

Abstract
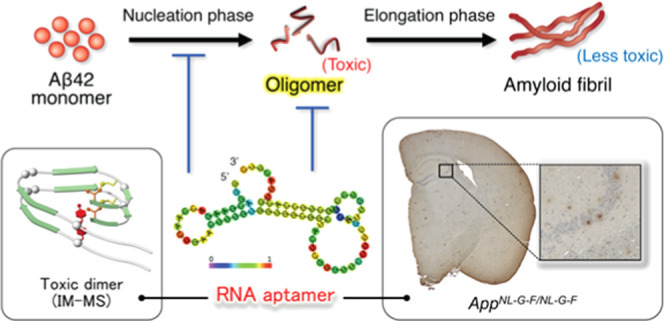
RNA aptamers have garnered attention for diagnostic applications due to their ability to recognize diverse targets. Oligomers of 42-mer amyloid β-protein (Aβ42), whose accumulation is relevant to the pathology of Alzheimer’s disease (AD), are among the most difficult molecules for aptamer recognition because they are prone to aggregate in heterogeneous forms. In addition to designing haptens for in vitro selection of aptamers, the difficulties involved in determining their effect on Aβ42 oligomerization impede aptamer research. We previously developed three RNA aptamers (E22P-AbD4, -AbD31, and -AbD43) with high affinity for protofibrils (PFs) derived from a toxic Aβ42 dimer. Notably, these aptamers recognized diffuse staining, which likely originated from PFs or higher-order oligomers with curvilinear structures in a knock-in AppNL-G-F/NL-G-F mouse, carrying the Arctic mutation that preferentially induced the formation of PFs, in addition to a PS2Tg2576 mouse. To determine which oligomeric sizes were mainly altered by the aptamer, ion mobility–mass spectrometry (IM–MS) was carried out. One aptamer, E22P-AbD43, formed adducts with the Aβ42 monomer and dimer, leading to suppression of further oligomerization. These findings support the utility of these aptamers as diagnostics for AD.
1. Introduction
Nucleic acid aptamers are molecular recognition tools for variable targets and offer advantages over antibodies with respect to diversity, production costs, and the need for animal experiments, among other reasons.1 However, difficulties associated with in vitro selection, characterization, and validation have hampered further research into aptamer development. In particular, the design of hapten molecules depends on empirical rules governing several parameters, such as the separation methods and initial template design.2 Metastable or aggregative molecules are considered among the most difficult molecules to generate aptamers exemplified amyloidogenic proteins such as 40-mer amyloid β-protein (Aβ40) fibrils,3−5 Aβ40 oligomers,6,7 transmissible prion,8,9 β-2 microglobulin fibrils,10 and the oligomers derived from the 42-mer amyloid β-protein (Aβ42).11
Aβ40 and Aβ42, whose accumulation is relevant to the pathology of Alzheimer’s disease (AD) based on long careful consideration,12,13 are generated from the Aβ precursor protein (APP).14,15 Aβ42 and Aβ40 aggregates are neurotoxic. The tendency of Aβ42 to aggregate and exhibit neurotoxicity is higher than that of Aβ40.16 Based on accumulated knowledge, metastable Aβ42 oligomers play a critical role in neuronal death and cognitive dysfunction.17,18 The RNA aptamers (Figure 1A: E22P-AbD4, -AbD31, and -AbD43) developed by Murakami and colleagues11 recognize protofibrils (PFs), which possess a curvilinear structure.19 E22P-Aβ42, which resembles the toxic conformer of Aβ42, was identified by Irie and colleagues.20 E22P-Aβ42 has demonstrated greater neurotoxicity than wild-type Aβ42 as it can form a turn structure at Glu22 and Asp23 (Figure 1A).21−23 E22P-V40DAP-Aβ42 dimer (Figure 1A) with a covalent linker at Val40 in the C-terminal hydrophobic core, which plays an important role in oligomer formation,24 can form PFs following incubation.25
Figure 1.

RNA aptamers targeting PFs. (A) The toxic conformer of Aβ42 forms a turn at Glu22 and Asp23, and the E22P mutation can enhance the ratio of the toxic conformer. E22P-V40DAP-Aβ42 dimer has the l,l-2,6-diaminopimeric acid (DAP) covalent linker at Val40, forming protofibrils (PFs) upon incubation. (B) Sequences of the aptamers (E22P-AbD4, -AbD31, and -AbD43) and oligonucleotides (SPB1 and TBA) used in this study. Bold letters indicate random regions of the aptamers, which were selected in the systematic evolution of ligands by an exponential enrichment (SELEX) procedure.
One aptamer, E22P-AbD43, delayed the nucleation phase of Aβ42 estimated by the thioflavin-S fluorescence test and suppressed its associated neurotoxicity toward SH-SY5Y human neuroblastoma cells.11 However, there was no information concerning the oligomer size to be targeted by the aptamer. Ion mobility–mass spectrometry (IM–MS) combined with native ionization techniques have enabled us to identify the distribution of the Aβ42 oligomer under nearly native conditions.26,27 The present study illustrates the assessment of antioligomer RNA aptamers by IM–MS. Based on the immunostaining results of the diffuse staining originating from PFs by E22P-AbD43 in a transgenic mouse model of AD (PS2Tg2576),11 further validation of the RNA aptamers was performed using not only PS2Tg2576 but also an APP knock-in AppNL-G-F/NL-G-F mouse model developed by Saido and colleagues.28 AppNL-G-F/NL-G-F mice possess the Arctic mutation that is prone to induce PF formation from Aβ.29,30
2. Results and Discussion
2.1. Analysis of RNA Aptamer Sequence
Based on previous CD and FT-IR studies of E22P-AbD43, the formation of a G-quadruplex structure may influence their binding to PFs.11 The three aptamers (E22P-AbD4, -AbD31, and -AbD43) showed a slightly higher guanine content than the average (25%) (Figure 1B). Consecutive guanine residues in the G-quadruplex are held in a square G-tetrad planar via Hoogsteen hydrogen bonding.31 The G-score deduced from quadruplex-forming G-rich sequences (QGRS) Mapper32 supported the possible existence of the G-quadruplex in E22P-AbD4 (highest G-score = 17) and E22P-AbD31 (highest G-score = 16) (Figure 2A). These values of E22P-AbD4 and -AbD31 are nearly comparable to SPB133 (highest G-score = 21) and TBA33 (highest G-score = 20) as typical examples of DNA oligonucleotide forming G-quadruplex (Figure 2B). These indicate the preferable formation of the G-quadruplex in E22P-AbD4 and E22P-AbD31. Further analysis of E22P-AbD43 for the potential formation of G-quadruplex such as NMR will be needed because of unsuccessful calculation of the G-score (data not shown).
Figure 2.

Sequences forming a G-quadruplex structure of (A) the aptamers (E22P-AbD4, -AbD31, and -AbD43) and (B) oligonucleotides (SPB1 and TBA) with a high G-score, indicating potential probability based on calculation from QGRS Mapper. The sequences that start from GG are shown. The G-score indicates the likelihood of forming a stable G-quadruplex (see Section 4). Underlines indicate consecutive guanine sequences. In (A), the putative sequence of E22P-AbD43 was not obtained.
2.2. RNA Aptamers Recognize PFs in AD Models of APP Transgenic Mouse and APP Knock-In Mouse
In addition to E22P-AbD43 observed in the previous study,11 we newly performed histochemical analysis for E22P-AbD4 and E22P-AbD31 using a transgenic mouse model for AD (PS2Tg2576), carrying the human wild-type Aβ sequence.34 Both E22P-AbD4 and E22P-AbD31 recognized diffuse staining of Aβ mainly in the cerebral cortex and hippocampus regions (Figure 3A) like E22P-AbD43.11 The immunoreactivity of E22P-AbD31 was slightly stronger than that of E22P-AbD4. By counting the numbers of aggregates, we found that the diffuse staining in both the cerebral cortex and hippocampus were exclusively recognized by both the aptamers (Figure 3B). As shown in Figure S1A (Supporting Information), senile plaques were observed in PS2Tg2576 by an anti-Aβ-N-terminus antibody (82E1)35 similarly to the previous reports.11,34
Figure 3.

Histochemical analysis of PS2Tg2576 mouse brains using RNA aptamers. (A) Representative micrographs were obtained after treatment with E22P-AbD4 and -AbD31 (400 nM). High-magnification images (scale bar = 50 μm) of the area (scale bar = 500 μm) inside the rectangles of the hippocampus are shown within each picture. Arrowheads indicate diffuse staining. (B) Comparison of the numbers of diffuse staining and senile plaques stained in three parts in the cortex and hippocampus of (A), respectively.
The diffuse staining, which was less fuzzy compared with “diffuse plaques”,36 detected by these aptamers may have originated from PFs or higher-order oligomers with curvilinear structures derived from Aβ. To further verify reactivity toward Aβ oligomers by the aptamers, a 4-month-old knock-in mouse (AppNL-G-F/NL-G-F) that harbors the APP-related Swedish and Beyreuther/Iberian mutations with the Arctic mutation within the Aβ gene sequence was used. Saido and colleagues reported that 4-month-old AppNL-G-F/NL-G-F mice exhibited subcortical amyloidosis28 and that these phenotypes are consistent with the pathology of human Arctic mutation carriers.37 As shown in Figure 4A, the diffuse staining was immunostained by all of the aptamers. Counting the numbers of aggregates also confirmed the detection of PF-related diffuse staining both in the area of the cortex and in hippocampus by the aptamers including E22P-AbD43 (Figure 4B). The shape of the diffuse staining appeared to be similar to observations of PS2Tg2576 (Figure 3A). In contrast, there were almost no aggregates with a dense core probed by 82E1 in AppNL-G-F/NL-G-F (Figure S1B). This result does not largely contradict the report by Latif-Hernandez et al., who reported minor staining by the 6E10 monoclonal antibody specific for Aβ1-16.38 82E1 reacted with diffuse staining in AppNL-G-F/NL-G-F like the aptamers, possibly because the N-termini of the PFs were exposed.
Figure 4.

Histochemical analysis of AppNL-G-F/NL-G-F mouse brains using RNA aptamers. (A) Representative micrographs were obtained after treatment with E22P-AbD4, -AbD31, and -AbD43 (400 nM). High-magnification images (scale bar = 50 μm) of the area (scale bar = 500 μm) inside the rectangles of the hippocampus are shown within each picture. Arrowheads indicate diffuse staining. (B) Comparison of the numbers of diffuse staining and senile plaques stained in three parts in the cortex and hippocampus of (A), respectively.
2.3. RNA Aptamer Formed Adducts with Aβ42 Oligomer as Determined by IM–MS Analysis
To characterize the early oligomeric profile of Aβ42 by RNA aptamers, ion mobility–mass spectrometry (IM–MS) was carried out. Avoiding the disruption of noncovalent interactions among Aβ oligomers by not using organic solvents enabled us to observe the near-native status of Aβ oligomers in the presence of aggregation inhibitors.39 After deconvolution based on the observed mass, peaks corresponding to oligomeric orders of Aβ42 and Aβ42-RNA adducts were assigned to the series of multivalent ions depending on their drift time (Table S1). n denotes an integer corresponding to the number of units coexisting in the solution [n = 1, 2, 3, ... denotes monomer (Mon), dimer (Dim), trimer (Tri), ..., respectively]. The Aβ42 dimer and trimer peaks were apparently found after dissolution with the buffer (Figure 5A). These adducts were not detectable after incubation for 1 h at 37 °C due to further aggregation of Aβ42 (data not shown). In contrast, the addition of E22P-AbD43, whose binding ability to PFs was the strongest among the three aptamers,11 induced the disappearance of these oligomer peaks, and the corresponding adduct peaks of the monomer and dimer with RNA were observed (Figure 5B), meaning that the formation of dimers, which are fundamental subunits of Aβ42 oligomers (n ≥ 3), was suppressed.
Figure 5.
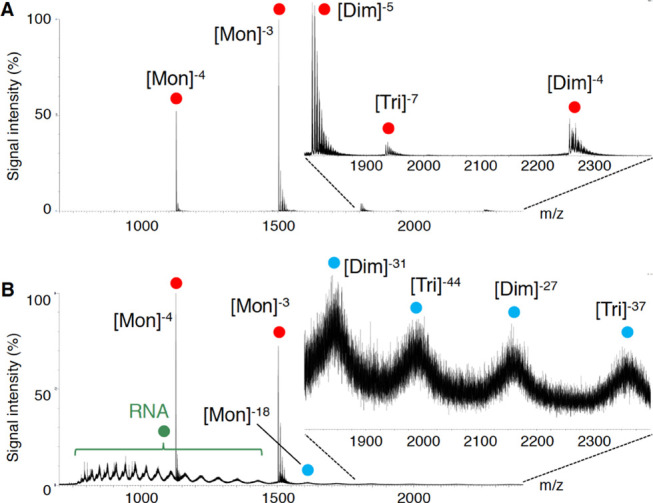
IM–MS analysis of Aβ42 in the absence (A) and presence (B) of E22P-AbD43. NanoESI-TOF-MS of Aβ42 (40 μM) either with or without 80 μM E22P-AbD43. Peaks for Aβ42 alone and for the Aβ42–E22P-AbD43 complex are indicated with red and blue circles, respectively. Green circles indicate RNA clusters.
3. Conclusions
To the best of our knowledge, we are the first to demonstrate the accumulation of diffuse staining deduced from PFs in AppNL-G-F/NL-G-F mouse by anti-Aβ42 oligomer RNA aptamers and that in particular E22P-AbD43 prevented further oligomerization of Aβ42 by directly interfering with the monomer or dimer unit using IM–MS. This interference may be associated with the formation of the G-quadruplex. Further studies will be required to clarify the structural information of RNA for its specific binding to the target and further characterization of affinities to other amyloidogenic proteins.
E22G-Aβ4230 and E22G-Aβ4029 preferably formed PFs in vitro. The levels of PFs reportedly correlated with the impairment of spatial learning in Arctic AD transgenic mice.40 The animal study on Arctic-mutant APP Tg lines suggested that the PF-related oligomeric species (>30-mer) could contain Aβ*56 (ca. 12-mer) as a nonfibrillar Aβ assembly.41 These findings suggest that diffuse staining detected by the aptamers could contain PFs. The age of 4 months in AppNL-G-F/NL-G-F may correspond to the initiation of subtle disease-related behavioral changes.38 Clinical studies have suggested that the accumulation of PFs is a promising biomarker of even mild cognitive impairment before the onset of AD.42 The humanized monoclonal antibody (BAN2401) recognizing PFs is also a promising drug candidate according to a phase 2 randomized trial.43 Recently, the involvement of neuronal membrane damage in the neurotoxicity induced from high-order oligomers of Aβ42 such as PFs was reported.44 Further experiments to shorten the length and to incorporate the modified base into the RNA aptamers for their passage across the blood–brain barrier toward Aβ imaging application are currently underway.
4. Materials and Methods
4.1. Preparation of RNA Aptamers
The aptamers were obtained using an in vitro selection method known as the systematic evolution of ligands by exponential enrichment (SELEX) using a membrane filter methodology, as reported previously.11 Using the ssDNA (Eurofins; Tokyo, Japan) of the RNA aptamer, RiboMAX Large-Scale RNA Production System-T7 (Promega, Madison, WI) was used to generate the RNA transcript. After phenol–chloroform extraction and desalting using Illustra MicroSpin G-25 columns (GE Healthcare), the integrity of RNA was confirmed by electrophoresis using 6% Tris–borate–ethylenediaminetetraacetic acid (EDTA)–urea acrylamide gels (Invitrogen) and stained by SYBR Green (TaKaRa). RNA quantification was performed using a UV BioPhotometer (Eppendorf). The RNA aptamer was denatured at 90 °C for 10 min and renatured rapidly on ice for 10 min for refolding before use in the following studies.
4.2. QRGS Mapping
Prediction of putative quadruplex-forming G-rich sequences (QGRS) in nucleotide sequences was performed using a Web server (http://bioinformatics.ramapo.edu/QGRS/index.php). This is a scoring system that calculates the probability of forming a stable G-quadruplex. The putative G-quadruplexes are identified using the following motif: GxNy1GxNy2GxNy3Gx, in which x = number of guanine tetrads in the G-quadruplex and y1, y2, y3 = length of gaps meaning the length of the loops that connect the guanine tetrads. The G-score is determined based on the following principles: (1) shorter loops are more common than longer loops, (2) G-quadruplexes have loops roughly equal in size, and (3) the greater the number of guanine tetrads, the more stable the G-quadruplex. The highest possible G-score is 105 in the case of 30-nt oligonucleotides, although the computed values depend on the maximum length.32
4.3. Histochemical Staining
All experimental procedures were performed, as previously described,11 in accordance with specified guidelines for the care and use of laboratory animals and were approved by the Animal Care and Use Committee of Chiba University.
Five micrometer thick coronal paraffin-embedded sections were prepared from 4% paraformaldehyde-fixed brain hemispheres of 6-month-old PS2Tg2576 mice34 and 4-month-old AppNL-G-F/NL-G-F knock-in mice.28 After deparaffinization and hydration, the slices were autoclaved at 120 °C for 20 min to allow antigen activation. To inactivate the endogenous peroxidase, brain sections were soaked in methanol with 0.1% H2O2 at room temperature for 30 min. After washing with ice-cold phosphate-buffered saline (PBS) plus potassium (PBS-K; 10 mM sodium phosphate, 140 mM NaCl, pH 7.4) containing 0.02% Tween-20 (PBS-T), blocking was performed in a blocking buffer, PBS-T with 10 μg/mL bovine serum albumin (Nacalai, Kyoto, Japan) and 10 μg/mL yeast tRNA (Nacalai) at room temperature for 60 min. The biotinylated aptamer (400 nM) diluted in RNase-free water (Invitrogen, Carlsbad, CA) with 1 mM EDTA was added at room temperature for 60 min, followed by reaction with horseradish peroxidase (HRP)-conjugated avidin by the VECTASTAIN ABC HRP Kit (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. Alternatively, the sections were treated with 82E1 (1 μg/mL) and diluted with PBS-T at room temperature for 60 min, followed by reaction with the biotinylated secondary antibody for 30 min at room temperature before incubation with HRP-conjugated avidin by the VECTORSTAIN ABC HRP Kit (Vector) for 30 min at room temperature. To visualize the signals, brain sections were treated with 3,3′-diaminobenzidine (Dojindo, Kumamoto, Japan) at 37 °C for 12 h (RNA aptamer) or at room temperature for 12 min (82E1). Nuclei were stained with hematoxylin reagent (Wako). Brain sections were mounted with Permount (FALMA, Tokyo, Japan) after dehydration and soaking in xylene.
4.4. Ion Mobility–Mass Spectrometry (IM–MS)
Aβ42 was dissolved in 0.1% NH4OH to a concentration of 400 μM and RNA was dissolved in nuclease-free water (Promega, Madison, WI) to a concentration of 400 μM. Next, the Aβ42 and RNA solutions were diluted 10-fold and 5-fold, respectively, in 25 mM ammonium acetate (pH 7.4). The resulting solution (40 μM Aβ42, 80 μM RNA) was centrifuged for 4 min at 2000g (4 °C) before infusion into an MS apparatus using a glass capillary (Nanoflow Probe Tip, Waters). Mass spectra and ion mobility experiments were accomplished on a SYNAPT G2-Si HDMS (Waters) using a nanoelectrospray as an ionization source, as reported previously.39 The instrument was operated in negative ion mode with a capillary voltage of 1.0 kV, a sample cone voltage of 10 V, and a source temperature of 50 °C. For the ion mobility measurement, nitrogen gas was used in the ion mobility cell, and the cell pressure was maintained at approximately 2.95 mbar with a wave velocity of 300–1000 m/s and a wave height of 10–40 V. Data acquisition and processing were performed with the MassLynx (V4.1) and DriftScope (V2.8) software supplied with the instrument. The CsI cluster ions were used for the m/z scale as a calibrator.
Acknowledgments
We thank Dr. Takaomi C. Saido and Dr. Takashi Saito at RIKEN Brain Science Institute for providing AppNL-G-F/NL-G-F mice. This study was supported in part by the JSPS KAKENHI, grant numbers 22603006 to K.M., 16H06194 to K.M., and 26221202 to K.I. and K.M.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02134.
Representative micrograph of brain histochemical staining of the PS2Tg2576 mouse and the AppNL-G-F/NL-G-F mouse using anti-N-terminus of the Aβ (82E1) antibody (Figure S1). The list of calculated and observed masses of Aβ42 and Aβ42 treated with E22P-AbD43 in IM–MS measurements (Table S1) (PDF)
Author Present Address
∥ Laboratory of Pharmaceutical Therapy and Neuropharmacology, Faculty of Pharmaceutical Sciences, University of Toyama, Toyama 930-0194, Japan.
Author Present Address
⊥ Aging Stress Response Research Project Team, National Center for Geriatrics and Gerontology, Obu 474-8511, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou J.; Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discovery 2017, 16, 440 10.1038/nrd.2017.86. [DOI] [PubMed] [Google Scholar]
- McKeague M.; McConnell E. M.; Cruz-Toledo J.; Bernard E. D.; Pach A.; Mastronardi E.; Zhang X.; Beking M.; Francis T.; Giamberardino A.; Cabecinha A.; Ruscito A.; Aranda-Rodriguez R.; Dumontier M.; DeRosa M. C. Analysis of in vitro aptamer selection parameters. J. Mol. Evol. 2015, 81, 150–161. 10.1007/s00239-015-9708-6. [DOI] [PubMed] [Google Scholar]
- Ylera F.; Lurz R.; Erdmann V. A.; Furste J. P. Selection of RNA aptamers to the Alzheimerʼs disease amyloid peptide. Biochem. Biophys. Res. Commun. 2002, 290, 1583–1588. 10.1006/bbrc.2002.6354. [DOI] [PubMed] [Google Scholar]
- Takahashi T.; Tada K.; Mihara H. RNA aptamers selected against amyloid β-peptide (Aβ) inhibit the aggregation of Aβ. Mol. Biosyst. 2009, 5, 986–991. 10.1039/b903391b. [DOI] [PubMed] [Google Scholar]
- Rahimi F.; Murakami K.; Summers J. L.; Chen C. H.; Bitan G. RNA aptamers generated against oligomeric Aβ40 recognize common amyloid aptatopes with low specificity but high sensitivity. PLoS One 2009, 4, e7694 10.1371/journal.pone.0007694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukakoshi K.; Abe K.; Sode K.; Ikebukuro K. Selection of DNA aptamers that recognize α-synuclein oligomers using a competitive screening method. Anal. Chem. 2012, 84, 5542–5547. 10.1021/ac300330g. [DOI] [PubMed] [Google Scholar]
- Chakravarthy M.; AlShamaileh H.; Huang H.; Tannenberg R. K.; Chen S.; Worrall S.; Dodd P. R.; Veedu R. N. Development of DNA aptamers targeting low-molecular-weight amyloid-β peptide aggregates in vitro. Chem. Commun. 2018, 54, 4593–4596. 10.1039/C8CC02256A. [DOI] [PubMed] [Google Scholar]
- Weiss S.; Proske D.; Neumann M.; Groschup M. H.; Kretzschmar H. A.; Famulok M.; Winnacker E. L. RNA aptamers specifically interact with the prion protein PrP. J. Virol. 1997, 71, 8790–8797. 10.1128/JVI.71.11.8790-8797.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A.; Kirby L.; Sayer N.; Wellesley R.; Disterer P.; Sylvester I.; Gill A.; Hope J.; James W.; Tahiri-Alaoui A. Characterization of 2’-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Biol. Chem. 2003, 278, 39697–39705. 10.1074/jbc.M305297200. [DOI] [PubMed] [Google Scholar]
- Bunka D. H.; Mantle B. J.; Morten I. J.; Tennent G. A.; Radford S. E.; Stockley P. G. Production and characterization of RNA aptamers specific for amyloid fibril epitopes. J. Biol. Chem. 2007, 282, 34500–34509. 10.1074/jbc.M703679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K.; Obata Y.; Sekikawa A.; Ueda H.; Izuo N.; Awano T.; Takabe K.; Shimizu T.; Irie K. An RNA aptamer with potent affinity for a toxic dimer of amyloid β42 has potential utility for histochemical studies of Alzheimerʼs disease. J. Biol. Chem. 2020, 295, 4870–4880. 10.1074/jbc.RA119.010955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. P.; Clark I. A.; Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimerʼs disease. Acta Neuropathol. Commun. 2014, 2, 135 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D.; Jackson R.; Paul G.; Shi J.; Sabbagh M. Why do trials for Alzheimerʼs disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Invest. Drugs 2017, 26, 735–739. 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G. G.; Wong C. W. Alzheimerʼs disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Simms G.; Weinman N. A.; Multhaup G.; McDonald B. L.; Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 4245–4249. 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimerʼs amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Tsuji M. Protofibrils of Amyloid-β are important targets of a disease-modifying approach for Alzheimerʼs disease. Int. J. Mol. Sci. 2020, 21, 952 10.3390/ijms21030952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Nakayama T.; Ono K.; Itami M.; Takahashi R.; Teplow D. B.; Yamada M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 5835–5840. 10.1073/pnas.1524807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie K. New diagnostic method for Alzheimerʼs disease based on the toxic conformation theory of amyloid β. Biosci., Biotechnol., Biochem. 2020, 84, 1–16. 10.1080/09168451.2019.1667222. [DOI] [PubMed] [Google Scholar]
- Morimoto A.; Irie K.; Murakami K.; Masuda Y.; Ohigashi H.; Nagao M.; Fukuda H.; Shimizu T.; Shirasawa T. Analysis of the secondary structure of β-amyloid (Aβ42) fibrils by systematic proline replacement. J. Biol. Chem. 2004, 279, 52781–52788. 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- Murakami K.; Irie K.; Ohigashi H.; Hara H.; Nagao M.; Shimizu T.; Shirasawa T. Formation and stabilization model of the 42-mer Aβ radical: implications for the long-lasting oxidative stress in Alzheimerʼs disease. J. Am. Chem. Soc. 2005, 127, 15168–15174. 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- Masuda Y.; Uemura S.; Ohashi R.; Nakanishi A.; Takegoshi K.; Shimizu T.; Shirasawa T.; Irie K. Identification of physiological and toxic conformations in Aβ42 aggregates. ChemBioChem 2009, 10, 287–295. 10.1002/cbic.200800411. [DOI] [PubMed] [Google Scholar]
- Irie Y.; Murakami K.; Hanaki M.; Hanaki Y.; Suzuki T.; Monobe Y.; Takai T.; Akagi K. I.; Kawase T.; Hirose K.; Irie K. Synthetic models of quasi-stable amyloid β40 oligomers with significant neurotoxicity. ACS Chem. Neurosci. 2017, 8, 807–816. 10.1021/acschemneuro.6b00390. [DOI] [PubMed] [Google Scholar]
- Murakami K.; Tokuda M.; Suzuki T.; Irie Y.; Hanaki M.; Izuo N.; Monobe Y.; Akagi K.; Ishii R.; Tatebe H.; Tokuda T.; Maeda M.; Kume T.; Shimizu T.; Irie K. Monoclonal antibody with conformational specificity for a toxic conformer of amyloid β42 and its application toward the Alzheimerʼs disease diagnosis. Sci. Rep. 2016, 6, 29038 10.1038/srep29038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J. E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimerʼs disease. Nat. Chem. 2009, 1, 326–331. 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kłoniecki M.; Jablonowska A.; Poznanski J.; Langridge J.; Hughes C.; Campuzano I.; Giles K.; Dadlez M. Ion mobility separation coupled with MS detects two structural states of Alzheimerʼs disease Aβ1-40 peptide oligomers. J. Mol. Biol. 2011, 407, 110–124. 10.1016/j.jmb.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Saito T.; Matsuba Y.; Mihira N.; Takano J.; Nilsson P.; Itohara S.; Iwata N.; Saido T. C. Single App knock-in mouse models of Alzheimerʼs disease. Nat. Neurosci. 2014, 17, 661–663. 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Nilsberth C.; Westlind-Danielsson A.; Eckman C. B.; Condron M. M.; Axelman K.; Forsell C.; Stenh C.; Luthman J.; Teplow D. B.; Younkin S. G.; Naslund J.; Lannfelt L. The ʼArcticʼ APP mutation (E693G) causes Alzheimerʼs disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Hashimoto T.; Wakutani Y.; Urakami K.; Nakashima K.; Condron M. M.; Tsubuki S.; Saido T. C.; Teplow D. B.; Iwatsubo T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. 10.1074/jbc.M608220200. [DOI] [PubMed] [Google Scholar]
- Burge S.; Parkinson G. N.; Hazel P.; Todd A. K.; Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikin O.; DʼAntonio L.; Bagga P. S. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T.; Noda Y.; Ito G.; Maeda M.; Shimizu T. Presenilin-2 mutation causes early amyloid accumulation and memory impairment in a transgenic mouse model of Alzheimerʼs disease. J. Biomed. Biotechnol. 2011, 2011, 617974 10.1155/2011/617974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y.; Mori T.; Maeda M.; Kinoshita N.; Sato K.; Yamaguchi H. Aβ N-terminal-end specific antibody reduced β-amyloid in Alzheimer-model mice. Biochem. Biophys. Res. Commun. 2004, 325, 384–387. 10.1016/j.bbrc.2004.10.039. [DOI] [PubMed] [Google Scholar]
- Shepherd C. E.; Gregory G. C.; Vickers J. C.; Halliday G. M. Novel ʼinflammatory plaqueʼ pathology in presenilin-1 Alzheimerʼs disease. Neuropathol. Appl. Neurobiol. 2005, 31, 503–511. 10.1111/j.1365-2990.2005.00667.x. [DOI] [PubMed] [Google Scholar]
- Kalimo H.; Lalowski M.; Bogdanovic N.; Philipson O.; Bird T. D.; Nochlin D.; Schellenberg G. D.; Brundin R.; Olofsson T.; Soliymani R.; Baumann M.; Wirths O.; Bayer T. A.; Nilsson L. N.; Basun H.; Lannfelt L.; Ingelsson M. The Arctic AβPP mutation leads to Alzheimerʼs disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol. Commun. 2013, 1, 60 10.1186/2051-5960-1-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif-Hernandez A.; Shah D.; Craessaerts K.; Saido T.; Saito T.; De Strooper B.; Van der Linden A.; DʼHooge R. Subtle behavioral changes and increased prefrontal-hippocampal network synchronicity in APP(NL-G-F) mice before prominent plaque deposition. Behav. Brain Res. 2019, 364, 431–441. 10.1016/j.bbr.2017.11.017. [DOI] [PubMed] [Google Scholar]
- Murakami K.; Yoshioka T.; Horii S.; Hanaki M.; Midorikawa S.; Taniwaki S.; Gunji H.; Akagi K. I.; Kawase T.; Hirose K.; Irie K. Role of the carboxy groups of triterpenoids in their inhibition of the nucleation of amyloid β42 required for forming toxic oligomers. Chem. Commun. 2018, 54, 6272–6275. 10.1039/C8CC03230K. [DOI] [PubMed] [Google Scholar]
- Lord A.; Englund H.; Soderberg L.; Tucker S.; Clausen F.; Hillered L.; Gordon M.; Morgan D.; Lannfelt L.; Pettersson F. E.; Nilsson L. N. Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimerʼs disease transgenic mice. FEBS J. 2009, 276, 995–1006. 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng I. H.; Scearce-Levie K.; Legleiter J.; Palop J. J.; Gerstein H.; Bien-Ly N.; Puolivali J.; Lesne S.; Ashe K. H.; Muchowski P. J.; Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007, 282, 23818–23828. 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Lannfelt L.; Moller C.; Basun H.; Osswald G.; Sehlin D.; Satlin A.; Logovinsky V.; Gellerfors P. Perspectives on future Alzheimer therapies: amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimerʼs disease. Alzheimers Res. Ther. 2014, 6, 16. 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbasi J. Promising Results in 18-Month Analysis of Alzheimer Drug Candidate. JAMA 2018, 320, 965. 10.1001/jama.2018.13027. [DOI] [PubMed] [Google Scholar]
- Yasumoto T.; Takamura Y.; Tsuji M.; Watanabe-Nakayama T.; Imamura K.; Inoue H.; Nakamura S.; Inoue T.; Kimura A.; Yano S.; Nishijo H.; Kiuchi Y.; Teplow D. B.; Ono K. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. 10.1096/fj.201900604R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.