

ABSTRACT
Mammalian cells, including neurons, use macroautophagy (here ‘autophagy’) to degrade damaged proteins and organelles, and recycle nutrients in response to starvation and other forms of cell stress. The basic cellular machinery responsible for autophagy is highly conserved from yeast to mammals. However, evidence for specific adaptations to more complex organisms and in highly differentiated cells (e. g. neurons) remains limited. RILP (Rab interacting lysosomal protein) mediates retrograde transport of late endosomes (LEs) in nonneuronal mammalian cells. We have now found that RILP plays additional important, fundamental roles in neuronal autophagosome (AP) transport, and, more surprisingly, in AP biogenesis, and cargo turnover as well. RILP accomplishes these tasks via sequential interactions with key autophagosomal components — ATG5 and LC3 — as well as the microtubule motor protein cytoplasmic dynein (Figure 1A). We found further that RILP expression and behavior are controlled by MTOR kinase, linking RILP to a potentially wide range of physiological and pathophysiological functions.
KEYWORDS: Autophagosome biogenesis, dynein, MTOR regulation, neuronal autophagy, retrograde transport, RILP, sequestosome 1/p62
RILP is an α-helical coiled-coil-containing protein that, through its N-terminal domain, binds cytoplasmic dynein, and through its C-terminal domain, RAB7-GTP (Figure 1A). We have now found [1] three C-terminal LC3-interacting regions (LIRs) near to, but distinct from, the RAB7-binding site. Using high-resolution live imaging in neurons, we found that RILP is recruited to, and transports in a retrograde manner not only LEs, but LC3-positive autophagosomes (APs) as well (Figure 1B). We found the LIRs to be necessary and sufficient for AP binding, whereas the RAB7 site has no role in this function. RILP depletion potently inhibits retrograde axonal transport of both LC3-positive APs and RAB7-positive LEs.
Figure 1.
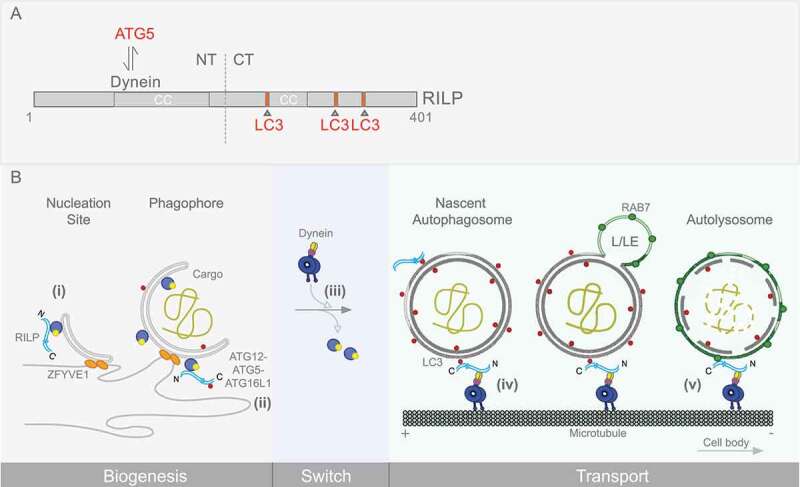
RILP domain organization and sequential roles in the autophagy pathway. (A) RILP domain organization and interactors are shown. The RILP N terminus interacts with cytoplasmic dynein, while the C terminus binds the late endosomal marker, RAB7. Our work also shows RILP to interact with ATG5 on phagophore membranes, and with LC3 on autophagosomes. Three C-terminal LC3-interacting regions (LIRs) were identified, and are depicted. (B) RILP roles at different stages in autophagy progression are shown. Nucleation, expansion of the phagophore membrane and maturation of the autophagosome are depicted. RILP is detected on nascent as well as more fully formed phagophore membranes, associated with ATG5 (i, ii). RILP persists on mature autophagosomes, linked through LC3 (iii, iv). RILP remains associated with AP-LE membranes through LC3 and RAB7 (v), until lysosomal fusion. This pathway is activated by MTOR regulation of RILP expression and distribution, and is required for autophagic clearance.
A particularly surprising finding was that RILP also participates in AP biogenesis. This is suggested by a marked decrease in AP number in cells subjected to RILP RNAi or expression of LIR-mutant RILP. Each treatment also dramatically reduces AP turnover, as evidenced by cytoplasmic accumulation of LC3- and ubiquitin-positive SQSTM1/p62 (sequestosome 1) aggregates.
How RILP might contribute to AP biogenesis was puzzling. However, the decrease in AP number by RILP depletion or LIR mutation is accompanied by a marked increase in the number of phagophores, the incompletely formed precursors of mature APs. These results suggest that reduced AP number might reflect a role for RILP in a key step in phagophore maturation and closure. We found that ATG5 binds RILP directly, through its N-terminal dynein-binding domain (Figure 1B). Furthermore, expression of this region of RILP is, alone, sufficient to block phagophore maturation. The underlying mechanism for this additional new RILP role remains uncertain. We speculate, however, that RILP linkage of ATG5 and LC3 might promote or allow efficient incorporation of LC3 into phagophore membranes, an issue for further investigation. We also found that RILP- and ATG5-positive phagophore membranes in axons are typically devoid of dynein, and are immotile. Recombinant ATG5 can also be displaced from RILP by dynein in in vitro experiments. These results suggest that ATG5 and dynein compete for a common site within the RILP N-terminal domain, a potentially important clue to the physiological purpose of this system (Figure 1B, discussed below).
Finally, we obtained clear evidence that RILP expression is controlled by MTOR kinase, a master regulator of autophagy. In particular, MTOR inhibition using Torin1 increases RILP mRNA and RILP protein levels. RILP recruitment to LC3-positive APs also seems to be enhanced. Together, our results identify a substantial new array of MTOR-dependent autophagosome behaviors uniquely controlled by RILP, and distinguish RILP from other factors such as MAPK8IP1/JIP1 and SNAPIN previously implicated in AP transport.
Our results together indicate that RILP is capable of binding APs from the phagophore stage through maturation to LC3- and RAB7-dually positive amphisomes, apparently persisting from AP formation through to the late degradative stage. Competitive dynein versus ATG5 binding may have important functional consequences. We find dynein absent from ATG5-positive structures. This observation may imply that ATG5 prevents dynein recruitment to phagophore membranes, to preclude premature initiation of transport along microtubules. Only upon phagophore membrane closure would the departure of the ATG5 complex allow RILP to recruit dynein for retrograde transport toward the lysosomal compartment, typically located in the cell soma. This RILP coordination of AP biogenesis with transport differs from, and may be uniquely more efficient, than mechanisms involving constitutive transport of autophagosomes.
An appealing further role for RILP might be in fusion of mature APs with LEs. Although SNARE proteins such as STX17 (syntaxin 17) and SNAP29 have already been implicated in AP-LE fusion, the ability of RILP to bind to both LC3- and RAB7-positive membranes seems to make a RILP role in their coalescence worth examining.
Finally, a RILP role in SQSTM1/p62 clearance has important implications for neurodegenerative diseases including Alzheimer, Parkinson and amyotrophic lateral sclerosis, where a marked accumulation of SQSTM1/p62-tagged ubiquitin aggregates in neurons and glial cells is a major pathological feature. We speculate that targeted ectopic expression of RILP in these diseased neurons might serve to upregulate the autophagy pathway, promoting aggregate clearance. RILP behavior in these diseases is not characterized, and a better understanding of its effects might provide critical therapeutic insights. Finally, the MTOR-regulated RILP-dynein mechanism described here might well account for the effects of starvation on endo-lysosomal distribution reported in earlier studies, suggesting broad relevance for this pathway.
In summary, RILP, through its multiple interactors, appears to play an unusual quasi-catalytic role in the autophagy pathway to coordinate distinct stages and to enhance autophagic efficiency. Further investigation into RILP behavior in vivo may help identify roles for this protein in the causality and treatment of neurological diseases.
Funding Statement
This work was supported by the National Institute of General Medical Sciences [R01GM102347]; National Institutes of Health [P01GM105536-01A1-Reg].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Khobrekar NV, Quintremil S, Dantas TJ, et al. The dynein adaptor RILP controls neuronal autophagosome biogenesis, transport, and clearance. Dev Cell. 2020;53:141–153.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]