

Abstract
The telomere is the specialized nucleoprotein complex at the end of the chromosome. Its highly conserved 5′‐TTAGGG‐3′ repeats and shelterin protein complexes form a protective loop structure to maintain the integrity and stability of linear chromosomes. Although human somatic cells gradually shorten telomeres to undergo senescence or crisis, cancer cells activate telomerase, or the recombination‐based mechanism to maintain telomeres and exhibit immortality. As the most frequent non‐coding mutations in cancer, gain‐of‐function mutations in the promoter region of the telomerase catalytic subunit, TERT, trigger telomerase activation. Promoter methylation and copy number gain are also associated with the enhanced TERT expression. Although telomerase inhibitors were pioneered from telomere‐directed therapeutics, their efficacies are limited to cancer with short telomeres and some hematological malignancies. Other therapeutic approaches include a nucleoside analog incorporated to telomeres and TERT promoter‐driven oncolytic adenoviruses. Tankyrase poly(ADP‐ribose) polymerase, a positive regulator of telomerase, has been rediscovered as a target for Wnt‐driven cancer. Meanwhile, telomeric nucleic acids form a higher‐order structure called a G‐quadruplex (G4). G4s are formed genome‐wide and their dynamics affect various events, including replication, transcription, and translation. G4‐stabilizing compounds (G4 ligands) exert anticancer effects and are in clinical investigations. Collectively, telomere biology has provided clues for deeper understanding of cancer, which expands opportunities to discover innovative anticancer drugs.
Keywords: cell immortality, G‐quadruplex, telomere, TERT promoter, Wnt/β‐catenin signaling
Starting from the chromosome ends, telomeres, and their functional modulators have brought new facets to our strategies for anticancer drug discovery. Promising approaches beyond targeting cell immortality include TERT/telomerase hijacking, Wnt signal inhibition, and stabilization of G‐quadruplexes.
1. INTRODUCTION
Telomeres are distinguished from DNA double‐strand breaks (DSBs), which otherwise induce cell cycle checkpoint, homologous recombination (HR), non‐homologous end‐joining, and cell senescence/death (Figure 1). Telomeric DNA consists of 5′‐TTAGGG‐3′ repeats in vertebrates, and bind to the protein complexes called shelterin. 1 Among the shelterin components (TRF1/TRF2/RAP1/TIN2/TPP1/POT1), TRF2 and POT1 play major roles in end‐capping. Mechanistically, the telomeric 3′‐overhang/G‐tail forms a lasso‐like “t‐loop” structure under the control of CDK‐mediated TRF2 phosphorylation, 2 and prevents the DNA damage response. In fission yeast, telomere loss causes lethality, whereas viable cells arise with all 3 chromosomes circularized. 3 These cells cannot produce viable spores, and it is possible that eukaryotic chromosomes need to be linear, not circular, for meiotic recombination.
Figure 1.
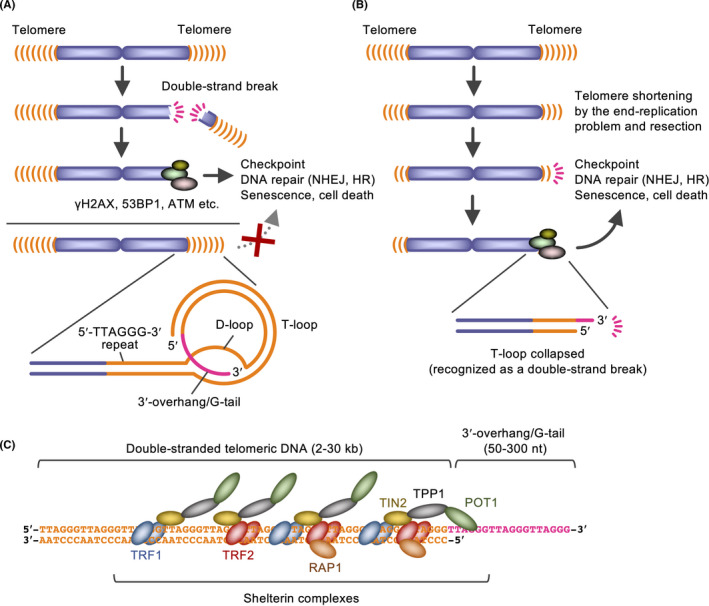
Functions, structure, and components of telomeres. A, Telomeres form t‐loops and protect chromosome ends from the DNA damage response. HR, homologous recombination; NHEJ, non‐homologous end‐joining. B, Telomeres gradually shorten according to the end‐replication problem and resection, which defines the cell replicative capacity. C, Shelterin complexes. TRF1/TRF2 and POT1 directly bind the double‐stranded and single‐stranded telomeric repeats, respectively
Linear chromosomes have another issue called the end‐replication problem; the classical replication machinery cannot completely replicate the DNA ends. Accordingly, telomeres gradually shorten at each replication in human somatic cells. Eventually, dysfunctional telomeres are marked by DNA damage response proteins, including γH2AX, 53BP1 and phosphorylated ATM, and the p53‐dependent response is induced. 4 Consequently, the cell undergoes replicative senescence, characterized by cell cycle arrest, flattened cell morphology, and senescence‐associated secretory phenotype. 5 Thus, telomere shortening prevents unlimited growth of cells (eg, pre‐cancerous cells that obtained oncogenic mutations). Meanwhile, telomeres are the buffer zone against the end‐replication problem: the telomeric sequence does not encode any gene, and its erosion will not cause loss of genomic information.
2. MECHANISMS AND IMPLICATIONS FOR ALTERED TELOMERE DYNAMICS IN CANCER
2.1. Telomerase confers cell immortality, a hallmark of cancer
Cells with infinite replicative capacity solve the end‐replication problem by activating the telomere‐synthesizing enzyme, telomerase. Telomerase holoenzyme consists of a catalytic subunit, TERT, and a template RNA, TR/TERC (Figure 2A,B). While TR is ubiquitously expressed, TERT is the limiting factor for telomerase activation. Ectopic TERT expression in fibroblasts maintains the telomere length and extends the replicative capacity or immortalizes the cells. 6 As well as proliferative germline cells and some reproductive cells, 85%‐95% of human cancer cells possess telomerase activity. 7 TERT has an RNA‐dependent RNA polymerase activity, which is also implicated for cancer progression. 8 TERT expression is mediated by various transcription factors, including MYC, SP‐1, E2F, and AP1. In addition, estrogen receptor α interacts with TERT promoter and induces its transcription. 9
Figure 2.
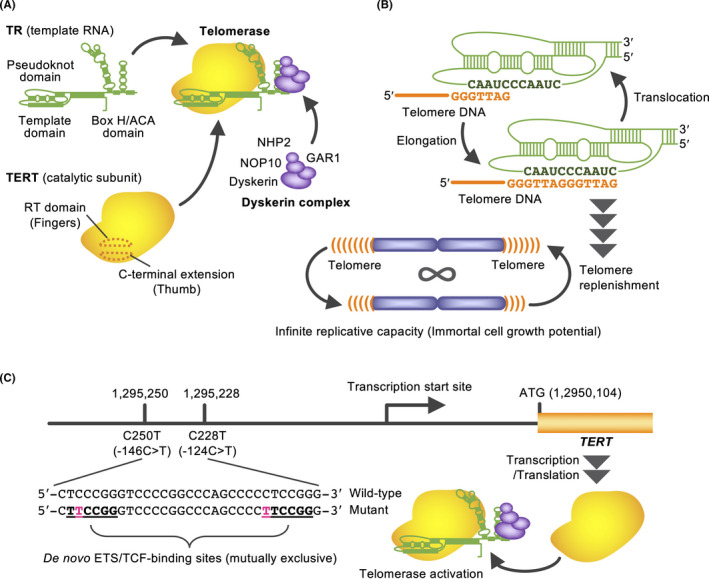
Telomerase‐mediated telomere synthesis and TERT promoter mutations. A, Telomerase components. Because telomerase uses RNA as a template for telomeric DNA synthesis, it is classified as a reverse transcriptase. B, The template region of TR anneals the telomeric 3′‐overhang, and telomerase repeats the strand synthesis and translocation. C, Gain‐of‐function mutations in the TERT promoter that produce ETS/TCF‐binding sites and cause TERT transcription
Recent cancer genome analyses have identified gain‐of‐function mutations in TERT promoter. 10 Mutually exclusive C228T/C250T mutations produce binding motifs for ETS/TCF family transcription factors and activate TERT (Figure 2C). These mutations are the most commonly observed non‐coding somatic mutations in cancer. For example, 83% of primary glioblastoma, 67% of melanoma, and 59% of bladder cancer harbor the TERT promoter mutations. The frequency of these mutations tends to be higher in cells that originally exhibited lower self‐renewal activity. 11 In addition to the TERT promoter mutations, TERT promoter methylation, and copy number gain of TERT are also associated with upregulation of TERT expression. 12
2.2. Telomere paradox in cancer and a potential role of the telomeric non‐coding RNA
Without telomerase, longer telomeres would be advantageous for the replicative lifespan of cells. Once telomerase is reactivated, however, cancer cells often maintain telomeres shorter than those of normal cells. 12 , 13 There would be reasons for this paradoxical phenomenon (Figure 3). First, a longer telomere has more TRF1s, which suppress telomerase access. This protein‐counting mechanism is conserved from yeast to human. 14 Second, length would not matter if t‐loops are intact. Third, shortened telomeres could easily induce genomic alterations, including aneuploidy, translocations and chromothripsis, 15 which are advantageous to cancer evolution. In fact, cancer with short telomeres exhibits poor prognosis. 16 Another explanation is that telomeres that are too long are disadvantageous to cancer. When human cancer cells with artificially elongated telomeres were injected into immunodeficient mice, the resulting tumors exhibited tissue reorganization, including duct‐like structure formation, downregulation of N‐cadherin (a poor prognostic factor), and repression of interferon‐stimulated genes (ISGs). 17 In those tumors, telomere‐elongated cancer cells expressed higher levels of the telomeric non‐coding RNA, TERRA. Because TERRA‐mimicking oligonucleotides inhibit ISG upregulation in three‐dimensional culture of cancer cells, 18 TERRA may repress ISGs in the telomere‐elongated tumors. Given that ISGs are implicated in cancer progression, 19 , 20 cancer cells may maintain short telomeres to allow ISG expression. 21 Furthermore, the telomere position effect modulates gene expression near telomeres and at long distances. 12 , 22 , 23
Figure 3.
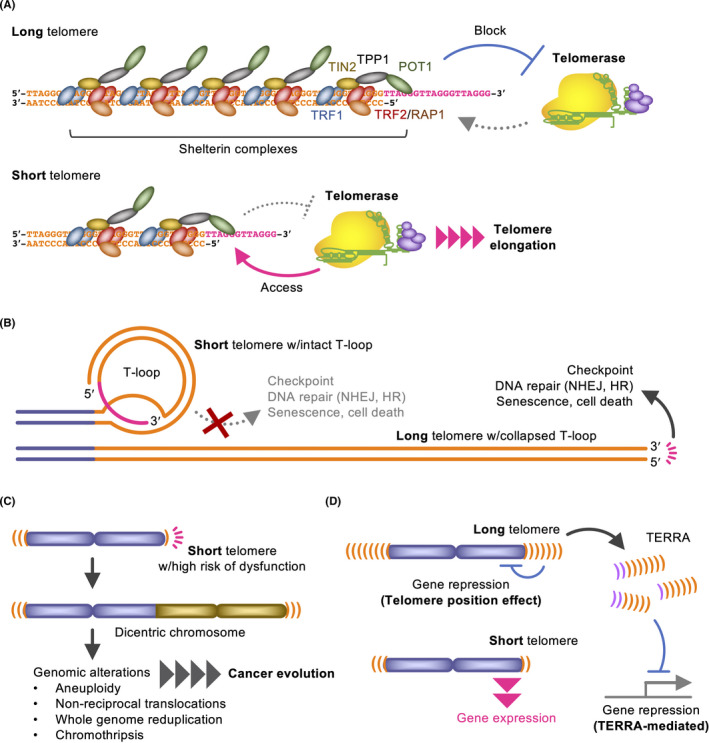
Possible implications for shortened telomeres in cancer. A, The protein‐counting mechanism blocks unlimited telomere elongation. B, Loop integrity but not the repeat length is important for the end‐protection. The TRF2 dominant‐negative mutant abolishes the 3′‐overhang and promptly decaps telomeres even if they are sufficiently long (lower). C, Short telomeres may easily become dysfunctional, this promotes genomic alterations and cancer evolution. D, Telomere length modulates gene expression by the telomere position effect and TERRA expression
2.3. ALTernative way to cell immortality without telomerase
Telomerase‐independent HR maintains telomeres in 5%‐15% of cancer cells. 24 , 25 This mechanism is called alternative lengthening of telomeres (ALT) and characterized by telomere length heterogeneity and formation of the nuclear promyelocytic leukemia (PML) bodies. ALT is more commonly observed in cancers from mesenchymal and neuroepithelial cell origins, including osteosarcoma, soft tissue sarcoma, and astrocytoma. Ectopic expression of TERT in ALT cells allows the co‐existence of telomerase‐mediated telomere maintenance and ALT, whereas telomerase‐positive cells have a factor that represses ALT. 26 At the genetic level, TERT promoter mutations are mutually exclusive with ALT‐associated loss‐of‐function mutations, including ATRX and DAXX, which work for chromatin remodeling at telomeres. 27 It has been postulated that ATRX/DAXX dysfunction induces loss of heterochromatin at telomeres, resulting in a recombination‐permissive status. ALT cells exhibit a reduced ability to release replication protein A from the single‐stranded telomere DNA, which presumably facilitates recruitment of ATM‐ and Rad3‐Related (ATR) kinase and telomeric HR. As a potential therapeutic strategy, it has been reported that ATR kinase inhibitors, such as VE‐821, preferentially inhibit the growth and induce apoptosis of ALT cells. 28
3. TELOMERASE AS A THERAPEUTIC TARGET AND BEYOND
3.1. Telomerase inhibitors induce telomere shortening and crisis in cancer cells
The first proof‐of‐concept for telomerase‐targeted therapy was established by a dominant‐negative mutant TERT, which causes telomere erosion and apoptosis of cancer cells. 29 Telomerase inhibitors, such as imetelstat/GRN163L, BIBR1532, and MST‐312, shorten telomeres and induce senescence/apoptosis in telomerase‐positive cancer cells. 30 , 31 , 32 , 33 The first and only telomerase inhibitor under clinical development is imetelstat, which is a lipid‐conjugated N3′→P5′ thio‐phosphoramidate oligonucleotide with complementary sequence to TR (Figure 4). However, the anticancer effects of telomerase inhibitors must await the emergence of critically shortened telomeres after continuous drug treatment. Accordingly, cancer cells with shorter telomeres are more sensitive to telomerase inhibitors. 34 , 35 Imetelstat can be administered to pediatric brain cancer patients for only 13 d on average. 36 Among the adverse effects, thrombocytopenia is frequent and major cause of discontinuation. Still, this drug has efficacies against myelofibrosis 37 and essential thrombocythemia, 38 and a clinical study is recruiting patients of myelodysplastic syndromes. In experimental settings, acquired resistance to telomerase inhibition is caused by enhanced access of the residual telomerase activity to shortened telomeres 32 or activation of ALT. 39
Figure 4.
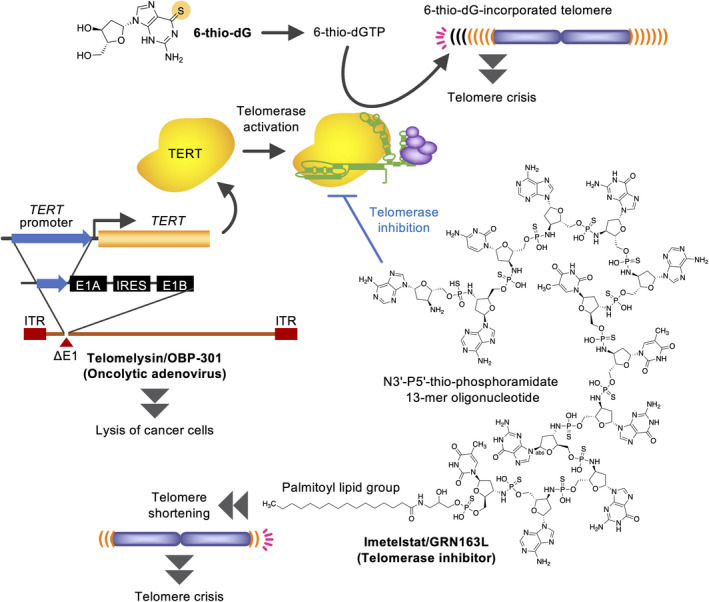
Telomerase‐targeted cancer therapeutics. 6‐Thio‐2′‐deoxyguanosine (6‐thio‐dG) is a mimetic of 2'‐deoxyguanosine, in which the oxygen atom of guanine is substituted with a sulfur atom. This compound is incorporated into telomeres in a telomerase‐dependent manner, which causes immediate crisis in telomerase‐positive cells (top). TERT promoter is used to construct oncolytic adenoviruses (lower left). Telomerase inhibitors shorten telomeres and eventually induce crisis in cancer (lower right). For example, imetelstat consists of a N3′‐P5′‐thio‐phosphoramidate 13‐mer oligonucleotide, which is complementary to the template region of TR, and a 5′ palmitoyl (C16) lipid group for enhanced cell permeability. IRES, internal ribosome entry site; ITR, inverted terminal repeat
3.2. 6‐Thio‐2′‐deoxyguanosine hijacks telomerase to induce telomere dysfunction
Instead of its inhibition, telomerase activity may be also used for producing anticancer impacts. The nucleoside analog 6‐thio‐2′‐deoxyguanosine (6‐thio‐dG) is incorporated into telomeres by telomerase (Figure 4). 6‐Thio‐dG‐incorporated telomeres induce DNA damage response and senescence or crisis only in telomerase‐positive cells. 40 Because this mechanism does not involve the end‐replication problem, its efficacy emerges rapidly. In mouse xenograft models, 6‐thio‐dG induces telomere dysfunction and inhibits tumor growth without significant side effects. 6‐Thio‐dG has been effective against various cancers, including NRAS‐driven melanoma, BRAF inhibitor/immunotherapy‐resistant melanoma, therapy‐resistant lung cancer, and pediatric brain cancer in preclinical settings. 41 In 6‐thio‐dG‐resistant cancer cells, SLC43A3, an equilibrative nucleobase transporter, is downregulated and is thus proposed as a biomarker for the drug sensitivity. 42
To date, the relationship between types of TERT gene abnormalities and the effects of telomere‐directed therapeutics remain speculative. For example, TERT promoter mutations and methylation are associated with shorter telomeres compared with other types of TERT alteration, 12 suggesting that these types of tumors might be more sensitive to telomerase inhibitors. In contrast, copy number gain of TERT is predicted to correlate with the highest telomerase activity among various TERT alterations. 12 Accordingly, TERT‐amplified tumors might be more susceptible to the antiproliferative effect of 6‐thio‐dG because this compound is incorporated into telomeres in a telomerase‐dependent manner.
3.3. Adenoviral gene therapies that induce telomerase promoter‐driven oncolytic activities
Telomelysin/OBP‐301 is a recombinant adenovirus, in which adenoviral E1A/E1B expression is driven by TERT promoter (Figure 4). 43 This adenovirus is selectively propagated in TERT‐positive cells and efficiently kills them, including esophageal, gastric, and colorectal cancers. Telomelysin also inhibits lymph node metastasis and enhances the efficacy of ionizing radiation in orthotopic colorectal and esophageal cancer xenografts, respectively. Cancer cells killed by OBP‐502, a telomelysin variant for mouse cells, release ATP and HMGB1 protein, which recruit CD8‐positive lymphocytes and inhibit Foxp3‐positive lymphocyte infiltration into tumors. Accordingly, OBP‐502 enhances the anticancer effect of an anti‐PD‐1 antibody. 44 Furthermore, OBP‐702, a p53‐expressing telomelysin variant, inhibits migration, invasion, and orthotopic xenograft tumor growth of pancreatic ductal adenocarcinoma cells more potently than telomelysin. 45
Other TERT promoter‐driven oncolytic adenoviruses include those driven by modified TERT promoters, which contain additional SP‐1/MYC‐binding sites and are combined with E2F promoter and hypoxia response elements. 46 These adenoviruses are efficiently replicated in cancer cells and exhibit anticancer efficacy. In addition, TERT promoter‐driven activation of the CRISPR/Cas9 system is used for targeting the HRAS gene in bladder cancer cells. 47
3.4. Tankyrase as a positive regulator for telomerase and Wnt signaling
The efficiency of telomere shortening by a telomerase inhibitor decreases when telomeres are shortened because the residual telomerase activity easily accesses the shortened telomeres. 32 This paradoxical issue is alleviated by blocking tankyrase, a member of poly(ADP‐ribose) polymerase (PARP) family (Figure 5A,B). 32 Tankyrase has 2 homologs (TNKS/PARP‐5a and TNKS2/PARP‐5b) and has been identified as a TRF1‐binding protein. 48 It recognizes TRF1 at the ankyrin repeat cluster regions, 49 and PARylated TRF1 dissociates from telomeres and are ubiquitinated for proteasomal degradation. 48 , 50 PARP inhibitors that block tankyrase‐mediated PARylation retain more TRF1s on telomeres and fasten telomere shortening by MST‐312. 32 Intriguingly, murine (Mus musculus) and rat TRF1s lack tankyrase‐binding motifs and are not PARylated by tankyrase. 51 Given that mice and rats have much longer telomeres (up to 150 kb) than humans (about 10 kb at birth) and activate telomerase in somatic tissues, tankyrase may not be necessary for these rodent telomerases.
Figure 5.
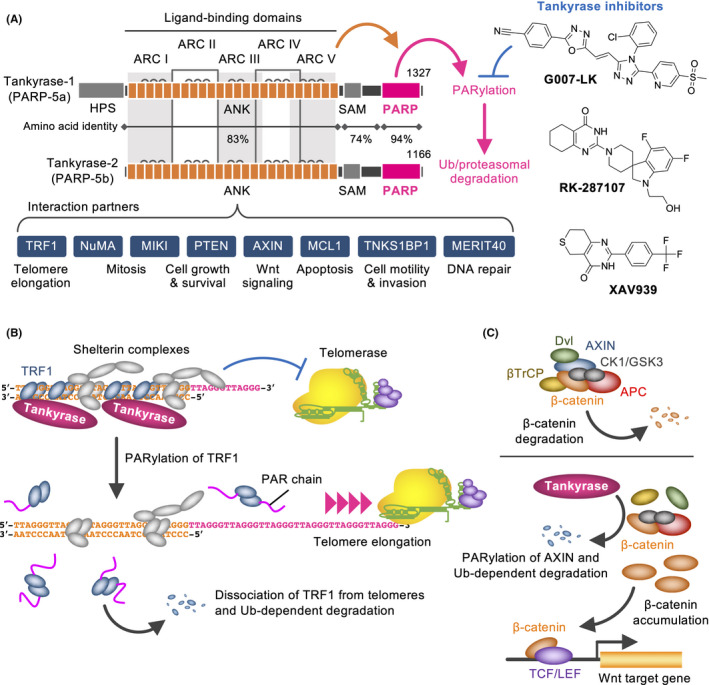
Tankyrase as a therapeutic target for cancer. A, Structures, partners and functions of tankyrases (left). Representative tankyrase inhibitors are also shown (right). ANK, ankyrin repeats; ARC, ankyrin repeat cluster; HPS, His‐Pro‐Ser motif; SAM, sterile α motif; PARP, poly(ADP‐ribose) polymerase domain. B, Tankyrase PARylates TRF1, resulting in promotion of telomerase access and telomere elongation. Ub, ubiquitin. C, APC destruction complex induces Ub‐dependent β‐catenin degradation (upper). Tankyrase PARylates AXIN and its Ub‐dependent degradation. This causes β‐catenin accumulation and enhances target gene expression (lower)
Apart from human TRF1, tankyrase‐binding proteins include NuMA, MIKI, MCL1, TNKS1BP1, AXIN1/2, PTEN, and MERIT40. Tankyrase PARylates them, which affects proliferation, mitosis, apoptosis, motility, invasion, and DNA repair. Among such functions, most striking is the positive regulation of Wnt/β‐catenin signaling. Tankyrase PARylates AXIN, a negative regulator for Wnt/β‐catenin signaling 52 (Figure 5C). PARylated AXIN is ubiquitinated by RNF146 E3 ligase and subjected to proteasomal degradation. 53 Tankyrase inhibitors, such as XAV939, G007‐LK, and RK‐287107, block AXIN PARylation, which in turn stabilizes AXIN and degrades β‐catenin. 52 , 54 , 55 Accordingly, tankyrase inhibitors downregulate Wnt/β‐catenin signaling and block colorectal cancer cell growth in xenograft models. APC loss‐of‐function mutations are potential predictive biomarkers of tankyrase inhibitors, 56 , 57 whereas β‐catenin/CTNNB1 gain‐of‐function mutations confer the drug resistance. 58 Because Wnt/β‐catenin signaling works for intestinal epithelial cells, continuous administration of tankyrase inhibitors may cause intestinal toxicity. 54 Regardless, tankyrase inhibitors target CD44‐positive colorectal cancer stem cells through c‐KIT repression and exhibit promising antitumor activities in combination with irinotecan. 59
4. G‐QUADRUPLEX: A PARADIGM SHIFT FROM THE LENGTH TO SHAPE OF TELOMERES
4.1. Biological significance of G‐quadruplex and its connection with cancer
The free energies required for histone association with telomeric DNA are 10‐15 times higher than average DNAs. The reason for such G‐rich repeats being conserved is elusive. Intriguingly, telomeric DNA/RNA can form a non‐canonical nucleic acid structure called G‐quadruplex (G4) (Figure 6A). G4 comprises stacks of planar G‐quartets, each formed by 4 guanines through Hoogsteen hydrogen bonding. 60 G4‐forming sequences exist in a genome‐wide manner, consisting of 4 G‐tracts with loop sequences between the G‐tracts.
Figure 6.
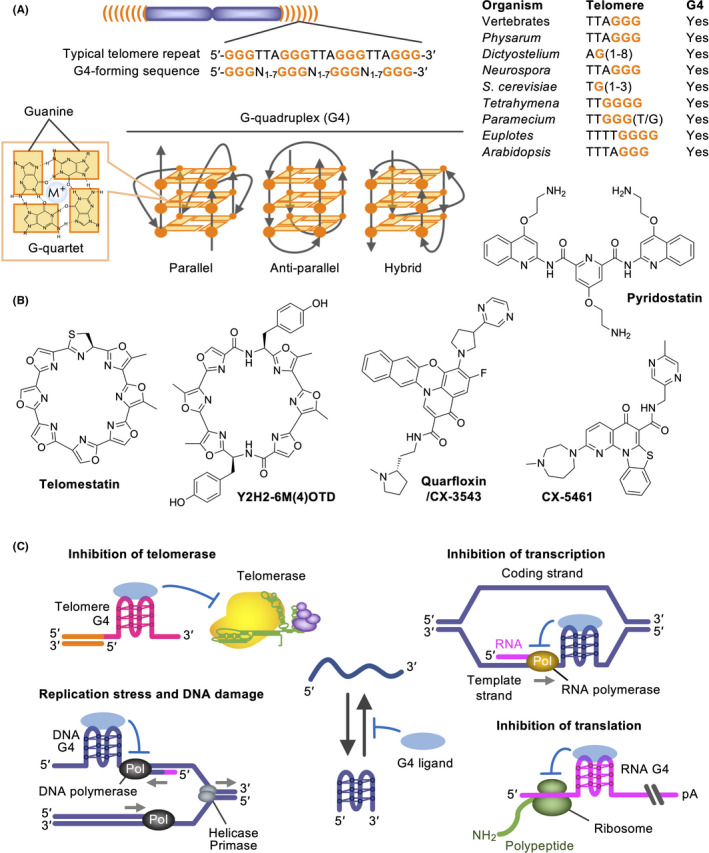
G‐quadruplexes as therapeutic targets for cancer. A, G‐quadruplex (G4)‐forming sequences, such as telomeric repeats of various organisms, and G4 conformations. B, G4 ligands, which recognize and stabilize G4s. C, Consequences of G4 stabilization. It is postulated that G4 ligands exert anticancer effects through telomeric and non‐telomeric DNA damage induction and transcriptional/translational perturbation of cancer‐related genes
G4s affect replication, transcription, mRNA splicing, translation, and epigenetics. Although G4s stall DNA replication forks, G4 formed at the origin G‐rich repeated element contributes to replication origin activity. 61 G4s in transcription sites bidirectionally regulate transcription, presumably by recruiting transcription factors or inhibiting the progression of RNA polymerase II. 62 , 63 G4s on mRNA repress translation by blocking the progression of ribosomes or the recruitment of translation initiation factors. 64 , 65 , 66
Tumor tissues exhibit elevated G4 formation compared with normal tissues. 67 Because dysfunction of G4 helicases, such as Werner syndrome protein (WRN) and Bloom syndrome protein (BLM), causes genome instability, 68 G4s may accelerate cancer genome evolution. Of note, putative G4‐forming sequences and G4s are enriched in proto‐oncogenes and cancer‐related loci. 69 , 70 Upregulation of eIF4A, a translation initiation factor with helicase activity, facilitates oncogene translation, including MYC, MYB, NOTCH1, MDM2, and BCL2, by unwinding G4s on the 5′ UTR of mRNAs, and promotes T‐cell acute lymphoblastic leukemia. 66 Furthermore, G4s in TERRA are implicated for ISG repression. 18 These observations suggest a functional linkage between altered G4 dynamics and carcinogenesis.
4.2. G‐quadruplex ligands as novel anticancer therapeutic drugs
G4 ligands are chemical compounds that stabilize G4s (Figure 6B). Telomestatin, a natural G4 ligand from Streptomyces anulatus, binds G4s and inhibits telomerase activity. 71 Telomestatin removes TRF2 and POT1 from telomeres and causes telomere dysfunction in cancer cells. 72 , 73 Telomestatin especially inhibits the growth of glioma stem cells by inducing replication stress and DNA damage. 74 , 75 Y2H2‐6M(4)‐oxazole telomestatin derivative inhibits the growth of glioma stem cells and glioblastoma cells in vivo. 76 Other G4 ligands, pyridostatin, quarfloxin/CX‐3543 and CX‐5461, cause synthetic lethality in BRCA1/2‐deficient 77 , 78 and ATRX‐deficient cancer cells. 79 CM03, another G4 ligand, inhibits the growth of pancreatic xenograft tumors. 80 This ligand represses the genes that have putative G4‐forming sequences and are frequently upregulated in pancreatic cancer.
Together, the anticancer impacts of G4 ligands involve their DNA damaging activities and abilities to alter cancer‐related gene expression (Figure 6C). Because G4s in proto‐oncogenes repress their translation, 66 those stabilized by G4 ligands may also contribute to therapeutic efficacy. Among various G4 ligands, quarfloxin and CX‐5461 are being clinically investigated. As exemplified by the CX‐5461 trial, which recruits patients with BRCA1/2 or HR deficiency germline aberrations, it is important to set biomarkers to predict the patients who will benefit from treatment.
5. CONCLUSIONS
The advancement of cancer genome analyses has revealed detailed genomic landscapes of cancer. This knowledge and expanding repertoire of molecularly targeted drugs have opened the door to cancer precision medicine. Although telomerase‐mediated cell immortality is a general hallmark of cancer, at least in cultures, anticancer impacts of telomerase inhibitors are limited on those with very short telomeres. In contrast, the nucleoside substrate analog and TERT promoter‐driven oncolytic adenoviruses seem to be broadly applicable to telomerase‐positive cancers. Furthermore, tankyrase inhibitors are cutting edge seeds that target the yet undruggable Wnt pathway. G4 ligands are intriguing drug seeds that target the shape of nucleic acids, although the precise mechanisms for the efficacy await further studies. In conclusion, starting from the chromosome ends, telomeres and their functional modulators have brought new facets to our strategies for anticancer drug discovery. The time is coming to harvest these fruits for cancer patients.
CONFLICTS OF INTEREST
I received a research grant from the Nippon Foundation.
ACKNOWLEDGMENTS
I would like to thank Yukiko Muramatsu for survey assistance, and the laboratory members for their discussions. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (19H03523 and 20H04789), grants from Practical Research for Innovative Cancer Control (20ck0106476h0002) and the Translational Research Program; Strategic Promotion for Practical Application of Innovative Medical Technology, the Japan Agency for Medical Research and Development, and from the Nippon Foundation. Funding for the open access charge: JSPS/19H03523.
Seimiya H. Crossroads of telomere biology and anticancer drug discovery. Cancer Sci. 2020;111:3089–3099. 10.1111/cas.14540
REFERENCES
- 1. de Lange T. Shelterin‐mediated telomere protection. Annu Rev Genet. 2018;52(1):223‐247. [DOI] [PubMed] [Google Scholar]
- 2. Sarek G, Kotsantis P, Ruis P, et al. CDK phosphorylation of TRF2 controls t‐loop dynamics during the cell cycle. Nature. 2019;575:523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naito T, Matsuura A, Ishikawa F. Circular chromosome formation in a fission yeast mutant defective in two ATM homologues. Nat Genet. 1998;20:203‐206. [DOI] [PubMed] [Google Scholar]
- 4. d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, et al. A DNA damage checkpoint response in telomere‐initiated senescence. Nature. 2003;426:194‐198. [DOI] [PubMed] [Google Scholar]
- 5. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life‐span by introduction of telomerase into normal human cells. Science. 1998;279:349‐352. [DOI] [PubMed] [Google Scholar]
- 7. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787‐791. [DOI] [PubMed] [Google Scholar]
- 8. Yasukawa M, Ando Y, Yamashita T, et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat Commun. 2020;11:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyo S, Takakura M, Fujiwara T, Inoue M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008;99:1528‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell RJA, Rube HT, Xavier‐Magalhaes A, et al. Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res. 2016;14:315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self‐renewal. Proc Natl Acad Sci U S A. 2013;110:6021‐6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiba K, Lorbeer FK, Shain AH, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two‐step mechanism. Science. 2017;357:1416‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shore D, Bianchi A. Telomere length regulation: coupling DNA end processing to feedback regulation of telomerase. EMBO J. 2009;28:2309‐2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gay‐Bellile M, Romero P, Cayre A, et al. ERCC1 and telomere status in breast tumours treated with neoadjuvant chemotherapy and their association with patient prognosis. J Pathol Clin Res. 2016;2:234‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirashima K, Migita T, Sato S, Muramatsu Y, Ishikawa Y, Seimiya H. Telomere length influences cancer cell differentiation in vivo. Mol Cell Biol. 2013;33:2988‐2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirashima K, Seimiya H. Telomeric repeat‐containing RNA/G‐quadruplex‐forming sequences cause genome‐wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015;43:2022‐2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferon‐related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490‐18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khodarev NN, Roach P, Pitroda SP, et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One. 2009;4:e5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okamoto K, Seimiya H. Revisiting telomere shortening in cancer. Cells. 2019;8(2):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baur JA, Zou Y, Shay JW, Wright WE. Telomere position effect in human cells. Science. 2001;292:2075‐2077. [DOI] [PubMed] [Google Scholar]
- 23. Robin JD, Ludlow AT, Batten K, et al. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014;28:2464‐2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319‐330. [DOI] [PubMed] [Google Scholar]
- 25. Heaphy CM, Subhawong AP, Hong S‐M, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608‐1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perrem K, Colgin LM, Neumann AA, Yeager TR, Reddel RR. Coexistence of alternative lengthening of telomeres and telomerase in hTERT‐transfected GM847 cells. Mol Cell Biol. 2001;21:3862‐3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flynn RL, Cox KE, Jeitany M, et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347:273‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hahn WC, Stewart SA, Brooks MW, et al. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164‐1170. [DOI] [PubMed] [Google Scholar]
- 30. Dikmen ZG, Gellert GC, Jackson S, et al. In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res. 2005;65:7866‐7873. [DOI] [PubMed] [Google Scholar]
- 31. Damm K, Hemmann U, Garin‐Chesa P, et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001;20:6958‐6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T. Tankyrase 1 as a target for telomere‐directed molecular cancer therapeutics. Cancer Cell. 2005;7:25‐37. [DOI] [PubMed] [Google Scholar]
- 33. Seimiya H, Oh‐hara T, Suzuki T, et al. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST‐312, MST‐295, and MST‐199. Mol Cancer Ther. 2002;1:657‐665. [PubMed] [Google Scholar]
- 34. Chiappori AA, Kolevska T, Spigel DR, et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non‐small‐cell lung cancer. Ann Oncol. 2015;26:354‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujiwara C, Muramatsu Y, Nishii M, et al. Cell‐based chemical fingerprinting identifies telomeres and lamin A as modifiers of DNA damage response in cancer cells. Sci Rep. 2018;8:14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salloum R, Hummel TR, Kumar SS, et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: a pediatric brain tumor consortium study. J Neurooncol. 2016;129:443‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908‐919. [DOI] [PubMed] [Google Scholar]
- 38. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373:920‐928. [DOI] [PubMed] [Google Scholar]
- 39. Hu J, Hwang S, Liesa M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148:651‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6‐thio‐2'‐deoxyguanosine. Cancer Discov. 2015;5:82‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sugarman ET, Zhang G, Shay JW. In perspective: An update on telomere targeting in cancer. Mol Carcinog. 2019;58:1581‐1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mender I, Batten K, Peyton M, et al. SLC43A3 Is a biomarker of sensitivity to the telomeric DNA damage mediator 6‐Thio‐2'‐Deoxyguanosine. Cancer Res. 2020;80:929‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fujiwara T. Multidisciplinary oncolytic virotherapy for gastrointestinal cancer. Ann Gastroenterol Surg. 2019;3:396‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanaya N, Kuroda S, Kakiuchi Y, et al. Immune modulation by telomerase‐specific oncolytic adenovirus synergistically enhances antitumor efficacy with Anti‐PD1 antibody. Mol Ther. 2020;28:794‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koujima T, Tazawa H, Ieda T, et al. Oncolytic virus‐mediated targeting of the ERK signaling pathway inhibits invasive propensity in human pancreatic cancer. Mol Ther Oncolytics. 2020;17:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Y, Hong J, Oh JE, Yoon AR, Yun CO. Potent antitumor effect of tumor microenvironment‐targeted oncolytic adenovirus against desmoplastic pancreatic cancer. Int J Cancer. 2018;142:392‐413. [DOI] [PubMed] [Google Scholar]
- 47. Huang X, Zhuang C, Zhuang C, Xiong T, Li Y, Gui Y. An enhanced hTERT promoter‐driven CRISPR/Cas9 system selectively inhibits the progression of bladder cancer cells. Mol Biosyst. 2017;13:1713‐1721. [DOI] [PubMed] [Google Scholar]
- 48. Hsiao SJ, Smith S. Tankyrase function at telomeres, spindle poles, and beyond. Biochimie. 2008;90:83‐92. [DOI] [PubMed] [Google Scholar]
- 49. Seimiya H, Muramatsu Y, Smith S, Tsuruo T. Functional subdomain in the ankyrin domain of tankyrase 1 required for poly(ADP‐ribosyl)ation of TRF1 and telomere elongation. Mol Cell Biol. 2004;24:1944‐1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang W, Dynek JN, Smith S. TRF1 is degraded by ubiquitin‐mediated proteolysis after release from telomeres. Genes Dev. 2003;17:1328‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muramatsu Y, Ohishi T, Sakamoto M, Tsuruo T, Seimiya H. Cross‐species difference in telomeric function of tankyrase 1. Cancer Sci. 2007;98:850‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang S‐M, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614‐620. [DOI] [PubMed] [Google Scholar]
- 53. DaRosa PA, Wang Z, Jiang X, et al. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP‐ribosyl)ation signal. Nature. 2015;517:223‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lau T, Chan E, Callow M, et al. A novel tankyrase small‐molecule inhibitor suppresses APC mutation‐driven colorectal tumor growth. Cancer Res. 2013;73:3132‐3144. [DOI] [PubMed] [Google Scholar]
- 55. Mizutani A, Yashiroda Y, Muramatsu Y, et al. RK‐287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018;109:4003‐4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tanaka N, Mashima T, Mizutani A, et al. APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol Cancer Ther. 2017;16:752‐762. [DOI] [PubMed] [Google Scholar]
- 57. Schatoff EM, Goswami S, Zafra MP, et al. Distinct colorectal cancer‐associated APC mutations dictate response to tankyrase inhibition. Cancer Discov. 2019;9:1358‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mashima T, Taneda Y, Jang M‐K, et al. mTOR signaling mediates resistance to tankyrase inhibitors in Wnt‐driven colorectal cancer. Oncotarget. 2017;8:47902‐47915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jang MK, Mashima T, Seimiya H. Tankyrase inhibitors target colorectal cancer stem cells via AXIN‐dependent downregulation of c‐KIT tyrosine kinase. Mol Cancer Ther. 2020;19:765‐776. [DOI] [PubMed] [Google Scholar]
- 60. Sen D, Gilbert W. Formation of parallel four‐stranded complexes by guanine‐rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364‐366. [DOI] [PubMed] [Google Scholar]
- 61. Prorok P, Artufel M, Aze A, et al. Involvement of G‐quadruplex regions in mammalian replication origin activity. Nat Commun. 2019;10:3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gray LT, Vallur AC, Eddy J, Maizels N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat Chem Biol. 2014;10:313‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Johnson JE, Cao K, Ryvkin P, Wang LS, Johnson FB. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G‐quadruplex forming potential. Nucleic Acids Res. 2010;38:1114‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Arora A, Dutkiewicz M, Scaria V, Hariharan M, Maiti S, Kurreck J. Inhibition of translation in living eukaryotic cells by an RNA G‐quadruplex motif. RNA. 2008;14:1290‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kumari S, Bugaut A, Huppert JL, Balasubramanian S. An RNA G‐quadruplex in the 5' UTR of the NRAS proto‐oncogene modulates translation. Nat Chem Biol. 2007;3:218‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wolfe AL, Singh K, Zhong YI, et al. RNA G‐quadruplexes cause eIF4A‐dependent oncogene translation in cancer. Nature. 2014;513:65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Biffi G, Tannahill D, Miller J, Howat WJ, Balasubramanian S. Elevated levels of G‐quadruplex formation in human stomach and liver cancer tissues. PLoS One. 2014;9:e102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Paeschke K, Bochman ML, Garcia PD, et al. Pif1 family helicases suppress genome instability at G‐quadruplex motifs. Nature. 2013;497:458‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887‐3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hänsel‐Hertsch R, Beraldi D, Lensing SV, et al. G‐quadruplex structures mark human regulatory chromatin. Nat Genet. 2016;48:1267‐1272. [DOI] [PubMed] [Google Scholar]
- 71. Shin‐ya K, Wierzba K, Matsuo K, et al. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123:1262‐1263. [DOI] [PubMed] [Google Scholar]
- 72. Tahara H, Shin‐Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G‐Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3' telomeric overhang in cancer cells. Oncogene. 2006;25:1955‐1966. [DOI] [PubMed] [Google Scholar]
- 73. Gomez D, Wenner T, Brassart B, et al. Telomestatin‐induced telomere uncapping is modulated by POT1 through G‐overhang extension in HT1080 human tumor cells. J Biol Chem. 2006;281:38721‐38729. [DOI] [PubMed] [Google Scholar]
- 74. Miyazaki T, Pan Y, Joshi K, et al. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G‐quadruplex and inhibition of the proto‐oncogene, c‐Myb. Clin Cancer Res. 2012;18:1268‐1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hasegawa D, Okabe S, Okamoto K, Nakano I, Shin‐ya K, Seimiya H. G‐quadruplex ligand‐induced DNA damage response coupled with telomere dysfunction and replication stress in glioma stem cells. Biochem Biophys Res Commun. 2016;471:75‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nakamura T, Okabe S, Yoshida H, et al. Targeting glioma stem cells in vivo by a G‐quadruplex‐stabilizing synthetic macrocyclic hexaoxazole. Sci Rep. 2017;7:3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zimmer J, Tacconi EMC, Folio C, et al. Targeting BRCA1 and BRCA2 deficiencies with G‐Quadruplex‐interacting compounds. Mol Cell. 2016;61:449‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xu H, Di Antonio M, McKinney S, et al. CX‐5461 is a DNA G‐quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun. 2017;8:14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Y, Yang J, Wild AT, et al. G‐quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX‐deficient malignant glioma. Nat Commun. 2019;10:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Marchetti C, Zyner KG, Ohnmacht SA, et al. Targeting multiple effector pathways in pancreatic ductal adenocarcinoma with a G‐quadruplex‐binding small molecule. J Med Chem. 2018;61:2500‐2517. [DOI] [PMC free article] [PubMed] [Google Scholar]