

Abstract
Central nervous system (CNS) disorders represent a broad spectrum of brain ailments with short- and long-term disabilities, and nanomedicine-based approaches provide a new therapeutic approach to treating CNS disorders. A variety of potential drugs have been discovered to treat several neuronal disorders; however, their therapeutic success can be limited by the presence of the blood-brain barrier (BBB). Furthermore, unique immune functions within the CNS provide novel target mechanisms for the amelioration of CNS diseases. Recently, various therapeutic approaches have been applied to fight brain-related disorders, with moderate outcomes. Among the various therapeutic strategies, nanomedicine-based immunotherapeutic systems represent a new era that can deliver useful cargo with promising pharmacokinetics. These approaches exploit the molecular and cellular targeting of CNS disorders for enhanced safety, efficacy, and specificity. In this review, we focus on the efficacy of nanomedicines that utilize immunotherapy to combat CNS disorders. Furthermore, we detailed summarize nanomedicine-based pathways for CNS ailments that aim to deliver drugs across the BBB by mimicking innate immune actions.
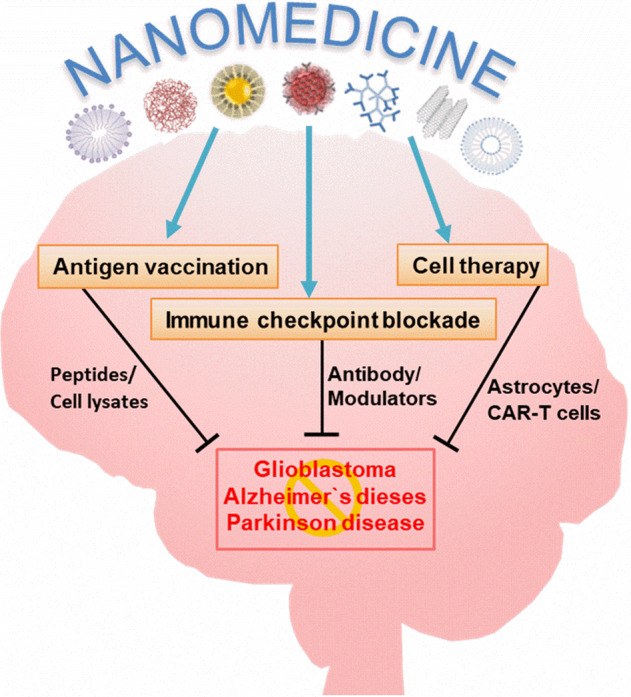
Overview of how nanomedicines can utilize multiple immunotherapy pathways to combat CNS disorders.
Keywords: central nervous system disorders, blood–brain barrier, nanomedicine, immunotherapy
Introduction
Central nervous system (CNS) diseases comprise a diverse range of severe neurological conditions, which mostly lack effective treatments [1]. Gliomas and glioblastoma are inherent brain tumors that originate from neuroglial ancestor cells [2]. The most common primary tumor within the CNS in adults is glioblastoma, which accounts for 50% of all gliomas and 15% of primary brain tumors. In recent decades, the US Food and Drug Administration (FDA) has approved only two drugs, namely, temozolomide (TMZ) and the antiangiogenic drug bevacizumab, after successful clinical trials for glioma therapy [3, 4]. Despite these therapeutic advances, individuals with glioblastoma have a median survival rate of less than 2 years [5, 6]. Alzheimer’s disease (AD) and Parkinson’s disease (PD) are more common neurodegenerative disorders that affect millions of people worldwide [7, 8]. FDA-approved medicines for AD include cholinesterase inhibitors (e.g., donepezil and rivastigmine) and N-methyl-D-aspartate (NMDA) receptor antagonists (e.g., memantine); however, these have limited effects on severe cognitive impairment and are unable to stop disease progression [9, 10]. Only one type of drug is approved for PD [i.e., Xadago (safinamide)]; however, this drug improves motor symptoms but does not address the underlying PD pathology [11]. The significant hurdles in this field are halting disease progression and addressing symptoms and pathology following late-stage diagnosis. These obstacles have led to the development of a new generation of therapeutic approaches.
Currently, methods to treat CNS disorders include radiotherapy, chemotherapy, gene therapy, immunotherapy, and surgery, with each therapeutic strategy having its benefits and limitations [12, 13]. Immunotherapy harnesses the host’s immune system by either enhancing or suppressing innate immune responses to target disease cells [14, 15]. Immunotherapeutic methods are classified as either active or passive immunotherapy [16]. Active immunotherapeutic strategies are designed to induce an immune response through nanomedicines, tumor vaccines, and nonspecific immune stimulants [17]. Passive immunotherapy acts as a promoter of antitumor effects by introducing antibodies or lymphocytes from immune cells into patients [18]. However, the unique immune microenvironment of the CNS needs to be considered when engaging in immunotherapeutic strategies [19–22]. The CNS immune system can be activated, distinct, and adaptive, as evidenced by the existence of microglia, astrocytes, and pericytes as antigen-presenting cells (APCs), the complement components, and the expression of Toll-like receptors (TLRs) [23–25]. Despite the dynamic and adaptive nature of the immune system, some infiltrating tumors contain nonenhancing regions, even with a disturbed blood–brain barrier (BBB), which limits therapeutic drug contact [26]. Furthermore, the powerful adaptive capabilities of glioblastoma, as well as its relative lack of immunogenicity, its production of a suppressive tumor microenvironment (TME), and intratumoral heterogeneity, make the delivery of drugs across the BBB a significant challenge. Indeed, some researchers have suggested that the CNS may be considered an “immunologically inactive” site, with a different and well-balanced environment employing the prime expression of immunosuppressive mediators [27].
Engaging surrogate immunosuppression via multiple mechanisms by brain tumors is a major obstacle that limits the efficacy of immunotherapies. In the diseased state, highly expressed markers (CD4+, CD25+, and FOXP3+) in regulatory T cells (Tregs) modulate the immunologically cold TME [28]. Glioblastomas are usually considered “cold TMEs” and are associated with either immune-desert tumors or only peripheral invasion of immune cells, which make them uniquely immunoprivileged tumors. Moreover, immune-inhibitory cytokines (interleukin 6, IL-6 and interleukin 10, IL-10) and secreted factors such as transforming growth factor-beta (TGF-β) also contribute to a cold TME [29]. In addition, inactive Tregs trigger the overexpression of programmed cell death protein-1 (PD-1), which helps tumors escape CNS immune responses and contributes to a cold TME [30–32]. Thus, brain immunity is maintained by a series of highly regulated checkpoints. Instructing the immune system to recognize disease sites has transformed immunotherapy in several ways [33]. One is immune checkpoint inhibition, whereby drugs (mostly antibodies) interrupt overexpressed immune checkpoints that have been co-opted by tumors, thus exposing cancer cells and, ultimately, activating immune-cell responses against the tumors (e.g., melanoma) [34]. Alternatively, genomically mutated targeted agents (i.e., chimeric antigen receptor T cells (CAR T cells)), which have been modified to recognize better and treat the patient’s cancer, can initiate immune responses [35]. Despite decades of scientific research, the treatment of CNS disorders with immunotherapy remains a major clinical challenge. For instance, nonresponsive tumors with fewer mutations and neoantigens exhibit a lower response to immune checkpoints and CAR T-cell therapy [36–39]. Indeed, the localization and morphologic resemblance of nonmalignant cells with neuroglial cells can make immunotherapy extremely challenging [40–44]. In addition, the inability to deliver therapeutically relevant doses to the disease location is a major hurdle in achieving an improved outcome for CNS diseases.
Other obstacles in the delivery of therapeutics to the CNS include penetration of the BBB and the blood–brain tumor barrier (BBTB), the short circulation time of medications in the blood, and clearance of drugs by the kidneys [45–48]. Occasionally, the payload to the CNS can be limited by a natural defense system of efflux pumps, such as multidrug-resistant protein [49] and convection-enhanced diffusion [50]. In response to these challenges, an active and functionally targeted nanomedicine-based approach is being widely examined in developing CNS therapeutics [51, 52]. Nanomedicine is an emerging field that utilizes nanoscale materials for a diverse range of applications in diagnosis and disease therapeutics [53–55]. Effective nanomedicines for CNS diseases need to cross the BBB efficiently, and various parameters need to be optimized (e.g., shape, size, functional surface chemistry, circulating half-life, structural stability, permeability, and extravasation) [56]. In addition, the surface conjugation chemistry and associated targeting capability need to be kept at a high level via multiple receptor-facilitated interactions. The main pathways that facilitate BBB penetration include receptor-mediated transcytosis (RMT) [57], adsorption-mediated transcytosis [58], and cell-mediated transport (stem cells and immune cells, macrophages, and monocytes) [47, 59].
To date, several nanoscale materials have been employed as nanomedicines, ranging from organic, inorganic, polymeric, and carbon-based materials to extracellular vesicles, liposomes, red blood cell membranes [60, 61], and metal nanostructures [28, 52, 62–66]. Drug-carrying nanomaterials substantially improve the pharmacokinetics and biodistribution relative to free drugs within the CNS [67–69]. In this review, we detail the efficacy of nanomedicine-based immunotherapies for CNS-associated diseases such as brain cancers and other neurodegenerative disorders, such as AD and PD. We have evaluated data from preclinical experimental models that target pathology associated with glioma, AD (tauopathies or amyloid-β), and PD (α-synucleinopathy or Lewy bodies). Here, we outline the current knowledge of immunology, which contributes to disease progression, describes recent breakthroughs in this field, and highlights unanswered questions. Finally, with this knowledge, we speculate on the potential future applications of immune-based nanomedicine approaches for CNS disorders.
The BBB
The brain is considered the most protected organ within the body. The brain has many defensive shields, including the skull, meninges, cerebrospinal fluid, BBB, and BBTB [45]. All of these factors can play a role in protecting the brain from external and internal injuries and in preventing disease. Nevertheless, in a disease state, these protective shields also decrease the accessibility of therapeutic agents to the brain. The BBB is a physiological barrier composed of tightly restricted cerebral capillary endothelium, pericytes, astrocytes, and basal membranes, with infrequent endocytosis and transcytosis [46]. Because of the highly expressed P glycoproteins on cerebral endothelial cells, it actively effluxes most therapeutics [70]. Owing to the very tight and small opening in these connective cells, the BBB prevents almost all types of macromolecules and large proportions of small molecules (including anticancer drugs) from entering the CNS, particularly into the brain [71, 72]. Therefore, the BBB is considered responsible for the failure of most current therapies [61]. Being of a lipid-like nature, epithelial cells and membrane systems such as abluminal and luminal membranes have the potential to transport substrates into the brain [47, 56, 73]. For instance, the transcellular lipophilic pathway is responsible for crossing numerous lipophilic molecules (≤400 Da), and oxygen, carbon dioxide, nicotine, steroid hormones, and alcohol can quickly diffuse and penetrate the BBB [47].
Various types of BBB models have been established, from simpler monolayers of brain endothelial cells (BECs) to more sophisticated spheroid and chip style models, as shown in Fig. 1. The simplest BBB models can be established by isolation of BECs that are grown to make a single monolayer at the abluminal side of transwell inserts [74]. Transwell systems build vertical side-by-side diffusion systems and are the most common and widely used cell-based in vitro models of the BBB [49]. Another type of BBB model utilizes astrocytes and pericytes that are guided to develop co-culture transwell systems of gel matrices containing extracellular matrix proteins [75]. This type of model provides higher transepithelial resistance, greater barrier strength, and lower permeability than single BEC models. However, it is also associated with several limitations of the implementation of shear stress and requires more expensive and difficult procedures. Recent BBB models use a combination of three different types of cells developing from either porcine, bovine, or rodent origin [76]. More complicated types of 3D-designed models have also been developed, which are made of spheroid or microfluidic models and are a closer representation of the in vivo environment [77]. Pericytes, astrocytes, and BECs can freely self-assemble into ball-shaped cellular aggregates when deprived of any framework materials. The microfluidic systems afford physiologic shear stress to BECs that further create a laminar flow via a computer-controlled pumping mechanism [78]. These types of models are beneficial because they better resemble the in vivo environment but require a difficult and expensive setup to establish [79].
Fig. 1. Representation of the common in vitro BBB cellular models for CNS diseases.

Reproduced with permission from [79]. Copyright 2015 Wolters Kluwer—Medknow
Methods for modeling CNS disorders
Experimental models of diseases are critical for a better understanding of pathogenesis and to assess the potential efficacy of novel therapeutic approaches. Tumor cells are sourced from either in vitro tumor tissue or tumor cell populations, which are usually collected from rodent models, or are patient-derived [80, 81]. Cultured tumor cells are then implanted directly into the brain of the host animal model by intracranial or subcutaneous injection to develop glioblastoma models [82]. The most widely implemented method in animal models is the subcutaneous or intracranial injection of tumor cells into rodents. Subcutaneously injected patient-derived xenograft models grow quickly but are not very confined within the subcutaneous space, making this model relatively different from the TME [83]. Intracranial injection for tumor implantation is superior because of the direct implantation of tumor cells into the brain rather than the degradation of some tumor cells via subcutaneous injection [84]. Models can also be established by other means, such as plasmid-based and viral vector (viral delivery of genetic material)-based mutated genetic material delivery [85, 86]. Breeding animals that carry germline mutations that cause the development of brain tumors are also preestablished in vivo rodent models.
Until now, most experimental animal models developed for AD are transgenic mice that overexpress human genes associated with the establishment of amyloid plaques and/or neurofibrillary tangles of hyperphosphorylated Tau protein [87]. Physiological models are developed via the action of neurotoxins, which are used to induce the accumulation of amyloid plaques in AD models. Similarly, PD models are classified as induced via neurotoxic or genetic means and involve the degeneration of dopaminergic neurons in the substantia nigra [88]. Neurotoxin models generally produce a rapid and robust cell loss in the substantia nigra and provoke motor symptoms as well as behavioral changes. In contrast, genetic-based models exhibit variable α-synuclein pathology along with some physical symptoms, such as cell loss and motor symptoms. α-Synuclein is the major constituent causing PD and related dementia with Lewy bodies [89]. The aggregation of α-synuclein occurs in physically linked neurons throughout the brain, along with loss of DA. Genetic mutations can be demonstrated by employing transgenic animals or be induced by viral transfection. Some examples of commonly used animal models for CNS disorders are mentioned in Table 1.
Table 1.
Summary of established animal models for glioblastoma, Alzheimer’s Disease, and Parkinson’s Disease
| Disease | Model | Characteristics | Refs. |
|---|---|---|---|
| GBM | Xenograft/HT1080 (human cell line) | IDH1 mutant (MGG152) | [210] |
| Xenograft/LNT-229 and LN-308 (human cell line) | IDH1 R132C mutant (HT1080) | [211] | |
| Xenograft/BT111 (TIC), BT116 (TIC) | Unmethylated MGMT (BT111) and (BT116) | [212] | |
| Xenograft and allograf/LN-319 (human cell line) GL261 (mouse cell line) | (LN-319) ErbB2 Expression (GL261) | [213] | |
| Xenograft/U251 (human cell line) | N/A | [214] | |
| Allograf GL261-Luc (mouse cell line) | N/A | [215] | |
| AD | PDAPP | Plaque-associated reactive astrocytes, general microgliosis | [216] |
| Tg2576 | Plaque-associated astrocytes and microgliosis | [217] | |
| APP23 | Fibrillar plaque-associated astrocytes and microgliosis and general astrocytosis | [218] | |
| J20 | General astrogliosis and microgliosis | [219] | |
| TgSwDI | CAA-associated astrocytes and microgliosis | [220] | |
| PD | MPTP Neurotoxin: inhibition of complex I | No α-synuclein aggregates, rapid and strong dopaminergic neurodegeneration, strong motor deficit | [221] |
| 6-OHDA Neurotoxin: inhibition of complex I and oxidative stress | No α-synuclein aggregates, rapid and strong dopaminergic neurodegeneration, strong asymmetric motor deficits | [222] | |
| SNCA transgenic rodents point mutations (A53T, A30P, and E46K) and overexpression of α-synuclein (ASO) | Mice and rats: widespread α-synuclein aggregation, no dopaminergic neurodegeneration, and some motor deficits | [223, 224] | |
| α-synuclein preformed fibrils seeding of α-synuclein aggregates | Mice, rats, and NHP: widespread α-synuclein aggregation, mild dopaminergic neurodegeneration, and lack of motor deficits in rats and NHP, some in mice | [225, 226] | |
| UCH-L1 I93M mutation | Mice: no α-synuclein aggregates, dopaminergic neurodegeneration, and mild motor deficits | [227] |
IDH isocitrate dehydrogenase, MGMT methylguanine-DNA methyltransferase, ErbB2 erythroblastic oncogene B2, NHP non-human primates, MPTP 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, 6-OHDA 6-hydroxydopamine, UCH-L1 ubiquitin carboxy-terminal hydrolase
Nanomedicine-based immunotherapy for glioblastoma
The cargo-based delivery of therapeutic agents, which induce an immune response, is a topic of high interest in glioma treatment, as highlighted in a recent article, “glioblastoma is ‘hot’ for personalized vaccines” [90]. A detailed schematic illustration of the immune response in glioblastoma is shown in Fig. 2.
Fig. 2. Optimal design of nanoparticle-based delivery of drugs, cargoes/or adjuvants, and immunotherapy of glioblastoma.

Diverse types of nanoscale materials can serve as vehicles for targeted delivery of tumor-cytotoxic nanomedicines
Immunotherapy for glioblastoma relies on the dendritic cell (DC)-mediated presentation of tumor-associated peptides, antigens, or epitopes (used in vaccination therapy) [91]. These tumor lysate-derived constituents are administered directly or through CAR T cells for adaptive immune organization. Then, cytotoxic T lymphocytes (CTLs) are activated after interacting with DCs via the major histocompatibility complex (MHC) class II–T-cell receptor (TCR) (signal 1) and CD80/CD86-CD28 (signal 2). Upon activation, CTLs interrogate glioblastoma-associated antigens expressed on MHC class I molecules and destroy tumor cells [92, 93]. However, glioblastoma cells frequently avoid obliteration through enhanced expression of immune checkpoint ligands, such as programmed death-ligand 1 (PD-L1) receptors. These ligand receptors bind with complementary receptors on CTLs via PD-1 and cause the suppression of CTL activation. Immune checkpoint blockade therapy with monoclonal antibodies can disrupt this communication [94, 95]. Moreover, the inhibitory immune checkpoint blockade of cytotoxic T lymphocyte protein 4 (CTLA-4) also stimulates the interaction of CD28 with CD80 and CD86 on DCs and promotes T-cell priming. Glioblastoma-associated antigens, containing IL-13 receptor subunit-α2 and epidermal growth factor receptor (EGFR) variant III, are also expressed on the tumor cell surface and can self-regulate MHC class I [96]. These tumor-associated antigens are exploited as specific targets of genetically modified CAR T-cell therapies [97]. A summary of the advantages and disadvantages of various immunotherapy-based strategies for the treatment of glioblastoma is presented in Table 2.
Table 2.
Comparison of multiple nanomedicine-based immunotherapies for glioblastoma
| Immunotherapy | Advantages | Disadvantages |
|---|---|---|
| Immune checkpoint inhibition |
Enhanced PD-L expression in glioblastoma Can overcome glioblastoma immune evasion Development of novel immune checkpoints The slow occurrence of side effects Combination of immunotherapy with radiotherapy or chemotherapy is more effective |
Complex immune evasion strategies Responsive evaluations Immune-related side events The balance between self-tolerance and autoimmunity |
| Vaccination therapy |
Multitude characterized Elicits potent, robust, and specific immune responses Available for most patients Ability to combine multiple targets into a cocktail vaccine Lowered risk of immune escape Safe, multivalent, and patient-specific Fully defined composition |
Reduced immune response due to central tolerance if expressed by normal tissues Available for a subset of patients Possible immune evasion (growth of tumor cells that lack antigen expression) Instability of peptides in vivo being rapidly degraded by peptidases High production costs |
| CAR T-cell therapy |
MHC-independent Overcomes tumor MHC molecule downregulation Potent in recognizing any cell-surface antigen (protein, carbohydrate, or glycolipid) Applicable to a broad range of patients and T-cell populations Production of large numbers of tumor-specific cells in a moderately short period |
Capable of targeting only cell-surface antigens Lethal toxicity due to cytokine storm reported Difficulties of target selection Most mutations occur in intracellular proteins |
Current obstacles in glioma therapy, such as lack of specificity, poor BBB penetrance, low circulation life, and limited targeting ability, can be easily addressed by nanomedicine-based immunotherapy [98]. The major disadvantages of the current research frontier using simple nanocarrier systems for therapeutic agents and cancer vaccines (various antigens and immunomodulatory agents isolated from astrocyte lysates) are that they are incapable of inducing cytotoxic antitumor T-cell responses and merely deliver the drug to the tumor [99]. Designing more precise and effective targeting cancer vaccine carriers can be achieved by incorporating a combination of various immunomodulatory agents and antigens. Cancer vaccines have the potential to target and modify the operation of T cells directly. This approach could also provide cargo for biological compounds and molecules such as immune checkpoint inhibitors, modulators of the suppressive TME, nucleic acids, RNAi, and other adjuvants [100–102]. In addition, DC-based vaccines in immunotherapy can be employed to increase the production of interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α) to alter the immune response in glioblastoma [103]. Herein, we will describe the various targeting steps used in nanomedicines to activate the immune cascade in cell-mediated immunity to treat glioblastoma and CNS tumors. Kateb et al. [104] developed multiwalled carbon nanotubes that simultaneously deliver DNA and short interfering RNA (siRNA) to specific cell types via biomolecular interactions with brain cells such as BV2 microglia and GL261 glioma. The siRNA-modified multiwalled carbon nanotubes (pMWCNTs-siRNA) may provide a nanovector for specifically targeting, stimulating, and improving the antitumor response of GL261 glioma. Antigen-modified nanoparticles are easily internalized by APCs to stimulate a robust immunostimulatory cascade against brain cancer cells [105]. Due to their inert nature, nanoplatforms are considered biodegradable and nontoxic, making these particles advantageous for immunogenetic therapy of brain tumors.
The upregulation of low accruement antigen expression in glioma APCs can be used for active glioma immunotherapy. For this purpose, a hybrid “clusterbomb” nanovaccine was developed with high antigen loading capacity on the surface of zinc oxide (ZnO) and triblock-copolymer nanoparticles (MPSDP–ZnO/Ag) [106]. The MPSDP–ZnO/Ag with a 180 nm hydrodynamic size has a spherical shape and is positively charged (+12.0 mV). The targeting nanosystems entered the DCs via receptor-mediated endocytosis. As shown in Fig. 3a, the presence of ZnO (3–5 nm) nanoparticles as the adjuvant in a multifunctional nanovaccine further promotes cellular and humoral immunity. MPSDP–ZnO/Ag enhanced both Ag-specific CD8+ and CD4+ T-cell responses, which brought on vigorous cellular and humoral immune responses in both in vitro and in vivo glioma therapy. The triplex-reduction-responsive nanovaccines allow the cluster to “bomb” within the APCs by stimulating CD8+ CTLs and the secretion of specific antibodies in response to cytokines. This multifunctional nanoplatform enhances drug accumulation and increases the survival rate of tumor-bearing mice [106]. Similarly, Liu et al. [107] used C6 glioma tumor-derived exosomes combined with natural killer T-cell activators such as α-galactosylceramide (α-GalCer) for treating glioblastoma-bearing rat models. The small size (30–50 nm) of tumor-derived exosomes with α-GalCer on a DC-based vaccine exhibited potent effects in glioblastoma immunotherapy, with a prolonged survival rate from 15 days (control) to 35 days.
Fig. 3. Nanomedicines based immunotherapies of glioma tumors.

a Schematic illustration of the hybrid ‘clusterbomb’ nanovaccines MPSDP–ZnO/Ag, which activates the cytotoxicity of regulatory T cells (Tregs) in the brain with F98npEGFRvIII-bearing rats. Reproduced with permission from [106]. Copyright 2019 Royal Society of Chemistry. b The schematic diagram of the synthesis and characterization of NICs. c The ELISA method used for validation of concurrent conjugation and activity of anti-msTfR and anti-CTLA-4/anti-PD-1 expression within a single platform and combined P/anti-CTLA-4 d and P/anti-PD-1, respectively. d The anticipated mechanism of local brain immune activation by NIC therapy, which utilizes synergistic treatment with anti-CTLA-4 and anti-PD-1 mAbs when the nanoimmune drug crosses the BBB. 1, 2, and 3 are different pathways and immune checkpoints that are activated or suppressed via NIC. Reproduced with permission from [111]. Copyright 2019 Nature Publishing Group
Recently, several therapeutic approaches have been designed to promote synergistic effects with immune checkpoint inhibition for the treatment of glioma. For instance, Qiao et al. [108] fabricated reactive oxygen species (ROS)-responsive polymer nanoparticles coloaded with small iron oxide nanoparticles, a TMZ drug, and siRNA against TGF-β, which is an immunosuppressive cytokine. Collectively, this was named ALBTA, angiopep-2 LipoPCB (TMZ + BAP/siTGF-β) with an average size of 135.9–51.3 nm, and the zeta potentials were changed from 3.18 to 51.3 mV after siRNA modification. Angiopep-2 was modified through thiol linkage on the surface of the nanoparticles to allow the nanoparticles to cross the BBB. This polymer nanoparticle facilitated the development of the immunosuppressive microenvironment and significantly enhanced the overall survival of tumor-bearing mice. Sun et al. [109] targeted the expression of tumor vascular laminin-411 (α4β1γ1), which is correlated with some cancer stem cell markers (e.g., Notch, CD133, Nestin, c-Myc) that are associated with a shorter survival time of glioblastoma patients. Sun et al. [109] developed a nanobioconjugate which inhibits laminin-411, allowing it to cross the BBB and target the TME. The inhibition of cancer stem cell markers leads to prolonged animal survival in mice carrying intracranial glioblastoma [109].
Furthermore, another study used a folate-targeted polymeric micelle that delivered the antiglioma drug TMZ as well as siRNA targeting B-cell lymphoma 2 (BCL2) into orthotopic glioma-bearing rat models [110]. Intracranial injection of this therapeutic nanocarrier carrying combined TMZ and anti-BCL2 cargo inhibited tumor growth and extended the survival time in tumor-bearing rats [110]. Recently, Galstyan et al. [111] demonstrated the application of nanoscale immunoconjugates (NICs), which are natural biopolymer scaffolds made of poly(β-L-malic acid, PMLA), covalently attached to an anti-CTLA-4 or anti-PD-1 targeting moiety (Fig. 3b-d). These NICs are capable of crossing the BBB via RMT by modification with transferrin or Angiopep-2 peptide. NICs were applied to intracranial GL261 glioblastoma-bearing moieties, causing an increase in CD8+ T cells, NK cells, and macrophages as well as a decrease in Tregs in the TME of the brain. Hence, the NICs activated the local immune system and sustained the survival of intracranial GBM GL261-bearing mice. This suggests that tumor-targeted polymer-conjugated NICs serve as checkpoint inhibitors for potential glioblastoma treatment and activate systemic and local brain tumor immune responses, prolonging survival in mouse models.
Nanotechnology, in combination with stimulated CAR T-cell therapy, represents a new frontier for treating solid tumor oncology. For instance, Zhang et al. [112] developed TME-facilitated iRGD-lipid nanoparticles that increased the function and perseverance of transferred CAR T cells into glioma cell lines and mouse models. These lipid nanocarriers deliver a combination of immune-modulatory agents, which (1) remove protumor cell populations (“releasing the brakes”) and (2) stimulate essential antitumor effector cells (“stepping on the gas”). Recently, Shi et al. [113] developed polymersomes decorated with angiopep-2 cargoes of anti-PLK1 siRNA. These carriers increased BBB permeability but were also targeted to glioblastoma in mouse models, which significantly enhanced their antiglioblastoma activity [113].
Nanomedicine-based combination therapies for glioblastoma
The complexity of glioblastoma incites researchers to develop many therapeutic strategies for treatments. Conventional single-glioblastoma therapy is often hampered by reoccurrences, short-term survival, and other regularity issues. In addition, drug-resistant glioblastoma therapy is also a major concern because of severe side effects, which demands suitable drug combinations and delivery systems. Therefore, an appealing alternative solution is combination therapy, which boosts the immune response in addition to the delivery of conventional therapies. Currently, combinations of therapeutic approaches in addition to immunotherapy are considered; these include chemotherapy, photodynamic therapy (PDT), and gene therapy. Herein, various combination strategies with immunotherapy are presented, and we discuss the advantages and disadvantages of these approaches.
Chemoimmunotherapy (CIT)
Glioblastoma and glioma exist as a complex immunosuppressive TME that overwhelms endogenic antitumor immune activity and results in upregulated immune tolerance [114]. Chemotherapy and radiotherapy are commonly used treatment alternatives for glioblastoma, which involve the delivery of chemotherapeutic agents. Though the intention of chemotherapy is to destroy tumorous cells, this approach can also damage noncancerous cells and cause severe side effects [92]. To combat this problem, immune-stimulating nonmethylated oligonucleotides (such as CpG), which can enhance long-term immunity, have recently been synthesized [115]. Lollo et al. have combined chemotherapy with immune-stimulating CpG-mediated immunotherapy to achieve the maximum therapeutic index against glioblastoma and reduce long-term relapse [116]. They developed a multifunctional lipid nanocapsule cargo system that systematically codelivered the chemotherapy drug paclitaxel (PTX) and the immunostimulant CpG agent to glioma tumors. The survival rate of orthotopic GL261 glioma mice was significantly prolonged following administration of PTX/CpG coloaded lipid nanocapsules compared with single-loaded PTX in the same system without CpG. This suggests that a combined chemo/immune therapy approach may be more effective than single chemotherapy for glioma treatment.
Recent studies have shown that specific tumor cells can escape immune system elimination via immune checkpoint pathways [117]. Indoleamine 2,3-dioxygenase (IDO) is considered an essential immune checkpoint receptor that is highly expressed in brain tumors. It is an attractive and important immune-therapeutic target for many researchers working on brain cancers [118, 119]. Importantly, 1-methyltryptophan (1MT), a specific competitive IDO inhibitor, slows tumor cell growth [120]. 1MT activates effector CD8+ T cells, and antigen-presenting DCs also inhibit immunosuppressive regulatory CD4+ T cells [121]. However, 1MT alone cannot be considered an efficient targeting agent to prevent progression, as it is only capable of retarding tumor cell growth [122]. Therefore, some research groups have combined 1MT with other drugs (e.g., doxorubicin, DOX) to increase the targeting efficiency [123]. For instance, Kuang et al. [124] developed a nanostrategy that codeliveres 1MT with the chemotherapeutic agent DOX on mesoporous silica nanoparticles. These nanoparticles were modified with the tumor-targeting/penetrating peptide CRGDK/RGPD/EC (iRGD) to improve the therapeutic application in orthotopic glioma [124]. With the help of peptide modification, DOX@MSN-SS-iRGD&1MT (DOX/1MT) was able to penetrate the BBB and deliver DOX/1MT precisely to tumor sites [124]. DOX/1MT treatment increased the number of cytotoxic CD3+ and CD8+ T cells and decreased the number of CD4+ T cells [124]. In addition, it also enhanced the expression levels of antitumor cytokines (e.g., IFNα/β, IFN-γ, TNF, and IL-17) and reduced the levels of the protumor proteins p-STAT3 and IL-10 in the brain tumor area, leading to a significant prolongation of the overall survival rate [124]. These results indicate that combination chemo/immunotherapy promotes local antiglioma cells as well as systemic antitumor immune responses.
Kadiyala et al. [125] developed nanodiscs modified with high-density lipoprotein-mimicking nanoformulations (i.e., sHDL nanodiscs) that not only deliver glioma tumor-specific antigens but also carry adjuvants and bioactive compounds. The sHDL nanodiscs are designed as CIT delivery vehicles for CpG deoxynucleotides, a TLR9 agonist, in combination with a chemotherapeutic, i.e., docetaxel (DTX), to target gliomas, as shown in Fig. 4. The nanodiscs were self-assembled by the synthetic apolipoprotein-I peptide that helps to target the tumor in the GL26 syngeneic mouse glioma model. The DTX-sHDL-CpG nanodiscs (a discoidal shape with an average size of 10 − 12 nm) not only carry drugs to the tumor site but also activate antitumor immune responses [125]. Thus, tumor relapse is restricted by enhancing the delivery of bioactive compounds to immune cells linked with glioma. The transport of cargo in DTX-sHDL-CpG nanodiscs into the tumor mass promotes tumor regression and enhances antitumor CD8+ T-cell responses in the brain immunosuppressive TME [125]. The combination of DTX-sHDL-CpG treatme7nt with radiation therapy, which is the standard treatment for glioblastoma, leads to tumor deterioration and enhanced median survival by up to 80% in glioma-bearing animals [125].
Fig. 4. The immune-mediated antiglioma mechanism utilized in docetaxel-loaded CpG-sHDL (high-density lipoprotein) nanodiscs, CpG oligodeoxynucleotide expressed by TRL9 ligand of immune cells.

a DTX-sHDL-CpG is formulated by incubating lipid-DTX (docetaxel) with CpG and preformed sHDL. b Intratumoral delivery of DTX-sHDL-CpG nanodiscs in combination with radiation results in Chemo-immuno-antiglioma activity. HDL-mimicking nanodiscs deliver the DTX payload to tumor cells in the TME, to suppress their microtubule depolymerization, resulting in mitotic cell cycle arrest in the G2/M phase (cell differentiation phases) and tumor cell death. In addition, radiation induces double-stranded DNA breaks, also leading to tumor cell death. Dying tumor cells express regulatory T cells (CRT) on their surface and are engulfed by antigen-presenting DCs and macrophages. Reprinted with permission from [125]. Copyright 2019, American Chemical Society
Targeting Galectin-1 in addition to chemotherapy can also improve outcomes in glioma models. Gal-1 is a 14 kDa lectin present in the TME [126] and is responsible for the enhanced migration of glioma cells and their resistance against chemotherapy (i.e., TMZ) [127]. Radiotherapy leads to the upregulation of Gal-1, which can cause a suppressed immune response [128]. Thus, Gal-1 is a crucial molecule in shaping the TME in glioma. Intranasal siRNA targeting Gal-1 transport (i.e., siGal-1) encourages a remarkable change in the TME, for example, reduced myeloid suppressor cells and Tregs, and increases CD4+ and CD8+ T cells during glioblastoma progression. Galectin-1 targeting siRNA (siGal-1) loaded in an intranasal chitosan nanoparticle can also improve the efficiency and reliability of chemotherapy combined with immune checkpoint inhibition to improve life expectancy in tumor-bearing mice [129]. Intranasal siGal-1 delivery changes in the TME of GL261 tumor-bearing mice by reducing myeloid suppressor cells and Tregs, along with enlarged CD4+ and CD8+ T cells. siGal-1 can silence Gal-1 expression in the TME and exhibits the synergistic effect of combination therapy, increasing the median survival rate from 32 days (single chemotherapy used in TMZ-treated mice) to 53 days.
The CIT approaches provide several advantages, such as reduced drug resistance, multiple mechanisms, reduced metastasis, and recurrence. However, due to the limitation of nonspecificity to tumor cells, along with the lack of unique antigens and the unavailability of suitable timing for immunotherapy, such treatment is extremely dangerous in clinical practice.
Photoimmunotherapy (PIT)
PIT is a newly reported glioblastoma therapy that takes advantage of a photosensitizer conjugated with targeted monoclonal antibodies. Under the photoactivation effect in PDT, enhanced generation of ROS [130] and inflammatory factors (TNF-α and IFN-γ) is combined to boost antitumor immunity in glioblastoma [131]. PIT involves antibody-targeted delivery with a nontoxic photosensitizer and light sources to activate a cytotoxic state for glioblastoma therapy [132]. Recently, the light-based near-infrared photoimmunotherapy (NIR-PIT) procedure has emerged as a new class of therapy for brain cancers, which permits the specific, image-guided, and spatiotemporally controlled elimination of tumor cells [133]. A near-infrared photosensitizer and target-specific antibody conjugates are used to selectively induce cell death in cancer cells and activate the host antitumor immune response [134, 135]. NIR-PIT has shown promising results in preclinical research [136, 137] and has recently entered clinical trials for inoperable head and neck cancer. Jing et al. formulated a conjugate of the photosensitizer IR700 with an AC133 monoclonal antibody for glioblastoma treatment [138]. The AC133 antibody recognizes CD133 specifically on human cancer stem cells, which enhances targeting specificity to orthotopic AC133+ brain tumors [138]. Treatment with AC133-targeted NIR-PIT induces strong shrinkage of both subcutaneous and invasively growing brain tumors and extends the median survival time by more than 18 days [138].
Glioblastomas can be differentially driven by alterations (amplification, deletion, or missense mutations) in the EGFR, and genetic mutations in glioblastoma can be induced by altering EGFR expression on tumor surfaces [139]. Burley et al. conjugated the photosensitizer IR700DX with an EGFR-specific affibody (ZEGFR-03115) for glioblastoma treatment [132]. Compared with the control group (ZTaq-IR700DX), the EGFR-specific antibody ZEGFR-03115-IR700DX showed a sixfold higher tumor specificity, which significantly reduced tumor growth in U87-MGvII, I bearing mice over 18 days [132]. However, because of the limited fabrication ratio of the photosensitizer-to-antibody, a sufficient amount of photo-immunoconjugates (PIC) was not delivered to the tumor cells to eradicate glioblastoma [132]. Huang et al. employed a poly(lactic-co-glycolic acid) (PLGA) system to overcome the insufficient delivery of PIC to tumor cells [140]. The photo-immunoconjugate benzoporphyrin derivative-cetuximab was bound to PLGA nanoparticles through click coupling (Fig. 5) [140]. The photo-immunoconjugate nanoparticle (PIC-NP) of 100 nm in diameter (PDI ≤ 0.11) and slightly negative (−4 mV) surface charge significantly increased the intratumoral accumulation of PIC, resulting in enhanced PIT-mediated tumor volume reduction compared with “standard” immunoconjugates [140]. PIC-NPs also increased light-activated cytotoxicity in EGFR-overexpressing U87 cells [140]. Importantly, the implementation of nanotechnology improved overall treatment outcomes compared with conventional PIC.
Fig. 5. Synthetic diagram of photo-immunoconjugate nanoparticles (PIC-NPs).

a The illustration of PIC synthesis by conjugation of BPD derivative photosensitizers to PEGylated cetuximab via carbodiimide crosslinker chemistry. b showing the various stoichiometry of BPD reacted with cetuximab species. c Transmission electron microscopy (TEM) image of poly (ethylene glycol)-poly(lactic-co-glycolic acid) (PEG-PLGA) polymeric nanoparticles prepared via nanoprecipitation method. Scale bar 100 nm. d Schematic representation of PIC-NP synthesis via copper-free click chemistry. Azide-containing FKR560 dye-loaded PLGA nanoparticles were reacted with the dibenzocyclooctyne (DBCO)-containing PICs to form PIC-NPs. e Covalent conjugation of PICs onto 80 nm PEG-PLGA NPs resulted in the formation of monodispersed PIC-NPs around 100 nm in diameter. Adapted with permission from [140]. Copyright 2018 WILEY‐VCH Verlag GmbH & Co
Overall, PIT has demonstrated improved tumor cell targeting compared with classic PDT due to the specificity of the antibodies conjugated to the surface of the nanoparticles. PIT can also enhance the delivery of photosensitizers into tumors, increasing light-activated cytotoxicity and tumor reduction. Studies have also reported negligible damage to noncancerous cells, which increases the safety of this approach. However, to date, the minimum ratio of photosensitizer to antibody that efficiently delivers the photosensitizer by PIC still requires improvement [141], and this approach for glioblastoma is still in its infancy.
Photoactivated nanoformulations exhibit very enhanced targeting ability and reduced tumor metastasis and recurrence; however, they are also associated with a restricted ratio of photosensitizers to antibodies, which makes them difficult in clinical use.
Gene immunotherapy (GIT)
Gene therapy has emerged as a novel cancer treatment by explicitly regulating oncogenes [142–145]. Immunomodulatory gene therapy is designed to restore the function of defective or absent genes for the treatment of glioblastoma. A combination of immunomodulatory and gene therapy is designed to produce a tumor environment optimized for the induction of an effective antitumor immune response [146]. In glioblastoma GIT, targeted delivery of cytokines into the TME can achieve higher local concentrations of immunostimulatory molecules, such as IL-2, IL-4, IL-12, IFN-γ, and IFN-β, without creating side effects [147]. In patients with newly diagnosed resectable glioblastoma, administration of adenovirus-mediated gene therapy containing the prodrug converting enzyme sitimagene ceradenovec followed by intravenous administration of the antiviral drug ganciclovir increases the mortality rate in patients, but does not increase overall survival [148]. Interestingly, gene therapy is being proposed as a valuable adjuvant for current glioblastoma treatment [149–151]. Supporting this approach, Speranza et al. utilized a nonreplicating adenovirus containing the HSV TK gene (AdV-tk) to enhance anti-PD-1 efficacy in syngeneic glioblastoma-bearing mouse models [152]. AdV-tk upregulated IFN signaling and enhanced PD-L1 levels, while cytotoxic CD8+ T cells were allowed to accumulate in tumors. The combination of gene therapy with immunotherapy significantly increased the percentage of long-term survival in glioblastoma-bearing animals from 30%–50% (treatment with single agents) to 88% [152].
Despite the promise of gene combination therapy for glioblastoma, this approach is still hindered by an inefficient delivery system to tumor sites [153, 154]. Recently, nanoparticles have overcome these shortfalls to deliver gene-mediated immunotherapeutic agents to brain tumors [155]. Erel-Akbaba et al. [156] developed a solid lipid nanoparticle (SLN) decorated with the cyclic peptide iRGD (CCRGDKGPDC) to deliver siRNAs against both EGFR and PD-L1 (Fig. 6). When the cyclic peptide iRGD is conjugated to nanoparticles, this enables the nanoparticles to cross the BBB and enhances the targeting ability of this therapy in glioblastoma-bearing mice. Moreover, the SLN possesses a strong degree of lipophilicity and a positive charge that facilitates BBB penetration. After binding with siRNA, the hydrodynamic size of the f(SLN)–iRGD:siRNA complex was measured to be 24.1 nm with a positive charge around the complex. Nanoparticle f(SLN)–iRGD:siRNA treatment led to the downregulation of PD-L1 and EGFR expression levels by 8.6% and 54.7%, respectively [156]. Furthermore, the median survival of the mice treated with f(SLN)–iRGD:siRNA combined with radiation increased to 38 days from 21 days (control group) [156]. To date, gene therapy has mostly been applied to enhance systemic therapy for glioma treatment, but it can also significantly regulate immunosuppressive signals. Some oncolytic viruses, such as talimogene laherparepvec, are FDA approved and are already in phase III clinical trials for the treatment of melanoma patients [157], facilitating their implementation in glioma combination treatment approaches. Indeed, synergistic use of these approaches can dramatically improve immune responses induced by immunotherapy. However, this approach requires more research to optimize combinations of therapies due to the lack of specific genes and antigens as treatment targets, and the route of delivery still represents a barrier for glioma treatment.
Fig. 6.

a Schematic route for the delivery of gene combination therapy using siRNA loaded nanoparticles for brain tumor treatment. b The chemical structure of the DSPE-PEG (2000)-DBCO, Esterquat, and iRGD peptide, and the construction of the nanoparticles SLN, f(SLN), f(SLN)–iRGD, and f(SLN)–iRGD:siRNA. Adapted with permission from [156]. Copyright 2019 American Chemical Society
The GIT approach provides several outermost advantages of indirect antitumor effects and reduced drug resistance in glioblastoma patients, but it is also associated with a lack of suitable and specific genes and antigens for glioblastoma.
Nanomedicine-based immunotherapy for neurodegenerative disorders
Neurodegenerative disorders are characterized by the tenacious and progressive loss of neuronal subtypes. Common neurodegenerative disorders include AD and PD. Herein, we summarize nanomedicine-based immunotherapeutic approaches for AD and PD.
Alzheimer’s disease (AD)
AD is characterized by the accumulation of amyloid-beta (Aβ) peptide within extracellular senile plaques and the presence of neurofibrillary tangles of hyperphosphorylated tau protein as insoluble aggregates inside neurons of the CNS [158, 159]. Individuals with AD suffer from progressive memory loss, executive function impairment, and language deterioration and eventually die from AD due to significant neuronal loss [160]. Approved drugs for AD include acetyl-cholinesterase inhibitors (e.g., donepezil and rivastigmine), nicotinic acetylcholine receptor allosteric modulators (e.g., galantamine), and NMDA receptor antagonists (e.g., memantine); however, these drugs do not stop disease progression or death in AD patients [161]. The Aβ peptide is generated by sequential cleavage of amyloid precursor protein (APP) by β secretase 1 (BACE1) and γ-secretase (APP) [162, 163]. The exact origin of Aβ, how it accumulates in the brain, and its role in AD is not understood [164]. The deposition of Aβ is hypothesized to trigger a pathological and distinctive immune response in AD [165]. Interestingly, microglial cells possess innate mechanisms for removing protein aggregates and debris from the brain, and this process can be utilized in immune-based approaches for AD [166]. Immunization strategies for AD can be classified into two main forms: active and passive immunization-based therapy [167]. Active immunization induces a humoral immune response and is long-lasting, as it influences immunological memory in patients [168]. While active vaccines have several benefits (e.g., fewer administrations are needed to be compared with passive immunotherapy and can induce a polyclonal response against multiple epitopes, thus increasing the efficacy of the drug), the production of these vaccines is not cost-effective, and these vaccines rely on the patient’s own immune response, which varies among individuals [168, 169].
On the other hand, passive immunization comprises the direct injection of monoclonal antibodies antiamyloid-β antibodies) or fragments deprived of necessitating the immune system to generate an antibody response against amyloid-β or improve memory deficits in AD mouse models [170]. Passive immunization has been shown to have beneficial effects on neuronal and synaptic plasticity function [171]. However, due to the difficulty in selecting appropriate antigen targets, the expense of requiring repeated injections, BBB penetration, and triggering of the immune response to the antibodies that are injected make it complicated for AD patients [172].
Both active and passive immunization may cause the activation of microglial cells and subsequent clearance of Aβ, similar to that observed after stroke or injection of Aβ into the brain [173]. Indeed, a reduction in Aβ deposits is associated with higher microglial cell activity in AD mouse models [174–176]. The clearance of Aβ by resident microglial cells can occur via several routes (Fig. 7) [170]. The upper section of Fig. 7 shows the inducers of microglial activation. The binding of various ligands to many cell-surface immune receptors, such as CD14, adheres lipopolysaccharide and protollin-like components, which can activate microglial cells. Some TLRs (e.g., TLR2 and TLR4) also attach to Protollin components. TCRs interact with MHC class II molecules, and CD40 binds to a CD40 ligand on the surface of T cells and astrocytes, whereas complement receptors bind to components such as C1q and Fc receptors. These activated microglial cells can then attack Aβ-specific antibodies. The lower section in Fig. 7 indicates the activated microglial cells that carry numerous scavenging receptors (SRs), e.g., integrin-αβ, CD36, CD47, SR-A, and SR-BI, which are responsible for the phagocytosis of Aβ oligomers and fibrils. These phagocytic ligations bind to cell-surface expression receptors such as heparan sulfate proteoglycans [177], proteinase inhibitor (serpin)–enzyme complex receptors (SEC-R) [178] and insulin receptors that are also expressed on activated microglial cells, leading to their complete dissolution. In addition, various Aβ-degrading enzymes, such as metalloproteases, insulin-degrading enzyme, and gelatinase A, are also expressed by activated microglia and are responsible for degrading Aβ [179, 180].
Fig. 7.
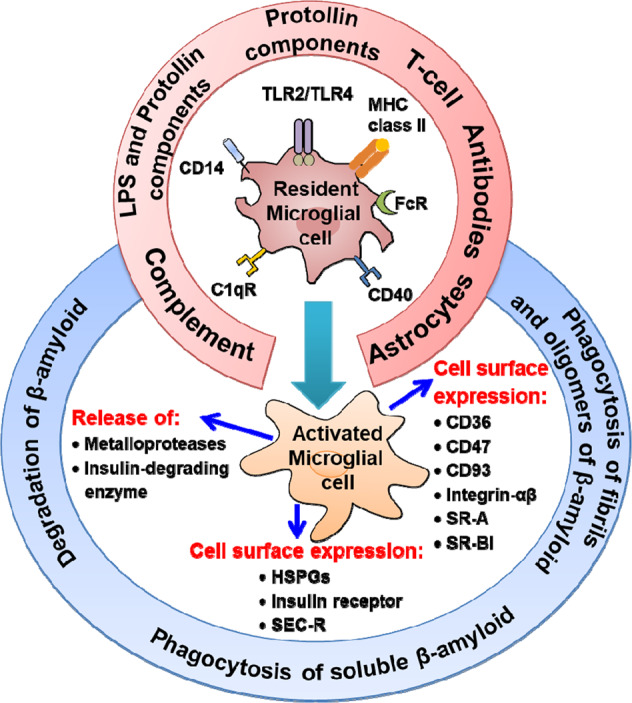
Clearance mechanisms of amyloid-β based on immunotherapeutic approaches for AD pathology
Active and passive immunotherapies have been considered the most acceptable approaches used to clear toxic Aβ aggregates in AD owing to the amyloid hypothesis. Another approach to remove solubilized Aβ aggregates is immune modulation, but this is still in earlier stages of an investigation. Three possible mechanisms of immune-mediated Aβ removal are proposed: Aβ solubilization by antibody binding; Aβ phagocytosis by microglia; and Aβ extraction from the brain via plasma antibodies, also named the “sink” hypothesis. To date, most research has focused on a reduction in Aβ, and relatively few reports have targeted Tau conformers. Importantly, histological analyses in AD brains demonstrate synergism between these pathologies [181–184]. One issue that needs to be addressed in this field is the potential toxicity associated with Aβ and Tau immunotherapy following the binding of Aβ and Tau owing to the cellular uptake of antibodies, which destabilizes the microtubules, resulting in cytoskeletal integrity and high intervention with axonal transport [184].
Nanoparticle-based AD treatment has been explored recently in the literature. Liu et al. reported on AD treatment application by zwitterionic poly(carboxybetaine) (PCB)-based nanoparticles coloaded with fingolimod, siSTAT3, and zinc oxide, henceforth named MCPZFS nanoparticles or MCPZFS NPs [185]. The diameter of the MCPZF NPs was between 84.0 and 89.4 nm. A mannose analog 4-aminophenyl α-d-mannopyranoside (Man) was added to the nanoparticles to facilitate BBB penetration and improve the targeting of microglia [185]. MCPZFS nanoparticles significantly improve microglial priming by reducing the level of proinflammatory mediators and promoting the secretion of brain-derived neurotrophic factor (Fig. 8) [185]. Importantly, MCPZFS nanoparticles increase Aβ recruitment into microglia, which substantially enhances Aβ phagocytosis, and Aβ degradation is changed from the conventional lysosomal/autophagy pathway to the proteasomal pathway in the presence of MCPZFS nanoparticles [185]. This system elevated numerous key proinflammatory cytokines, such as interleukin 1-β (IL-1β), IFN-γ, IL-6, and interleukin 17A, in the brains of APP or presenilin 1-transgenic mice. A reduction in Aβ burden, neuron damage, memory deficits, and neuroinflammation in the brain was observed following MCPZFS NP administration. In addition, immunization can be used to produce the polyclonal response in which multiple epitopes on the target protein surface are recognized and accessed through affinity antibodies [186]. However, the immune response depends on the host immune system; thus, the antibody-based responses in various patients may vary from individual to individual [187].
Fig. 8. The immune-modulating effect of a zwitterionic poly(carboxybetaine) (PCB)-based nanoparticles (Man-PCBPB/ZnO/fingolimod/siSTAT3 NPs: MCPZFS) for AD therapy.

a Structural composition and preparation of the MCPZFS for AD therapy. b The mechanism of MCPZFS BBB permeability and binding with Aβ for endocytosed into microglia cells. Reproduced with permission from [185]. Copyright 2019, WILEY‐VCH Verlag GmbH & Co
Parkinson’s disease (PD)
Neuropathological hallmarks of PD include the accumulation of α-synuclein protein aggregates and intracellular inclusions known as Lewy bodies [188]. Significant dopamine cell loss in the substantia nigra is also a hallmark of PD [189, 190]. Recent advances in our understanding of immune system function in PD suggest that using nanomedicine-based immunotherapies could provide a novel mechanism to boost the immune response against α-synucleinopathies and Lewy body accumulation [191]. In PD, the accumulation of α-synuclein leads to the activation of microglia and fosters neuroinflammation, which can facilitate the progressive degeneration of dopaminergic neurons [192]. It has been hypothesized that inflammation-induced changes in microglia impair both their efficacy in taking up extracellular α-synuclein and their ability to degrade it [192]. Microglial activation increases the release of proinflammatory cytokines, such as IL-1β, IL-6, INF-γ, and TNF-α [193]. Proinflammatory cytokines can increase the production of oxidative stress proinflammatory markers (i.e., ROS and nitric oxide, NO) in astrocytes. Numerous neuroinflammatory measures, including microgliosis, astrocytosis, and infiltration of T-leukocytes, are evident in the midbrain of PD patients and are also observed in PD rodent models [194].
Current treatments for PD (e.g., pharmacotherapy, deep brain stimulation) do not halt disease progression and often have significant side effects [195]. There is considerable interest in the increased clearance of α-synuclein with drugs that promote autophagy and prevent the seeding and prion-like spreading of α-synuclein [196, 197]. Recent preclinical research suggests that new immunotherapy-based approaches that are packaged into nanoparticles can show promise for PD. Alternatively, a decrease in α-synuclein aggregation can be achieved via small molecules [198]. Clearance of α-synuclein can also be induced with drugs (e.g., potentially neuroprotective agents such as isradipine, caffeine, and nicotine; antioxidants such as glutathione and N-acetylcysteine), peptides (e.g., PD01A and PD03A), antibodies (e.g., PRX002 and BIIB054), and conformational stabilizers (NPT200-11) [196, 197]. Some immunotherapy studies demonstrate that vaccination against α-synuclein reduces α-synuclein accumulation by activating autophagy [199] or microglial pathways [200]. Passive or active immunization strategies with monoclonal antibodies that bind the C-terminus epitopes of α-synuclein can reverse behavioral deficits in PD rodent models, as well as inhibit α-synuclein accumulation within neurons [201] and microglial cells [202]. TLRs can also stimulate innate immune responses through mitogen-activated protein kinase and nuclear factor-kappa B signaling pathways. After internalizing α-synuclein, microglial cells can additionally release the proinflammatory cytokines TNFα and IL-6 and eventually activate innate immune checkpoints, which can further induce TLR stimulation responses (e.g., TLR2/1- and TLR7-mediated responses) [203]. Therefore, the TLR-mediated immune response induced by microglia has a significant impact on activating the assault of sporadic α-synuclein-related neuropathologies and microglia-mediated neuroinflammation.
Several studies have utilized fluorescent quantum dots (graphene quantum dots, GQDs) to inhibit α-synuclein pathogenesis and amyloidosis [204, 205]. Fig. 9 shows the BBB permeability of GQDs due to their small size (~5–20 nm) and antibody-modified surfaces. Seven days of treatment with GQDs in a PD mouse model rescued neuronal death and synaptic loss reduced Lewy body and Lewy neurite formation, ameliorated mitochondrial dysfunction, and prevented the neuron-to-neuron transmission of α-synuclein pathology provoked by α-synuclein preformed fibrils [205]. Interestingly, it seems that GQDs interact with α-synuclein fibrils, stopping their development and even promoting their disaggregation. Therefore, proper amendments in the biosafety of such nanomedicines might be a valuable addition to the treatment of PD and other neurogenerative diseases.
Fig. 9. GQDs as nanomedicine for α-synuclein fibrillization and their disaggregation process.

a Schematic representation of α-synuclein fibrillization (5 mg/mL α-synuclein monomers) and disaggregation (5 mg/mL α-synuclein fibrils) before and after the addition of GQDs (5 mg/mL). b Transmission electron microscopy images show α-synuclein fibrillization before and after the addition of GQDs. c In vitro quantification of the effect of GQDs on α-synuclein preformed fibrils (PFF)-induced neuronal death via phosphorylated α-synuclein (p-α-synuclein) immunofluorescence. d Illustration of the microfluidic device component for the transmission of pathologic α-synuclein. e, f Kinetics measurements of α-synuclein fibrils after incubation with GQDs via thioflavin T (ThT)-based fluorescence and turbidity assays, respectively. g, h Kinetics measurement and turbidity assays for α-synuclein fibrillization via ThT-based fluorescence, respectively. Reproduced with permission from [205]. Copyright 2018 Nature Publishing group
Outlook and conclusion
Recent advances in multidisciplinary sciences, including medicine, biochemistry, protein engineering, and materials science, have given rise to innovative nanoscale targeting techniques. These techniques have the potential to revolutionize immunotherapy for CNS disorders. Indeed, in the glioblastoma treatment field, researchers have recently focused on new developmental therapeutic strategies that specifically target tumor resistance to improve clinical outcomes. Novel approaches in nanobiotechnology can also enhance the delivery of immune therapies for CNS-related diseases (e.g., antibodies, cytotoxic drugs, vaccine antigens, siRNAs, etc.). These nanomedicines are capable of dynamically targeting immune cells and releasing their cargo within the TME. Recently developed nanotechnologies can transport synergistic drug combinations that improve the patient’s innate and adaptive immunity to combat glioblastoma. The efficacy of these approaches is further improved by the use of biodegradable (water-soluble hydrogels and matrices) and stimuli-sensitive nanomaterial (i.e., pH and temperature)-based nanoparticles. In addition, some nanoimmunomodulators are designed to be taken up by macrophages and DCs to activate the immune response. Surface modification of nanocarrier systems has been designed to include selective targeting ligands (e.g., proteins, peptides, and aptamers) or cell membrane-derived vesicles to facilitate barrier crossing and improve targeting efficiency. Consideration needs to be given to the use of immunotherapy in cancer patients, as engaging a compromised immune system can increase the risk of adverse side effects. Further research will investigate how nanodelivery strategies can activate innate and adaptive cellular immune responses for the treatment of glioblastoma [206, 207].
AD is another area in which nanobased medicines could significantly improve treatment efficacy by precise regulation of AD-specific pathological pathways in targeted locations. The combination of immunotherapy for AD with nanomedicines or vaccines is also an exciting and burgeoning field. While current nanomedicines or vaccine strategies can have issues of toxicity, tolerance, and efficacy, the development of more biocompatible nanoscale materials is an area of increasing research interest [208, 209].
Because of the unavailability of representative biomarkers on PD brains, nanomedicines that can target-specific PD pathology are lacking. Recently, the discovery of a novel nanocarrier system for the delivery of immunotherapeutics for PD may also help to improve PD pathology. However, because α-synuclein can adopt various conformational changes (e.g., due to environmental influences or interpatient variability), this can make it exceptionally challenging to develop nanomedicine-based immunotherapy targeting α-synuclein [196]. The development of highly regulated pharmacokinetic and biocompatible nanoscale materials could bring PD immunotherapy from bench to bedside.
The challenges that must be overcome by immunotherapy include differential mechanisms of action owing to the interindividual variable immune response and cold serological response to the antigen in older patients with weakened immune systems. Moreover, metal-based therapeutics (titanium oxides, GQDs, ZnO, silver, and gold nanoparticles), despite their advantageous benefits, can induce oxidative damage in the brain. In addition, the dose, adjuvants, administration route, and clinical protocols need to be precisely monitored to bring the immunotherapy into clinical practice. Even though neurodegenerative ailments share a distinctive malfunctioning of the immune response that could help as a therapeutic target, the finetuning of appropriate surface modifications should be carefully considered for individual diseases to exploit the effectiveness of immunotherapies and decrease the risk of side effects. Finally, such therapies need to be employed at the earliest stage of the disease to increase their potential therapeutic value, which represents a challenge in the handling of CNS disorders.
Acknowledgements
This work was supported by National Natural Science Foundation of China (31600809, 31800841, U1604177, and U1804139), the National Key Technologies R&D Program of China (2018YFA0209800), Australian Endeavour Fellowship (No. 69172018), Mason Foundation National Medical Program (MAS2017F034), the National Health and Medical Research Council (NHMRC) Project grant (GNT1166024), and Dementia Fellowship (GNT1111611).
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Sumaira Hanif, Pir Muhammad
Contributor Information
Rong-jun Qian, Email: qrjqqx@163.com.
Meng Zheng, Email: mzheng@henu.edu.cn.
Bing-yang Shi, Email: bs@henu.edu.cn.
References
- 1.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20:1429–38. doi: 10.1093/neuonc/noy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zanganeh S, Georgala P, Corbo C, Arabi L, Ho JQ, Javdani N, et al. Immunoengineering in glioblastoma imaging and therapy. Wiley Interdiscip Rev. 2019;11:1575. doi: 10.1002/wnan.1575. [DOI] [PubMed] [Google Scholar]
- 5.Perry JR, Laperriere N, O’Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. 2017;376:1027–37. doi: 10.1056/NEJMoa1611977. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 7.Amiri H, Saeidi K, Borhani P, Manafirad A, Ghavami M, Zerbi V. Alzheimer’s disease: pathophysiology and applications of magnetic nanoparticles as MRI theranostic agents. ACS Chem Neurosci. 2013;4:1417–29. doi: 10.1021/cn4001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Prim. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 9.Behrens S, Rattinger GB, Schwartz S, Matyi J, Sanders C, DeBerard MS, et al. Use of FDA approved medications for Alzheimer’s disease in mild dementia is associated with reduced informal costs of care. Int Psychogeriatr. 2018;30:1499–507. doi: 10.1017/S104161021800011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrmann N, Chau SA, Kircanski I, Lanctot KL. Current and emerging drug treatment options for Alzheimer’s disease. Drugs. 2011;71:2031–65. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Ellis JM, Fell MJ. Current approaches to the treatment of Parkinson’s disease. Bioorg Med Chem Lett. 2017;27:4247–55. doi: 10.1016/j.bmcl.2017.07.075. [DOI] [PubMed] [Google Scholar]
- 12.Hou LC, Veeravagu A, Hsu AR, Victor C. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus. 2006;20:E3. doi: 10.3171/foc.2006.20.4.2. [DOI] [PubMed] [Google Scholar]
- 13.Madajewicz S, Chowhan N, Tfayli A, Roque C, Meek A, Davis R, et al. Therapy for patients with high grade astrocytoma using intraarterial chemotherapy and radiation therapy. Cancer. 2000;88:2350–6. doi: 10.1002/(sici)1097-0142(20000515)88:10<2350::aid-cncr20>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Till SJ, Francis JN, Nouri-Aria K, Durham SR. Mechanisms of immunotherapy. J Allergy Clin Immunol. 2004;113:1025–34. doi: 10.1016/j.jaci.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Jiang W, Nam J, Moon JJ, Kim BY. Immunomodulating nanomedicine for cancer therapy. Nano Lett. 2018;18:6655–9. doi: 10.1021/acs.nanolett.8b02340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–93. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg SA, Dudley ME, Restifo NP. Cancer immunotherapy. N Engl J Med. 2008;359:1072. doi: 10.1056/NEJMc081511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krol S, Macrez R, Docagne F, Defer G, Laurent S, Rahman M, et al. Therapeutic benefits from nanoparticles: the potential significance of nanoscience in diseases with compromise to the blood brain barrier. Chem Rev. 2012;113:1877–903. doi: 10.1021/cr200472g. [DOI] [PubMed] [Google Scholar]
- 21.Su W, Gao C, Wang P, Huang J, Qian Y, Guo L, et al. Correlation of circulating T lymphocytes and intracranial hypertension in intracerebral hemorrhage. World Neurosurg. 2017;107:389–95. doi: 10.1016/j.wneu.2017.07.179. [DOI] [PubMed] [Google Scholar]
- 22.Carpentier AF, Meng Y. Recent advances in immunotherapy for human glioma. Curr Opin Oncol. 2006;18:631–6. doi: 10.1097/01.cco.0000245321.34658.f4. [DOI] [PubMed] [Google Scholar]
- 23.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 24.Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther. 2016;16:1265–75. doi: 10.1080/14712598.2016.1214266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci. 2017;38:291–304. doi: 10.1016/j.tips.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2017;20:184–91. doi: 10.1093/neuonc/nox175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen MD, Julien J-P, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- 28.Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x. [DOI] [PubMed] [Google Scholar]
- 29.Reardon DA, Wucherpfennig K, Chiocca EA. Immunotherapy for glioblastoma: on the sidelines or in the game? Discov Med. 2017;24:201–8. [PubMed] [Google Scholar]
- 30.Lowther DE, Goods BA, Lerner BA, Raddassi K, van Dijk D. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight. 2016;1:e85935. doi: 10.1172/jci.insight.85935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 32.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24:4175–86.. doi: 10.1158/1078-0432.CCR-17-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res. 2014;20:3651–9. doi: 10.1158/1078-0432.CCR-13-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904–11. doi: 10.1002/cncr.30642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian JM, Yu JB, Kluger HM, Chiang VL. Timing and type of immune checkpoint therapy affect the early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer. 2016;122:3051–8. doi: 10.1002/cncr.30138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berghoff AS, Venur VA, Preusser M, Ahluwalia MS. Immune checkpoint inhibitors in brain metastases: from biology to treatment. Am Soc Clin Oncol Edu Book. 2016;36:e116–e22. doi: 10.1200/EDBK_100005. [DOI] [PubMed] [Google Scholar]
- 40.Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134:521–35. doi: 10.1007/s00401-017-1769-8. [DOI] [PubMed] [Google Scholar]
- 41.Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 42.Wen PY, Huse JT. 2016 World Health Organization classification of central nervous system tumors. Continuum. 2017;23:1531–47. doi: 10.1212/CON.0000000000000536. [DOI] [PubMed] [Google Scholar]
- 43.Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ, Uhm JH, et al. Central nervous system tumors. Mayo Clin. Proc. 2007;82:1271–86. doi: 10.4065/82.10.1271. [DOI] [PubMed] [Google Scholar]
- 44.Tomitaka A, Kaushik A, Kevadiya BD, Mukadam I, Gendelman HE, Khalili K, et al. Surface-engineered multimodal magnetic nanoparticles to manage CNS diseases. Drug Discov Today. 2019;24:873–82. doi: 10.1016/j.drudis.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutkin A, Cohen ZR, Peer D. Harnessing nanomedicine for therapeutic intervention in glioblastoma. Expert Opin Drug Deliv. 2016;13:1573–82.. doi: 10.1080/17425247.2016.1200557. [DOI] [PubMed] [Google Scholar]
- 46.Chakroun RW, Zhang P, Lin R, Schiapparelli P, Quinones‐Hinojosa A, Cui H. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdiscip Rev. 2018;10:e1479. doi: 10.1002/wnan.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Liu L. Modern methods for delivery of drugs across the blood–brain barrier. Adv Drug Deliv Rev. 2012;64:640–65. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–72. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rauf MA, Rehman FU, Zheng M, Shi B. The strategies of nanomaterials for traversing blood-brain barrier. In: Xue X, editor. Nanomedicine in brain diseases: principles and application. Singapore; Springer Singapore: 2019. p. 29–57.
- 50.Lonser RR, Sarntinoranont M, Morrison PF, Oldfield EH. Convection-enhanced delivery to the central nervous system. J Neurosurg. 2015;122:697–706. doi: 10.3171/2014.10.JNS14229. [DOI] [PubMed] [Google Scholar]
- 51.Ozdemir-Kaynak E, Qutub AA, Yesil-Celiktas O. Advances in glioblastoma multiforme treatment: new models for nanoparticle therapy. Front Physiol. 2018;9:170. doi: 10.3389/fphys.2018.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng M, Tao W, Zou Y, Farokhzad OC, Shi B. Nanotechnology-based strategies for siRNA brain delivery for disease therapy. Trends Biotechnol. 2018;36:562–75. doi: 10.1016/j.tibtech.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 53.Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc ChemRes. 2011;44:1029–38. doi: 10.1021/ar200019c. [DOI] [PubMed] [Google Scholar]
- 54.Athar M, Das AJ. Therapeutic nanoparticles: state-of-the-art of nanomedicine. Adv Mater Rev. 2014;1:25–37. [Google Scholar]
- 55.Rehman FU. Nanomedicine: why it still taking long from “bench to bedside”? Biomed Lett. 2018;4:1–13. [Google Scholar]
- 56.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 57.Banks WA. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15:275. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 58.Oller-Salvia B, Sánchez-Navarro M, Giralt E, Teixido M. Blood–brain barrier shuttle peptides: an emerging paradigm for brain delivery. Chem Soc Rev. 2016;45:4690–707. doi: 10.1039/c6cs00076b. [DOI] [PubMed] [Google Scholar]
- 59.Ali IU, Chen X. Penetrating the blood–brain barrier: promise of novel nanoplatforms and delivery vehicles. ACS Nano. 2015;9:9470–4. doi: 10.1021/acsnano.5b05341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y, Zou Y, Feng C, Lee A, Yin J, Chung R, et al. Charge conversional biomimetic nanocomplexes as a multifunctional platform for boosting orthotopic glioblastoma RNAi therapy. Nano Lett. 2020;20:1637–46. doi: 10.1021/acs.nanolett.9b04683. [DOI] [PubMed] [Google Scholar]
- 61.He W, Zou Y, Zheng M, Shi B. Cell-derived biomimetic drug delivery systems for cancer therapy. Sci Sin. 2019;49:1203–12. [Google Scholar]
- 62.Tayyba T. Role of extracellular vesicles in human diseases. Biomed Lett. 2019;5:67–8. [Google Scholar]
- 63.Kim BY, Rutka JT, Chan WC. Nanomedicine. N Engl J Med. 2010;363:2434–43. doi: 10.1056/NEJMra0912273. [DOI] [PubMed] [Google Scholar]
- 64.Zheng M, Liu Y, Wang Y, Zhang D, Zou Y, Ruan W, et al. ROS-responsive polymeric siRNA nanomedicine stabilized by triple interactions for the robust glioblastoma combinational RNAi therapy. Adv Mater. 2019;31:1903277. doi: 10.1002/adma.201903277. [DOI] [PubMed] [Google Scholar]
- 65.Zou Y, Liu Y, Yang Z, Zhang D, Lu Y, Zheng M, et al. Effective and targeted human orthotopic glioblastoma xenograft therapy via a multifunctional biomimetic nanomedicine. Adv Mater. 2018;30:1803717. doi: 10.1002/adma.201803717. [DOI] [PubMed] [Google Scholar]
- 66.Wang S, Li C, Qian M, Jiang H, Shi W, Chen J, et al. Augmented glioma-targeted theranostics using multifunctional polymer-coated carbon nanodots. Biomaterials. 2017;141:29–39. doi: 10.1016/j.biomaterials.2017.05.040. [DOI] [PubMed] [Google Scholar]
- 67.Pridgen EM, Langer R, Farokhzad OC. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine. 2007;2:669–80. doi: 10.2217/17435889.2.5.669. [DOI] [PubMed] [Google Scholar]
- 68.Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 69.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lesniak MS, Brem H. Targeted therapy for brain tumours. Nat Rev Drug Discov. 2004;3:499. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 71.Strazielle N, Ghersi-Egea J. Physiology of blood–brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharmacol. 2013;10:1473–91. doi: 10.1021/mp300518e. [DOI] [PubMed] [Google Scholar]
- 72.Zhang TT, Li W, Meng G, Wang P, Liao W. Strategies for transporting nanoparticles across the blood–brain barrier. Biomater Sci. 2016;4:219–29. doi: 10.1039/c5bm00383k. [DOI] [PubMed] [Google Scholar]
- 73.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abbott NJ, Dolman DE, Yusof SR, Reichel A. In vitro models of CNS barriers. In: Drug delivery to the brain. New York: Springer, Springer-Verlag; 2014. p 163–97.
- 75.DeStefano JG, Jamieson JJ, Linville RM, Searson PC. Benchmarking in vitro tissue-engineered blood–brain barrier models. Fluids Barriers CNS. 2018;15:32. doi: 10.1186/s12987-018-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Toh Y-C, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, et al. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 2007;7:302–9. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- 77.Urich E, Patsch C, Aigner S, Graf M, Iacone R, Freskgård PO. Multicellular self-assembled spheroidal model of the blood brain barrier. Sci Rep. 2013;3:1500. doi: 10.1038/srep01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wager TT, Liras JL, Mente S, Trapa P. Strategies to minimize CNS toxicity: in vitro high-throughput assays and computational modeling. Expert Opin Drug Metab Toxicol. 2012;8:531–42. doi: 10.1517/17425255.2012.677028. [DOI] [PubMed] [Google Scholar]
- 79.Ruck T, Bittner S, Meuth S. Blood-brain barrier modeling: challenges and perspectives. Neural Reg Res. 2015;10:889–91. doi: 10.4103/1673-5374.158342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humpel C. Organotypic brain slice cultures: a review. Neurosci. 2015;305:86–98. doi: 10.1016/j.neuroscience.2015.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell. 2015;60:307–18. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res. 2016;22:1185–96. doi: 10.1158/1078-0432.CCR-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffman RM. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer. 2015;15:451–2. doi: 10.1038/nrc3972. [DOI] [PubMed] [Google Scholar]
- 84.Miyai M, Tomita H, Soeda A, Yano H, Iwama T, Hara A. Current trends in mouse models of glioblastoma. J Neuro Oncol. 2017;135:423–32. doi: 10.1007/s11060-017-2626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oldrini B, Curiel-García Á, Marques C, Matia V, Uluçkan Ö, Graña-Castro O, et al. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nat Commun. 2018;9:1–16. doi: 10.1038/s41467-018-03731-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell. 2016;30:578–94. doi: 10.1016/j.ccell.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Banik A, Brown RE, Bamburg J, Lahiri DK, Khurana D, Friedland RP, et al. Translation of pre-clinical studies into successful clinical trials for alzheimer’s disease: what are the roadblocks and how can they be overcome? J Alzheimer’s Dis. 2015;47:815–43. doi: 10.3233/JAD-150136. [DOI] [PubMed] [Google Scholar]
- 88.Blesa J, Przedborski S. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat. 2014;8:155. doi: 10.3389/fnana.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henderson MX, Cornblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ, et al. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci. 2019;22:1248–57. doi: 10.1038/s41593-019-0457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baratta MG. Glioblastoma is ‘hot’for personalized vaccines. Nat Rev Cancer. 2019;19:129. doi: 10.1038/s41568-019-0118-8. [DOI] [PubMed] [Google Scholar]
- 91.Swartz AM, Shen SH, Salgado MA, Congdon KL, Sanchez-Perez L. Promising vaccines for treating glioblastoma. Expert Opin Biol Ther. 2018;18:1159–70.. doi: 10.1080/14712598.2018.1531846. [DOI] [PubMed] [Google Scholar]
- 92.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–42.. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 93.Rajaraman S, Canjuga D, Ghosh M, Codrea MC, Sieger R, Wedekink F, et al. Measles virus-based treatments trigger a pro-inflammatory cascade and a distinctive immunopeptidome in glioblastoma. Mol Theroncolytics. 2019;12:147–61. doi: 10.1016/j.omto.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol. 2017;18:51. doi: 10.1007/s11864-017-0492-y. [DOI] [PubMed] [Google Scholar]
- 95.Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of.glioblastoma. Nat Rev Neurol. 2015;11:504. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK, et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat Commun. 2017;8:1913. doi: 10.1038/s41467-017-01392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–53. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV. Engineering challenges for brain tumor immunotherapy. Adv Drug Deliv Rev. 2017;114:19–32. doi: 10.1016/j.addr.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115:11109–46. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lou J, Zhang L, Zheng G. Advancing cancer immunotherapies with nanotechnology. Adv Ther. 2019;2:1800128. [Google Scholar]
- 101.Mahjub R, Jatana S, Lee SE, Qin Z, Pauli G, Soleimani M, et al. Recent advances in applying nanotechnologies for cancer immunotherapy. J Control Release. 2018;288:239–63. doi: 10.1016/j.jconrel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 102.Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175–96. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parajuli P, Sloan AE. Dendritic cell-based immunotherapy of malignant gliomas. Cancer Investig. 2004;22:405–16. doi: 10.1081/cnv-200034909. [DOI] [PubMed] [Google Scholar]
- 104.Kateb B, Van Handel M, Zhang L, Bronikowski MJ, Manohara H, Badie B. Internalization of MWCNTs by microglia: possible application in immunotherapy of brain tumors. NeuroImage. 2007;37:S9–S17. doi: 10.1016/j.neuroimage.2007.03.078. [DOI] [PubMed] [Google Scholar]
- 105.Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Nanoparticle delivery systems in cancer vaccines. Pharmacol Res. 2011;28:215–36. doi: 10.1007/s11095-010-0241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen Q, Yang J, Liu R, Liu L, Zhang J, Shen S, et al. Hybrid ‘clusterbombs’ as multifunctional nanoplatforms potentiate brain tumor immunotherapy. Mater Horiz. 2019;6:810–6. [Google Scholar]
- 107.Liu H, Chen L, Liu J, Meng H, Zhang R, Ma L, et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017;411:182–90. doi: 10.1016/j.canlet.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 108.Qiao C, Yang J, Shen Q, Liu R, Li Y, Shi Y, et al. Traceable nanoparticles with dual targeting and ROS response for RNAi-based immunochemotherapy of intracranial glioblastoma treatment. Adv Mater. 2018;30:1705054. doi: 10.1002/adma.201705054. [DOI] [PubMed] [Google Scholar]
- 109.Sun T, Patil R, Galstyan A, Klymyshyn D, Ding H, Chesnokova A, et al. Blockade of a laminin-411–notch axis with CRISPR/Cas9 or a nanobioconjugate inhibits glioblastoma growth through tumor-microenvironment cross-talk. Cancer Res. 2019;79:1239–51. doi: 10.1158/0008-5472.CAN-18-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peng Y, Huang J, Xiao H, Wu T, Shuai X. Codelivery of temozolomide and siRNA with polymeric nanocarrier for effective glioma treatment. Int J Nanomed. 2018;13:3467. doi: 10.2147/IJN.S164611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R, et al. Blood–brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun. 2019;10:1–13. doi: 10.1038/s41467-019-11719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang F, Stephan SB, Ene CI, Smith TT, Holland EC, Stephan MT. Nanoparticles that reshape the tumor milieu create a therapeutic window for effective t-cell therapy in solid malignancies. Cancer Res. 2018;78:3718–30. doi: 10.1158/0008-5472.CAN-18-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi Y, Jiang Y, Cao J, Yang W, Zhang J, Meng F, et al. Boosting RNAi therapy for orthotopic glioblastoma with nontoxic brain-targeting chimaeric polymersomes. J Control Release. 2018;292:163–71. doi: 10.1016/j.jconrel.2018.10.034. [DOI] [PubMed] [Google Scholar]
- 114.Lollo G, Vincent M, Ullio-Gamboa G, Lemaire L, Franconi F, Couez D, et al. Development of multifunctional lipid nanocapsules for the co-delivery of paclitaxel and CpG-ODN in the treatment of glioblastoma. Int J Pharm. 2015;495:972–80. doi: 10.1016/j.ijpharm.2015.09.062. [DOI] [PubMed] [Google Scholar]
- 115.Badie B, Berlin JM. The future of CpG immunotherapy in cancer. Immunotherapy. 2013;5:1–3. doi: 10.2217/imt.12.148. [DOI] [PubMed] [Google Scholar]
- 116.Bastiancich C, Bozzato E, Luyten U, Danhier F, Bastiat G, Préat V. Drug combination using an injectable nanomedicine hydrogel for glioblastoma treatment. Int J Pharm. 2019;559:220–7. doi: 10.1016/j.ijpharm.2019.01.042. [DOI] [PubMed] [Google Scholar]
- 117.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Löb S, Königsrainer A, Rammensee H-G, Opelz G, Terness P. Inhibitors of indoleamine-2, 3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. 2009;9:445. doi: 10.1038/nrc2639. [DOI] [PubMed] [Google Scholar]
- 119.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon K-S, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18:6110–21. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2, 3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 121.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The indoleamine 2, 3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer. 2014;2:21. doi: 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 123.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2, 3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 124.Kuang J, Song W, Yin J, Zeng X, Han S, Zhao YP, et al. iRGD modified chemo‐immunotherapeutic nanoparticles for enhanced immunotherapy against glioblastoma. Adv Funct Mater. 2018;28:1800025. [Google Scholar]
- 125.Kadiyala P, Li D, Nuñez FM, Altshuler D, Doherty R, Kuai R, et al. High-density lipoprotein-mimicking nanodiscs for chemo-immunotherapy against glioblastoma multiforme. ACS Nano. 2019;13:1365–84. doi: 10.1021/acsnano.8b06842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Verschuere T, Van Woensel M, Fieuws S, Lefranc F, Mathieu V, Kiss R, et al. Altered galectin-1 serum levels in patients diagnosed with high-grade glioma. J Neuro Oncol. 2013;115:9–17. doi: 10.1007/s11060-013-1201-8. [DOI] [PubMed] [Google Scholar]
- 127.Toussaint LG, Nilson AE, Goble JM, Ballman KV, James CD, Lefranc F, et al. Galectin-1, a gene preferentially expressed at the tumor margin, promotes glioblastoma cell invasion. Mol Cancer. 2012;11:32. doi: 10.1186/1476-4598-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Welsh JW, Seyedin SN, Cortez MA, Maity A, Hahn SM. Galectin-1 and immune suppression during radiotherapy. Clin Cancer Res. 2014;20:6230–2. doi: 10.1158/1078-0432.CCR-14-2702. [DOI] [PubMed] [Google Scholar]
- 129.Van Woensel M, Mathivet T, Wauthoz N, Rosière R, Garg AD, Agostinis P, et al. Sensitization of glioblastoma tumor micro-environment to chemo-and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci Rep. 2017;7:1217. doi: 10.1038/s41598-017-01279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qian W, Qian M, Wang Y, Huang J, Chen J, Ni L, et al. Combination glioma therapy mediated by a dual-targeted delivery system constructed using OMCN–PEG–Pep22/DOX. Small. 2018;14:1801905. doi: 10.1002/smll.201801905. [DOI] [PubMed] [Google Scholar]
- 131.Li F, Cheng Y, Lu J, Hu R, Wan Q, Feng H. Photodynamic therapy boosts anti‐glioma immunity in mice: a dependence on the activities of T cells and complement C3. J Cell Biochem. 2011;112:3035–43. doi: 10.1002/jcb.23228. [DOI] [PubMed] [Google Scholar]
- 132.Burley TA, Mączyńska J, Shah A, Szopa W, Harrington KJ, Boult JK, et al. Near-infrared photoimmunotherapy targeting EGFR—shedding new light on glioblastoma treatment. Int J Cancer. 2018;142:2363–74.. doi: 10.1002/ijc.31246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685. doi: 10.1038/nm.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ogawa M, Tomita Y, Nakamura Y, Lee MJ, Lee S, Tomita S, et al. Immunogenic cancer cell death selectively induced by near infrared photoimmunotherapy initiates host tumor immunity. Oncotarget. 2017;8:10425. doi: 10.18632/oncotarget.14425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qian M, Chen L, Du Y, Jiang H, Huo T, Yang Y, et al. Biodegradable mesoporous silica achieved via carbon nanodots-incorporated framework swelling for debris-mediated photothermal synergistic immunotherapy. Nano Lett. 2019;19:8409–17. doi: 10.1021/acs.nanolett.9b02448. [DOI] [PubMed] [Google Scholar]
- 136.Ogata F, Nagaya T, Nakamura Y, Sato K, Okuyama S, Maruoka Y, et al. Near-infrared photoimmunotherapy: a comparison of light dosing schedules. Oncotarget. 2017;8:35069. doi: 10.18632/oncotarget.17047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nagaya T, Okuyama S, Ogata F, Maruoka Y, Knapp DW, Karagiannis SN, et al. Near infrared photoimmunotherapy targeting bladder cancer with a canine anti-epidermal growth factor receptor (EGFR) antibody. Oncotarget. 2018;9:19026. doi: 10.18632/oncotarget.24876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jing H, Weidensteiner C, Reichardt W, Gaedicke S, Zhu X, Grosu AL, et al. Imaging and selective elimination of glioblastoma stem cells with theranostic near-infrared-labeled CD133-specific antibodies. Theranostics. 2016;6:862. doi: 10.7150/thno.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA. 1990;87:4207–11. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang HC, Pigula M, Fang Y, Hasan T. Immobilization of photo-immunoconjugates on nanoparticles leads to enhanced light-activated biological effects. Small. 2018;14:1800236. doi: 10.1002/smll.201800236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Inglut CT, Baglo Y, Liang BJ, Cheema Y, Stabile J, Woodworth GF, et al. Systematic evaluation of light-activatable biohybrids for anti-Glioma photodynamic therapy. J Clin Med. 2019;8:1269. doi: 10.3390/jcm8091269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rosenberg SA. Immunotherapy and gene therapy of cancer. Cancer Res. 1991;51:5074s–9s. [PubMed] [Google Scholar]
- 143.Chou S-T, Patil R, Galstyan A, Gangalum PR, Cavenee WK, Furnari FB, et al. Simultaneous blockade of interacting CK2 and EGFR pathways by tumor-targeting nanobioconjugates increases therapeutic efficacy against glioblastoma multiforme. J Control Release. 2016;244:14–23. doi: 10.1016/j.jconrel.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kozielski KL, Ruiz-Valls A, Tzeng SY, Guerrero-Cázares H, Rui Y, Li Y, et al. Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials. 2019;209:79–87. doi: 10.1016/j.biomaterials.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang S, Reinhard S, Li C, Qian M, Jiang H, Du Y, et al. Antitumoral cascade-targeting ligand for IL-6 receptor-mediated gene delivery to glioma. Mol Ther. 2017;25:1556–66.. doi: 10.1016/j.ymthe.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang S, Huang R. Non-viral nucleic acid delivery to the central nervous system and brain tumors. J Gene Med. 2019;21:e3091. doi: 10.1002/jgm.3091. [DOI] [PubMed] [Google Scholar]
- 147.Kamran N, Calinescu A, Candolfi M, Chandran M, Mineharu Y, Asad AS, et al. Recent advances and future of immunotherapy for glioblastoma. Expert Opin Biol Ther. 2016;16:1245–64. doi: 10.1080/14712598.2016.1212012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Westphal M, Ylä-Herttuala S, Martin J, Warnke P, Menei P, Eckland D, et al. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–33. doi: 10.1016/S1470-2045(13)70274-2. [DOI] [PubMed] [Google Scholar]
- 149.Lozada-Delgado EL, Grafals-Ruiz N, Vivas-Mejía PE. RNA interference for glioblastoma therapy: Innovation ladder from the bench to clinical trials. Life Sci. 2017;188:26–36. doi: 10.1016/j.lfs.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Candolfi M, Kroeger KM, Muhammad A, Yagiz K, Farrokhi C, Pechnick RN, et al. Gene therapy for brain cancer: combination therapies provide enhanced efficacy and safety. Curr Gene Ther. 2009;9:409–21. doi: 10.2174/156652309789753301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim SS, Harford JB, Moghe M, Slaughter T, Doherty C, Chang EH. A tumor-targeting nanomedicine carrying the p53 gene crosses the blood-brain barrier and enhances anti-PD-1 immunotherapy in mouse models of glioblastoma. Int J Cancer. 2019;145:2535–46. doi: 10.1002/ijc.32531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Speranza M-C, Passaro C, Ricklefs F, Kasai K, Klein SR, Nakashima H, et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 2017;20:225–35.. doi: 10.1093/neuonc/nox139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bhaskaran V, Nowicki MO, Idriss M, Jimenez MA, Lugli G, Hayes JL, et al. The functional synergism of microRNA clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat Commun. 2019;10:442. doi: 10.1038/s41467-019-08390-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hajj KA, Whitehead KA. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat Rev Mater. 2017;2:17056. [Google Scholar]
- 155.Schneider T, Becker A, Ringe K, Reinhold A, Firsching R, Sabel BA. Brain tumor therapy by combined vaccination and antisense oligonucleotide delivery with nanoparticles. J Neuroimmunol. 2008;195:21–7. doi: 10.1016/j.jneuroim.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 156.Erel-Akbaba G, Carvalho LA, Tian T, Zinter M, Akbaba H, Obeid PJ, et al. Radiation-induced targeted nanoparticle-based gene delivery for brain tumor therapy. ACS Nano. 2019;13:4028–40.. doi: 10.1021/acsnano.8b08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Blennow K, Hampel H, Zetterberg H. Biomarkers in amyloid-β immunotherapy trials in Alzheimer’s disease. Neuropsychopharmacology. 2014;39:189. doi: 10.1038/npp.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Riemenschneider M, Wagenpfeil S, Vanderstichele H, Otto M, Wiltfang J, Kretzschmar H, et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt–Jakob disease from other dementias. Mol Psychiatry. 2003;8:343. doi: 10.1038/sj.mp.4001220. [DOI] [PubMed] [Google Scholar]
- 160.Foidl BM, Humpel C. Can mouse models mimic sporadic Alzheimer’s disease? Neural Reg Res. 2020;15:401. doi: 10.4103/1673-5374.266046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Andrieux K, Couvreur P. Nanomedicine as a promising approach for the treatment and diagnosis of brain diseases: the example of Alzheimer’s disease. Ann Pharm Fr. 2013;71:225–33. doi: 10.1016/j.pharma.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 162.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–70. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 163.Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–53. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 164.Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50:397–409. doi: 10.1016/j.phrs.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 165.McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobio Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 166.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 167.Gu H, Dodel R, Farlow M, Du Y. Advances in the development of antibody-based immunotherapy against prion disease. Antib Technol J. 2014;4:45. [Google Scholar]
- 168.Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer’s disease. Alzheimer’s Res Ther. 2014;6:7. doi: 10.1186/alzrt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jadhav S, Avila J, Schöll M, Kovacs GG, Kövari E, Skrabana R, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun. 2019;7:22. doi: 10.1186/s40478-019-0664-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 171.Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6:108–19. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wisniewski T, Goñi F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85:1162–76. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Akiyama H, McGeer PL. Specificity of mechanisms for plaque removal after Aβ immunotherapy for Alzheimer disease. Nat Med. 2004;10:117. doi: 10.1038/nm0204-117. [DOI] [PubMed] [Google Scholar]
- 174.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 175.Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde M, McConlogue L, et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 176.Wyss-Coray T, Yan F, Lin AHT, Lambris JD, Alexander JJ, Quigg RJ, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA. 2002;99:10837–42. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bussini S, Meda L, Scarpini E, Clementi E, Conti G, Tiriticco M, et al. Heparan sulfate proteoglycan induces the production of NO and TNF-α by murine microglia. Immun Ageing. 2005;2:11. doi: 10.1186/1742-4933-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Miners JS, Barua N, Kehoe PG, Gill S, Love S. Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70:944–59. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- 179.Townsend M, Mehta T, Selkoe DJ. Soluble Aβ inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–12. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- 180.Nalivaeva NN, Beckett C, Belyaev ND, Turner AJ. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J Neurochem. 2012;120:167–85. doi: 10.1111/j.1471-4159.2011.07510.x. [DOI] [PubMed] [Google Scholar]
- 181.Murphy GM, Jr, Eng LF, Ellis WG, Perry G, Meissner LC, Tinklenberg JR. Antigenic profile of plaques and neurofibrillary tangles in the amygdala in Down’s syndrome: a comparison with Alzheimer’s disease. Brain Res. 1990;537:102–8. doi: 10.1016/0006-8993(90)90345-c. [DOI] [PubMed] [Google Scholar]
- 182.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 183.Ribé EM, Pérez M, Puig B, Gich I, Lim F, Cuadrado M, et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis. 2005;20:814–22. doi: 10.1016/j.nbd.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 184.Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett AD, Dineley K, et al. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J Nanoneurosci. 2015;35:4857–68. doi: 10.1523/JNEUROSCI.4989-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Liu R, Yang J, Liu L, Lu Z, Shi Z, Ji W, et al. An “Amyloid‐β cleaner” for the treatment of Alzheimer’s disease by normalizing microglial dysfunction. Adv Sci. 2020;7:1901555. doi: 10.1002/advs.201901555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Albert M, Mairet-Coello G, Danis C, Lieger S, Caillierez R, Carrier S, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain. 2019;142:1736–50. doi: 10.1093/brain/awz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.De Genst E, Messer A, Dobson CM. Antibodies and protein misfolding: from structural research tools to therapeutic strategies. Biochim Biophys Acta. 2014;1844:1907–19. doi: 10.1016/j.bbapap.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 188.Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, et al. Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42:77–94. doi: 10.1111/nan.12297. [DOI] [PubMed] [Google Scholar]
- 189.Halliday GM, Ophof A, Broe M, Jensen PH, Kettle E, Fedorow H, et al. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain. 2005;128:2654–64. doi: 10.1093/brain/awh584. [DOI] [PubMed] [Google Scholar]
- 190.Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–66. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein—a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10:92. doi: 10.1038/nrneurol.2013.275. [DOI] [PubMed] [Google Scholar]
- 192.Zhang G, Wang T, Xia Y, Wan F, Ma K, Guo XF, et al. New perspectives on roles of alpha-synuclein in Parkinson’s disease. Front Aging Neurosci. 2018;10:370. doi: 10.3389/fnagi.2018.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29:1690–701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lotankar S, Prabhavalkar KS, Bhatt LK. Biomarkers for Parkinson’s disease: recent advancement. Neurosci Bull. 2017;33:585–97. doi: 10.1007/s12264-017-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhao L, Wang Z. MicroRNAs: Game changers in the regulation of α-synuclein in Parkinson’s disease. Parkinson’s Dis. 2019;2019:1743183. doi: 10.1155/2019/1743183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurol. 2013;14:38. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Valera E, Masliah E. Therapeutic approaches in Parkinson’s disease and related disorders. J Neurochem. 2016;139:346–52. doi: 10.1111/jnc.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pujols J, Peña-Díaz S, Lázaro DF, Peccati F, Pinheiro F, González D, et al. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc Natl Acad Sci USA. 2018;115:10481–6. doi: 10.1073/pnas.1804198115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathologica. 2014;127:861–79. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10. doi: 10.1186/s13024-015-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 202.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, et al. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–69. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, et al. Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE. 2013;8:e79160. doi: 10.1371/journal.pone.0079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Aderibigbe B, Naki T, Owonubi S. Graphene-based materials for brain targeting. In: Handbook of graphene: biomaterials. Beverly, Massachuset: Scrivener Publishing LLC; 2019. p. 225.
- 205.Kim D, Yoo JM, Hwang H, Lee J, Lee SH, Yun SP, et al. Graphene quantum dots prevent α-synucleinopathy in Parkinson’s disease. Nat Nanotechnol. 2018;13:812–8. doi: 10.1038/s41565-018-0179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ghosh D, Nandi S, Bhattacharjee S. Combination therapy to checkmate glioblastoma: clinical challenges and advances. Clin Transl Med. 2018;7:33. doi: 10.1186/s40169-018-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Field KM, Simes J, Nowak AK, Cher L, Wheeler H, Hovey EJ, et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro Oncol. 2015;17:1504–13. doi: 10.1093/neuonc/nov104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu Y-H, Giunta B, Zhou H-D, Tan J, Wang Y-J. Immunotherapy for Alzheimer disease—the challenge of adverse effects. Nat Rev Neurol. 2012;8:465. doi: 10.1038/nrneurol.2012.118. [DOI] [PubMed] [Google Scholar]
- 209.Haanen J, Carbonnel F, Robert C, Kerr K, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28:iv119–42. doi: 10.1093/annonc/mdx225. [DOI] [PubMed] [Google Scholar]
- 210.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28:773–84. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Szabo E, Schneider H, Seystahl K, Rushing EJ, Herting F, Weidner KM, et al. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro Oncol. 2016;18:1242–52. doi: 10.1093/neuonc/now043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sharpe MA, Livingston AD, Gist TL, Ghosh P, Han J, Baskin DS. Successful treatment of intracranial glioblastoma xenografts with a monoamine oxidase B-activated pro-drug. EBioMedicine. 2015;2:1122–32. doi: 10.1016/j.ebiom.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108:djv375. doi: 10.1093/jnci/djv375. [DOI] [PubMed] [Google Scholar]
- 214.Garros-Regulez L, Aldaz P, Arrizabalaga O, Moncho-Amor V, Carrasco-Garcia E, Manterola L, et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets. 2016;20:393–405. doi: 10.1517/14728222.2016.1151002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013;33:6245–56. doi: 10.1523/JNEUROSCI.3672-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Elfenbein H, Rosen R, Stephens S, Switzer R, Smith Y, Pare J, et al. Cerebral ß-amyloid angiopathy in aged squirrel monkeys. Histol Histopathol. 2007;22:155–67. doi: 10.14670/HH-22.155. [DOI] [PubMed] [Google Scholar]
- 218.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimer’s Dement. 2015;11:700–9. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Boutajangout A, Wisniewski T. Tau-based therapeutic approaches for Alzheimer’s disease-a mini-review. Gerontology. 2014;60:381–5. doi: 10.1159/000358875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 222.Schober A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318:215–24. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- 223.Fleming SM, Fernagut PO, Chesselet MF. Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx. 2005;2:495–503. doi: 10.1602/neurorx.2.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cannon JR, Geghman KD, Tapias V, Sew T, Dail MK, Li C, et al. Expression of human E46K-mutated α-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp Neurol. 2013;240:44–56. doi: 10.1016/j.expneurol.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Volpicelli‐Daley LA, Kirik D, Stoyka LE, Standaert DG, Harms AS. How can rAAV‐α‐synuclein and the fibril l‐synuclein models advance our understanding of Parkinson’s disease? J Neurochem. 2016;139:131–55.. doi: 10.1111/jnc.13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger α‐synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 227.Setsuie R, Wang YL, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem Int. 2007;50:119–29. doi: 10.1016/j.neuint.2006.07.015. [DOI] [PubMed] [Google Scholar]