

Abstract
Freshwater mussels (order Unionida) are among the world’s most biodiverse but imperiled taxa. Recent unionid mass mortality events around the world threaten ecosystem services such as water filtration, nutrient cycling, habitat stabilization, and food web enhancement, but causes have remained elusive. To examine potential infectious causes of these declines, we studied mussels in Clinch River, Virginia and Tennessee, USA, where the endemic and once-predominant pheasantshell (Actinonaias pectorosa) has suffered precipitous declines since approximately 2016. Using metagenomics, we identified 17 novel viruses in Clinch River pheasantshells. However, only one virus, a novel densovirus (Parvoviridae; Densovirinae), was epidemiologically linked to morbidity. Clinch densovirus 1 was 11.2 times more likely to be found in cases (moribund mussels) than controls (apparently healthy mussels from the same or matched sites), and cases had 2.7 (log10) times higher viral loads than controls. Densoviruses cause lethal epidemic disease in invertebrates, including shrimp, cockroaches, crickets, moths, crayfish, and sea stars. Viral infection warrants consideration as a factor in unionid mass mortality events either as a direct cause, an indirect consequence of physiological compromise, or a factor interacting with other biological and ecological stressors to precipitate mortality.
Subject terms: Freshwater ecology, Infectious diseases, Limnology, Pathogens, Virology
Introduction
Freshwater mussels (order Unionida) are important members of freshwater biomes, providing ecosystem services such as water filtration, nutrient cycling and deposition, physical habitat stabilization, and food web enhancement1. Mussels filter-feed on bacteria, suspended algae, detritus, phytoplankton and zooplankton2, removing suspended particulate matter from the water column and from interstitial spaces within the substrate. During periods of low summer discharge in small rivers, mussel assemblages are capable of circulating water as it flows over them, leading to multiple cycles of filtration3 that can strongly influence ecosystem processes, even at moderate mussel densities4. Unionids are also gaining attention for their ability to filter out chemical contaminants and water-borne pathogens5–7.
Unfortunately, the order Unionida contains an exceptional number of imperiled taxa. Among North America’s 298 recognized unionid species8, > 70% are considered endangered, threatened, or vulnerable9, with 23 species having gone extinct from the Southeastern United States alone. Historically, habitat destruction (e.g., river impoundments), pollution, sedimentation, over-harvest for commercial use (most notably, pearl harvest and manufacture of shirt buttons from shells ca. 1850–1950)10, and competition from invasive species (e.g. the Asian clam Corbicula fluminea, zebra mussel Dreissena polymorpha, and quagga mussel D. bugensis)11 have greatly reduced or extirpated many native mussel fauna. These threats have been present since the early twentieth century, mirroring trends in human development and land use12.
Since the late 1970s, episodic mass mortality events have been documented in unionids throughout their range, including catastrophic mortality (> 90% population declines) in some cases12. Unlike the aforementioned gradual declines, many mass mortality events in freshwater mussels have not been directly attributed to any specific environmental changes or events12. Furthermore, mass mortality events often affect only a single species of mussel within a broad ecological community. Environmental factors (e.g. chemical spills, extreme weather events) would be expected to affect many or all unionid species, in addition to other invertebrates and fishes13. A meta-analysis of the causes of mussel population declines found that only 48% of studies could attribute declines to any particular cause, and over 75% of studies cited multiple causes without substantial evidence of mechanisms14.
The Clinch River watershed in southwestern Virginia and northeastern Tennessee is one of the most ecologically important and biodiverse freshwater systems in North America15. With 46 extant species of freshwater mussels (20 of which are federally listed as endangered) and over 100 species of fish (5 of which are federally listed as either threatened or endangered), the Clinch River supports the highest concentration of extant federally listed aquatic species in the USA16. Long-term quantitative monitoring has shown that mussel richness and abundance in the upper river in Virginia steadily fell from 1979 to 2014, with densities at some sites declining as much as 95%16. In contrast, mussel densities in the lower river in Tennessee increased from 1979 to 201417. Several studies have examined Clinch River water and sediment quality and their effects on freshwater mussel assemblage in an attempt to explain this “zone of decline,” but few direct links to water quality, sediment, or physical habitat quality have been identified18.
Beginning in summer 2016, field biologists began documenting mass die-offs of mussels within the “healthy” reach of the lower Clinch River19. Mortality episodes were characterized by large numbers of recently dead or dying mussels on the surface of the river substrate in late summer and fall. Field surveys, collection of shells from freshly dead mussels, and comparisons to known species assemblage patterns demonstrated that the pheasantshell (Actinonaias pectorosa) comprised a disproportionate (to their relative abundance within the community) and overwhelming majority of affected individuals17. These mortality events resulted in population declines of approximately 50–90% of pheasantshells at monitoring sites throughout the lower river. For example, at one monitoring site (Kyle’s Ford), data from yearly quantitative surveys documented a loss of 85.4% of the pheasantshell population from 2016 to 2019, translating to a loss of approximately 80,000 individuals from this 200-m reach of the Clinch River19. Remarkably, similar mass mortality has not been observed in the other species of mussels inhabiting the same areas of the river. Moreover, since 2016, mass mortality of pheasantshells has occurred in upstream sites originally considered unaffected19. Pheasantshell are large-bodied and abundant, historically comprising over 50% of the Clinch River’s mussel biomass16. Thus, there is great concern that this decline, if unchecked, could permanently alter the Clinch River’s ecology and irreversibly affect the ecosystem services that its mussels provide.
Here, we describe a multi-year investigation into the Clinch River pheasantshell die-off focusing on infection, which has been cited as a potential—even likely—cause for unionid die-offs13,20,21 but has remained understudied22. This study is part of a broader collaborative effort to investigate potential causes for pheasantshell die-offs in the Clinch River and elsewhere23. We focus on viral causes because of (1) the specificity of the die-off for pheasantshells, (2) the apparent upstream spread of pheasantshell mortality between 2016 and 2019, (3) lack of evidence for bacterial or eukaryotic etiological agents24,25, and (4) lack of evidence of changes in physical characteristics of the environment that might explain the die-off17. Moreover, viruses are known to cause epidemic mortality in marine bivalves26,27, and Lea plague virus can decimate farmed populations of Chinese triangleshell (Hyriopsis cumingii) freshwater mussels used for production of freshwater pearls28,29. We took advantage of advances in metagenomic technologies for detecting and characterizing unknown viruses and viral communities, which have proven useful for elucidating the invertebrate “virosphere”30,31. By applying these methodologies alongside a rigorous case–control study design in which we compared affected and unaffected animals during two consecutive years (2017 and 2018), were able to examine which constituents of the pheasantshell virome might be associated with disease.
Results
Sampling
We collected and analyzed samples from 58 pheasantshells from the Clinch River, including 26 cases (11 from 2017 and 15 from 2018) and 32 controls (8 from 2017 and 24 from 2018) at 6 sites (Fig. 1; Table S1). During sampling, we chose as cases mussels that were on the surface of the substrate, gaping, slow to respond to tactile stimuli, and able to close their valves only weakly, and we chose as controls mussels that were firmly buried in the substrate, fast to respond to tactile stimuli, and able to close their valves strongly. In 2017, we sampled in October and November 2017 during an active mass mortality event. In 2018, we began sampling in August, before mortality was observed, and we continued sampling during September and October when mass mortality did occur. Prolonged flood conditions immediately after the October 2018 sampling event prevented further sample collection in 2018.
Figure 1.

Map of sampling locations. The map was created using ArcMap version 10.4.1 (Esri, Redlands, California, USA; https://support.esri.com/en/products/desktop/arcgis-desktop/arcmap/10-4-1).
Viromics and statistical analyses
Metagenomic sequencing of 58 pheasantshells from the Clinch River yielded an average of 1,921,287.6 sequence reads per hemolymph sample (standard deviation 1,127,991.5) with an average length of 118.3 nucleotides (standard deviation 10.6), after length and quality trimming. De novo assembly of these reads yielded 20,058 contiguous sequences (contigs) averaging 1,671 nucleotides in length (range 856–92,913). From these data, we identified 17 viruses of varied genomic compositions and taxonomic classifications (Table 1). Most viruses are only distantly related to known viruses phylogenetically, but many are related to viruses of freshwater and marine mollusks and other invertebrates (Fig. S1). Mussels identified as cases harbored an average of 4.4 (standard error = 0.66) viruses, whereas mussels identified as controls harbored an average of 3.2 (SE = 0.27) viruses, and this difference was statistically significant (t = 1.839; df = 56; P = 0.0356). Average loads of all viruses were 0.135 log10 viral reads per million total reads per kilobase of target sequence (vRPM/kb) for cases and 0.064 log10 vRPM/kb for controls, and this difference was also statistically significant (t = 3.706; df = 54; P = 0.0003). Frequency distributions of viral richness and viral load were right-skewed for cases but less so for controls, with some cases having exceptionally high viral richness and viral loads (Fig. 2).
Table 1.
Viruses identified in Clinch River pheasantshells.
| ID1 | Virus name | Accession | Genome | Closest relative (source, location, year, accession)2 | Family3 | Genus3 | %ID (aa)2 |
|---|---|---|---|---|---|---|---|
| A | Clinch densovirus 1 | MT341473 | ssDNA (linear) | Periplaneta fuliginosa densovirus (cockroach, China, 1990, AF192260) | Parvoviridae | Ambidensovirus | 63.7 |
| B | Clinch narna-like virus 1 | MT341474 | ssRNA(+) | Sanxia narna-like virus 2 (shrimp, China, 2014, KX883567) | Unclassified | Unclassified | 45.4 |
| C | Clinch noda-like virus 1 | MT341475 | ssRNA(+) | Hubei noda-like virus 2 (freshwater shellfish, China, 2014, KX883205) | Unclassified | Unclassified | 51.9 |
| D | Clinch picorna-like virus 1 | MT341476 | ssRNA(+) | Marine RNA virus SF-2 (wastewater, USA, 2010, NC_043518) | Marnaviridae | Locarnavirus | 41.9 |
| E | Clinch CRESS virus 1 | MT341477 | ssDNA (circular) | CRESS virus (minnow, USA, 2017, MH616916) | Unclassified | Unclassified | 61.7 |
| F | Clinch picorna-like virus 2 | MT341478 | ssRNA(+) | Hubei picorna-like virus 4 (freshwater shellfish, China, 2014, NC_033087) | Unclassified | Unclassified | 65.8 |
| G | Clinch picorna-like virus 3 | MT341479 | ssRNA(+) | Wenzhou picorna-like virus 7 (shrimp, China, 2013, NC_032842) | Unclassified | Unclassified | 55.7 |
| H | Clinch circular virus 1 | MT341480 | ssDNA (circular) | Blackfly DNA virus 6 (black flies, New Zealand, 2015, MK433220) | Unclassified | Unclassified | 70.1 |
| I | Clinch calicivirus 1 | MT341481 | ssRNA(+) | Bat calicivirus (bat, USA, 2009, MH259583) | Caliciviridae | Calicivirus | 80.2 |
| J | Clinch circular virus 2 | MT341482 | ssDNA (circular) | Bat circovirus (bat, China, 2013, KJ641738) | Circoviridae | Unclassified | 97.5 |
| K | Clinch dicistro-like virus 1 | MT341483 | ssRNA(+) | Beihai picorna-like virus 105 (snails, China, 2014, NC_032604) | Unclassified | Unclassified | 79.1 |
| L | Clinch tombus-like virus 1 | MT341484 | ssRNA(+) | Hubei tombus-like virus 15 (centipede, China, 2013, NC_033009) | Tombusviridae | Unclassified | 63.8 |
| M | Clinch sobemo-like virus 1 | MT341485 | ssRNA(+) | Beihai sobemo-like virus 25 (razor shell, China, 2014, NC_032895) | Luteoviridae | Unclassified | 65.6 |
| N | Clinch dicistro-like virus 2 | MT341486 | ssRNA(+) | Hypsignathus monstrosus dicistrovirus (bat, Republic of the Congo, 2015, MH310078) | Dicistroviridae | Unclassified | 63.0 |
| O | Clinch picobirnavirus 1 | MT341487 | dsRNA (segmented) | Pink-eared duck picobirnavirus (duck, Australia, 2017, MK204418) | Picobirnaviridae | Picobirnavirus | 64.1 |
| P | Clinch picobirna-like virus 1 | MT341488 | ssRNA(+) | Shahe picobirna-like virus 1 (freshwater isoptera, China, 2013, KX884156) | Unclassified | Unclassified | 76.5 |
| Q | Clinch totivirus 1 | MT341489 | ssRNA(+) | Drosophila melanogaster totivirus (fruit fly, USA, 2009, NC_013499) | Totiviridae | Unclassified | 96.0 |
Figure 2.
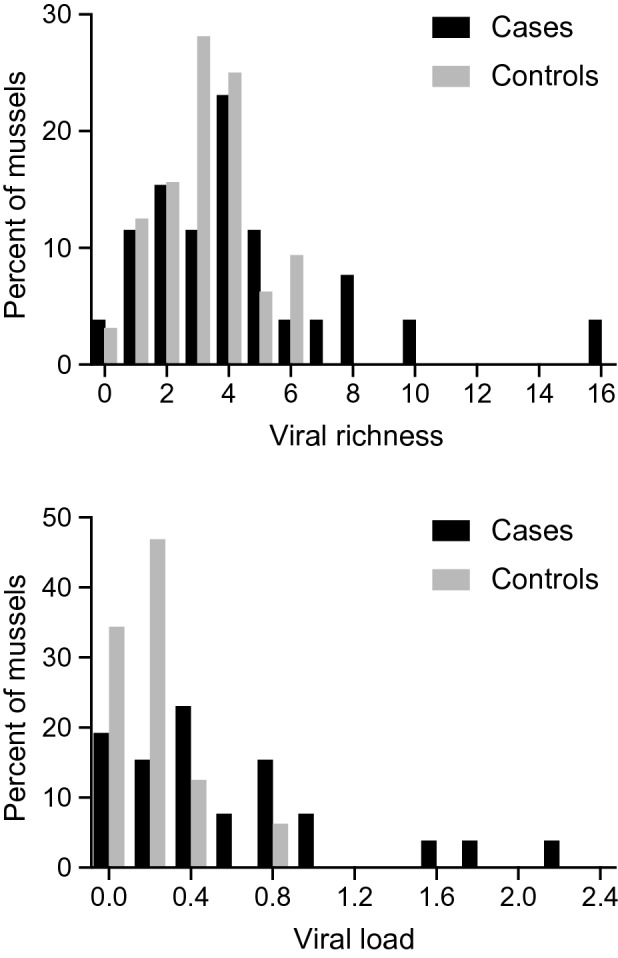
Frequency distribution of viral richness (number of viruses) and viral load (log10 viral reads per 106 total reads per kilobase of target sequence) in Clinch River pheasantshell cases and controls.
Individual viruses varied markedly in their prevalence, load, and association with case or control status (Table 2). In univariate analyses, five viruses (Clinch densovirus 1, Clinch narna-like virus 1, Clinch noda-like virus 1, Clinch picorna-like virus 1, and Clinch CRESS virus 1) showed significantly higher prevalence and/or viral load in cases than in controls (Table 2). Two of these viruses were relatively rare: Clinch narna-like virus 1 was found in 6 cases and 1 control and Clinch noda-like virus 1 was found in 3 cases and 0 controls. Two other of these viruses (Clinch picorna-like virus 1 and Clinch CRESS virus 1) had higher viral loads in cases than in controls but showed no significant differences in prevalence between cases and controls. Thus, Clinch densovirus 1 was the only virus for which both prevalence and load were significantly higher in cases than in controls (odds ratio (OR) = 4.30, 95% confidence interval (CI) 1.42–13.0; P = 0.0084, and Mann–Whitney U = 40, P = 0.0035, respectively). The remaining 12 viruses showed no statistically significant differences in prevalence or load between cases and controls overall or within years (Table 2; Fig. 3).
Table 2.
Univariate statistical associations between clinical classification (case or control) and prevalence and loads of viruses in Clinch River pheasantshells.
| ID1 | Virus name | Individuals infected | Prevalence (%)2 | Viral load (Log10vRPM/kb)3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | OR (95% CI) | P | Cases | Controls | U | P | |||
| A | Clinch densovirus 1 | 29 | 69.2 | 34.4 | 4.30 (1.42, 13) | 0.0084 | 1.057 | 0.396 | 40 | 0.0035 |
| B | Clinch narna-like virus 1 | 7 | 23.1 | 3.1 | 9.30 (1.041, 83.12) | 0.0267 | 0.601 | 0.074 | n/a | n/a |
| C | Clinch noda-like virus 1 | 3 | 11.5 | 0.0 | 9.68 (0.4771, 196.4) | 0.0360 | 0.512 | 0.000 | n/a | n/a |
| D | Clinch picorna-like virus 1 | 36 | 61.5 | 62.5 | 0.96 (0.3306, 2.788) | 0.9999 | 0.992 | 0.225 | 78 | 0.00415 |
| E | Clinch CRESS virus 1 | 32 | 57.7 | 53.1 | 1.20 (0.4241, 3.413) | 0.4676 | 0.911 | 0.544 | 80 | 0.03785 |
| F | Clinch picorna-like virus 2 | 18 | 34.6 | 28.1 | 1.35 (0.443, 4.132) | 0.4017 | 0.628 | 0.289 | 21 | 0.0939 |
| G | Clinch picorna-like virus 3 | 3 | 11.5 | 0.0 | 9.68 (0.4771, 196.4) | 0.0843 | 0.657 | 0.000 | n/a | n/a |
| H | Clinch circular virus 1 | 15 | 34.6 | 18.8 | 2.29 (0.6908, 7.619) | 0.1423 | 0.737 | 0.592 | 17 | 0.13605 |
| I | Clinch calicivirus 1 | 14 | 23.1 | 25.0 | 0.90 (0.2675, 3.028) | 0.9999 | 0.630 | 0.254 | 6 | 0.1725 |
| J | Clinch circular virus 2 | 42 | 69.2 | 75.0 | 0.75 (0.2363, 2.38) | 0.8435 | 0.930 | 0.946 | 207 | 0.8308 |
| K | Clinch dicistro-like virus 1 | 2 | 7.7 | 0.0 | 6.63 (0.3045, 144.5) | 0.1966 | 0.638 | 0.000 | n/a | n/a |
| L | Clinch tombus-like virus 1 | 4 | 11.5 | 3.1 | 4.04 (0.3948, 41.41) | 0.2314 | 0.567 | 0.471 | n/a | n/a |
| M | Clinch sobemo-like virus 1 | 4 | 3.8 | 9.4 | 0.39 (0.03779, 3.956) | 0.7774 | 0.895 | 0.092 | n/a | n/a |
| N | Clinch dicistro-like virus 2 | 3 | 7.7 | 3.1 | 2.58 (0.221, 30.2) | 0.4213 | 0.859 | 0.101 | n/a | n/a |
| O | Clinch picobirnavirus 1 | 1 | 3.8 | 0.0 | 3.82 (0.1494, 97.84) | 0.4483 | 1.048 | 0.000 | n/a | n/a |
| P | Clinch picobirna-like virus 1 | 1 | 3.8 | 0.0 | 3.82 (0.1494, 97.84) | 0.4483 | 1.473 | 0.000 | n/a | n/a |
| Q | Clinch totivirus 1 | 1 | 3.8 | 0.0 | 3.82 (0.1494, 97.84) | 0.4483 | 1.727 | 0.000 | n/a | n/a |
1Letters refer to Table 1, Figs. 3, and S1.
2Percentage of mussels within each group (case or control) with reads mapping to each virus, plus odds ratios and 95% confidence intervals. P values (statistically significant values in bold) were calculated using Fisher's exact tests.
3Log10 reads mapping to each virus per million total reads per kilobase of target sequence, Mann–Whitney U statistics, and associated P values (infected mussels only).
Figure 3.

Heatmap of viral loads in Clinch River pheasantshells. Data are log10 viral reads per 106 total reads per kilobase of target sequence for each virus separately (viruses A–Q) and for all viruses combined (All). Data are presented separately for cases and controls in 2017 and 2018. Raw data on viral loads are presented in Table S2.
Based on the results described above, we conducted multivariate statistical analyses that included five viruses (Clinch densovirus 1, Clinch narna-like virus 1, Clinch noda-like virus 1, Clinch picorna-like virus 1, and Clinch CRESS virus 1) because of their significantly higher prevalence and/or load in cases than in controls. In the resulting general linear model (GLM) relating clinical status to viral infection and ecological variables, only two significant factors emerged: infection with Clinch densovirus 1 [P = 0.004, adjusted OR (95% CI) = 11.18 (2.12–58.92)] and mussel shell length [P = 0.043, adjusted OR (95% CI) = 1.09 (1.00–1.17)]. In the GLM relating clinical status to viral load and ecological variables, the only significant factor identified was Clinch densovirus 1 load [P = 0.0287, adjusted OR (95% CI) = 24.56 (1.39, 432.52)]; no other viruses and no ecological factors were significant. The general linear model relating viral richness to ecological factors (site, sampling date, and length) had no significant terms.
Because of the strong associations of Clinch densovirus 1 prevalence and load with morbidity, we examined associations between Clinch densovirus 1 and the presence and load of other viruses using Fisher's exact tests and Student’s t tests, respectively. Infection with Clinch densovirus 1 was associated with a higher frequency of infection with Clinch circular virus 1 (odds ratio = 5.9 [95% CI 1.33–37.6] Fisher's 1-tailed exact P = 0.007) and with a higher load of Clinch CRESS virus 1 (t = 2.527; df = 26.185; P = 0.0179); however, no other significant associations were detected.
The genome of Clinch densovirus 1 (GenBank accession number MT341473) is 5,429 bases long and contains 5 open reading frames (ORFs 1–5) of lengths 735, 1,671, 1,620, 807, and 759 nucleotides in the typical arrangement of members of subfamily Densovirinae, encoding putative non-structural and structural proteins, which are transcribed by host cellular machinery through alternative mRNA splicing and leaky scanning32,33. The Clinch densovirus 1 coding genome is also flanked by inverted terminal repeats characteristic of members of this viral subfamily32. The amino acid sequence difference between Clinch densovirus 1 and its closest relative, periplaneta fuliginosa densovirus, a member of the genus Ambidensivirus (Table 1), is 63.7% within the non-structural protein NS1. This degree of divergence exceeds the 85% relatedness threshold accepted by the International Committee on the Taxonomy of Viruses as a species demarcation criterion within the genus Ambidensovirus34.
Discussion
Clinch River pheasantshells host a diverse virome. Three of the 17 viruses we identified (Clinch picorna-like virus 1, Clinch CRESS virus 1, and Clinch circular virus 2; Table 1) are likely members of the “normal” pheasantshell virome. Such viruses would be expected to infect mussels at high prevalence (> 50% in these cases) and load, but without association with clinical disease. Three other viruses infected pheasantshells at moderate prevalence (between 20 and 50%) but also showed no association between case and control status (Clinch picorna-like virus 2, Clinch circular virus 1, and Clinch calicivirus 1). The other viruses we identified all occurred at low prevalence (sometimes in only one animal) and may be hypoendemic, sporadic, or derived from the environment. For example, the picobirnavirus detected in one case sample from 2018 is part of a group of viruses shed in the feces of mammals such as cows and marmots35. Although hemolymph, like mammalian blood, is not directly connected to the environment36, filter feeding bivalves can remove viral pathogens from suspension in the water column37,38.
Among the five viruses with prevalence or loads associated with case status by univariate analyses (Table 1), only Clinch densovirus 1 had both higher prevalence and load in cases than in controls, and these associations were the strongest observed in the study. In multivariate analyses, the other four viruses fell out as non-significant with respect to both prevalence and load, as did all other factors except for mussel shell length, which was retained in the GLM examining prevalence. Clinch densovirus 1 is therefore the only of the 17 viruses identified that, when other variables are accounted for, is associated with disease in Clinch River pheasantshells.
Densoviruses are members of the viral family Parvoviridae, subfamily Densovirinae, and can be highly host-specific and lethal39. Mass mortality in invertebrates is a well-characterized consequence of densovirus infection, with examples including shrimp40, silkworms41, cockroaches42, mosquitos43, crickets44, moths45, and crayfish46. In fact, so lethal are some densoviruses that they have been used commercially as powerful bioinsecticides47. Common signs of densovirus infection include lethargy, anorexia, development of tumors, flaccidity, and death39. Notably, sea star-associated densovirus, also a member of the genus Ambidensovirus, is the putative cause of mass mortality in another benthic invertebrate, the sunflower sea star (Pycnopodia helianthoides)48. Sea star wasting disease is characterized by loss of turgor (a “deflated” appearance), behavioral changes, and rapid degradation leading to death48.
Henley et al.24 conducted a histological study of moribund Clinch River pheasantshells collected during the beginning of the die-off in 2016 from the Kyle’s Ford sampling site. This study documented internal organ damage, including pervasive necrosis, but was unable to link any measured factor (including parasitic trematode infestation and bacterial infection), to mortality. Certain of the histologic lesions documented, however, would be consistent with densovirus infection, as described in other invertebrates (see above). Ultimately, experimental infection and studies of pathogenesis will be necessary to resolve any causal relationship between phesantshell mussel mortality and infection with Clinch densovirus 1, as has been attempted in the case of sea star wasting disease48 and cherax quadricarinatus densovirus46.
In this light, we caution that our results, while suggestive, do not demonstrate direction of causality. For example, a preceding diseased state may render mussels more susceptible to infection with Clinch densovirus 1. We also note that we characterized viruses from hemolymph, because it is useful for bivalve health assessment and can be obtained non-lethally36,49, but other tissues may host different viruses. Our focus on hemolymph may also account for our finding of only relatively small viruses (similar to vertebrate blood). Other (and perhaps larger) viruses may have tropisms for different tissues (e.g. mantle, gill, gonads), and these tissues also warrant investigation. Quantitative polymerase chain reaction assays could be developed to measure the tissue-specific loads of viruses determined by epidemiology and metagenomics to be linked to disease states, including Clinch densovirus 1 but not dismissing other viruses (discovered and as-yet undiscovered). Such assays could also be applied to environmental samples (e.g. water or sediment) to investigate viral transmission and persistence.
Should infection with Clinch densovirus 1 or other pathogens ultimately be a cause of pheasantshell mass mortality, this result would not exclude the possibility of “upstream drivers.” Infectious diseases are often proximate causes of mortality while also being caused by other factors themselves. For example, introductions of exotic species and their pathogens, climate change, and ecologically induced physiological stressors have all been implicated as predisposing factors for infectious disease in wildlife50. Determining proximate causes is nevertheless important for management and conservation. For example, vaccines, probiotics, or controlled exposure to pathogens to induce resistance might be effective in conditioning mussels in captive rearing facilities, where many species are bred for restoration efforts51.
Overall, our results show that, while diverse, the virome of Clinch River pheasantshells contains only one virus, Clinch densovirus 1, showing a strong and consistent association with disease. Mass mortality events in freshwater mussels are unfortunately accelerating worldwide12. Studying other species of mussels in other geographic locations using both epidemiology and metagenomics could help reveal whether infection with viruses or other agents is a generalized characteristic of unionid mass mortality events. The resulting information would be important for conserving and managing this remarkable complex of imperiled species.
Methods
Field sampling
We sampled pheasantshells in 2017 and 2018. We collected moribund mussels (cases) and apparently healthy mussels (controls) during mortality events using swim-through searches of shoals. We focused on the months of September, October and November of each year because these were the months in which mass mortality was observed, although we added a sampling event in August 2018 in anticipation of a mortality event. At four sites along the river (Frost Ford, Kyle’s Ford, Wallen’s Bend, and Sycamore Island; Fig. 1), we first located moribund individuals (lying on the surface with shells gaping and minimally responsive to tactile stimuli). We then located apparently healthy individuals (buried in the substrate, siphoning normally, with tightly closed shells and strongly resistant to being opened) at the same sites and from two additional upstream sites (Speers Ferry and Artrip) where no mortality had been observed.
We sampled hemolymph because it is useful for health and disease assessment in freshwater bivalves and can be collected non-lethally36,49 and because (similar to vertebrate blood) it is not directly exposed to the physical environment, unlike other accessible organs (e.g. foot, mantle, gill). We first gently opened the valves of each animal with a sterile pediatric nasal speculum. We then disinfected the outer surface of the anterior adductor muscle with 70% isopropyl alcohol and extracted up to 1.0 ml hemolymph (depending on the size of the mussel) from the anterior adductor muscle sinus using a 1 ml tuberculin syringe. We placed hemolymph immediately in sterile tubes on dry ice in the field then stored samples at − 80 °C until further analysis. For each individual, we noted its general appearance, recorded the strength and speed of its response to tactile stimuli (opening the valves and application of isopropanol), and measured the length of its shell using digital calipers. We marked animals with FPN glue-on shellfish tags (Hallprint, Hindmarsh Valley, Australia) to prevent re-sampling during successive sampling events and then returned animals to the shoals from which they were collected.
Metagenomic sequencing and bioinformatics
We processed hemolymph for metagenomic sequencing for virus discovery as described previously52. Briefly, we clarified hemolymph by centrifugation at 10,000×g for 10 min and used the QIAamp MinElute virus kit (Qiagen, Hilden, Germany) to extract total nucleic acids from the supernatant. We then converted RNA to double-stranded cDNA using random hexamers and prepared libraries using the Nextera XT DNA sample preparation kit (Illumina, San Diego, California, USA), after which we sequenced the libraries on an Illumina MiSeq instrument (V3 chemistry, 600 cycle kit; Illumina, San Diego, California, USA). Using CLC Genomics Workbench version 11.0 (CLC bio, Aarhus, Denmark), we first trimmed low-quality bases (phred quality score < 30) and discarded short reads (< 75 bp). We then conducted de novo assembly using the native CLC assembler with a word size of 50 and a bubble size of 5,000 and analyzed both contigs and unassembled reads for nucleotide-level (blastn) and protein-level (blastx) similarity to viruses in the GenBank database. For each mussel, we measured its infection status (positive or negative) for each virus and, for infected mussels, vRPM/kb, which is a metagenomic measure of viral load that adjusts for sequencing depth and target sequence length and is correlated with quantitative real-time PCR52.
We inferred phylogenetic relationships among newly identified virus sequences and published sequences of the most closely related viruses in the GenBank database using viral replicase (polymerase) genes when available. We first aligned sequences using a codon-based version of the Prank algorithm53 and applied the Gblocks algorithm54 to remove regions with poor alignment, as implemented in the computer program TranslatorX55. We then inferred maximum likelihood phylogenetic trees from the resulting alignments using PhyML 3.056, with 1,000 bootstrap replicates to assess statistical confidence in clades. We used FigTree v1.4.4 to display final trees.
Statistical analyses
We used a multi-tiered statistical approach to examine associations between viral infection, load, and richness (total number of viruses infecting a mussel) and clinical status (cases versus controls). First, we used Fisher’s exact tests and Mann–Whitney U tests to assess univariate statistical differences between cases and controls with respect to these measures. Based on the results of these univariate analyses (Table 2), we constructed general linear models to investigate the combined influence of viruses and other predictor variables (shell length, sampling location, and date of sampling) on clinical status (case or control). We conducted all statistical analyses using R software57.
Ethics statement
We obtained biological samples in accordance with all federal, state, and local laws and policies.
Supplementary information
Acknowledgements
We thank the U.S. Fish and Wildlife Service (USFWS) Virginia Field Office for logistic support during field sampling, and the USFWS La Crosse Fish Health Center, USFWS Midwest Fisheries Center, and U.S. Geological Survey (USGS) National Wildlife Health Center for assistance with sample processing and analyses, and especially Sara Erickson for invaluable assistance with sample collection and associated logistics. This work was supported by USFWS (Award Number G18AC00334), USGS (Award Number 140F0518P0079), and by the University of Wisconsin-Madison through the John D. MacArthur Professorship Chair. The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views of the U.S. Fish and Wildlife Service. The use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author contributions
J.C.R., E.L., D.W., S.K., J.P., and T.L.G. designed the study; J.C.R., R.A., and T.L.G. performed field work. C.D.D and T.L.G. performed laboratory experiments; J.C.R., C.D.D. and T.L.G. analyzed the data; J.C.R. and T.L.G. wrote the manuscript. All authors reviewed and improved the final manuscript.
Data availability
All data generated during the current study are available in GenBank (accession numbers MT341473–MT341489) or are included in this published article and its Supplementary Information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-71459-z.
References
- 1.Vaughn CC. Ecosystem services provided by freshwater mussels. Hydrobiologia. 2018;810:15–27. doi: 10.1007/s10750-017-3139-x. [DOI] [Google Scholar]
- 2.Christian AD, Smith BN, Berg DJ, Smoot JC, Findlay RH. Trophic position and potential food sources of 2 species of unionid bivalves (Mollusca:Unionidae) in 2 small Ohio streams. Freshw. Sci. 2004;23:101–113. [Google Scholar]
- 3.Vaughn CC. Biodiversity losses and ecosystem function in freshwaters: emerging conclusions and research directions. Bioscience. 2010;60:25–35. doi: 10.1525/bio.2010.60.1.7. [DOI] [Google Scholar]
- 4.Howard JK, Cuffey KM. The functional role of native freshwater mussels in the fluvial benthic environment. Freshw. Biol. 2006;51:460–474. doi: 10.1111/j.1365-2427.2005.01507.x. [DOI] [Google Scholar]
- 5.Izumi T, Yagita K, Izumiyama S, Endo T, Itoh Y. Depletion of Cryptosporidium parvum oocysts from contaminated sewage by using freshwater benthic pearl clams (Hyriopsis schlegeli) Appl. Environ. Microbiol. 2012;78:7420–7428. doi: 10.1128/AEM.01502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ismail NS, Müller CE, Morgan RR, Luthy RG. Uptake of contaminants of emerging concern by the bivalves Anodonta californiensis and Corbicula fluminea. Environ. Sci. Technol. 2014;48:9211–9219. doi: 10.1021/es5011576. [DOI] [PubMed] [Google Scholar]
- 7.Ismail NS, et al. Improvement of urban lake water quality by removal of Escherichia coli through the action of the bivalve Anodonta californiensis. Environ. Sci. Technol. 2015;49:1664–1672. doi: 10.1021/es5033212. [DOI] [PubMed] [Google Scholar]
- 8.Williams JD, et al. A revised list of the freshwater mussels (Mollusca: Bivalvia: Unionida) of the United States and Canada. Freshw. Mollusk Biol. Conserv. 2017;20:33. doi: 10.31931/fmbc.v20i2.2017.33-58. [DOI] [Google Scholar]
- 9.Lydeard C, et al. The global decline of nonmarine mollusks. Bioscience. 2004;54:321. doi: 10.1641/0006-3568(2004)054[0321:TGDONM]2.0.CO;2. [DOI] [Google Scholar]
- 10.Haag WR. North American Freshwater Mussels: Natural History, Ecology, and Conservation. Cambridge: Cambridge University Press; 2012. [Google Scholar]
- 11.Strayer DL. Effects of alien species on freshwater mollusks in North America. J. N. Am. Benthol. Soc. 1999;18:74–98. doi: 10.2307/1468010. [DOI] [Google Scholar]
- 12.Haag WR. Reassessing enigmatic mussel declines in the United States. Freshw. Mollusk Biol. Conserv. 2019;22:43–60. doi: 10.31931/fmbc.v22i2.2019.43-60. [DOI] [Google Scholar]
- 13.Goldberg TL, Dunn CD, Leis E, Waller DL. A novel picornalike virus in a Wabash Pigtoe (Fusconaia flava) from the Upper Mississippi River, USA. Freshw. Mollusk Biol. Conserv. 2019;22:81–84. [Google Scholar]
- 14.Downing JA, Van Meter P, Woolnough DA. Suspects and evidence: a review of the causes of extirpation and decline in freshwater mussels. Anim. Biodivers. Conserv. 2010;33:151–185. [Google Scholar]
- 15.Zipper CE, et al. Freshwater mussel population status and habitat quality in the Clinch Rver, Virginia and Tennessee, USA: a featured collection. J. Am. Water Resour. Assoc. 2014;50:807–819. doi: 10.1111/jawr.12220. [DOI] [Google Scholar]
- 16.Jones J, et al. Clinch River freshwater mussels upstream of Norris Reservoir, Tennessee and Virginia: a quantitative assessment from 2004 to 2009. J. Am. Water Resour. Assoc. 2014;50:820–836. doi: 10.1111/jawr.12222. [DOI] [Google Scholar]
- 17.Jones JW, et al. Collapse of the Pendleton Island mussel fauna in the Clinch River, Virginia: setting baseline conditions to guide recovery and restoration. Freshw. Mollusk Biol. Conserv. 2018;21:36–56. [Google Scholar]
- 18.Cope, W. G. & Jones, J. W. Recent precipitous declines of endangered freshwater mussels in the Clinch River: an in situ assessment of water quality stressors related to energy development and other land-use. 244 (U.S. Fish and Wildlife Service, Southwestern Virginia Field Office, 2016).
- 19.Richard JC. Clinch River mussel die-off. Ellipsaria. 2018;20:1–3. [Google Scholar]
- 20.Neves, R. J. Proceedings of the Workshop on Die-offs of Freshwater Mussels in the United States (U.S. Fish and Wildlife Service, Upper Mississippi River Conservation Committee, 1986).
- 21.Starliper CE, Powell J, Garner JT, Schill WB. Predominant bacteria isolated from moribund Fusconaia ebena ebonyshells experiencing die-offs in Pickwick Reservoir, Tennessee River, Alabama. J. Shellfish Res. 2011;30:359–366. doi: 10.2983/035.030.0223. [DOI] [Google Scholar]
- 22.Grizzle JM, Brunner CJ. Infectious diseases of freshwater mussels and other freshwater bivalve mollusks. Rev. Fish. Sci. 2009;17:425–467. doi: 10.1080/10641260902879000. [DOI] [Google Scholar]
- 23.Leis E, et al. Building a response network to investigate potential pathogens associated with unionid mortality events. Ellipsaria. 2018;20:44–45. [Google Scholar]
- 24.Henley WF, Beaty BB, Jones JW. Evaluations of organ tissues from Actinonaias pectorosa collected during a mussel die-off in 2016 at Kyles Ford, Clinch River, Tennessee. J. Shellfish Res. 2019;38:681. doi: 10.2983/035.038.0320. [DOI] [Google Scholar]
- 25.Leis E, Erickson S, Waller D, Richard J, Goldberg T. A comparison of bacteria cultured from unionid mussel hemolymph between stable populations in the Upper Mississippi River basin and populations affected by a mortality event in the Clinch River. Freshw. Mollusk Biol. Conserv. 2019;22:70–80. [Google Scholar]
- 26.Garcia C, et al. Ostreid herpesvirus 1 detection and relationship with Crassostrea gigas spat mortality in France between 1998 and 2006. Vet. Res. 2011;42:73. doi: 10.1186/1297-9716-42-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arzul I, Corbeil S, Morga B, Renault T. Viruses infecting marine molluscs. J. Invertebr. Pathol. 2017;147:118–135. doi: 10.1016/j.jip.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Sufang D, Yimin X, Jie W. Studies on the mussel Hyriopsis cumingii plague. I. a new viral infectious disease. Acta Hydrobiol. Sin. 1986;26:308–312. [Google Scholar]
- 29.Zhong L, Xiao T-Y, Huang J, Dai L-Y, Liu X-Y. Histopathological examination of bivalve mussel Hyriopsis cumingii lea artificially infected by virus. Acta Hydrobiol. Sin. 2011;35:666–671. [Google Scholar]
- 30.Shi M, et al. Redefining the invertebrate RNA virosphere. Nature. 2016;540:539–543. doi: 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- 31.Zhang YZ, Shi M, Holmes EC. Using metagenomics to characterize an expanding vrosphere. Cell. 2018;172:1168–1172. doi: 10.1016/j.cell.2018.02.043. [DOI] [PubMed] [Google Scholar]
- 32.Bergoin M, Tijssen P. Molecular biology of Densovirinae. Contrib. Microbiol. 2000;4:12–32. doi: 10.1159/000060329. [DOI] [PubMed] [Google Scholar]
- 33.Mietzsch M, Penzes JJ, Agbandje-McKenna M. Twenty-five years of structural parvovirology. Viruses. 2019 doi: 10.3390/v11040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotmore SF, et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019;100:367–368. doi: 10.1099/jgv.0.001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganesh B, Masachessi G, Mladenova Z. Animal picobirnavirus. Virus Dis. 2014;25:223–238. doi: 10.1007/s13337-014-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafson LL, et al. Evaluation of a nonlethal technique for hemolymph collection in Elliptio complanata, a freshwater bivalve (Mollusca: Unionidae) Dis. Aquat. Organ. 2005;65:159–165. doi: 10.3354/dao065159. [DOI] [PubMed] [Google Scholar]
- 37.Lees D. Viruses and bivalve shellfish. Int. J. Food Microbiol. 2000;59:81–116. doi: 10.1016/S0168-1605(00)00248-8. [DOI] [PubMed] [Google Scholar]
- 38.Faust C, Stallknecht D, Swayne D, Brown J. Filter-feeding bivalves can remove avian influenza viruses from water and reduce infectivity. Proc. R. Soc. B. 2009;276:3727–3735. doi: 10.1098/rspb.2009.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fédière, G. in Parvoviruses. From Molecular Biology to Pathology and Therapeutic Uses. Contributions to Microbiology. Vol. 4 (eds S. Faisst & J. Rommelaere) 1–11 (Karger, 2000).
- 40.Kalagayan H, et al. IHHN virus as an etiological factor in runt-deformity syndrome (RDS) of juvenile Penaeus vannamei cultured in Hawaii. J. World Aquacult. Soc. 1991;22:235–243. doi: 10.1111/j.1749-7345.1991.tb00740.x. [DOI] [Google Scholar]
- 41.Ito K, Kidokoro K, Shimura S, Katsuma S, Kadono-Okuda K. Detailed investigation of the sequential pathological changes in silkworm larvae infected with Bombyx densovirus type 1. J. Invertebr. Pathol. 2013;112:213–218. doi: 10.1016/j.jip.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Jiang H, et al. Genetic engineering of Periplaneta fuliginosa densovirus as an improved biopesticide. Arch. Virol. 2007;152:383–394. doi: 10.1007/s00705-006-0844-6. [DOI] [PubMed] [Google Scholar]
- 43.Ledermann JP, Suchman EL, Black WC, Carlson JO. Infection and pathogenicity of the mosquito densoviruses AeDNV, HeDNV, and APeDNV in Aedes aegypti mosquitoes (Diptera: Culicidae) J. Econ. Entomol. 2004;97:1828–1835. doi: 10.1093/jee/97.6.1828. [DOI] [PubMed] [Google Scholar]
- 44.Szelei J, et al. Susceptibility of North-American and European crickets to Acheta domesticus densovirus (AdDNV) and associated epizootics. J. Invertebr. Pathol. 2011;106:394–399. doi: 10.1016/j.jip.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Kouassi N, et al. Pathogenicity of diatraea saccharalis densovirus to host insets and characterization of its viral genome. Virol. Sin. 2007;22:53–60. doi: 10.1007/s12250-007-0062-8. [DOI] [Google Scholar]
- 46.Bowater R, et al. A parvo-like virus in cultured redclaw crayfish Cherax quadricarinatus from Queensland, Australia. Dis. Aquat. Organ. 2002;50:79–86. doi: 10.3354/dao050079. [DOI] [PubMed] [Google Scholar]
- 47.Johnson RM, Rasgon JL. Densonucleosis viruses ('densoviruses') for mosquito and pathogen control. Curr. Opin. Insect. Sci. 2018;28:90–97. doi: 10.1016/j.cois.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hewson I, et al. Densovirus associated with sea-star wasting disease and mass mortality. Proc. Natl. Acad. Sci. USA. 2014;111:17278–17283. doi: 10.1073/pnas.1416625111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fritts AK, Peterson JT, Hazelton PD, Bringolf RB. Evaluation of methods for assessing physiological biomarkers of stress in freshwater mussels. Can. J. Fish. Aquat. Sci. 2015;72:1450–1459. doi: 10.1139/cjfas-2014-0564. [DOI] [Google Scholar]
- 50.Cunningham AA, Daszak P, Wood JLN. One health, emerging infectious diseases and wildlife: two decades of progress? Philos. Trans. R. Soc. Lond. B. 2017 doi: 10.1098/rstb.2016.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patterson MA, et al. Freshwater Mussel Propagation for Restoration. Cambridge: Cambridge University Press; 2018. [Google Scholar]
- 52.Toohey-Kurth K, Sibley SD, Goldberg TL. Metagenomic assessment of adventitious viruses in commercial bovine sera. Biologicals. 2017;47:64–68. doi: 10.1016/j.biologicals.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 53.Löytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014;1079:155–170. doi: 10.1007/978-1-62703-646-7_10. [DOI] [PubMed] [Google Scholar]
- 54.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 55.Abascal F, Zardoya R, Telford M. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 57.R Core Team. R: A language and environment for statistical computing, version 3.6.3. https://www.R-project.org (R Foundation for Statistical Computing, Vienna, 2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated during the current study are available in GenBank (accession numbers MT341473–MT341489) or are included in this published article and its Supplementary Information files.