

Abstract
The Far UpStream Element (FUSE)-binding protein-interacting repressor (FIR), a c-myc transcriptional suppressor, is alternatively spliced removing the transcriptional repression domain within exon 2 (FIRΔexon2) in colorectal cancers. SAP155 is a subunit of the essential splicing factor 3b (SF3b) subcomplex in the spliceosome. This study aims to study the significance of the FIR–SAP155 interaction for the coordination of c-myc transcription, pre-mRNA splicing, and c-Myc protein modification, as well as to interrogate FIRΔexon2 for other functions relating to altered FIR pre-mRNA splicing. Knockdown of SAP155 or FIR was used to investigate their reciprocal influence on each other and on c-myc transcription, pre-mRNA splicing, and protein expression. Pull down from HeLa cell nuclear extracts revealed the association of FIR, FIRΔexon2, and SF3b subunits. FIR and FIRΔexon2 were coimmunoprecipitated with SAP155. FIR and FIRΔexon2 adenovirus vector (Ad–FIR and Ad–FIRΔexon2, respectively) were prepared to test for their influence on c-myc expression. FIR, SAP155, SAP130, and c-myc were coordinately upregulated in human colorectal cancer. These results reveal that SAP155 and FIR/FIRΔexon2 form a complex and are mutually upregulating. Ad–FIRΔexon2 antagonized Ad–FIR transcriptional repression of c-myc in HeLa cells. Because FIRΔexon2 still carries RRM1 and RRM2 and binding activity to FUSE, it is able to displace repression competent FIR from FUSE in electrophoretic mobility shift assays, thus thwarting FIR-mediated transcriptional repression by FUSE. Thus aberrant FIRΔexon2 production in turn sustained c-Myc expression. In conclusion, altered FIR and c-myc pre-mRNA splicing, in addition to c-Myc expression by augmented FIR/FIRΔexon2–SAP155 complex, potentially contribute to colorectal cancer development.
Introduction
c-Myc plays a critical role in cell proliferation and tumorigenesis. The c-myc proto-oncogene is activated in various tumors, and its ectopic expression generally induces transformation. The expression of c-Myc is tightly regulated in all stages of macromolecular biosynthesis, but how its regulation is coordinated between transcription, pre-mRNA splicing, and c-Myc protein modification remains largely unexplored. The Far UpStream Element (FUSE) is a sequence required for the proper transcriptional regulation of the human c-myc gene (1). The FUSE is located 1.5 kb upstream of the c-myc promoter P1 and recognized by the FUSE-binding protein (FBP); FBP is a transcription factor that stimulates c-myc expression through FUSE (2-4). Yeast 2-hybrid analysis revealed that FBP binds to a protein with transcriptional inhibitory activity termed the FBP-interacting repressor (FIR); by suppressing the TFIIH/p89/XPB helicase, FIR represses c-myc transcription (5). Cells from XPB and XPD patients are defective in FIR repression, indicating that mutations in TFIIH impair c-myc transcriptional regulation by FIR, which contributes to tumor development (6). In addition, FIRΔexon2, an exon 2 lacking splice variant of FIR devoid of c-myc repression activity, is frequently found in human primary colorectal cancers but not in the corresponding noncancerous epithelium, indicating that a dominant, repression-defective FIR could be generated by altered pre-mRNA splicing in cancers (7). Thus, FIRΔexon2 expression has the potential to promote tumor development by disabling authentic c-myc repression of FIR; thereby, high levels of c-Myc will sustain growth and often promote apoptosis by increasing the cell division rate, with associated genomic instability and checkpoint-driven apoptosis as consequences.
Splicing factor 3b (SF3b) is a subcomplex of the U2 small nuclear ribonucleoprotein (snRNP) in the spliceosome. SF3b consists of SAP130, SAP145, SAP155, and p14 subunits in nearly equimolar stoichiometry quantities (8). The p14 subunit, cross-linked to the branch point adenosine of pre-mRNA introns within the spliceosome, interacts stably with SAP155, and thus, SF3b is required for intron recognition (9). PUF60, a splicing variant of FIR, is a splicing factor-associated protein (8, 10). However, how FIR directly or indirectly interacts with U2 snRNPs is not yet known. Recently, SAP155 was found to directly bind to PUF60 (11, 12), but this was not determined in the case of FIR. In addition, 2 natural chemical derivatives, spliceostatin A (SSA; ref. 13) and pladienolide (14), inhibit SF3b and thereby induce an antitumor effect. These results imply that SF3b normally promotes tumors.
In this study, the SAP155-mediated regulation of FIR/FIRΔexon2 expression was investigated along with its pre-mRNA splicing in c-myc gene expression. We examined SF3b expression in excised human colorectal cancer tissues, effects of SAP155 knockdown or SSA treatment on FIR pre-mRNA splicing, and total protein expression of FIRs in terms of c-Myc repression. We found that SAP155 is required for FIR expression and vice versa, and that SAP155 regulates alternative splicing of FIR. Both FIR and FIRΔexon2 were pulled down with SAP155. Importantly, FIRΔexon2 could markedly enhance c-Myc expression, whereas FIR suppresses c-Myc expression. Thus, sustained FIR/FIRΔexon2–SAP155 interaction affects the well-established functions of FIR and SAP155, and hence, interferes with transcription and alternative splicing of c-myc gene, respectively. In addition, the reason why FIR, FIRΔexon2, and SAP155 are activated in colorectal cancers is also discussed.
Materials and Methods
Excised human tumor samples
Tissues from 34 cases of primary colorectal cancer were surgically excised at Chiba University Hospital. The tumor samples were obtained from tumor epithelium immediately after surgical excision, and corresponding nontumor epithelial samples were taken 5 to 10 cm away from the tumor. Two pathologists microscopically confirmed all tissue samples as adenocarcinomas. All excised tissues were immediately placed in liquid nitrogen and stored at −80°C until analysis. Written informed consent was obtained from each patient before surgery.
Cell culture
HeLa, HCT116, and 293T cell lines were purchased from American Type Culture Collection (ATCC) and stored in the liquid nitrogen before use. Cells were grown at 37° C in 5% CO2 in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FBS (Invitrogen) and 1% penicillin–streptomycin (Invitrogen).
Protein extraction from tissue samples and immunoblotting
Proteins from whole-cell extracts were dissolved in a sample buffer as described previously (7). The amount of protein in the supernatant was measured by a protein assay (Bio-Rad). The proteins were separated by electrophoresis on polyacrylamide gels of suitable concentration and transferred to a polyvinylidene fluoride membrane (Millipore) using a tank transfer apparatus (Bio-Rad). The membrane was blocked with 5% skim milk in PBS for 1 hour.
Western blot and antibodies
Protein extracts were separated by electrophoresis on a 7.5% to 15% Perfect NT Gel (DRC). Proteins were then transferred to polyvinylidene fluoride membranes (Millipore) using a tank transfer apparatus (Bio-Rad). The membranes were blocked with 0.5% skim milk in PBS. The primary mouse monoclonal antibody against FIR C-terminus (6B4) was prepared by Dr Nozaki (15). Briefly, the synthetic peptide was used as immunization antigen (Supplementary Fig. S2B; ref. 16). Anti-FIR rabbit polyclonal antibodies were prepared or purchased from Japan BioService. Other primary and secondary antibodies are listed in Supplementary Table S1. Antigens on the membrane were detected with enhanced chemiluminescence detection reagents (GE Healthcare, UK Ltd). The intensity of each band was measured by NIH Image.
Plasmids
Full-length FIR cDNA was cloned into a p3xFLAG-CMV-14 vector (Sigma) to introduce a FLAG-tag at the carboxyl terminus, and FIRΔexon2 cDNA was cloned into a pcDNA 3.1 plasmid (Invitrogen). Myc-tag at the carboxyl terminus was prepared by PCR with suitable primers, including restriction enzyme sites (BglII and XbaI; Supplementary Table S2) through FIR- or FIRΔexon2-FLAG-tag vector. Plasmids were prepared by CsCl ultracentrifugation or using the Endofree Plasmid Maxi Kit (Qiagen) and DNA sequences were verified.
Stable transfection
Cells were transfected with plasmids using Lipofectamine 2000 reagents (Gibco BRL). For stable transfection, 5 × 104 cells were transfected with the above FIR-FLAG or pcDNA3.1-FIRΔexon2 plasmids and transferred to 10-cm dishes 48 hours after transfection. The complete medium contained 400 μg/mL geneticin in addition to IMDM, 10% FBS, and 1% penicillin–streptomycin. The complete medium with geneticin was replaced every 4 days until geneticin-resistant colonies appeared. At least 30 clones were screened by immunoblotting and immunostaining with anti-FLAG and anti-FIR antibodies (6B4) to find FIR-FLAG expressing clones for FIR-FLAG stably expressing cells, or with anti-c-Myc antibody to examine c-Myc expression for FIRΔexon2 stably expressing cells.
Extraction of nuclear protein and immunoprecipitation (pull-down assay)
Cells (~1 × 108) were resuspended in 5 mL cold buffer [50 mmol/L phosphate (pH-8.0), 20 mmol/L NaCl, 1 mmol/L DTT, 0.1% NP-40, protease inhibitor cocktail (Roche Diagnostics)] and left on ice for 15 minutes. The cells were then homogenized in a Dounce homogenizer or vigorously vortexed twice for 15 seconds before being centrifuged for 5 minutes at 100 ×g. After the pellet was washed twice with the same cold buffer, it was solubilized in lysis buffer [50 mmol/L phosphate buffer (pH 8.0), 150 mmol/L NaCl, 1 mmol/L DTT, 0.1% NP-40, and protease inhibitor cocktail] and then centrifuged for 1 hour at 20,000 ×g. The supernatant nuclear proteins were then used in a Western blot.
For immunoprecipitation by anti–FLAG antibody-conjugated beads, the nuclear fraction (NF) was reacted with magnetic Magnosphere MS300/carboxyl beads (Como Bio) precoated with anti-mouse IgG to reduce nonspecific protein binding and then reacted with anti-FLAG antibody for 1 hour at 4°C. After immunoprecipitation, the IgG and anti-FLAG antibody–conjugated beads were washed 5 times with 50 mmol/L phosphate buffer and the bound proteins were eluted with extraction buffer [40 mmol/L Tris-HCl (pH 6.8), 1% SDS, 1 mmol/L DTT] for 1 hour at 60°C. The immunoprecipitates were then analyzed by gel-based liquid chromatography-mass spectrometry (GeLC-MS) and protein identification (17). For immunoprecipitation by anti-SAP155 antibody–conjugated beads, Dynabeads ProteinG (Invitrogen) was prepared by same procedures as anti-FLAG antibody. After immunoprecipitation with NF, anti–SAP155 antibody-conjugated beads were washed 5 times with 100 mmol/L Glycine (WAKO Pure Chemical Industries Ltd.; pH 2.0) for 10 minutes at 4°C.
FIR binding protein identification
Exhaustive screening of FIR binding proteins was carried out using 2 independent methods. One was GeLC-MS (18-20) via Flag-conjugated bead pull down with LC-MS. Digested peptides were injected into a 0.3 × 5-mm L-trap column and a 0.1 × 150-mm L-column2 (Chemicals Evaluation and Research Institute, Saitama, Japan) attached to a NanoSpace high-performance liquid chromatography (HPLC) pump (Shiseido Fine Chemicals) and Magic 2002 splitter (AMR). The flow rate of the mobile phase was 500 nL/min. The solvent composition of the mobile phase was programmed to change in 60-minute cycles with varying mixing ratios of solvent A (2% v/v CH3CN and 0.1% v/v HCOOH) to solvent B (90% v/v CH3CN and 0.1% v/v HCOOH): 5% to 45.5% B 35 minute, 45.5% to 90% B 4 minute, 90% B 0.5 minute, 90%–5% B 1 minute, 5% B 20 minute. Purified peptides were introduced from HPLC to LTQ XL (Thermo Scientific), an ion-trap mass spectrometer, via an attached PicoTip (New Objective). The Mascot search engine (Matrixscience) was used to identify proteins from the mass and tandem mass spectra of the peptides. Peptide mass data were matched by searching the Human International Protein Index database [IPI, July. 2009, 80412 entries, European Bioinformatics Institute (Cambridge, UK)] using the MASCOT engine. Database search parameters were peptide mass tolerance 1.2 Da; fragment tolerance, 0.6 Da; enzyme set to trypsin, allowing up to one missed cleavage; variable modifications, methionine oxidation. Identification data (MASCOT dat file) were organized by Scaffold 3.0.2 software (Proteome Software, Inc.). The minimum criteria for protein identification were protein and peptide thresholds set to 95.0% (Scaffold's probability threshold filter) and number of unique peptides set to 2. The methods for the direct nanoflow liquid chromatography-tandem mass spectrometry (LC/MS-MS) system with FIR-FLAG transiently transfected 293T nuclear extracts have been described previously (21).
Immunocytochemistry
The FIR-FLAG stably expressing HeLa cells were grown on coverslips overnight and then subjected to immunocytochemistry as described previously (7). The primary mouse monoclonal anti-FLAG (Santa Cruz Biotechnology) and primary polyclonal antibodies against SAP155 were diluted 1:500 and 1:200, respectively, in the blocking buffer. The coverslips were incubated at room temperature for 1 hour. After washing with PBS, the secondary antibodies Alexa Fluor 488–conjugated goat anti-rabbit or 594-conjugated goat anti-mouse IgG secondary antibody (Molecular Probes) was applied at a dilution of 1:1,000. DNA was counter-stained with 4′, 6-diamidino-2-phenylindole (DAPI) III (Vysis, Abbott Park) and cells were observed under an immunofluorescence microscope (Leica QFISH; Leica Microsystems). Other primary and secondary antibodies used in this study are listed in Supplementary Table S1.
siRNA against FIR or SAP155
FIR and SAP155 siRNA duplexes were purchased from Sigma Aldrich. The target sequences for FIR siRNA and SAP155 siRNA oligonucleotides are listed (Supplementary Table S2). Transient transfection of siRNA was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were cultured for 72 hours at 37° C in a CO2 incubator.
Reverse transcriptase PCR and quantitative real-time PCR
Total RNA was extracted from HeLa cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from total RNA by the first strand cDNA Synthesis Kit for reverse transcriptase PCR (RT-PCR; Roche). Using the cDNA as a template, FIR cDNA was amplified with suitable primers by RT-PCR (Supplementary Table S2). Glyceraldehyde-3-phosphate dehydrogenase cDNA was amplified and used as the control. The PCR product was loaded on a 2.5% agarose gel (Promega), purified with the Gel Extraction Kit (Qiagen) and cloned using the pGEM-T Easy vector system (Promega) for DNA sequencing.
Quantitative real-time PCR (qRT-PCR) for c-myc or FIR cDNA was done using the LightCycler (Roche) in 20 μL of reaction mixture consisting of a master mixture (LightCycler FastStart DNA Master SYBR Green I) that contained FastStart Taq DNA polymerase, dNTPs, and buffer (LightCycler DNA Master hybridization probes; Roche), 3.0 mmol/L MgCl2, 0.5 μmol/L each of sense and antisense primers, and 1 μL of template cDNA in a LightCycler capillary. LightCycler software version 3.3 (Roche) was used for the analysis of real-time RT-PCR. Primer and probe sets for β-actin were purchased from Roche Diagnostics. Primers for RT-PCR and qRT-PCR, siRNAs were purchased and used simultaneously in accordance with the manufacturer's instructions (Nihon Gene Research Laboratories Inc.; Supplementary Table S2). Locations of primers and probes for real-time PCR in this study are indicated for FIR, FIRΔexon2, Δ3, and Δ4 cDNAs (Supplementary Fig. S2C).
Spliceostatin A, SF3b (SAP155) inhibitor, and adenovirus vectors
SSA was prepared as previously (13). The FIR and FIRΔexon2 adenovirus vector was also prepared (Supplementary Fig. S1). Briefly, recombinant adenoviral vectors expressing full-length human FIR proteins were constructed by homologous recombination in Escherichia coli using the AdEasy XL system (Stratagene). The HindIII-PmeI fragment of pcDNA3.1-FIR (/FIRΔexon2) or the HindIII-EcoRV fragment of pcDNA3.1-CMV-LacZ or pcDNA3.1-CMV-GFP was cloned into the HindIII-EcoRV site of pShuttle-CMV, generating pShuttle-CMV-FIR (otherwise FIRΔexon2), pShuttle-CMV-LacZ or pShuttle-CMV-GFP (as controls). The resultant shuttle vectors were linearized with PmeI digestion and subsequently cotransfected into E. coli BJ5183-AD-1. The recombinants were linearized with PacI digestion and transfected into the E1 transcomplementing 293 cell line to generate Ad-FIR (FIRΔexon2) and Ad-LacZ. The viruses were propagated in the adenovirus packaging 293 cell line (ATCC) and purified by double CsCl density gradient centrifugation followed by dialysis in 10 mmol/L Tris buffer (pH 8.0) with 10% glycerol. The virus titer was determined by conventional limiting dilution of 293 cells, that is, a plaque-forming assay was carried out with 293 cells (TCID50 method). The viruses were aliquoted and stored at −80°C until use. The recombinant adenovirus vectors were used to examine the effect to c-Myc expression.
FIR or FIRΔexon2 protein preparation
To reduce the dimerization, the C-terminal (95 amino acids)-truncated FIR (447 a.a.) or FIRΔexon2 (418 a.a), including FIR RRM1+RRM2 (Supplementary Fig. S2B and C), was cloned into NdeI/XhoI site of pET21b vector (Novagen; Merck chemicals) to introduce His-Tag at C-terminal and then was transfected to BL21-CodonPlus (DE3)-RIPL competent cells (Stratagene). Culture was carried out in TB medium (Invitrogen) in the presence of 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Expression was introduced with 0.5 mmol/L IPTG and then culture was continued at 30° C for 4 hours. Cells were harvested by centrifugation and disrupted by sonication in PBS. The supernatants from centrifugation were applied to HisTrap HP (GE Healthcare) and eluted by imidazole linear gradient. The eluted proteins were dialyzed to 50 mmol/L Tris/HCl, pH8.0, then applied HiTrap Q HP column (GE Healthcare). FIR or FIRΔexon2 was eluted NaCl linear gradient (0.2 to 1.0 mol/L). The eluted proteins were concentrated and loaded to HiLoad16/60 Superdex75pg gel filtration column (GE Healthcare), then eluted by 50 mmol/L Tris/HCl, 150 mmol/L NaCl, 10% glycerol, pH8.0 at 0.5 mL/min.
FUSE ssDNA oligonucleotides
All ssDNA oligonucleotides and 5′- or 3′-biotinylated ssDNA oligonucleotides pools were chemically synthesized (Nihon Gene Research Laboratories).
Electrophoresis mobility shift assay (gel shift assay)
FIR or FIRDexon2 protein binding assay with FUSE antisense ssDNA oligonucleotides was carried out by Light-Shift Chemiluminescent electrophoresis mobility shift assay (EMSA) Kit. EBNA-1 protein and EBNA-1 binding DNA sequence was employed as positive control (Thermo Scientific) according to company's instruction.
Statistical analysis
Comparison of SAP130, SAP145, SAP155, and FIR expression between cancer tissues and noncancer epithelium was evaluated using the Student t test. Correlation between FIR and SAP155 expression was evaluated using the Pearson product–moment correlation coefficients.
Results
Relationship among FIR, FIRΔexon2, and SF3b subunits expression in colorectal cancer tissues
PUF60 is an SF3b-associated protein (8, 12), and FIR is a splicing variant of PUF60 that lacks exon 5. Total FIR is increased in colorectal cancers (7). Recently, SSA (13) and pladienolide (14) have been reported to inhibit SF3b and induce a strong antitumor effect, implying that SF3b normally promotes tumors. Therefore, we examined SF3b expression in surgically excised human colorectal cancers (Fig. 1A and B). Total FIR and SF3b subunit levels were increased in colorectal cancers tissues compared with their corresponding nontumor epithelia (Fig. 1C). Furthermore, the levels of total FIR, SAP155, and SAP130 were positively correlated (Fig. 1C). However, the level of SAP145, another component with nearly equimolar subunit stoichiometry with the other essential subunits of SF3b, was paradoxically lower in cancer relative to SAP155 and SAP130 expression (Fig. 1A and B). c-myc, SAP155, SAP130, FIR, FIRΔexon2, and the ratio of FIRΔexon2/FIR were all increased at the mRNA level in colon cancer tissues (T) compared with corresponding noncancer epithelium (N; Fig. 1D). These results suggested that total FIRs, SAP155, or SAP130 tightly correlate with each other and may cooperate during tumor formation and progression, perhaps to elevate c-Myc.
Figure 1.

SAP155 and SAP130, SF3b subunits were activated in a positive correlation with FIR in colon cancer tissues. A, total protein lysates were prepared from 30 matched tumor samples (T) and adjacent nontumor epithelial tissue (N). FIR, SAP155, and SAP130 were activated, whereas SAP145 was downregulated in colorectal cancer tissues. β-Actin was used as an internal control. B, the intensity of each band was measured by NIH image, and the relative mean of SAP155, SAP130, FIR, and SAP145 protein levels between (T) and (N) with β-actin were calculated. Histogram indicating that SAP155, SAP130, and FIR expression levels in (T) were significantly higher than those in (N; P = 0.032, 0.005, and 0.001 from a t test, respectively). Inversely, SAP145 expression was significantly higher in (N) than in (T; P = 0.03 from a t test). Note, the P value of the SAP155 and SAP145 comparison is borderline acceptable. C, FIR–SAP155, FIR–SAP130, and SAP155–SAP130 expression in each colorectal cancer tissue sample was correlated between (T) and (N). The Pearson product–moment correlation coefficients (r) were 0.78 (T) and 0.95 (N) for FIR–SAP155, 0.66 (T) and 0.74 (N) for FIR–SAP130, and 0.80 (T) and 0.71 (N) for SAP155–SAP130. D, c-myc, FIR, FIRΔexon2, SAP155, and SAP130 mRNAs were all significantly activated in colon cancer tissues compared with the corresponding nontumor epithelium. c-myc and FIRΔexon2 mRNAs were definitely overexpressed, and the ratio of FIRΔexon2/FIR mRNA was increased.
FIR and FIRΔexon2 were coimmunoprecipitated and colocalized with SAP155, indicating FIR, FIRΔexon2, and SAP155 potentially forms a complex
By immunoprecipitation, SAP155 associated with FIR (Fig. 2A, top). Subcellular localization analysis of FIR and SAP155 showed that these 2 proteins colocalized in the nucleoplasm (Fig. 2A, bottom). To show whether SAP155, FIR, and FIRΔexon2 directly interact with each other, FLAG-tag or Myc-tag recombinant proteins were stably or transiently expressed in HeLa cells. Then reciprocal pull-down assays among those proteins were carried out (at least 3 replicates each). Association between FIR-SAP155 (Fig. 2B, top and bottom panels, arrowheads), FIRΔexon2 interacted strongly with SAP155 regardless of which partner was tagged (Fig. 2B, top and bottom, arrows), FIR also interacted more strongly with FIRΔexon2 (Fig. 2C, left, arrow), than with itself (Fig. 2C, left, arrowhead) and FIRΔexon2 also was strongly self-interacting (Fig. 2C, right, arrow). SAP155 was pull down with FIR- and FIRΔexon2-containing complexes, but SAP130 was not detected (Fig. 2C). Therefore, it is likely that FIR and SF3b are functionally linked. The dynamics and affinities among FIR, FIRΔexon2, and SAP155 and their relationship to cancer await further studies.
Figure 2.

FIR, FIRΔexon2, and SAP155 forms complex. A, proteins associated with anti-FLAG beads in the nuclear extracts of FIR–FLAG- or FIRΔexon2–FLAG stably expressing HeLa cells were analyzed with Western blot using anti-SAP155 or SAP130 antibodies. SAP155 and SAP130 were much less detectable in the FIRΔexon2–FLAG complex, 0.10 and 0.35, compared with the FIR–FLAG complex at 1.0, respectively (top). Immunofluorescent study showed that endogenous total FIRs (red) and SAP155 (green) were colocalized in the nucleoplasm. Both endogenous total FIRs and SAP155 were also colocalized in the nucleus (bottom). B, to examine the interaction between FIR or FIRΔexon2 and SAP155, FIRΔexon2 or FIR was pulled down with anti-SAP155–conjugated beads in nuclear extracted proteins (NE) of FIR/FIRΔexon2-FLAG stably (top) or transiently (bottom) expressing HeLa cells. The eluted proteins were immunoblotted with anti-FLAG antibody to examine the association between FIR/FIRΔexon2 and SAP155 (top). NE of FIR/FIRΔexon2-FLAG transiently expressing HeLa cells were prepared and treated with anti-SAP155–conjugated beads. The eluted proteins were immunoblotted with anti-FLAG antibody to examine the association between FIR/FIRΔexon2 and SAP155 (bottom). C, to examine the interaction between FIR-FIR, FIR-FIRΔexon2, or FIRΔexon2-FIRΔexon2, FIR-FLAG or FIRΔexon2-FLAG was pulled down with anti-FLAG–conjugated beads in NEs of FIR-Myc tag (top) or FIRΔexon2-Myc-tag (bottom) transiently expressing HeLa cells. NEs of FIR/FIRΔexon2-FLAG stably expressing HeLa cells were prepared and either FIR-Myc tag (top) or FIRΔexon2-Myc-tag (bottom) expressing vector was transfected. Then the eluted proteins were immunoblotted with anti-FLAG or anti-Myc antibody to examine the association between FIR-FIR, FIR-FIRΔexon2, or FIRΔexon2-FIRΔexon2, respectively. Note, SAP155 was apparently pulled down with FIR-FIR or FIR-FIRΔexon2 complex, but SAP130 was below detection level. IB, immunoblotting.
SAP155 regulates alternative splicing of FIR pre-mRNA and amounts of endogenous total FIR proteins
To gain insight into the physiologic relationship between FIR and SAP155, we first examined the effect of SAP155 knockdown by siRNA on the splicing and expression of FIR. Treatment of HCT116 or HeLa cells with SAP155 siRNA for 48 hours induced an altered splicing pattern of FIR (Fig. 3A, arrows) and a reduction in the total amount of endogenous FIRs. Novel FIR splicing variants, Δ3 (Exons 1, 3, and 6–12) and Δ4 (Exons 1, 6–12), were found (Fig. 3A and B, Supplementary Fig. S2A and C). Analysis using quantitative real-time PCR (qRT-PCR; Fig. 3A, bottom panels) showed that the ratio of Δ3 and Δ4 mRNA relative to FIR mRNA was increased by the SAP155 siRNA treatment in HCT116 and HeLa cells, and the ratio of FIRΔexon2 mRNA relative to FIR mRNA was increased in HeLa cells (Fig. 3A, bottom panels). SSA, an SF3b inhibitor, was used to verify the role of SAP155 in the alternative splicing of FIR mRNA. Although SAP155 expression was not suppressed by SSA, FIR pre-mRNA splicing was affected, just as was seen using SAP155 siRNA (Fig. 3B, arrows). SSA treatment generated the FIR splicing variants Δ3 and Δ4, revealed by DNA sequencing of RT-PCR products (Fig. 3B and Supplementary Fig. S2A and C). These results indicated that pattern of FIR splicing is sensitive to the level or function of SAP155.
Figure 3.
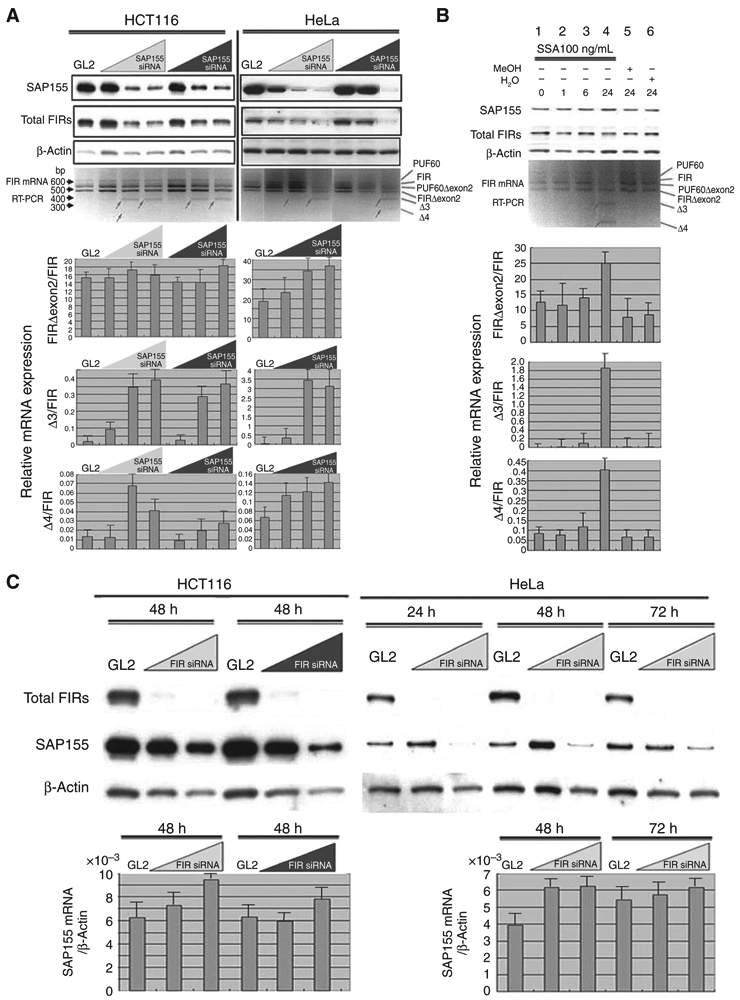
siRNA knockdown of SAP155 reduces FIR levels and vice versa. A, treatment with SAP155 siRNA for 48 hours suppressed FIR protein expression and FIR pre-mRNA splicing in HCT116 and HeLa cells. RT-PCR of full-length FIR cDNAs isolated from HeLa or HCT116 cells was carried out to amplify the aminoterminal regions. Note that novel FIR splicing variants, Δ3 and Δ4, were induced by SAP155 siRNA detected by RT-PCR (bottom, arrows). The expression levels of FIR and FIRΔexon2 mRNAs were quantified by qRT-PCR after SAP155 siRNA treatment for 48 hours in HCT116 and HeLa cells (bottom). The ratios of Δ3 and Δ4 mRNA relative to FIR mRNA were increased by the SAP155 siRNA treatment in HCT116 and HeLa cells, as well as the ratio of FIRΔexon2/FIR mRNA in HeLa cells. There was no significant alteration of the ratio of FIRΔexon2/FIR mRNA expression by SAP155 siRNA in HCT116 cells. B, effects of SSA, an SF3b inhibitor, on endogenous FIR expression and pre-mRNA splicing in HeLa cells were analyzed by qRT-PCR. Total FIRs expression was reduced by treatment with 100 ng/mL SSA for 24 hours. Altered FIR pre-mRNA splicing (lane 4) was detected by qRT-PCR in SSA-treated cells. Novel FIR splicing variants, Δ3 and Δ4, were accumulated by SSA treatment as well as by SAP155 siRNA, as shown in Fig. 1A. The ratios of FIRΔexon2, Δ3, and Δ4 mRNA relative to FIR mRNA were increased by SSA treatment. C, FIR siRNA suppressed SAP155 expression in HeLa and HCT116 cells for 48 hours. The ratio of SAP155/β-actin mRNA was not suppressed by FIR siRNA as determined by qRT-PCR.
So why did the knockdown of SAP155 by siRNAs reduce the amount of endogenous total FIR (Fig. 3A)? Is SAP155 required for endogenous FIR expression? To examine whether FIR and SAP155 interact with each other, we next tested whether FIR knockdown could also reduce SAP155 expression. FIR siRNA treatment reciprocally decreased the SAP155 protein level, but not the SAP155 mRNA level, in HeLa and HCT116 cells (Fig. 3C). These results indicated that FIR and SAP155 form a complex at the protein level but that FIR does not significantly affect the SAP155 mRNA level. For proper expression, FIR requires SAP155 at the RNA level, but SAP155 requires FIR at the protein level.
SAP155 and SAP130, but not SAP145, were coimunoprecipitated with FIR
The expression of FIR and SF3b subunits SAP155 and SAP130, but not SAP145, significantly correlated in colon cancer (Fig. 1C). FIR seems to have a significantly higher affinity for SAP155 by pull-down assay (Fig. 2B-2D), whereas SF3b consists of SAP130, SAP145, and SAP155 subunits in nearly equimolar stoichiometry amounts (8). To explore this discrepancy, we carried out exhaustive screening of FIR-binding proteins using 2 independent analyses. One was gel-enhanced LC/MS-MS (GeLC/MS-MS) analysis using FLAG-conjugated bead pull down with FIR– or FIRΔexon2–FLAG stably transfected HeLa cell nuclear extracts (refs. 18-20; Supplementary Table S3). The other was a direct nano-LC/MS-MS system with FLAG-conjugated bead pull down with FIR– or FIRΔexon2–FLAG transiently transfected 293T cell nuclear extracts (refs. 21-24; Supplementary Table S4). In both exhaustive screenings in HeLa cells, SAP155 was pulled down with FIR, but none of SAP155, SAP130, or SAP145 coimmunoprecipitated with FIRΔexon2. In 293T cells, SAP155 and SAP130 were pulled down with FIR and FIRΔexon2, respectively (Supplementary Table S5).
SF3b is a highly stable protein complex that remains intact at high ionic strengths (25, 26). If SF3b is highly stable, how does SAP155 encounter FIR and form stable complexes? We hypothesize that SAP155 encounters FIR before forming the SF3b complex. If this is true, the synthesis of authentic SF3b should be hindered because of the imbalance in SAP155, SAP145, and SAP130 proportions, and thus, SF3b dysfunction might occur in cancer cells. In this scenario, the elevated FIR–SAP155 complexes in colorectal cancers modify their predominant activities in nonneoplastic cells, FIR as a c-myc transcriptional repressor and SF3b as a splicing factor.
SeV/ΔF/FIR suppressed SSA-activated c-Myc, whereas Ad–FIRΔexon2 activated c-myc transcription and led to c-Myc overexpression
To determine whether the increase in c-myc is likely attributable to the reduced FIR activity in cells, the SeV/ΔF/FIR (27) was used in attempt to block the increase in c-Myc that results from treatment with either SAP155 siRNA or SSA. SeV/ΔF/FIR suppressed SSA-induced c-Myc activation (Fig. 4A, compare lane 2 with lane 1) but not basal c-Myc expression (Fig. 4A, compare lanes 4 to 3 and 6 to 5, respectively). These results were consistent with previous reports that FIR suppresses activated, not basal, c-myc transcription (5). Ad–FIRΔexon2 activates not only c-myc transcription but also c-Myc protein expression in HeLa cells (Fig. 4B). As expected, FIR antagonized the upregulation of MYC by FIRΔexon2 (Fig. 4C). How should we interpret this result? c-Myc mRNA and protein are not in a linear relationship because c-Myc increases the efficiency of translation, including its own mRNA mediated by coding region determinant-binding protein/insulin-like growth factor 2 mRNA binding protein 1 (CRD-BP/IGF2BP-1; refs. 28, 29). Notably, CRD-BP/IGF2BP-1 was coimmunoprecipitated with both FIR and FIRΔexon2 (Supplementary Table S5). These observations suggested that the increase in c-myc from either the SAP155 siRNA or SSA treatment is likely due to the reduced FIR or an imbalance between FIR and FIRΔexon2 in HeLa cells. Together, FIRΔexon2-FIR and SAP155 serves as molecular switches for c-myc expression.
Figure 4.
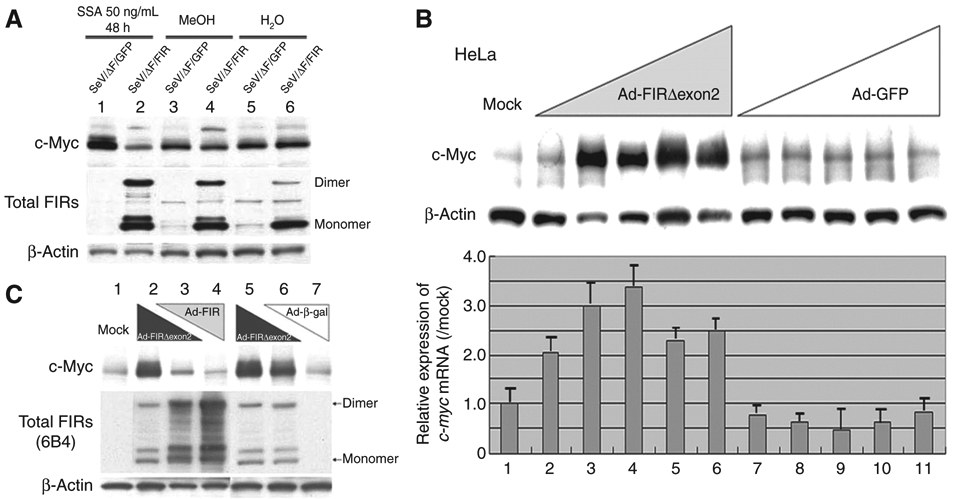
Ad–FIR suppressed SSA-activated c-Myc, whereas Ad–FIRΔexon2 activated c-myc transcription and led to c-Myc overexpression. A, c-Myc activation in 50 ng/mL SSA treatment for 48 hours was suppressed by enforced SeV/ΔF/FIR expression (lane 2). The effect of SeV/ΔF/FIR (27) was examined for whether the increase in c-Myc from either the SAP155 siRNA or SSA treatment is impaired by FIR. SeV/ΔF/FIR suppressed SSA-induced c-Myc activation (compare lane 2 with lane 1), but not basal c-Myc expression (compare lanes 4 and 6 with lanes 3 and 5, respectively). B, HeLa cells were treated with Ad–FIRΔexon2 for 48 hours, and whole cell proteins and total RNAs were extracted. Western blot analysis for c-Myc protein expression and qRT-PCR for c-myc mRNA were carried out. Ad–FIRΔexon2 activated c-Myc protein and c-myc mRNA in HeLa cells. Ad–GFP was used as a control vector. Mock shows that HeLa crude extract proteins are not subject to adenovirus vector treatment. Lanes 2, 7: 0.1 MOI; lanes 3, 8: 0.5 MOI; lanes 4, 9: 1 MOI; lanes 5, 10: 5 MOI; lanes 6, 11: 10 MOI of Ad–FIRΔexon2 or Ad–GFP, respectively. Ad–FIRΔexon2 apparently increased c-Myc more than 20 times over mock expression, whereas Ad–GFP did not affect c-Myc. The increase of c-myc mRNA was much less, 2 to 3 times that of c-Myc protein elevation by Ad–FIRΔexon2. C, FIR and Ad–FIRΔexon2 antagonized against c-Myc expression (compare lanes 3 and 6). Lane 1 (Mock): no adenovirus vector; Lane 2: 10 MOI of Ad–FIRΔexon2; Lane 3: 5 MOI of Ad–FIRΔexon2 and 5 MOI of Ad–FIR; Lane 4: 10 MOI of Ad–FIR; Lane 5: 10 MOI of Ad–FIRΔexon2 (same as lane 2); Lane 6: 5 MOI of Ad–FIRΔexon2 and 5 MOI of Ad–β-gal; Lane 7: 10 MOI of Ad–β-gal. Notably, c-myc mRNA was not significantly activated by Ad–FIRΔexon2 (data not shown). MOI, multiplicity of infection.
Reduction of SAP155 elevates c-Myc expression with FIR suppression in HeLa cells
As SAP155 knockdown reduced the expression of FIR, a c-myc transcriptional repressor, it is possible that c-Myc expression is regulated, at least partly, by SAP155. Indeed, SAP155 siRNA apparently increased c-Myc but decreased FIR protein expression in HeLa cells (Fig. 5A). Accordingly, c-Myc activation by SAP155 siRNA is likely to be indirect via FIR. SAP155 siRNA also reduced the level of SAP130 (Fig. 5B), suggesting that SAP155, SAP130, and FIR form a complex. Thus, to further examine the relationship between c-Myc protein expression and FIRΔexon2, we measured the ratio of FIRΔexon2/FIR mRNA during SAP155 knockdown (Fig. 5C) or SSA treatment (Fig. 5D). As anticipated, the ratio of FIRΔexon2/FIR mRNA correlated well with c-Myc protein expression in those cells. These findings indicated that the disturbance of FIR pre-mRNA splicing, and thus the ratio of FIRΔexon2/FIR mRNA, had an effect on not only c-myc gene transcription, but also on c-Myc protein levels during SAP155 knockdown or SSA treatment.
Figure 5.

The ratio of FIRΔexon2/FIR mRNA coincided well with c-Myc expression. A, treatment with SAP155 siRNA at the indicated concentrations for 48 hours induced an increase in c-myc mRNA, as determined by qRT-PCR, and protein and a decrease in FIR protein expression. FBP expression was unchanged by SAP155 siRNA. Knockdown of SAP155 by siRNA apparently increased c-Myc about 4 times more than mock expression. Notably, the increase of c-myc mRNA was, at most, 2 times more than GL2. B, knockdown of SAP155 by siRNA also decreased SAP130 expression in HeLa cells. C, interestingly, c-Myc activation was partly transcriptional because c-myc mRNA determined by qRT-PCR was increased along with SAP155 siRNA. The ratio of FIRΔexon2/FIR mRNA determined by qRT-PCR; FIRΔexon2/FIR mRNA coincided well with c-Myc protein expression level. D, SSA (100 ng/mL) was administered in a time-dependent manner to HeLa cells. MeOH: an equal amount of methanol that dissolves SSA was used as a control; H2O: an equal amount of H2O that dissolves SSA was used as another control. Interestingly, c-myc mRNA and c-Myc protein expression levels correlated well with the ratio of FIRΔexon2/FIR mRNA expression rather than with FIR or FIRΔexon2 mRNA expression alone.
c-myc gene intron was actively transcripted in colon cancer tissues
These results strongly suggested that FIR has a higher affinity with SF3b, SAP155, and SAP130. Hence, in cancer cells, it is likely that SAP155/FIR or SAP155/130/FIR form alternative complexes to the ordinary SF3b complex, and that this may disturb c-myc pre-mRNA splicing. Actually, c-myc gene intron 1 sequences were relatively more abundant in cancers in which the expression of the SF3b subunits was disturbed than in adjacent noncancer epithelia (Fig. 6A). Of note, unspliced c-myc mRNA was also overproduced in cancers (7). RT-PCR using a variety of primer sets showed that SAP155 siRNA increased total c-myc mature mRNA (Fig. 5A), as well as immature c-myc intron 1-containing mRNA (Fig. 6B) levels. These results confirmed that total c-myc transcription was activated by SAP155 knockdown. In addition, it is possible that SSA severely impairs c-myc pre-mRNA splicing (Fig. 6C). FIR siRNA also slightly inhibited c-myc pre-mRNA splicing (Fig. 6D). Collectively, disturbed FIR or SAP155 expression at least partly affected both c-myc gene transcription and splicing in human colon cancer tissues.
Figure 6.
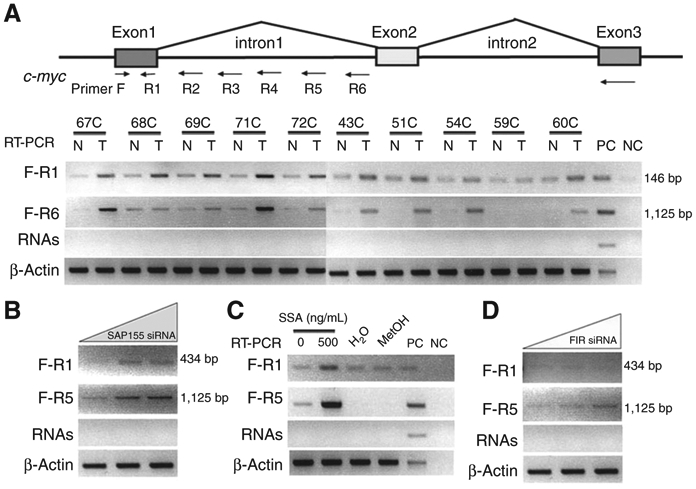
Endogenous c-myc gene splicing was altered in colon cancer tissues. Total RNA was extracted from 5 matched, operatively excised human tumor samples (T) and adjacent nontumor epithelial tissue (N). cDNA was synthesized from RNA. c-myc intron 1 was examined to determine the extent of transcription by RT-PCR, through changing the reverse primers located in c-myc intron 1 (R2–R6). A, primers for detection of transcribed c-myc intron 1 are listed in Supplementary Table S2. c-myc intron 1 was disturbed and transcribed in (T) but not in (N). PC: positive control (cDNA synthesized by HeLa cell total mRNA); NC: negative control (H2O). B, treatment with SSA (500 ng/mL) disturbed transcription of c-myc intron 1. Transcription of c-myc intron 1 was also apparently disturbed by siRNA against SAP155 (C) and FIR (D). RNAs: PCR was carried out with primers F-R1 and F-R6 with 1 μg of extracted total RNA as a template to exclude genomic DNA contamination.
FIRΔexon2ΔC interferes FIRΔC to bind to FUSE
FIRΔC- or FIRΔexon2ΔC-His tag proteins that deleted C-terminal 95 amino acids, containing RRM1 and RRM2, were purified (Fig. 7A). FIRΔexon2ΔC was found to interfere with FIRΔC binding to FUSE via EMSA (Fig. 7B). FIR and FIRΔexon2 form complex (Fig. 2C). These results indicated that formation of FIRΔexon2 competes with FIR for binding to FUSE; probably the protein–protein interaction interferes with DNA recognition by RRM1. A model of summarizing the consequences of FIRΔexon2's interactions with FIR and with SAP155/SAP130 is shown in Fig. 7C. FIR suppresses c-myc gene transcription and SAP155 regulates alternative splicing in noncancer cells (Fig. 7C, left), whereas FIRΔexon2 interferes with FIR binding to FUSE, resulting c-myc activation with potent SF3b dysfunction in cancer cells (Fig. 7C, right)
Figure 7.
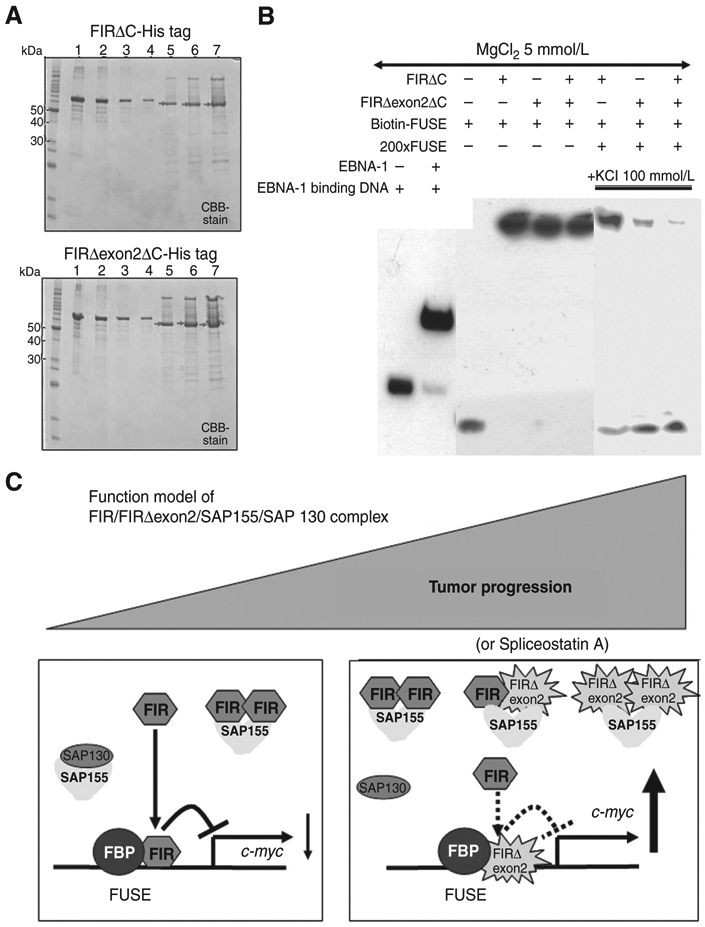
FIRΔexon2 potentially interferes with FIR to bind to FUSE along with tumor progression. A, C-terminal 95 amino acids deleted FIR- or FIRΔexon2-His tag protein, FIRΔC- or FIRΔexon2ΔC-His tag or with FIR-RRM1 and RRM2 (amino acids 103–297, exons 6–9), was prepared, purified, and was analyzed by SDS-PAGE. After affinity purification by HisTrapHP, recovered sample was dialyzed against 50 mmol/L Tris-HCl (pH9.0). Lane 1; 1,000 ng of bovine serum albumin (BSA), lane 2; 500 ng of BSA, lane 3; 250 ng of BSA, lane 4; 125 ng of BSA, lane 5; 2 μL lane 6; 4 μL, lane 7; 8 μL of purified FIR- (top) or FIRΔexon2-His tag (bottom) proteins. Concentration of FIRΔexon2-His tag proteins was estimated by band intensity using with BSA as a standard. B, EMSA revealed that FIRΔexon2ΔC interferes with FIRΔC to bind to FUSE. C, functional model of FIR/FIRΔexon2/SAP155/SAP130 complex along with tumor progression. FIR suppresses c-myc gene transcription and SAP155 engages in alternative splicing in noncancer cells, whereas potential FIR, FIRΔexon2, and SAP155 complex disturbed authentic FIR and SAP155 function simultaneously in cancer cells.
Discussion
The results of this study are summarized as follows: (i) FIRΔexon2 interacts with FIR and SAP155 (ii) siRNA knockdown of FIR reduces SAP155 levels and vice versa; (iii) increased levels of FIRΔexon2 and siRNA knockdown of SAP155 increased c-Myc levels; and (iv) FIRΔexon2 potently interferes with FIR binding to FUSE, yielding ineffective suppression of c-myc. In other words, augmented and sustained FIR/FIRΔexon2–SAP155 interaction modifies the functions of FIR and SAP155, thereby interfering with c-myc transcription and alternative splicing, respectively. In cancer cells, alternative splicing of FIR or c-myc pre-mRNA may also be disturbed (8). FIR/FIRΔexon2–SAP155 complex formation potently disturbs the proportion of SAP155, SAP130, and SAP145 in SF3b, thus altering FIRpre-mRNA and changing the ratio of FIRΔexon2 to FIR mRNA expression. SAP155 siRNA or SSA treatment induces transient c-Myc activation in HeLa cells (Fig. 5A, C, and D). Reduction of FIR increases c-myc expression, and overexpression of FIRΔexon2 leads to c-Myc protein activation. Thus, this process forms a novel bona fide molecular switch for c-myc gene expression.
Why were both FIR and FIRΔexon2 overexpressed in colorectal cancer tissues? Why was SAP155 activated in cancer tissues, although SAP155 siRNA or SSA treatment induced c-Myc. First reason might be that FIR is a c-Myc target. Also, the tight FIR/FIRΔexon2–SAP155 interaction disables established FIR and SAP155 functions disturbing the synthesis of normally spliced FIR mRNA. Second, FIRΔexon2 potently forms a heterodimer with FIR (Fig. 2B-D) and thus FIRΔexon2 interferes with FIR to bind to FUSE (Fig. 7B and C). These results strongly suggest that FIRΔexon2 antagonized FIR in c-myc transcriptional suppression and simultaneously interferes with SF3b in splicing during tumor progression. Third, genomic or somatic SAP155 is mutated in cancers (30, 31). How would these SAP155 somatic mutations affect to the regulation of c-Myc by FIR and its alternative splicing products? SAP155 somatic mutations observed in cancers accumulated at a specific site, supporting a gain-of-function of SAP155, and perhaps strengthening binding with FIR or FIRΔexon2. The FBP–FIR–FUSE system mediates c-myc transcriptional control because the RNA recognition motifs of FIR provide a platform for independent FUSE DNA and FBP protein binding, and this explains the structural basis of the reversibility of the FBP–FIR interaction (32, 33). Does FIRΔexon2 interfere with the dimerization of FIR on nucleic acid binding? What we do know is that the first RNA recognition motif (RRM) of FIR (amino acids 112–187-RRM1, Supplementary Fig. S2C) binds nucleic acids, and thus, it would be helpful to examine whether a conformational change would occur in FIRΔexon2 because FIR has been shown to dimerize upon nucleic acid binding (33).
At least one alternative splicing variant is present in 60% to 95% of all human genes (34). Does the FIR/FIRΔexon2–SAP155 complex affect alternative splicing in addition to FIR or the c-myc gene? Although further studies are required to answer this question, the disturbance of alternative splicing by SSA treatment has been found only in limited genes (35). Transcription affects splicing and vice versa, but much remains to be learned about how these processes are coupled (36-38). How does FIRΔexon2 activate c-myc? Does FBP–FIR or FIR–SAP155 binding differ among noncancer, transformed, and cancer cells? FBP was identified as a FIR- or FIRΔexon2-binding protein in 293T cells (Supplementary Tables S4 and S5), but it was below the detection levels in HeLa nuclear extracts (Supplementary Tables S3 and S4).
The existence of proteins binding uniquely to FIR or FIRΔexon2, as well as of proteins binding to both (Supplementary Fig. S4A) between FIR and FIRΔexon2 in HeLa cells (Supplementary Table S3) and 293T cells (Supplementary Table S5) suggests that this system can be tuned depending on the cell conditions. IGF2BP-1/CRD-BP, which meditates stabilization of c-myc mRNA and in turn transcriptionally regulated by c-myc (28, 29), was coimmunoprecipitated with both FIR and FIRΔexon2 (Supplementary Table S5). Accordingly, the balance of c-myc transcription, splicing patterns as well as c-myc mRNA stability can be adjusted differently in different cells or in tumors because authentic FIR function in c-myc transcriptional repression and SAP155 in alternative splicing were simultaneously disturbed by FIR/FIRΔexon2/SAP155 complex formation. Our study proposes that c-myc is regulated not only at the transcriptional but also by the efficiency of the splicing of its pre-mRNA. Further studies of agents that modify FIR/FIRΔexon2-SAP155 complex formation and SAP155 inhibitors (39) may provide clues to cancer therapy.
Supplementary Material
Acknowledgments
The authors thank Dr. Naohito Nozaki at the Department of Biochemistry and Molecular Biology, Kanagawa Dental College, Yokosuka, Kanagawa, for preparation of FIR monoclonal antibodies.
Grant Support
This study was supported in part by grant-in-aid 18591453 for priority areas in cancer research from the Ministry of Education, Science, Sports and Culture of Japan and "Seed Finding Programs" and "Mini-Feasibility Study Project" of JST (Japan Science and Technology) Agency to K. Matsushita, and Grant-in-Aid for Scientific Research (S) from the Japan Society for the Promotion of Science to R. Yashimoto and M. Yoshido.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Avigan MI, Strober B, Levens D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem 1990;265:18538–45. [PubMed] [Google Scholar]
- 2.Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, et al. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev 1994;8:465–80. [DOI] [PubMed] [Google Scholar]
- 3.Bazar L, Meighen D, Harris V, Duncan R, Levens D, Avigan M. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J Biol Chem 1995;270:8241–8. [DOI] [PubMed] [Google Scholar]
- 4.Michelotti GA, Michelotti EF, Pullner A, Duncan RC, Eick D, Levens D. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol Cell Biol 1996;16:2656–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, He L, Collins I, Ge H, Libutti D, Li J, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell 2000;5:331–41. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Akoulitchev S, Weber A, Ge H, Chuikov S, Libutti D, et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 2001;104:353–63. [DOI] [PubMed] [Google Scholar]
- 7.Matsushita K, Tomonaga T, Shimada H, Shioya A, Higashi M, Matsubara H, et al. An essential role of alternative splicing of c-myc suppressor FIR in carcinogenesis. Cancer Res 2006;66:1409–17. [DOI] [PubMed] [Google Scholar]
- 8.Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Lührmann R. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 2002;21:4978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spadaccini R, Reidt U, Dybkov O, Will C, Frank R, Stier G, et al. Biochemical and NMR analyses of an SF3b155-p14-U2AF-RNA interaction network involved in branch point definition during pre-mRNA splicing. RNA 2006;12:410–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hastings ML, Allemand E, Duelli DM, Myers MP, Krainer AR. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF (65). PLoS One 2007;2:e538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mollet I, Barbosa-Morais NL, Andrade J, Carmo-Fonseca M. Diversity of human U2AF splicing factors. FEBS J 2006;273:4807–16. [DOI] [PubMed] [Google Scholar]
- 12.Corsini L, Hothorn M, Stier G, Rybin V, Scheffzek K, Gibson TJ, et al. Dimerization and protein binding specificity of the U2AF homology motif of the splicing factor Puf60. J Biol Chem 2009;284:630–9. [DOI] [PubMed] [Google Scholar]
- 13.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 2007;3:576–83. [DOI] [PubMed] [Google Scholar]
- 14.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 2007;3:570–5. [DOI] [PubMed] [Google Scholar]
- 15.Kimura K, Nozaki N, Enomoto T, Tanaka M, Kikuchi A. Analysis of M phase-specific phosphorylation of DNA topoisomerase II. J Biol Chem 1996;271:21439–45. [DOI] [PubMed] [Google Scholar]
- 16.Page-McCaw PS, Amonlirdviman K, Sharp PA. PUF60: a novel U2AF65-related splicing activity. RNA 1999;5:1548–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kristjansdottir K, Wolfgeher D, Lucius N, Angulo DS, Kron SJ. Phosphoprotein profiling by PA-GeLC-MS/MS. J Proteome Res 2008;7:2812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomonaga T, Matsushita K, Ishibashi M, Nezu M, Shimada H, Ochiai T, et al. Centromere protein H is up-regulated in primary human colorectal cancer and its overexpression induces aneuploidy. Cancer Res 2005;65:4683–9. [DOI] [PubMed] [Google Scholar]
- 19.Seimiya M, Tomonaga T, Matsushita K, Sunaga M, Oh-Ishi M, Kodera Y, et al. Identification of novel immunohistochemical tumor markers for primary hepatocellular carcinoma; clathrin heavy chain and formiminotransferase cyclodeaminase. Hepatology 2008;48:519–30. [DOI] [PubMed] [Google Scholar]
- 20.Nishimori T, Tomonaga T, Matsushita K, Oh-Ishi M, Kodera Y, Maeda T, et al. Proteomic analysis of primary esophageal squamous cell carcinoma reveals downregulation of a cell adhesion protein, periplakin. Proteomics 2006;6:1011–8. [DOI] [PubMed] [Google Scholar]
- 21.Natsume T, Yamauchi Y, Nakayama H, Shinkawa T, Yanagida M, Takahashi N, et al. A direct nanoflow liquid chromatography-tandem mass spectrometry system for interaction proteomics. Anal Chem 2002;74:4725–33. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003;116:1679–88. [DOI] [PubMed] [Google Scholar]
- 23.Yanagida M, Hayano T, Yamauchi Y, Shinkawa T, Natsume T, Isobe T, et al. Human fibrillarin forms a sub-complex with splicing factor 2-associated p32, protein arginine methyltransferases, and tubulins alpha 3 and beta 1 that is independent of its association with pre-ribosomal ribonucleoprotein complexes. J Biol Chem 2004;279:1607–14. [DOI] [PubMed] [Google Scholar]
- 24.Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J 2004;23:1977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brosi R, Hauri HP, Krämer A. Separation of splicing factor SF3 into two components and purification of SF3a activity. J Biol Chem 1993;268:17640–6. [PubMed] [Google Scholar]
- 26.Das BK, Xia L, Palandjian L, Gozani O, Chyung Y, Reed R. Characterization of a protein complex containing spliceosomal proteins SAPs 49, 130, 145, and 155. Mol Cell Biol 1999;19:6796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura A, Matsushita K, Takiguchi Y, Shimada H, Tomonaga T, Matsubara H, et al. Synergistic effect of non-transmissible Sendai virus vector encoding the c-myc suppressor FUSE-binding protein-interacting repressor plus cisplatin in treatment of malignant pleural mesothelioma. Cancer Sci 2011;102:1366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noubissi FK, Nikiforov MA, Colburn N, Spiegelman VS. Transcriptional regulation of CRD-BP by c-myc: implications for c-myc functions. Genes Cancer 2010;10:1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J, et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature 2006;441: 898–901. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011. September 11 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011. October 13;365:1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cukier CD, Hollingworth D, Martin SR, Kelly G, Díaz-Moreno I, Ramos A. Molecular basis of FIR-mediated c-myc transcriptional control. Nat Struct Mol Biol 2010;17:1058–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crichlow GV, Zhou H, Hsiao HH, Frederick KB, Debrosse M, Yang Y, et al. Dimerization of FIR upon FUSE DNA binding suggests a mechanism of c-myc inhibition. EMBO J 2008;27:277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics 2006;7:325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furumai R, Uchida K, Komi Y, Yoneyama M, Ishigami K, Watanabe H, et al. Spliceostatin A blocks angiogenesis by inhibiting global gene expression including VEGF. Cancer Sci 2010;101:2483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zorio DA, Bentley DL. The link between mRNA processing and transcription: communication works both ways. Exp Cell Res 2004;296:91–7. [DOI] [PubMed] [Google Scholar]
- 37.Alexander R, Beggs JD. Cross-talk in transcription, splicing and chromatin: who makes the first call? Biochem Soc Trans 2010;38:1251–6. [DOI] [PubMed] [Google Scholar]
- 38.Matsushita K, Tomonaga T, Kajiwara T, Shimada H, Itoga S, Hiwasa T, et al. c-myc suppressor FBP-interacting repressor for cancer diagnosis and therapy. Front Bios 2009;14:3401–8. [DOI] [PubMed] [Google Scholar]
- 39.Hasegawa M, Miura T, Kuzuya K, Inoue A, Won KS, Horinouchi S,et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol 2011;6:229–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.