

ABSTRACT
The DUSP3 phosphatase regulates cell cycle, proliferation, apoptosis and senescence of different cell types, lately shown as a mediator of DNA repair processes. This work evaluated the impact of DUSP3 loss of function (lof) on DNA repair-proficient fibroblasts (MRC-5), NER-deficient cell lines (XPA and XPC) and translesion DNA synthesis (TLS)-deficient cells (XPV), after UV-radiation stress. The levels of DNA strand breaks, CPDs and 6-4-PPs have accumulated over time in all cells under DUSP3 lof, with a significant increase in NER-deficient lines. The inefficient repair of these lesions increased sub-G1 population of XPA and XPC cells 24 hours after UV treatment, notably marked by DUSP3 lof, which is associated with a reduced cell population in G1, S and G2/M phases. It was also detected an increase in S and G2/M populations of XPV and MRC-5 cells after UV-radiation exposure, which was slightly attenuated by DUSP3 lof due to a discrete increase in sub-G1 cells. The cell cycle progression was accompanied by changes in the levels of the main Cyclins (A1, B1, D1 or E1), CDKs (1, 2, 4 or 6), and the p21 Cip1 inhibitor, in a DUSP3-dependent manner. DUSP3 lof affected the proliferation of MRC-5 and XPA cells, with marked worsening of the XP phenotype after UV radiation. This work highlights the roles of DUSP3 in DNA repair fitness and in the fine control of regulatory proteins of cell cycle, essential mechanisms to maintenance of genomic stability.
KEYWORDS: DUSP3 phosphatase, NER pathway, genomic stability
1. Introduction
Xeroderma Pigmentosum (XP) is a rare genetic disorder that manifests in early childhood and causes hypersensitivity to sunlight. The most affected areas, with a high propensity for developing cancers following exposure to ultraviolet (UV) radiation, are the eyes [1] and skin (non-melanoma and melanoma) [2,3]. XP patients can also develop internal cancers (leukemia and brain cancer of the astrocytoma, medulloblastoma and sarcoma types) as well as severe neurological abnormalities [3]. XP is caused by mutations in the genetic group XPA-XPG, which is involved in the repair of DNA lesioned by UV radiation. XPA-XPG proteins are key components of the nucleotide excision repair (NER) pathway, the major mechanism for repairing lesions caused by UV [2]. Another important XP phenotype is related to mutation in the polh gene that transcribes a polymerase (Pol η) involved in Translesion DNA Synthesis (TLS). Pol η is specialized in the replication of DNA containing UV lesions [4] and incorporates nucleotides opposite the damaged templates, thereby allowing DNA synthesis via classical replicative polymerases with low error rates [5]. The lack of this “tolerance to damage” mechanism causes a deficiency called XP Variant (XPV) [6,7].
The foremost lesions caused by UV are cyclobutane pyrimidine dimers (CPD) and 6–4 pyrimidine–pyrimidinone (6-4-PP) that promote substantial distortions in the DNA helix, both recognized and repaired by the NER pathway. UV radiation activates the DNA damage response (DDR) pathway, which functionality depends on a fine balance between the phosphorylation/dephosphorylation of key proteins involved in recognition of the damage site, and which is necessary for the cell fate [8]. Some phosphatases may directly or indirectly influence this balance and affect DNA repair, such as the protein tyrosine phosphatase (PTP) SHP-1 [9], the serine/threonine phosphatases PP5 [10] and WIP1 [11], and more recently, DUSP3 [12]. DUSP3 or VHR (Vaccinia H1-Related Phosphatase) is a PTP belonging to the dual-specificity phosphatase (DUSP) group that exhibits dual-activity toward Tyr and Thr residues [13]. DUSP3 acts in cellular processes such as cell cycle arrest and senescence, apoptosis, cell adhesion and migration, respectively related to its substrates MAPKs (ERK, JNK, p38), STAT5, ERBB2 and FAK [14–17]. Interestingly, and more recently, DUSP3 was shown to physically interact with the nucleophosmin (NPM), nucleolin (NCL) and heterogeneous nuclear ribonucleoprotein (hnRNP C1/C2) [18] proteins, which play roles related to DNA repair through different pathways. NPM acts in BER, NCL in NHEJ, and hnRNP C1/C2 in HR and NHEJ; however, the three proteins act in NER pathway. Also, another link between these proteins and DNA repair is the p53 protein, a suggestive common pathway for DUSP3 action [19,20], as previously supported by findings of straight DUSP3 co-localization with p-p53 (S15) before and after IR exposure [21].
Considering these non-canonical influences in the maintenance of genome stability, in this work we permanently silenced DUSP3 in normal fibroblasts proficient in DNA repair (MRC-5) and compared them to NER-deficient cells (lacking XPA or XPC proteins, known as XPA or XPC cell lines, respectively), and also to TLS-deficient (but NER-proficient) cells (the XPV cell line). DUSP3-proficient cells were able to handle DNA damage caused by UVC or UVB radiation treatments, escape cell death and proliferate/survive, with clearly different abilities depending on the cell phenotype. However, DUSP3 loss of function (lof) caused inefficient repair of UV-induced lesions, which culminate in cell cycle deregulation and reduced proliferation, and collectively elicited significant negative impacts on the genomic stability of normal cells, as well as for XP lines that already carry a compromised phenotype for the repair of these lesions.
2. Results
To expand the biological functions of DUSP3 phosphatase in distinct DNA repair pathways, we focused on the NER-deficient XPA and XPC lines, and on the TLS-deficient XPV cell line in order to compare them with DNA repair proficient cells, the normal fibroblast MRC-5 line. Therefore, we first promoted DUSP3 lof by silencing the dusp3 gene in these four cell lines by using lentiviral shRNA technology. Three different shRNAs were able to reduce DUSP3 expression between 50% (shRNAs #1 and #3, named shDUSP3−) and approximately 95% (shRNA#2, named shDUSP3–-) (Figure S1A). The non-silencing (NS) shRNA carrying a scramble sequence did not change DUSP3 expression (Figure S1B). These cellular clones were characterized by assessing the expression level and phosphorylation status of the control protein ERK; DUSP3 silencing caused an increase in ERK phosphorylation, as expected for this classical DUSP3 substrate (Figure S1 C). Moreover, DUSP3 silencing caused changes in the morphology and migration of the four cell lines (Figure S1D), according to previously reported in literature [15]. We finished the clonal characterization by confirming that DUSP3 expression levels do not change after UVC or UVB radiation treatments (Figure S2). From now on, the most silenced DUSP3 clone (shDUSP3–-, hereinafter referred only as shDUSP3) will be used in all experiments of this work.
2.1. DUSP3 silencing affects proliferation and cell cycle progression of NER-proficient and NER-deficient cells changing levels of cell cycle regulatory proteins after UV stress
We measure the proliferative capacity of the MRC-5, XPA, XPC and XPV cells, proficient or deficient in DUSP3 phosphatase, by evaluating their proliferation in consecutive days after exposure to UVC radiation (Figure 1a). The growth curves showed that all DUSP3-proficient cell lines proliferate similarly for 7 d, except for MRC-5 and XPA cells with DUSP3 lof which exhibited a slightly reduced proliferation compared to XPC and XPV cells within 1, 3 and 7 consecutive d. However, after exposure to UVC treatments, the NER-deficient cells (XPA and XPC) did not show significant proliferation even for longer times, especially under DUSP3 silencing. On the other hand, the MRC-5 fibroblast cells and TLS-deficient XPV cell line (both NER-proficient) were not strongly affected by UV radiation and continue to proliferate, while DUSP3 silencing was only sensed by the normal MRC-5 cells.
Figure 1.
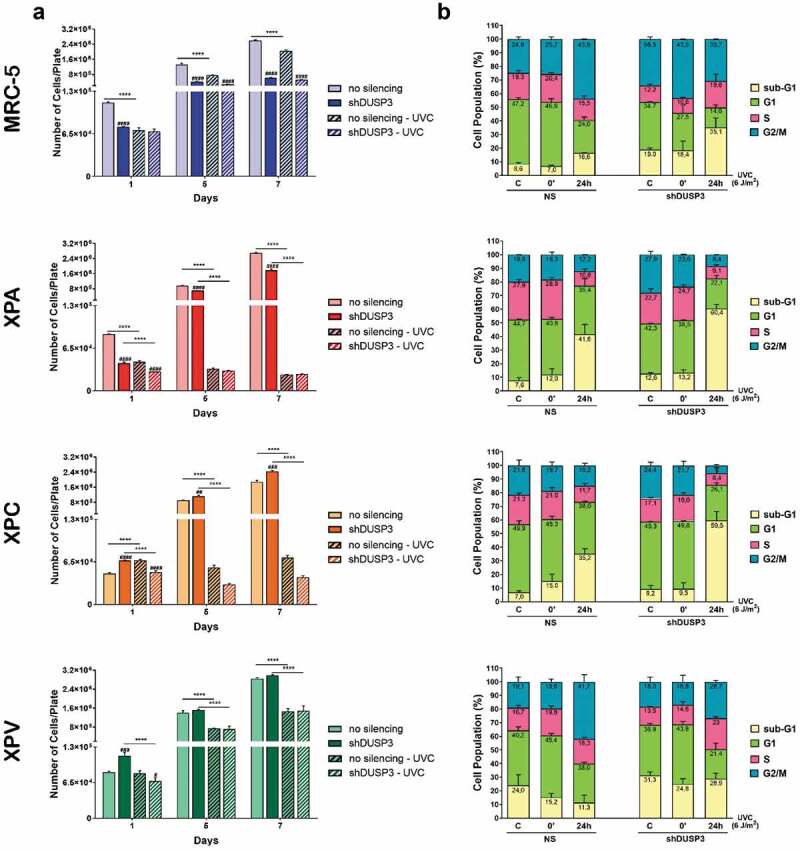
DUSP3 lof reduces cell proliferation and reorganizes cell cycle distribution after genotoxic stress. (A) Control cells (non-silenced, NS) or cells silenced for DUSP3 (shDUSP3) were plated at day “0” and exposed to UVC irradiation (4 J/m2) 24 hours after (day “1”); then cells were collected and counted in the subsequent days. Graphs are average of three independent experiments ± standard deviation. (B) MRC-5, XPA, XPC or XPV shDUSP3 cells, or their control (NS), were submitted to UVC radiation (6 J/m2) treatments and collected at 0ʹ and 24 hours later. Cells were fixed, permeabilized, stained with propidium iodide and analyzed in a cytometer. Graphs represent average of three independent experiments ± standard deviation.
Next, we decided to investigate the influence of DUSP3 in cell death and cell cycle progression using flow cytometry analysis within 24 hours after UV radiation, when all cells demonstrated proliferative responses at their first division cycle (Figure 1b). In general, for the four cell lines, particularly comparing control conditions (C or the 0ʹ after UVC) versus 24 h after UVC radiation, it is possible to observe an increase in the sub-G1 population, especially in DUSP3-silenced cells and mainly in the XPA and XPC cells, as well as in the control MRC-5 cell line, with only minor effects in XPV cells. This sub-G1 increase was allowed by a proportional reduction of cells in G1, S and G2/M phases, particularly for XPA and XPC cells. The more proliferative cells (MRC-5 and XPV) exhibited a different cell cycle distribution, showing increase in S phase cells and/or the maintenance of G2/M cell populations, both associated with G1 shortening (Figure 1b). These results highlight a trend observed in DNA repair proficient (MRC-5 and XPV) cells compared to NER-deficient cells (XPA and XPC): the lower the DNA repair capacity (and the greater the damage), the higher is the sub-G1 population (and the lower the S and G2/M cell population). This intriguing trend, which matches cell proliferation with genomic stability, seems to be kept unaltered in spite ofthe different cell phenotypes, and moreover, it seems to be dependent on DUSP3 proficiency.
Next, we carried out an extensive immunoblotting analysis of protein expression to evaluate the levels of the foremost regulatory proteins of cell cycle: Cyclins (A1, B1, D1, E1), CDKs (1, 2, 4, 6) and the p21Cip1 inhibitor, which may explain the observed cellular distribution throughout the cell cycle phases (Figure 2 and S3). Proteins which expression levels were significantly and statistically affected by the DUSP3 silencing and UV-radiation treatment are highlighted in red rectangles and their bands were quantified (Figure 2), while the uncut immunoblottings containing the complete kinetics can be seen in Supplementary Figure S3. Under DUSP3 silencing, the MRC-5 cells (MRC-5 shDUSP3) presented a decrease in CDK4 and CDK6, which probably caused the observed decrease in the G1 phase. These cells also presented a discrete increase in Cyclin A that can sustain the observed increase in S-phase cells, and an increase in the basal levels of p21Cip1 (non-irradiated control and 0ʹ after UVC), which is likely cause of the increased number of cells in the sub-G1 phase (Figure 1b).
Figure 2.
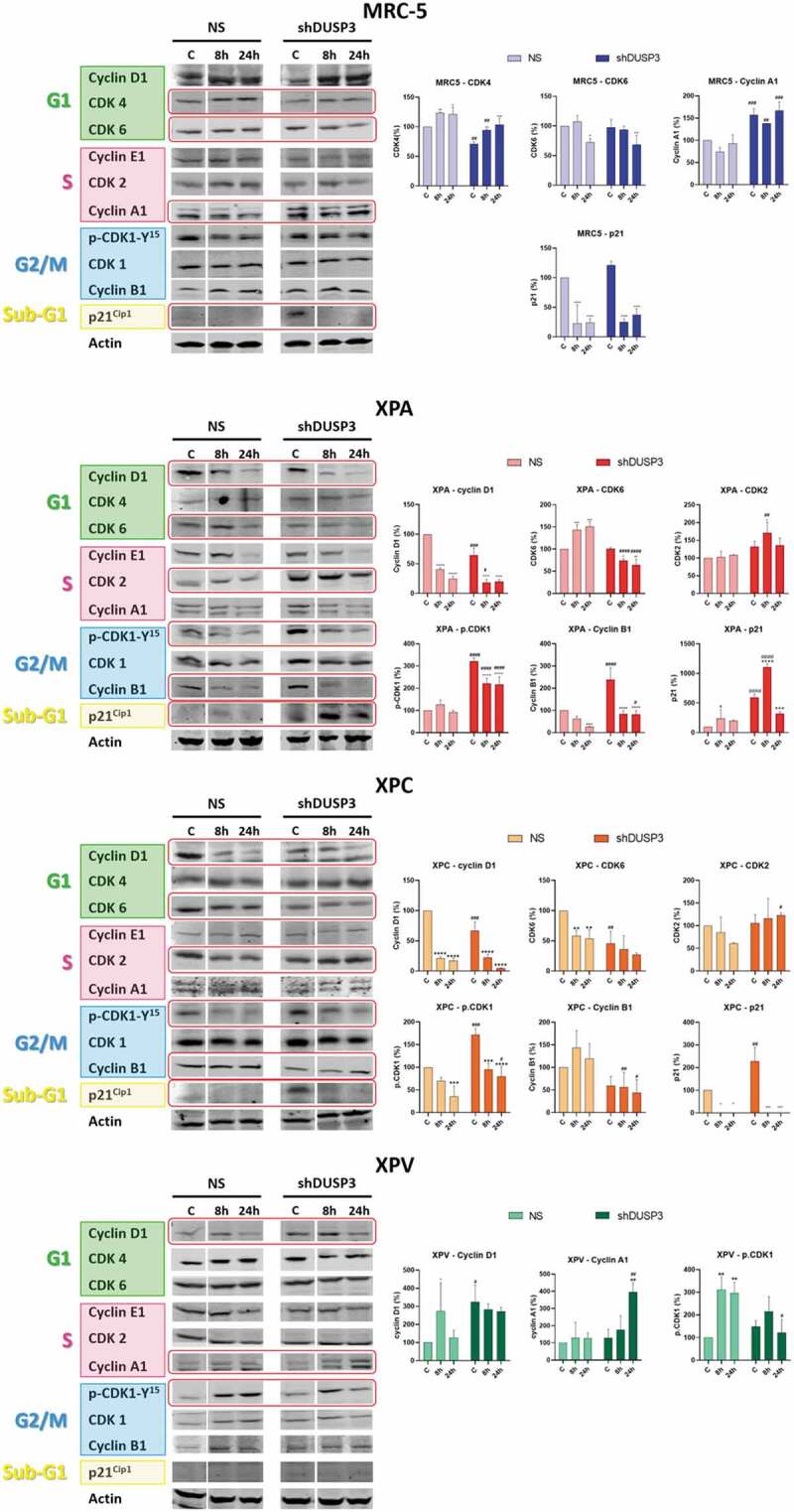
DUSP3 lof affects the expression of cell cycle regulatory proteins . The levels of Cyclins A1/B1/D1/E1, CDKs 1/2/4/6 and p21Cip1 were evaluated in all four (MRC-5, XPA, XPC and XPV) cells proficient (NS) or deficient for DUSP3 (shDUSP3) after treatment with UVC (6 J/m2) radiation. Cells were collected in different time points, lysed accordingly and the levels of protein expression were measured by immunoblottings (for each protein and cell line analyzed, all samples were loaded on the same gel/membrane). Proteins with levels affected by the DUSP3 silencing were quantified using Image Studio Software (Li-Cor) and the representative figures are highlighted in red. The bar graphs represent the average of three independent experiments ± standard deviation. ANOVA – */#: p < 0,05; ***/###: p < 0.001; **/##: p < 0.01; ****/####: p < 0.0001; *: relative to respective non-irradiated group (control); #: relative to NS (non-silencing) at the same treatment time point.
Since XPA and XPC cells presented more significant changes in the cell cycle under DUSP3 silencing, with or without UVC radiation treatment (Figure 1b), they also presented important variations in the expression of Cyclins and CDKs. In both NER-deficient shDUSP3 cells there was a significant decrease in Cyclin D1 and CDK6 in comparison to NS cells. This justifies the reduction in % of cells in G1 phase for the XPA and XPC shDUSP3 cells 24 h after UVC treatment. There was a significant increase in CDK2, but without a corresponding effect on the Cyclin A1 and E1 levels, which are probably in charge of the reduced S-phase cell population. Interestingly, a marked increase of phosphorylated CDK1-Y15 levels was also detected in DUSP3 silenced cells, accompanied by a reduction in Cyclin B1 levels (Figure 2). These results support a strong reduction of cells in G2/M phase of the cell cycle (Figure 1b). More importantly XPA cells, and, to a lesser extent, XPC cells, both exhibited elevated levels of p21Cip1 expression under DUSP3 silencing and especially after UVC exposure, which can be correlated with the arrested cell population in the sub-G1 state (see summary of results in Figure 3).
Figure 3.
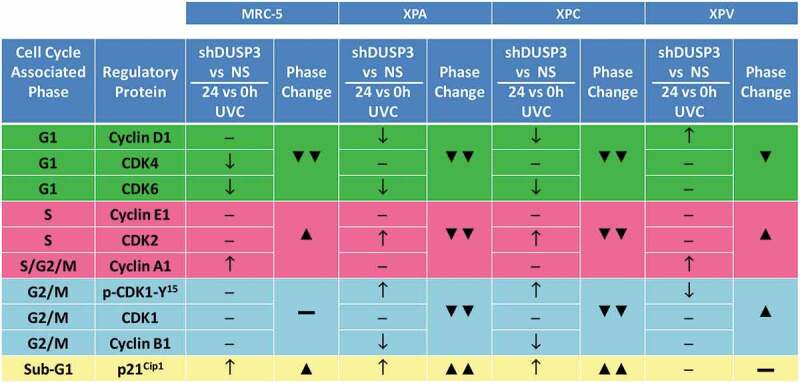
Summary box associating levels of cell cycle regulatory proteins with the percentage of cell population present in the different phases of the cellular cycle, before and after UV-irradiation of the four cell lines. Results obtained from Figures 1B and 2 showing cell cycle-associated phases and the corresponding regulatory proteins for each phase. It is empirically assumed for protein levels, comparing shDUSP3 versus NS cells in conditions of 24 hours versus 0 hours after UVC (shDUSP3 vs. NS/24 h vs. 0 h UVC), that: ↑ = increase or ↓ = decrease. It is empirically assumed as percentage of cells in cell cycle phases (phase change), that: ▲ = increase or ▼ = decrease. The levels of regulatory protein were estimated according to the quantification of immunoblotings (Figure 2 and Supplementary Figure S3).
Only few variations in cell cycle regulators were observed in XPV cells, which presented cell cycle progression and cellular proliferation very similar to the normal MRC-5 cells. For example, increased levels of Cyclin D1 were observed under DUSP3 silencing and UVC exposure compared to NS cells, suggesting a slight shortening of the G1 phase (Figure 1b), whereas a discrete increase in Cyclin A1 might sustain a higher S-phase population (Figure 2). Coincidently, a slight decrease of pCDK1-Y15 levels in XPV shDUSP3 cells was also detected, which coincides with a large cell population transitioning (entering and exiting) through the G2/M phase of the cell cycle (see the summary of results in Figure 3).
2.2. DUSP3 knockdown lowers the cells’ ability to repair specific UV-promoted lesions as well as UV indirect DNA damage
To investigate whether DUSP3 plays some role in repairing different lesions caused directly or indirectly by UV radiation, we started performing alkaline comet assays to assess the levels of single- and double-DNA strand breaks of cells submitted to UVC or UVB treatments (Figure 4 and Supplementary Figures S4 and S5). When MRC-5 cells were exposed to UVB, we observed that DUSP3 silencing caused a slight delay in DNA repair; however, after UVC exposure, these cells showed practically no repair of DNA up to 6 h after treatment, while NS cells had their DNA almost totally repaired. XPV cells presented a very similar behavior to MRC-5 fibroblasts, with a minor delay in DNA repair after either UVB or UVC exposure, However, a major accumulation of damage and a long delay in repair (6 hours of recovery) were also observed under DUSP3 silencing. These two NER-proficient cell lines were more susceptible to non-oxidative UVC-induced damage compared to more oxidative UVB damage, presenting higher levels of fragmented or unrepaired DNA even after 6 hours, and especially under DUSP3 lof. On the other hand, in the XPA NS cell line, the most sensitive to UV radiation due to a deficiency of the XPA protein [22], high levels of DNA damage were observed 6 h after UVC, and most notably after UVB exposure. However, in XPA shDUSP3 cells, the level of strand breaks damage was maintained high, particularly after UVB radiation treatment. The same effect of DUSP3 lof in the repair of these different UV-induced lesions was still observed in the XPC cell line, which is deficient in the GG-NER pathway. The damage accumulation found in XPC NS cells was strongly increased and maintained under DUSP3 lof, almost indistinctly after UVB or UVC radiation. To avoid doubts about the involvement of DUSP3 with the functioning of NER pathway, we also investigated the other active branch TCR-NER through additional comet assays using a CSB-deficient cell line. The latter was transiently silenced by siRNA-DUSP3 and further exposed to UVC radiation. Interestingly these cells did not completely repair the DNA damage up to 6 h after UVC exposure and, confirming our data from Figure 4, CSB-deficient cells with DUSP3 lof presented much higher accumulation of DNA strand breaks when compared to DUSP3-proficient cells (Supplementary Figure S6).
Figure 4.
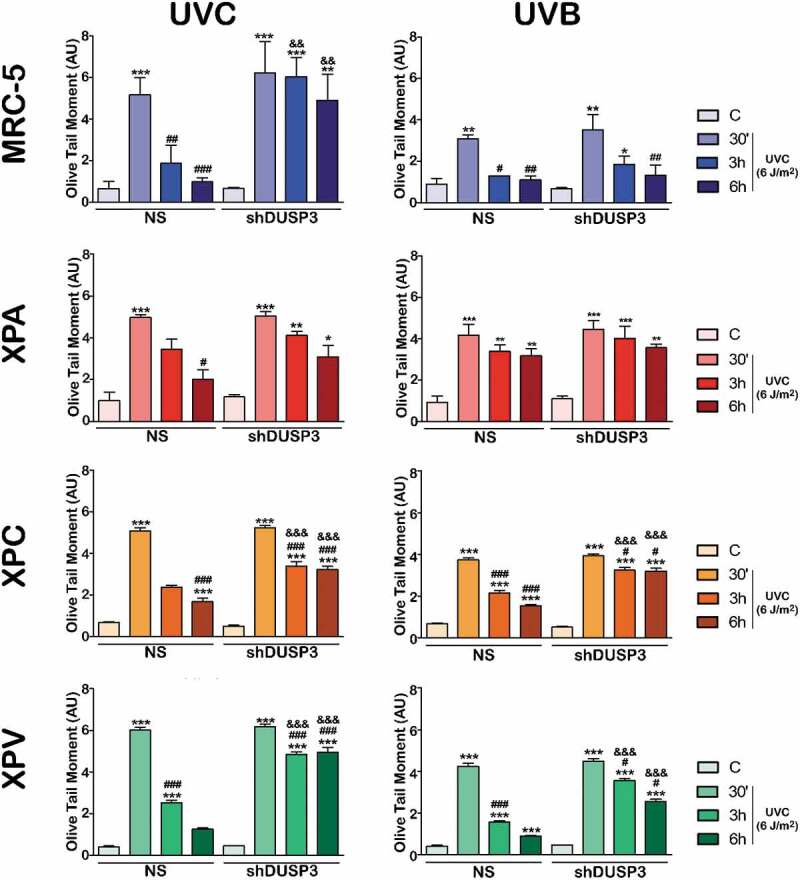
DUSP3 influences the global repair of DNA strand breaks after UV exposure. MRC-5, XPA, XPC and XPV cell lines were exposed to UVC (6 J/m2) (a) or UVB (50 J/m2) (b) radiation and collected at 30ʹ, 3 h and 6 h after. Alkaline comet assays were performed to provide the OTM index used to evaluate the levels of DNA damage and to infer the repair ability. The bars represent the averages of three independent experiments ± standard deviations. ANOVA – */#/&: p < 0.01; **/##/&&: p < 0.001; ***/###/&&&: p < 0.0001; *: relative to respective non-irradiated group (control); #: relative to respective initial DNA damage (30ʹ); &: relative to NS (non-silencing) at the same treatment time. (Representative images of comet tails used for quantification procedures are shown in the Supplementary Figures S4 and S5).
Since UV radiation directly incides on DNA to cause the formation of CPD and 6-4-PP lesions [23,24], we evaluated the influence of DUSP3 on NER repair by performing immuno slot-blots of DNA extracted from cells exposed to UVC or UVB by using antibodies that specifically recognize these lesions (Figures 5 and 6; Supplementary Figures S7 and S8). CPD removal from the genomic DNA of DUSP3-proficient MRC-5 and XPV cells (NS) is practically achieved within 72 hours. In the XPA and XPC cells, the CPD levels remained high and unremoved following 72 hours after UVC (Figure 5, left). More interesting were the results obtained in cells with DUSP3 lof, once both the NER-proficient (MRC-5 and XPV) and the NER-deficient (XPA) lines behaved similarly and showed a CPD accumulation up to 72 hours after UVC radiation, which was not completely repaired. Likewise, after UVB exposure, all four cell lines presented similar kinetics of CPD repair, albeit in some cases with lower amounts of damage probably due to differences in the dose and electromagnetic power of UVB compared to UVC (Figure 5 and Supplementary Figure S7).
Figure 5.
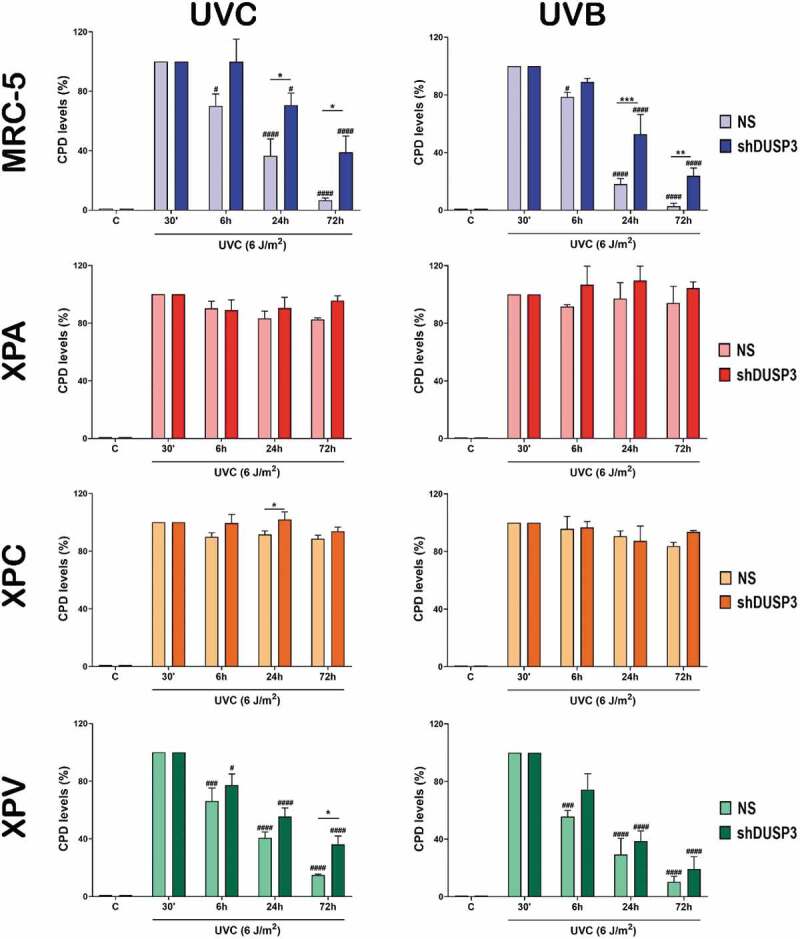
The cell's ability to repair CPD lesions caused by UV exposure is affected by the DUSP3 lof. Levels of CPD lesions were assessed by immuno slot-blotting the genome of MRC-5, XPA, XPC and XPV cells, shDUSP3 or NS, exposed to UVC or UVB treatments and collected at the indicated times. The results are representative of three independent experiments. ANOVA – */#: p < 0.05; ***/###: p < 0.001; ****/####: p < 0.0001; *: relative to NS at the same treatment time; #: relative to respective initial DNA damage (30ʹ). (Representative images of immuno slot-blots used for quantification procedures are shown in the Supplementary Figure S7).
Figure 6.
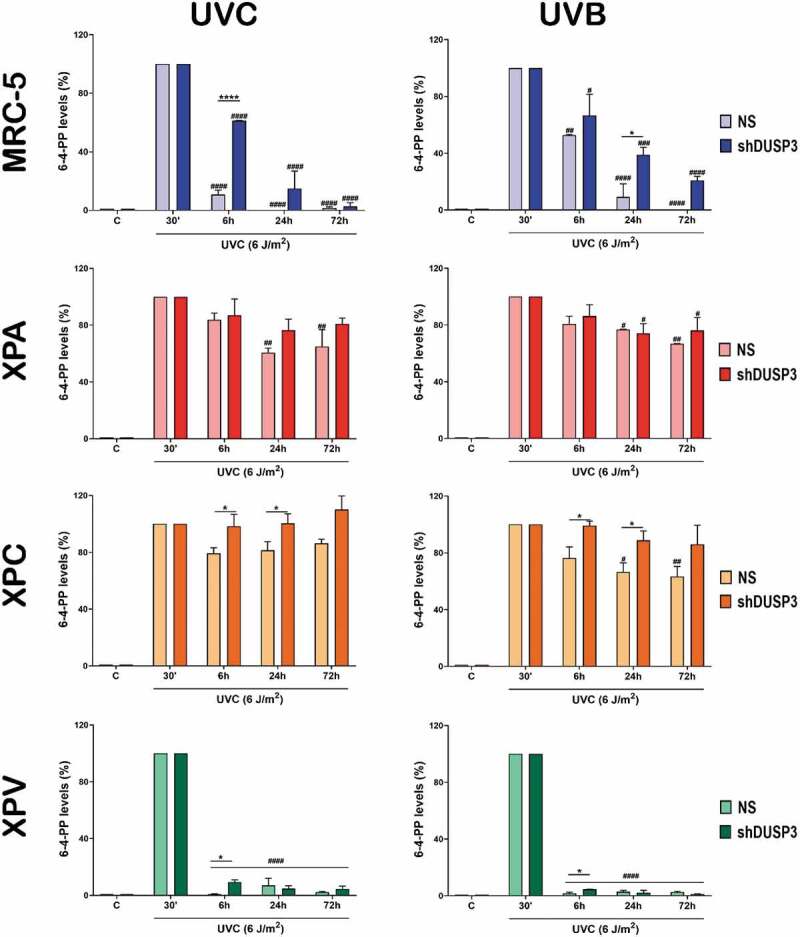
The repair of 6-4-PP lesions promoted by UV is also affected by the DUSP3 lof. Measurements of the 6-4-PPs were performed by immuno slot-blotting the genome of MRC-5, XPA, XPC and XPV cells, shDUSP3 or NS, after exposure to UVC or UVB treatments, according to the indicated times. The results are representative of three independent experiments. ANOVA – */#: p < 0.05; **/##: p < 0.01; ***/###: p < 0.001; ****/####: p < 0.0001; *: relative to NS at the same treatment time; #: relative to respective initial DNA damage (30ʹ). (Representative images of immuno slot-blots used for quantification procedures are shown in the Supplementary Figure S8).
We next measure the levels of 6-4-PP lesions through immuno slot-blots to probe the interdependence between NER pathway with DUSP3 deficiency in cells exposed to UVC or UVB treatments. Although this lesion is less predominant and removed more rapidly than CPDs, the MRC-5 cells again showed a quick and efficient repair of 6-4-PPs, which do not occur under DUSP3 lof and particularly under UVB exposure. Interestingly, the XPV cells presented the fastest repair of 6-4-PPs, with a very discrete delay under DUSP3 lof, which quickly returned to basal levels right after 6 h of either UVC or UVB treatments. Additionally, and as expected, XPA and XPC cells were not able to completely remove the 6-4-PP lesions even up to 72 h after UVC exposure. Nevertheless, DUSP3 lof led to a significant accumulation of these lesions, which were not repaired 72 h after. These effects persisted even when the cells were exposed to UVB, although they were less intense than observed with UVC, and particularly in XPA cells (Figure 6 and Supplementary Figure S8).
3. Discussion
DUSP3 is widely expressed and enzymatically active in various tissues; its deficiency drives cells to cell cycle arrest (G1/S and/or G2/M transitions) [17,25], accompanied or not by senescence [17]. This enzyme functions were recently correlated with DNA damage response (DDR) and DNA repair by the HR and NHEJ pathways [12]. This study aimed to verify the contribution of DUSP3 to genomic stability [19] through its influence on the NER pathway, the foremost active mechanism to repair UV-induced damage, and which dysfunction causes Xeroderma Pigmentosum disease [26]. Therefore, we associated DUSP3 lof with the deficiency of either XPA or XPC proteins in order to evaluate potential impacts on the removal of CPD and 6-4-PP lesions caused by UVC or UVB radiation [27,28]. We observed that NER-proficient cells (MRC-5 and XPV) delayed DNA damage repair after UV exposure more intensely under DUSP3 knockdown. They were also more susceptible to non-oxidative UVC-induced strand breaks compared to more oxidative UVB damage, but comparable to previously reported using ionizing radiation stress [12]. Similar DUSP3-dependent responses were found in NER-deficient cells (XPA and XPC), which ability to repair DNA fragmentations was also impaired (Figure 4). However, these cells were more susceptible to strand breaks induced by the more oxidative UV radiation (UVB) that knowingly causes other effects on DNA structure due to the production of reactive oxygen species (ROS), such as the formation of 8-oxo-7,8-dihydroguanine (8-oxo-Gua) [29], much less frequently caused by UVC [30]. NER-deficient cells usually exhibit DSBs on DNA-containing CPD sites due to collapse of replication forks [8] and DUSP3 seems to affect proteins involved in the establishment, recognition and/or repair of DNA strand breaks caused directly or indirectly by these different types of UV radiation [18–21].
To assess if DUSP3 is able to affect the NER pathway functioning we measured the levels of CPD and 6-4-PP in the cells genome [24,31]. DUSP3-proficient MRC-5 and XPV cells remove 6-4-PPs faster than CPDs, respectively, 24 hours and 72 hours after UVC and UVB exposure.. The highest detected CPD and 6-4-PP levels confirmed that DUSP3 lof significantly affects the ability of these cells to repair these UV-induced DNA damage, even knowing that CPD formation is about 3 times higher than of 6-4-PPs for UVC, and about 7 times higher for UVB radiation [32,33]. CPD distortion is known to remain in the genetic material up to 24 h after exposure to UVB and UVC [34], while 6-4-PPs are removed faster (~80% are removed within the first 4 hours, whereas ~60% of CPDs are repaired within the first 24 hours) [35]. On the other hand, DUSP3-proficient XPA and XPC cells [36,37] are not able to repair CPD and 6-4-PP lesions, which accumulate over time and cause dramatic distortions in genomic DNA. Interestingly, the amount of these lesions is still increased in XPA (more CPDs than 6-4-PPs) and XPC (more 6-4-PPs than CPDs) cells under DUSP3 lof, which are still enhanced by the reduced cell proliferation that XPA/XPC exhibit after UVC/UVB exposure (Figures 5 and 6). These results suggest that DUSP3 may be involved on the recognition of DNA damage site by the XPC-containing complex in the GG-NER branch [26], and also in the effective repair of UV-induced damage by the XPA protein through its direct interaction with NER core factors at the damage, independently of GG or TC branches [38]. Another important fact is that DUSP3 knockdown caused the same effect in CSB cells (Figure S2), which are deficient of the CSB protein that specifically acts as a damage sensor in the TC-NER branch [39]. Mutation or depletion of the CSB gene causes the Cockayne Syndrome, a disorder characterized by UV sensitivity and severe neurological manifestations leading to premature death [40]. Thus, we can assume that DUSP3 lof seems to also affect the NER pathway in the GG-NER branch.
The hypothesis that PTPs may also regulate genomic stability has lately strengthened. In addition to DUSP3 [12], other phosphatases [10,41–43] were shown to interfere with DNA repair after IR or UV radiation by dephosphorylating proteins belonging to different repair pathways. Our previous studies [18,21] show that DUSP3 interacts with and regulates nuclear proteins including NPM, hnRNP C1/C2, NCL, NBS1 and ATM/ATR, which are known to mediate different aspects of the DDR pathway. However, the precise mechanisms through which DUSP3 affects the NER pathway to remove specific UV-induced distortions on DNA will need further investigations. One of our hypotheses might be sustained by the fact that XP cells express high levels of p53 protein [44] and, while we observed DUSP3 knockdown did not alter p53 expression, its phosphorylation on Ser15 residue is strongly increased and hampers its association with other proteins, such as Mdm2 (for subsequent proteasome degradation of p53) or NPM (allowing a faster nucleolus-nucleoplasm translocation), consisting in a very promising mechanism regulated by DUSP3 and that is under extensive investigation by our group (Russo LC & Forti FL unpublished results, manuscript in preparation).
Another hypothesis we propose connects (direct or indirectly) DUSP3 with mechanisms of genomic stability by modulating the expression of cell cycle regulatory proteins (Cyclins, CDKs and CKIs) to control cell cycle and cellular proliferation (Figures 1 and 3). This was firstly evidenced in non-stressed HeLa S3 cells transiently silenced for DUSP3, which showed an altered expression of some cell cycle regulators [17]. Other evidence is that XPA silencing causes an increased expression of CDK2/Cyclin E and CDK1/Cyclin B complexes in the carcinogenesis of bladder cancer cells [45]. In NER-deficient cells under DUSP3 lof we observed an increase in CDK2 that was not accompanied by the levels of CDK1, Cyclin E1 and A1 (Figure 1), which altogether caused the dramatic reduction of cell population in S and G2/M phases (Figure 2, 3 and Supplementary S3). A third evidence supporting our hypothesis correlates ERK1/2 phosphorylation with a consequent increase in expression of Cyclin A2 and CDK2 in cervical cancer cells [46], results that are in agreement with our observations in XPA and XPC cells (at least for CDK2) under DUSP3 lof. Indeed DUSP3 knockdown increases ERK1/2 phosphorylation (Figure S1C) with consequent decreases in the cellular proliferation of NER-deficient cells [25].
Cell cycle progression is controlled by a temporal balance of phosphorylation/dephosphorylation events causing stabilization/degradation of many proteins until the cells exit mitosis (M) [47,48]. In fact, our findings showed that DUSP3 knockdown caused an increase of CDK1-Y15 phosphorylation in NER-deficient cell lines (XPA and XPC) after UVC exposure and also caused a further decrease in Cyclin B1 levels (Figure 2). As the Cyclin B1/CDK1 activity predominates cells enter M, but if this complex is not properly dephosphorylated cells cannot exit M [49,50]. The shedding of UVC light in cells cycling in the G1 phase has been shown to affect the repair of DNA lesions, leading to arrest in G2 phase [51]. Accordingly, our group previously reported that loss of DUSP3 activity in HeLa cells exposed to IR attenuates the phosphorylation of DDR proteins (ATM/CHK2), causing G2/M arrest [12]. Since UV causes activation of ATR/CHK1 it might be assumed that DUSP3 lof very likely will attenuate this response and will reduce phosphorylation of Cdc25C, the phosphatase responsible for dephosphorylation and inactivation of pCDK1-Y15, leading cells to failentering G2/M transition and not undergoing M [52].
The activity of the Cyclin D1/CDK4 complex is necessary for cell cycle progression through G1 phase and the completion of M, whereas loss a CDK4 causes G2 arrest after UVC exposure [53]. Therefore, cells with normal (or high) CDK4 expression and reduced levels of Cyclin D1, as for example XPA and XPC cells with DUSP3 lof, consequently do not effectively progress into G2/M and arrest at G1 (Figure 2). Also, Cyclin D1 protein is down-regulated after UV exposure to block G1 progression and thus allows cells to repair the DNA damage [54]. This was also observed in DUSP3-silencedXPA and XPC cells and, considering that Cyclin D1 interacts with two repair proteins (BRCA1 and PCNA) [54,55], its regulation by DUSP3 seems to impact on DNA repair. On the other hand, cells with high (or normal) levels of Cyclin D1 and constitutive CDK4 levels, such as DUSP3-silenced MRC-5 and XPV cells exposed to UV, were able to rapidly progress through G1 phase, efficiently reach G2/M and undergo M (Figures 1 and 2). Regarding the Cyclin E1-A1/CDK2 complex, which activity limits entry and progression through the S phase [56], we found normal levels of Cyclin E1 and CDK2 expression in MRC-5 and XPV cells under DUSP3 lof that are contrasted by high levels of Cyclin A1 (Figure 2). These results corroborate with the high percentage of cells in S phase and the proliferation exhibited by these two cell lines (Figure 2, 3 and S3).
The Cyclin E1/CDK2 complex activity was shown to be regulated by the cell cycle inhibitor p21Cip1, in conjunction with p53 [57]. Interestingly NER-deficient cells express high basal levels of p21Cip1 probably due to their high levels of p53 [44], which can even be increased after UV radiation [58], and that are remarkably affected by DUSP3 knockdown (Figure 2 and S3). Our data show DUSP3 controls p21Cip1 protein levels, with or without genotoxic stress, very likely by dephosphorylating proteins involved in p53 stability, as for example NPM [18]. p21Cip1 by itself can induce cell death through apoptosis either by the modulation of transcription or via caspases [59]. Therefore, the accumulation of XPA cells in sub-G1 after UV irradiation (Figure 1) is probably caused by both high levels of DNA damage (Figures 4, 5 and 6) and p21Cip1 expression (Figure 2), which were enhanced by DUSP3 lof. The subsequent p21Cip1 reduction after UV-radiation exposure was shown to be caused by its polyubiquitination and proteasome degradation [57]. The failure to degrade p21Cip1 compromises DNA repair [57], as observed for the XPA cell line DUSP3-silenced that is driven to death. Similarly, in MRC-5, XPC and XPV cells, the low levels of p21Cip1 in non-treated cells immediately increase after UVC exposure (0ʹ condition) and were enhanced under DUSP3 silencing, but rapidly declined (in 3 hours) due to p21Cip1 degradation (Figure 2). These results are in good agreement with the literature [17,57,60] and explain the differences in the levels of p21Cip1 found in XPA versus the other three cell lines: the DUSP3 lof increases p21Cip1 stability and/or p21Cip1 expression in a p53-dependent manner due to the increase in p53-Ser15 phosphorylation and its transcriptional activity (Russo LC & Forti FL, unpublished results, manuscript in preparation).
Last but not least, are the intriguing results obtained with the TLS-deficient (and NER-proficient) XPV cell line in which the lack of Pol η [61] does not impact in the NER activity [62]. Interestingly, it was shown that PCNA-p21Cip1 interaction impairs PCNA-Pol η association and inhibits Pol η foci assembly. Therefore, p21Cip1 is considered a negative regulator of PCNA partners in TLS and it is essential for the inhibition of DNA replication [63,64]. Here we show XPV cells with undetectable levels of p21Cip1 (Figure 2) independently of DUSP3 presence what suggests there is no obstacle for the recruitment of NER proteins, being these cells proficient or even more efficient in NER repair. This assumption is supported by our data showing CPD and (especially) 6-4-PP lesions are repaired faster in XPV than in other cells (Figure 4, 5, and 6). It is also reflected on proliferation assays in which XPV cells are almost not affected by UVC radiation and are able to quickly recover after stress and, more importantly, these cells are much less impacted by the DUSP3 lof (Figure 1). In fact, we only observed discrete variations in Cyclins and CDKs in XPV cells after UV radiation, with or without DUSP3, with a small reduction in pCDK1-Tyr15 levels, yielding a high number of cells transiting in G2/M phases (Figure 2 and S3) to promote mitosis entry and accelerated proliferation. These results also support our hypothesis about DUSP3 roles in DNA repair and raise another important question since they show that DUSP3 lof preferentially affects the repair, but not the proliferation of XPV cell line.
4. Conclusion
DUSP3 ameliorates the DNA repair of cells exposed to UV radiation, reducing the amount of direct (CDP and 6-4-PP) and indirect (strand breaks) lesions on the genetic material probably through the interaction and/or dephosphorylation of proteins biochemically indispensable for the NER pathway, as well as other DNA repair pathways, as for example, the NPM phosphoprotein. The proof-of-concept was the use of NER-deficient XP cells which fragile phenotype can still be affected by DUSP3 lof, making them more sensitive to UV light. Moreover, the DUSP3 silencing causes deregulation in the expression and/or stability of specific regulatory proteins of cell cycle, which is dependent on cell phenotype and stress conditions, and still has a direct impact on cell cycle progression and cellular proliferation. Therefore, this work confirms important functions and contributions of DUSP3 phosphatase in the genomic stability of cells through modulation of DNA repair and/or control of the cell cycle, although what mechanism is firstly and preferentially affected by the DUSP3 lof remains an open question.
5. Materials and methods
5.1. Cell culture and DUSP3 loss of function (lof) by lentiviral shRNA knockdown
MRC-5V1 (MRC-5), XP12RO (XPA), XP4 PA (XPC) and XP30RO (XPV) cells [36,37,65,66] were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, penicillin (100 U/mL) and streptomycin (100 g/mL) (Life Technologies) under incubation at 37°C and 5% CO2 atmosphere. The stable DUSP3 knockdown was performed with “Thermo Scientific Open Biosystems Expression GIPZ lentiviral shRNAmir” kit (Dharmacon, a Horizon Discovery Group) containing three sequences to target DUSP3 (#1: AGGTTATAGCCGCTCCCCA; #2: AGGTCCTTCATGCACGTCA; #3: AACGACACACAGGAGTTCA). As a negative control, a scramble sequence (Non-Silencing, named NS) with no homology to existing mRNA was used. In brief, 72 h after lentiviral transduction, cells were submitted to selection by puromycin (1 µg/mL) and the selection of resistant clones was accompanied by immunofluorescence of GFP-positive cells and by western blot analyses checking the levels of DUSP3 protein. One clone from each cell line exhibiting at least 90% DUSP3 knockdown (shDUSP3) and one containing scramble sequence (NS shRNA) were used in this work. All isolated stable clonal cell lines were cultured in medium containing 0.75 µg/mL puromycin.
5.2. UVB and UVC radiation treatments
The clonal cell lines NS or shDUSP3 were subjected to two different UV radiation treatments. Cells in PBS were exposed to UV radiation doses using specific lamps with wavelengths corresponding to UVB (50 kJ/m2) or UVC (6 J/m2). A VLX-3 W dosimeter (Vilber Lourmat, Eberhardzell, Baden-Württemberg, Germany) was used to keep the UVB or UVC lamps calibrated. Following each radiation treatment, the PBS was replaced with fresh medium for the indicated periods for further analyses [67].
5.3. Alkaline comet assays
NS or shDUSP3 clones (3x105 cells) were seeded on 35 mm plates) 24 h before UVB or UVC irradiation. After treatments, cells were collected at times of 0ʹ, 30ʹ, 3 h and 6 h by trypsinization, and immediately mixed with 0.5% low-melting agarose at 37ºC; this mixture was applied onto glass slides previously covered with a thin layer of 1.5% agarose. Proteins and cellular membranes were removed by incubation in lysis solution for 24 h at 4°C and the slides were subjected to electrophoresis (25 V/cm and 300 mA for 30 min), neutralized, dried and fixed as previously described [68]. The cell nucleus was stained with 2 μg/mL ethidium bromide and visualized under a fluorescence microscope (Olympus BX51). The results of DNA fragmentation were expressed as the olive tail moment (OTM) index obtained using the Komet 6.0 software (Andor Technology, Oxford, UK), from a total of 100 cells analyzed per sample [68].
5.4. Immuno slot-blot for CPD and 6-4-PP lesions
Slot-blot immunoassay using antibodies that specifically recognize UV-induced lesions (CPDs and 6-4-PPs) on genomic DNA is a very reliable and quantitative technique commonly used for studies involving UV damage and NER pathway [27]. Cells (30x104) were seeded in a 35 mm plate 24 h before the exposure to UVB or UVC radiation, and then maintained at 37°C until the collection time (0ʹ, 30ʹ, 6 h, 24 h, and 72 h) for the extraction of genomic DNA, as previously described [27]. DNA was quantified in Epoch equipment (Biotek) and samples of 75 ng (for CPDs) or 400 ng (for 6-4-PPs) were combined with salmon sperm DNA to a final amount of 1000 ng of DNA. The samples were boiled and transferred to a pre-hydrated (1 M ammonium acetate) nitrocellulose membrane through a vacuum system, which was blocked in 5% nonfat milk (PBS) for 18 h at 4ºC and then incubated with the primary antibodies for CPD (2 h at RT, 1:2000 in PBS containing 5% nonfat milk) or 6-4-PP (3 h at RT, 1:1000 dilution in PBS containing 5% nonfat milk) (Cosmo Bio Co., Ltd). After 6 washes (5 min, RT, 1% Tween 20 in PBS), the membranes were incubated with secondary antibodies IR Dye 680CW or 800CW (1:15,000; Li-Cor) for 1 h and revealed as described in section 5.6 [27].
5.5. Flow cytometry analyses
NS or shDUSP3 clonal cells (4 × 105 cells) were plated on 35mm dishes at 37°C for 24 h. Non-irradiated or UVC-irradiated cells (6 J/m2) were harvested by trypsinization, washed with PBS, and fixed with 70% ethanol in PBS. Samples were washed by centrifugation (830 x g, 12 min) and stained with propidium iodide (PI) buffer (10 μg/mL PI, 10 µg/mL RNAse, 0,1% triton X-100, 0,1% sodium citrate in PBS). A total of 30,000 events were read by an AttuneTM cytometer (Thermo Fisher Scientific) and analyzed using the Kaluza software (Beckman Coulter) [69].
5.6. Western blottings
For immunoblottings, cells were lysed with RIPA buffer (50 mmol/L Tris-HCl, pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mmol/L NaCl, 10 mmol/L MgCl2, 1 mM Na3VO4, 1 mM NaF, 2 μg/mL leupeptin, pepstatin, aprotinin and 1 mmol/L PMSF) (Sigma-Aldrich, Saint Louis, MO, USA) [33] and 50 μg of total protein was mixed with Laemmli sample buffer [34]. SDS-PAGE was performed at different concentrations of polyacrylamide gel and proteins were transferred to a nitrocellulose membrane (Merck-Millipore, Billerica, MA, USA). The membrane blocking was in 5% nonfat dry milk in TTBS buffer (25 mM Tris-HCl, pH 7.4, and 125 mM NaCl (TBS) containing 0.1% Tween 20) for 1 h at RT. Specific antibodies diluted in TTBS were used as follows: anti-DUSP3 (1:1000, BD Biosciences), anti-p53 (1:1000, Santa Cruz), anti-phospho-ERK1/2 (T202/Y204, 1:1000, Cell Signaling), anti-ERK1/2 (1:1000, Santa Cruz), anti-phospho-CDK1 (Y15, 1:1000, Cell Signaling), anti-CDK1 (1:1000, Santa Cruz), anti-CDK2 (1:1000, Santa Cruz), anti-CDK4 (1:1000, Santa Cruz), anti-CDK6 (1:1000, Santa Cruz), anti-Cyclin D1 (1:1000, Santa Cruz), anti-Cyclin E1 (1:1000, Santa Cruz), anti-Cyclin A1 (1:1000, Santa Cruz), anti-Cyclin B1 (1:1000, Santa Cruz), anti-p21 (1:1000, Santa Cruz) and anti-Actin (1:1000, Santa Cruz). The membranes were incubated with fluorescent secondary antibody IR Dye 680CW or 800CW (1:15,000; Li-Cor) and the bands were visualized by the Odyssey Infrared Imaging System and analyzed (quantified) using Image Studio software (Li-Cor, Bad Homburg, Germany).
5.7. Growth curves
Cells were plated in 35 mm dishes at a density of 4 × 104 cells per plate the day before and then irradiated with UVC or not. After that, cells were harvested for counting and again on the following seven consecutive days as briefly shown: the plates were washed with PBSA and 300 μL trypsin (Thermo) was added for 3 min at 37°C. The cells were re-suspended in 400 µL of PBS and fixed with 300 μL formaldehyde 37% (Cromoline, Diadema, SP, Brazil) and counted using a Z Series Coulter Counter (Beckman Coulter®). Three independent experiments were performed in triplicate [12].
5.8. Statistics
Graphs and statistical analyses were realized in GraphPad Prism 8. Results are expressed as the mean ± SEM as appropriate. Statistical comparisons between all groups were performed using ANOVA followed by a Tukey test. p < 0.0001 (***), p < 0.001 (**) or p < 0.01 (*) indicate significance.
Supplementary Material
Acknowledgments
The authors thank Prof. Carlos F. M. Menck, from the Institute of Biomedical Sciences - University of São Paulo, for the donation of anti-CPD and anti-6,4-PP antibodies. We thank Prof. Hugo A. Armelin and Dr. Maria Carolina E. Sabbaga for allowing us to access the facilities of CeTICS (Butantan Institute, São Paulo, Brazil), as well as Dr. Matheus Henrique S. Dias for supporting us with the Attune cytometer and cytometry analyses. We also thank Prof. Marisa Helena G. Medeiros, Prof. Bianca S. Zingales and the technician Marcelo Nunes Silva, from the Institute of Chemistry - University of São Paulo, for equipment usage and reagents exchange.
Funding Statement
This work was primarily supported by the Sao Paulo Research Foundation - FAPESP (Grants No 2015/03983-0 and 2018/01753-6) and the Brazilian National Research Council – CNPq (Grant No 402230/2016-7). LCR is founded by a PNPD fellowship from the Ministry of Education through the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (88887.136364/2017-00), and JOF is supported by a FAPESP PhD fellowship (Grant No 2017/16491-4)
Author contributions
LCR and JOF performed the study; FLF supervised and designed the study; LCR, JOF and FLF analyzed and interpreted the data; LCR and FLF wrote the manuscript.
Disclosure statement
The authors declare no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kaliki S, Jajapuram SD, Maniar A, et al. Ocular and periocular tumors in xeroderma pigmentosum: A study of 120 Asian Indian patients. Am J Ophthalmol. 2019 Feb;198:146–153. doi:10.1016/j.ajo.2018.10.011. Epub 2018 Oct 16. [DOI] [PubMed] [Google Scholar]
- [2].Mullenders LHF. Solar UV damage to cellular DNA: from mechanisms to biological effects. Photochem Photobiol Sci. 2018;17:1842–1852. [DOI] [PubMed] [Google Scholar]
- [3].Weon JL, Glass DA II.. Novel therapeutic approaches to xeroderma pigmentosum. Br J Dermatol. 2019 Aug;181(2):249–255. doi:10.1111/bjd.17253. Epub 2018 Nov 25. [DOI] [PubMed] [Google Scholar]
- [4].Yoon JH, Prakash L, Prakash S. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc Natl Acad Sci U S A. 2009;106:18219–18224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Annu Rev Biophys. 2015;44:207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hedglin M, Pandey B, Benkovic SJ. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. Elife. 2016 Oct 22;5. pii: e19788. doi:10.7554/eLife.19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koch SC, Simon N, Ebert C, et al. Molecular mechanisms of xeroderma pigmentosum (XP) proteins. Q Rev Biophys. 2016;49:e5. [DOI] [PubMed] [Google Scholar]
- [8].Rastogi RP, Richa KA, Tyagi MB, et al. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu W, Li L, Wang G, et al. KU70 inhibition impairs both non-homologous end joining and homologous recombination DNA damage repair through SHP-1 induced dephosphorylation of SIRT1 in adult T-cell leukemia-lymphoma cells. Cell Physiol Biochem. 2018;49:2111–2123. [DOI] [PubMed] [Google Scholar]
- [10].Kang Y, Cheong HM, Lee JH, et al. Protein phosphatase 5 is necessary for ATR-mediated DNA repair. Biochem Biophys Res Commun. 2011;404:476–481. [DOI] [PubMed] [Google Scholar]
- [11].Nguyen TA, Slattery SD, Moon SH, et al. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair (Amst). 2010;9:813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Torres TEP, Russo LC, Santos A, et al. Loss of DUSP3 activity radiosensitizes human tumor cell lines via attenuation of DNA repair pathways. Biochim Biophys Acta Gen Subj. 2017 Jul;1861(7):1879–1894. doi:10.1016/j.bbagen.2017.04.004. Epub 2017 Apr 4. [DOI] [PubMed] [Google Scholar]
- [13].Alonso A, Sasin J, Bottini N, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. [DOI] [PubMed] [Google Scholar]
- [14].Arnoldussen YJ, Lorenzo PI, Pretorius ME, et al. The mitogen-activated protein kinase phosphatase vaccinia H1-related protein inhibits apoptosis in prostate cancer cells and is overexpressed in prostate cancer. Cancer Res. 2008;68:9255–9264. [DOI] [PubMed] [Google Scholar]
- [15].Chen YR, Chou HC, Yang CH, et al. Deficiency in VHR/DUSP3, a suppressor of focal adhesion kinase, reveals its role in regulating cell adhesion and migration. Oncogene. 2017;36:6509–6517. [DOI] [PubMed] [Google Scholar]
- [16].Hoyt R, Zhu W, Cerignoli F, et al. Cutting edge: selective tyrosine dephosphorylation of interferon-activated nuclear STAT5 by the VHR phosphatase. J Immunol. 2007;179:3402–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rahmouni S, Cerignoli F, Alonso A, et al. Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence. Nat Cell Biol. 2006;8:524–531. [DOI] [PubMed] [Google Scholar]
- [18].Panico K, Forti FL. Proteomic, cellular, and network analyses reveal new DUSP3 interactions with nucleolar proteins in HeLa cells. J Proteome Res. 2013;12:5851–5866. [DOI] [PubMed] [Google Scholar]
- [19].Russo LC, Farias JO, Ferruzo PYM, et al. Revisiting the roles of VHR/DUSP3 phosphatase in human diseases. Clinics (Sao Paulo). 2018;73:e466s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Monteiro LF, Ferruzo PYM, Russo LC, et al. DUSP3/VHR: a druggable dual phosphatase for human diseases. Rev Physiol Biochem Pharmacol. 2019;176:1–35. doi: 10.1007/112_2018_12. [DOI] [PubMed] [Google Scholar]
- [21].Forti FL. Combined experimental and bioinformatics analysis for the prediction and identification of VHR/DUSP3 nuclear targets related to DNA damage and repair. Integr Biol (Camb). 2015;7:73–89. [DOI] [PubMed] [Google Scholar]
- [22].Marteijn JA, Lans H, Vermeulen W, et al. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–481. [DOI] [PubMed] [Google Scholar]
- [23].Lee JH, Hwang GS, Choi BS. Solution structure of a DNA decamer duplex containing the stable 3ʹ T.G base pair of the pyrimidine(6-4)pyrimidone photoproduct [(6-4) adduct]: implications for the highly specific 3ʹ T –> C transition of the (6-4) adduct. Proc Natl Acad Sci U S A. 1999;96:6632–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McAteer K, Jing Y, Kao J, et al. Solution-state structure of a DNA dodecamer duplex containing a Cis-syn thymine cyclobutane dimer, the major UV photoproduct of DNA. J Mol Biol. 1998;282:1013–1032. [DOI] [PubMed] [Google Scholar]
- [25].Cerignoli F, Rahmouni S, Ronai Z, et al. Regulation of MAP kinases by the VHR dual-specific phosphatase: implications for cell growth and differentiation. Cell Cycle. 2006;5:2210–2215. [DOI] [PubMed] [Google Scholar]
- [26].Sugasawa K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair (Amst). 2016;44:110–117. [DOI] [PubMed] [Google Scholar]
- [27].Russo LC, Minaya PY, Silva LE, et al. Assessing the roles of Rho GTPases in cell DNA repair by the nucleotide excision repair pathway. Methods Mol Biol. 2018;1821:319–338. [DOI] [PubMed] [Google Scholar]
- [28].Hu J, Adebali O, Adar S, et al. Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci U S A. 2017;114:6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ryter SW, Tyrrell RM. Singlet molecular oxygen ((1)O2): a possible effector of eukaryotic gene expression. Free Radic Biol Med. 1998;24:1520–1534. [DOI] [PubMed] [Google Scholar]
- [30].Cadet J, Sage E, Douki T. Ultraviolet radiation-mediated damage to cellular DNA. Mutat Res. 2005;571:3–17. [DOI] [PubMed] [Google Scholar]
- [31].Besaratinia A, Yoon JI, Schroeder C, et al. Wavelength dependence of ultraviolet radiation-induced DNA damage as determined by laser irradiation suggests that cyclobutane pyrimidine dimers are the principal DNA lesions produced by terrestrial sunlight. Faseb J. 2011;25:3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chandrasekhar D, Van Houten B. In vivo formation and repair of cyclobutane pyrimidine dimers and 6-4 photoproducts measured at the gene and nucleotide level in Escherichia coli. Mutat Res. 2000;450:19–40. [DOI] [PubMed] [Google Scholar]
- [33].Perdiz D, Grof P, Mezzina M, et al. Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis. J Biol Chem. 2000;275:26732–26742. [DOI] [PubMed] [Google Scholar]
- [34].Kobayashi N, Katsumi S, Imoto K, et al. Quantitation and visualization of ultraviolet-induced DNA damage using specific antibodies: application to pigment cell biology. Pigment Cell Res. 2001;14:94–102. [DOI] [PubMed] [Google Scholar]
- [35].Riou L, Eveno E, van Hoffen A, et al. Differential repair of the two major UV-induced photolesions in trichothiodystrophy fibroblasts. Cancer Res. 2004;64:889–894. [DOI] [PubMed] [Google Scholar]
- [36].Soufir N, Ged C, Bourillon A, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol. 2010;130:1537–1542. [DOI] [PubMed] [Google Scholar]
- [37].Satokata I, Tanaka K, Miura N, et al. Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res. 1992;273:193–202. [DOI] [PubMed] [Google Scholar]
- [38].de Vries A, van Oostrom CT, Hofhuis FM, et al. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995;377:169–173. [DOI] [PubMed] [Google Scholar]
- [39].Zhu Q, Wani AA. Nucleotide excision repair: finely tuned molecular orchestra of early pre-incision events. Photochem Photobiol. 2017;93:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Karikkineth AC, Scheibye-Knudsen M, Fivenson E, et al. Cockayne syndrome: clinical features, model systems and pathways. Ageing Res Rev. 2017;33:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Oghabi Bakhshaiesh T, Majidzadeh AK, Esmaeili R. Wip1: A candidate phosphatase for cancer diagnosis and treatment. DNA Repair (Amst). 2017;54:63–66. [DOI] [PubMed] [Google Scholar]
- [42].Naderali E, Khaki AA, Rad JS, et al. Regulation and modulation of PTEN activity. Mol Biol Rep. 2018 Dec;45(6):2869–2881. doi:10.1007/s11033-018-4321-6. Epub 2018 Aug 25. [DOI] [PubMed] [Google Scholar]
- [43].Huang CY, Hsieh FS, Wang CY, et al. Palbociclib enhances radiosensitivity of hepatocellular carcinoma and cholangiocarcinoma via inhibiting ataxia telangiectasia-mutated kinase-mediated DNA damage response. Eur J Cancer. 2018;102:10–22. [DOI] [PubMed] [Google Scholar]
- [44].Carvalho H, Ortolan TG, dePaula T, et al. Sustained activation of p53 in confluent nucleotide excision repair-deficient cells resistant to ultraviolet-induced apoptosis. DNA Repair (Amst). 2008;7:922–931. [DOI] [PubMed] [Google Scholar]
- [45].Zhi Y, Ji H, Pan J, et al. Downregulated XPA promotes carcinogenesis of bladder cancer via impairment of DNA repair. Tumour Biol. 2017;39:1010428317691679. [DOI] [PubMed] [Google Scholar]
- [46].Ming P, Cai T, Li J, et al. A novel arylbenzofuran induces cervical cancer cell apoptosis and G1/S arrest through ERK-mediated Cdk2/cyclin-A signaling pathway. Oncotarget. 2016;7:41843–41856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kamenz J, Ferrell JE Jr.. The Temporal Ordering Of Cell-Cycle Phosphorylation. Mol Cell. 2017;65:371–373. [DOI] [PubMed] [Google Scholar]
- [48].Swaffer MP, Jones AW, Flynn HR, et al. CDK substrate phosphorylation and ordering the cell cycle. Cell. 2016;167:1750–61 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cundell MJ, Hutter LH, Nunes Bastos R, et al. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J Cell Biol. 2016;214:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Goldberg ML. Cell cycle: abrogating interphase/M phase bistability. Curr Biol. 2018;28:R1342–R5. [DOI] [PubMed] [Google Scholar]
- [51].Wigan M, Pinder A, Giles N, et al. A UVR-induced G2-phase checkpoint response to ssDNA gaps produced by replication fork bypass of unrepaired lesions is defective in melanoma. J Invest Dermatol. 2012;132:1681–1688. [DOI] [PubMed] [Google Scholar]
- [52].Schmit TL, Ahmad N. Regulation of mitosis via mitotic kinases: new opportunities for cancer management. Mol Cancer Ther. 2007;6:1920–1931. [DOI] [PubMed] [Google Scholar]
- [53].Gabrielli BG, Sarcevic B, Sinnamon J, et al. A cyclin D-Cdk4 activity required for G2 phase cell cycle progression is inhibited in ultraviolet radiation-induced G2 phase delay. J Biol Chem. 1999;274:13961–13969. [DOI] [PubMed] [Google Scholar]
- [54].Jirawatnotai S, Hu Y, Livingston DM, et al. Proteomic identification of a direct role for cyclin d1 in DNA damage repair. Cancer Res. 2012;72:4289–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhong Q, Hu Z, Li Q, et al. Cyclin D1 silencing impairs DNA double strand break repair, sensitizes BRCA1 wildtype ovarian cancer cells to olaparib. Gynecol Oncol. 2018;152:157–165. [DOI] [PubMed] [Google Scholar]
- [56].Minella AC, Swanger J, Bryant E, et al. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol. 2002;12:1817–1827. [DOI] [PubMed] [Google Scholar]
- [57].Bendjennat M, Boulaire J, Jascur T, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. [DOI] [PubMed] [Google Scholar]
- [58].Al-Khalaf HH, Hendrayani SF, Aboussekhra A. ATR controls the p21(WAF1/Cip1) protein up-regulation and apoptosis in response to low UV fluences. Mol Carcinog. 2012;51:930–938. [DOI] [PubMed] [Google Scholar]
- [59].Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–56. [DOI] [PubMed] [Google Scholar]
- [60].Itoh T, Linn S. The fate of p21CDKN1A in cells surviving UV-irradiation. DNA Repair (Amst). 2005;4:1457–1462. [DOI] [PubMed] [Google Scholar]
- [61].Lehmann AR, Niimi A, Ogi T, et al. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst). 2007;6:891–899. [DOI] [PubMed] [Google Scholar]
- [62].Soria G, Belluscio L, van Cappellen WA, et al. DNA damage induced Pol eta recruitment takes place independently of the cell cycle phase. Cell Cycle. 2009;8:3340–3348. [DOI] [PubMed] [Google Scholar]
- [63].Gary R, Ludwig DL, Cornelius HL, et al. The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J Biol Chem. 1997;272:24522–24529. [DOI] [PubMed] [Google Scholar]
- [64].Soria G, Speroni J, Podhajcer OL, et al. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J Cell Sci. 2008;121:3271–3282. [DOI] [PubMed] [Google Scholar]
- [65].Huschtscha LI, Holliday R. Limited and unlimited growth of SV40-transformed cells from human diploid MRC-5 fibroblasts. J Cell Sci. 1983;63:77–99. [DOI] [PubMed] [Google Scholar]
- [66].Masutani C, Araki M, Yamada A, et al. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. Embo J. 1999;18:3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Espinha G, Osaki JH, Magalhaes YT, et al. Rac1 GTPase-deficient HeLa cells present reduced DNA repair, proliferation, and survival under UV or gamma irradiation. Mol Cell Biochem. 2015;404:281–297. [DOI] [PubMed] [Google Scholar]
- [68].Magalhaes YT, Farias JO, Monteiro LF, et al. Measuring the contributions of the Rho Pathway to the DNA damage response in tumor epithelial cells. Methods Mol Biol. 2018;1821:339–355. [DOI] [PubMed] [Google Scholar]
- [69].Russo LC, Araujo CB, Iwai LK, et al. A Cyclin D2-derived peptide acts on specific cell cycle phases by activating ERK1/2 to cause the death of breast cancer cells. J Proteomics. 2017;151:24–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.