dsRNA binding between CP AB and CC dimers. The EF loop (yellow) of a neighboring CP CC dimer around an icosahedral threefold symmetry axis allows unrestricted binding of the RNA helix (top), while the same loop at the fivefold symmetry axis appears to cause a steric clash that prevents RNA binding (bottom).
In the context of RNA binding, it is interesting to also note the Beihai14 CP, which has its N termini exposed to the interior of the particle. The N termini, which were disordered and not visible in the electron density, contain a string of positively charged and polar residues, which is consistent with their role in binding RNA. It can be envisioned that in the Beihai14 virus, the CP N termini are extended into the folded genome in a functionally similar manner as several ribosomal proteins have long extensions that penetrate into the organelle to organize and stabilize the RNA structure (33). It can be noted that several plant and animal ssRNA viruses have positively charged N-terminal CP extensions, which are involved in RNA binding (34); hence, the Beihai14 phage represents an interesting case of convergent evolution between two unrelated protein families.
Evolution of the CP fold
RNA viruses are the fastest-evolving life forms on Earth, which makes untangling their evolutionary past a nontrivial task. All RNA viruses are believed to be monophyletic and descended from an ancestral RdRp (2), which is also the only universally conserved protein in all RNA virus lineages. Compared to most other viral proteins, the capsid proteins are also fairly well conserved, and those of the single jelly roll architecture are ubiquitously found in plant, animal, and fungal RNA viruses (35). The ssRNA phage lineage, however, is believed to have separated very early from the other RNA viruses and evolved their own unique type of CP not found in any other modern viruses. The 22 novel ssRNA phage VLP structures now provide a window to the results of probably billions-of-years-long independent evolution of these proteins.
The present study has revealed that the structural diversity of the ssRNA phages extends significantly beyond the canonical MS2-like CP fold and redefines the core ssRNA phage CP architecture as only a four-stranded central β sheet and a single C-terminal α helix. Both elements make up the hydrophobic core of the CP dimer; the central β sheet additionally forms an RNA binding surface, and the α helices are important for dimerization. Evidently, any significant modification to these structures is so detrimental to the virus that none are observed even in the most highly diverged CPs. These constraints are not nearly as strict in the CP terminal regions, particularly in the N termini, where the high mutation rate of the ssRNA phages can be observed in full strength. The canonical MS2 CP fold topologically corresponds to an α-β sandwich, but a repeatedly observed theme in the novel VLP structures is a three-layered configuration where the C-terminal α helices are covered either by the N termini, like in the Wenzhou4, GQ-907, and AVE015-like CPs, or by long loops, as evidenced in some of the MS2-like proteins. However, these proteins otherwise do not appear to be closely related, which hints of convergence to this particular architecture from different starting points. Conversely, sequence alignment and structural superposition point to a recently shared evolutionary history of the EMS014 and ESE021 as well as the Beihai32 and Wenzhou4 CPs, and the prominent changes in the CP fold (swapped αB helices and rearranged N and C termini, respectively) appear to be relatively new acquisitions. It can be presumed that similar CP reorganizations with a certain regularity happen in all ssRNA phage lineages, which in turn suggests for some caution when trying to infer the evolutionary past of the ssRNA phages based solely on their capsid structure.
A recent phylogenetic reconstruction of ssRNA phages suggests of an ancient split into two lineages, of which the first is represented by the extant Cb5-like and AVE015-like viruses, while the other comprises the rest of the currently known ssRNA phages including MS2 (15). The phylogenetic analysis places the MS2-like and Cb5-like phage lineages at the opposite ends of the tree, but somewhat strikingly, their CPs follow the same canonical fold and their structures can be reasonably well superimposed (fig. S1). At the same time, some supposedly more closely related (e.g., ESE020-like and MS2-like, or AVE015-like and Cb5-like) phages present considerably larger structural differences in their CPs. A possible model to explain the current results would be to assume that before the ancient split into the two lineages, the ancestral ssRNA phage CP had a structure resembling the “canonical” CP fold with the N-terminal β-hairpin and that this architecture has survived relatively unchanged in the MS2-like, ESE020-like, and Cb5-like lineages. In the lineage leading to the GQ-907 phage, the AB loop of the ancestral CP can be envisioned to have increasingly extended to a point where it allowed the βA strand of one CP molecule to pair with the βB strand of the other monomer while maintaining essentially the same β-hairpin–like structure. Conversely, the AC-like phages might have emerged when the CP N terminus in a particular lineage started to become shorter, first losing the βA and then the βB strand, while the central β sheet simultaneously compensated the loss by gradually extending upward. In the lineages leading to the modern Beihai32-like and AP205-like phages, the CP C terminus might have started to extend and at some point could have substituted the βA strand, which became redundant and got lost, resulting in the observed circular permutation. The Beihai14 CP has diverged the most from others, but the long loop preceding the central β sheet still occupies broadly the same position as the N-terminal β-hairpin in MS2-like phages, suggesting that it too evolved from an ancestral MS2-like fold. Last, the AVE015-like CPs are the only proteins without a structure analogous to the N-terminal β-hairpin, but their N-terminal configuration might have emerged when the original β-hairpins unfolded and then rearranged with the βA strand switched to pair with the nearby βF strand.
MATERIALS AND METHODS
VLP production and purification
CP expression and VLP production and purification were done essentially as previously described (16). Briefly, CP coding sequences in pET24 bacterial expression vectors were obtained by gene synthesis, the constructs were transformed in Escherichia coli strain BL21(DE3), and the CP expression was done for 4 hours at 37°C or for 20 hours at 15°C. Cells were harvested by centrifugation, resuspended in lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 0.1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride] in a wet cell weight/buffer volume ratio of 1:4, lysed by sonication, and centrifuged for 30 min at 13,000g. The clarified lysate was loaded on a Sepharose 4 FF column (GE Healthcare) equilibrated with phosphate-buffered saline (PBS), the VLP-containing fractions were pooled and applied to a Fractogel DEAE (M) (Merck Millipore) ion exchange column, and the bound proteins were eluted with a linear 10 column volume gradient to PBS containing 1 M NaCl. The purest VLP-containing fractions were pooled and used for crystallization.
Crystallization and structure determination
Using Amicon Ultra 100K centrifugal filter units (Merck Millipore), the purified VLPs were transferred to a 20 mM tris-HCl (pH 8.0) buffer and concentrated to approximately 10 mg/ml with an assumption that A260 (absorbance at 260 nm) of 8.0 corresponds to a VLP concentration of 1 mg/ml. Initial crystallization trials were done with commercial or in-house formulated screens in 0.4-μl sitting drops using a Tecan EVO 75 liquid handling robot, with further optimization of crystal growth conditions as necessary. The final crystallization conditions of all VLPs are provided in table S2. Crystals were flash frozen in liquid nitrogen, cryoprotected with 30% glycerol if necessary, and x-ray diffraction data were collected at MAX-lab (Lund, Sweden) beamline I911-3, BESSY II (Berlin, Germany) beamline 14.1, or MAX IV (Lund, Sweden) beamline BioMAX. We were unable to solve the Beihai14 VLP structure using frozen crystals, and in this case, several datasets were collected from a single ~1.5-mm crystal mounted inside a capillary at room temperature and subsequently merged together. Diffraction data were scaled and merged using Mosflm (36) and Scala (37) from the CCP4 software suite (38) or using XDS (39) through the XDSAPP graphical user interface (40). VLP orientation in the unit cell with respect to the standard icosahedral orientation was determined using the locked self-rotation function in GLRF (41). The crystallographic asymmetric unit was deduced from the crystal symmetry and unit cell parameters and prepared from a model of bacteriophage MS2 [Protein Data Bank (PDB) ID: 2MS2]. The VLP position in the unit cell was either obvious from crystal symmetry or was determined by additional translation search. In case of a one-dimensional search, the VLP model was systematically translated along the axis of interest, an initial map calculated (see below) at each location and selected for the position with the lowest R factor. Two- and three-dimensional translation searches were performed in Phaser (42). A correctly oriented and positioned MS2 model was placed in the unit cell and examined for crystal contacts, followed by adjustment of the VLP diameter, if necessary. The placed MS2 model was then used for calculating Fcalc to 10-Å resolution with the program SFALL from the CCP4 software suite, and a 5-Å mask around the icosahedral asymmetric unit was generated using the program MAMA (43). The CCP4 program SIGMAA (44) was used to calculate weighted Fourier coefficients, which were used for calculating a map with the CCP4 program FFT. The map was averaged in the program AVE (45) using noncrystallographic symmetry and the previously prepared mask, and Fcalc structure factors from the averaged map were calculated with SFALL. The new Fcalc set was again used as an input for SIGMAA, and the averaging procedure was repeated for 10 more cycles. Fit of the averaged map to experimental data was monitored with the CCP4 program RSTATS at the end of each cycle. The final set of Fcalc structure factors was used as an input for phase extension. The procedure followed the same 10-cycle averaging protocol as described above, but after the end of the last cycle, higher-resolution data corresponding to a shell of one Miller index along the longest cell axis was added to the input data, followed by another 10 cycles of averaging; the procedure was repeated until all data to the resolution of the crystal had been included. The final icosahedrally averaged map was used for manual model building in COOT (46). The EMS014 subunit structure was solved by molecular replacement using the program PHASER with a polyalanine model of the ESE021 CP dimer as the search model. Refinement was done in PHENIX (47), and the model was validated using the tools provided in COOT and the MolProbity server (48).
Determination of VLP thermal stability
VLP samples at a concentration of 1 mg/ml in PBS buffer with or without 20 mM EDTA were heated for 15 min in a Veriti thermal cycler (Applied Biosystems) in a 5°C-increment step gradient and then loaded on an ethidium bromide containing 1% agarose gel. Electrophoresis was performed in 1× TAE buffer, after which the RNA was visualized under ultraviolet light and protein with subsequent staining with Coomassie blue. The thermal stability was defined as the highest temperature at which the VLP band was still detectable.
Supplementary Material
Acknowledgments
We thank the staff at the MAX-lab, MAX IV, and BESSY II synchrotrons for their assistance during our many data collection visits. Funding: This work was supported by the European Regional Development Fund through grant 1.1.1.1/16/A/104. Author contributions: J.R. designed the study, solved the crystal structures, analyzed and interpreted the results, and wrote the paper. I.L. supervised VLP production and purification, purified VLPs, and determined their thermal stability. G.K. and M.Š. crystallized VLPs. I.A. performed CP expression in bacteria. J.B. purified VLPs. K.T. designed and supervised the study, analyzed and interpreted the results, and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper, Supplementary Materials, or available in public databases. The three-dimensional structures have been deposited in the Protein Data Bank under accession codes 6YF7 (AC), 6YF9 (AVE002), 6YFA (AVE015), 6YFB (AVE016), 6YFC (AVE019), 6YFD (Beihai14), 6YFE (Beihai19), 6YFF (Beihai21), 6YFG (Beihai32), 6YFH (EMS014 VLP), 6YFI (EMS014 subunit), 6YFJ (ESE001), 6YFK (ESE007), 6YFL (ESE020), 6YFM (ESE021), 6YFN (ESE058), 6YFO (GQ-112), 6YFP (GQ-907), 6YFQ (NT-214), 6YFR (NT-391), 6YFS (PQ-465), 6YFT (Wenzhou1), and 6YFU (Wenzhou4). Additional data or materials related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eabc0023/DC1
REFERENCES AND NOTES
- 1.P. Pumpens, Single-Stranded RNA Phages: From Molecular Biology to Nanotechnology (CRC Press, 2020). [Google Scholar]
- 2.Wolf Y. I., Kazlauskas D., Iranzo J., Lucía-Sanz A., Kuhn J. H., Krupovic M., Dolja V. V., Koonin E. V., Origins and evolution of the global RNA virome. mBio 9, e02329-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valegård K., Liljas L., Fridborg K., Unge T., The three-dimensional structure of the bacterial virus MS2. Nature 345, 36–41 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Dai X., Li Z., Lai M., Shu S., Du Y., Zhou Z. H., Sun R., In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 541, 112–116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liljas L., Fridborg K., Valegård K., Bundule M., Pumpens P., Crystal structure of bacteriophage fr capsids at 3.5 Å resolution. J. Mol. Biol. 244, 279–290 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Golmohammadi R., Fridborg K., Bundule M., Valegård K., Liljas L., The crystal structure of bacteriophage Qβ at 3.5 Å resolution. Structure 4, 543–554 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Tars K., Bundule M., Fridborg K., Liljas L., The crystal structure of bacteriophage GA and a comparison of bacteriophages belonging to the major groups of Escherichia coli leviviruses. J. Mol. Biol. 271, 759–773 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Tars K., Fridborg K., Bundule M., Liljas L., The three-dimensional structure of bacteriophage PP7 from Pseudomonas aeruginosa at 3.7-Å resolution. Virology 272, 331–337 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Persson M., Tars K., Liljas L., The capsid of the small RNA phage PRR1 is stabilized by metal ions. J. Mol. Biol. 383, 914–922 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Plevka P., Kazaks A., Voronkova T., Kotelovica S., Dishlers A., Liljas L., Tars K., The structure of bacteriophage φCb5 reveals a role of the RNA genome and metal ions in particle stability and assembly. J. Mol. Biol. 391, 635–647 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Shishovs M., Rumnieks J., Diebolder C., Jaudzems K., Andreas L. B., Stanek J., Kazaks A., Kotelovica S., Akopjana I., Pintacuda G., Koning R. I., Tars K., Structure of AP205 coat protein reveals circular permutation in ssRNA bacteriophages. J. Mol. Biol. 428, 4267–4279 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Krishnamurthy S. R., Janowski A. B., Zhao G., Barouch D., Wang D., Hyperexpansion of RNA bacteriophage diversity. PLOS Biol. 14, e1002409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi M., Lin X.-D., Tian J.-H., Chen L.-J., Chen X., Li C.-X., Qin X.-C., Li J., Cao J.-P., Eden J.-S., Buchmann J., Wang W., Xu J., Holmes E. C., Zhang Y.-Z., Redefining the invertebrate RNA virosphere. Nature 540, 539–543 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Starr E. P., Nuccio E. E., Pett-Ridge J., Banfield J. F., Firestone M. K., Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. U.S.A. 116, 25900–25908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callanan J., Stockdale S. R., Shkoporov A., Draper L. A., Ross R. P., Hill C., Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci. Adv. 6, eaay5981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liekniņa I., Kalniņš G., Akopjana I., Bogans J., Šišovs M., Jansons J., Rūmnieks J., Tārs K., Production and characterization of novel ssRNA bacteriophage virus-like particles from metagenomic sequencing data. J. Nanobiotech. 17, 61 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pumpens P., Renhofa R., Dishlers A., Kozlovska T., Ose V., Pushko P., Tars K., Grens E., Bachmann M. F., The true story and advantages of RNA phage capsids as nanotools. Intervirology 59, 74–110 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Harrison S. C., Olson A. J., Schutt C. E., Winkler F. K., Bricogne G., Tomato bushy stunt virus at 2.9 Å resolution. Nature 276, 368–373 (1978). [DOI] [PubMed] [Google Scholar]
- 19.Tars K., Zeltins A., Liljas L., The three-dimensional structure of cocksfoot mottle virus at 2.7 Å resolution. Virology 310, 287–297 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Plevka P., Tars K., Zeltins A., Balke I., Truve E., Liljas L., The three-dimensional structure of ryegrass mottle virus at 2.9 A resolution. Virology 369, 364–374 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Asensio M. A., Morella N. M., Jakobson C. M., Hartman E. C., Glasgow J. E., Sankaran B., Zwart P. H., Tullman-Ercek D., A selection for assembly reveals that a single amino acid mutant of the bacteriophage MS2 coat protein forms a smaller virus-like particle. Nano Lett. 16, 5944–5950 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Luque A., Reguera D., The structure of elongated viral capsids. Biophys. J. 98, 2993–3003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cielens I., Ose V., Petrovskis I., Strelnikova A., Renhofa R., Kozlovska T., Pumpens P., Mutilation of RNA phage Qβ virus-like particles: From icosahedrons to rods. FEBS Lett. 482, 261–264 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Rumnieks J., Ose V., Tars K., Dislers A., Strods A., Cielens I., Renhofa R., Assembly of mixed rod-like and spherical particles from group I and II RNA bacteriophage coat proteins. Virology 391, 187–194 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Moody M. F., The shape of the T-even bacteriophage head. Virology 26, 567–576 (1965). [DOI] [PubMed] [Google Scholar]
- 26.Sicard A., Michalakis Y., Gutiérrez S., Blanc S., The strange lifestyle of multipartite viruses. PLOS Pathog. 12, e1005819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valegård K., Murray J. B., Stockley P. G., Stonehouse N. J., Liljas L., Crystal structure of an RNA bacteriophage coat protein–operator complex. Nature 371, 623–626 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Persson M., Tars K., Liljas L., PRR1 coat protein binding to its RNA translational operator. Acta Crystallogr. D Biol. Crystallogr. 69, 367–372 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Rumnieks J., Tars K., Crystal structure of the bacteriophage Qβ coat protein in complex with the RNA operator of the replicase gene. J. Mol. Biol. 426, 1039–1049 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Chao J. A., Patskovsky Y., Almo S. C., Singer R. H., Structural basis for the coevolution of a viral RNA–protein complex. Nat. Struct. Mol. Biol. 15, 103–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorzelnik K. V., Cui Z., Reed C. A., Jakana J., Young R., Zhang J., Asymmetric cryo-EM structure of the canonical Allolevivirus Qβ reveals a single maturation protein and the genomic ssRNA in situ. Proc. Natl. Acad. Sci. U.S.A. 113, 11519–11524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rolfsson Ó., Middleton S., Manfield I. W., White S. J., Fan B., Vaughan R., Ranson N. A., Dykeman E., Twarock R., Ford J., Kao C. C., Stockley P. G., Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J. Mol. Biol. 428, 431–448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein D. J., Moore P. B., Steitz T. A., The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 340, 141–177 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Rao A. L. N., Genome packaging by spherical plant RNA viruses. Annu. Rev. Phytopathol. 44, 61–87 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Krupovic M., Koonin E. V., Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. U.S.A. 114, E2401–E2410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W., iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans P. R., Scala. Joint CCP4 + ESF-EAMCB. Newslett. Protein Crystallogr. 33, 22–24 (1997). [Google Scholar]
- 38.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sparta K. M., Krug M., Heinemann U., Mueller U., Weiss M. S., XDSAPP2.0. J. Appl. Crystallogr. 49, 1085–1092 (2016). [Google Scholar]
- 41.Tong L., Rossmann M. G., Rotation function calculations with GLRF program. Methods Enzymol. 276, 594–611 (1997). [PubMed] [Google Scholar]
- 42.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.G. J. Kleywegt, T. A. Jones, Halloween ... masks and bones, in From First Map to Final model. Proceedings of the CCP4 Study Weekend, S. Bailey, R. Hubbard, D. Waller, Eds. (SERC Daresbury Laboratory, Daresbury, 1994), pp. 59–66. [Google Scholar]
- 44.Read R. J., Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A 42, 140–149 (1986). [Google Scholar]
- 45.Kleywegt G. J., Read R. J., Not your average density. Structure 5, 1557–1569 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Liebschner D., Afonine P. V., Baker M. L., Bunkóczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adams P. D., Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krissinel E., Henrick K., Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Holm L., Benchmarking fold detection by DaliLite v.5. Bioinformatics 35, 5326–5327 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eabc0023/DC1