

Abstract
The purpose of this review is to provide medical researchers, especially those without a bioinformatics background, with an easy-to-understand summary of the concepts and technologies used in microbiome research. First, we define primary concepts such as microbiota, microbiome, and metagenome. Then, we discuss study design schemes, the methods of sample size calculation, and the methods for improving the reliability of research. We emphasize the importance of negative and positive controls in this section. Next, we discuss statistical analysis methods used in microbiome research, focusing on problems with multiple comparisons and ways to compare β-diversity between groups. Finally, we provide step-by-step pipelines for bioinformatics analysis. In summary, the meticulous study design is a key step to obtaining meaningful results, and appropriate statistical methods are important for accurate interpretation of microbiome data. The step-by-step pipelines provide researchers with insights into newly developed bioinformatics analysis methods.
Keywords: Microbiome, Microbiota, Study design, Statistical analysis, Sample size, Bioinformatics analysis, Pipeline
Introduction
With the development of sequencing technologies and data analysis methods, several achievements in microbiome research have been made in recent years.[1–3] These include compelling discoveries in the field of medicine such as associations between the microbiome and metabolic diseases,[4–6] digestive diseases,[7–10] and cardiovascular diseases.[11] These developments and discoveries have increased the interest of physicians on microbiome research, with a dramatic increase in the number of publications in the field.[12]
In addition, microbiome analysis methods have improved rapidly due to the emergence of advanced technologies or pipelines, including Quantitative Insights Into Microbial Ecology (QIIME) 2[13] and multi-omics analyses,[1,9] which are broadly used in medical and non-medical research. However, understanding and mastering these technologies or pipelines are challenging, especially for medical researchers.
The purpose of this review is to provide researchers without a bioinformatics background with an easy-to-understand summary of the concepts and technologies used in microbiome research. In particular, we provide a detailed discussion on primary concepts, study design, sample collection, statistical methods, and bioinformatics analysis used in microbiome research.
Primary Concepts
Microbiota, microbiome, and related terms
Microbiota refers to the microorganisms that inhabit a specific site on/in the body, which consists of a wide variety of bacteria, archaea, viruses, fungi, and protozoans.[14,15] In medical research, microbiota refers to bacteria and archaea if samples are determined using 16S ribosomal RNA (rRNA) gene (also known as rDNA) sequencing. On the other hand, microbiome refers to the entire habitat, including the microorganisms, their genomes, and the surrounding environmental conditions.[14,15] Note, however, that the terms of microbiota and microbiome are sometimes used interchangeably. We recommend that the term microbiota should be used when referring purely to the microorganisms in your research, otherwise the term microbiome should be used [Figure 1]. For example, if a researcher would like to explore the relationship between the short-chain fatty acids in blood and feces and the microorganisms in gut, then the term microbiome should be used in this research. Metagenome is the collection of all genomes of the microbiota, which is obtained using shotgun metagenomic sequencing,[14] and metagenomics is the study of the metagenome.[12,14] Virome refers to the collection of all viruses in or on humans, including endogenous retroviruses, eukaryotic, and prokaryotic viruses.[16] The study of the virome is known as viromics or viral metagenomics.
Figure 1.
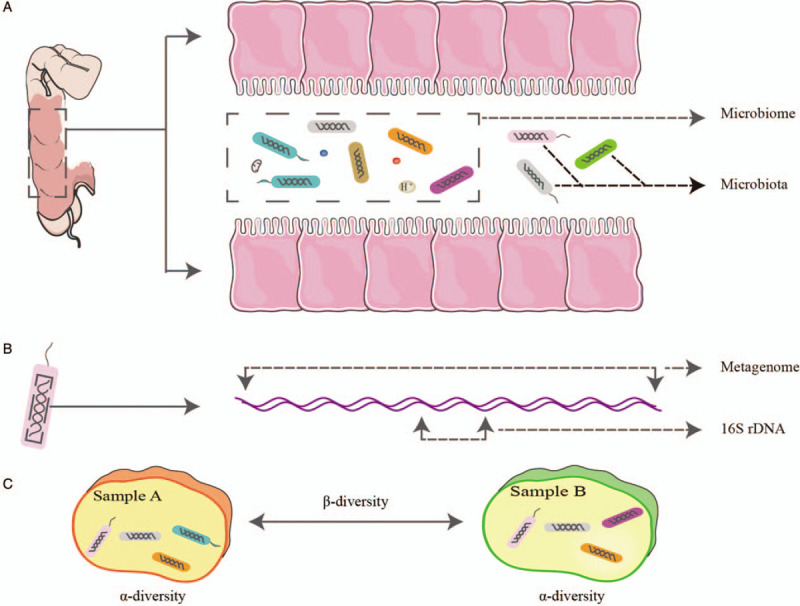
Definitions of microbiome, microbiota, metagenome, and 16S rDNA. (A) The concept of microbiome covers not only the microorganisms but also the surrounding environmental conditions. Microbiota only means the microorganisms. (B) Metagenome means all genomes of the microorganisms, while 16S rDNA only covers a segment of the genomes. (C) α-diversity measures the diversity within a sample, while β-diversity compares the difference between samples.
Bacterial taxonomy
In bacterial taxonomy, the most commonly used ranks or levels in their descending order are: phyla, classes, orders, families, genera, and species. For example, the taxonomic ranks for Escherichia coli, which is a very common bacteria in human intestines, are shown in Table 1.
Table 1.
Taxonomic ranks or levels in descending order.

Operational taxonomic units (OTUs) and amplicon sequence variants (ASVs)
The construction of an OTU table is important for marker gene (amplicon) data analysis.[17] OTU, which refers to a group of closely related sequences, is used to categorize bacteria based on sequence similarity. The similarity threshold of an OTU is typically defined as 97%.[18,19] That is, the marker-gene sequences which have the 97% similarity are considered as an OTU. However, the OTU method has apparent drawbacks. In particular, it imposes an arbitrary similarity threshold on OTU picking and misses subtle and real biological sequence variations.[20] The ASV has been developed recently to address these problems, which uses error profiles to resolve sequence data into exact sequence features. ASV has single-nucleotide resolution and has similar or better sensitivity and specificity than OTU.[20] It is important to note that the OTU or ASV is not equal to species. An OTU/ASV may include several species and vice versa.[21]
α-diversity
α-diversity refers to the diversity within a sample such as fecal, saliva, or bronchoalveolar lavage fluid sample.[15] There are three α-diversity indices often used in medical research: Chao 1 index, Shannon-Wiener index, and Simpson index.
The Chao 1 index, a metric of richness, estimates the total number of species in a sample.[22] It takes into account the following three factors: the number of species, the number of singleton taxa, and the number of doubleton taxa.[22] This means that it cannot reflect the abundance of the microbiota.
The Shannon-Wiener index combines richness and evenness.[23] It gives more weight to rare species,[22] which means that it is higher when the number of rare species increases. Its value generally does not exceed 5.0; the higher its value is, the more abundant is the α-diversity.[22]
The Simpson index also combines richness with evenness. However, in contrast to the Shannon-Wiener index, it puts more emphasis on common species. Its value ranges from 0 to almost 1; the higher its value is, the more abundant is the α-diversity.[22]
In the above indices, richness refers to the total number of species in a sample,[17,24] while abundance refers to the raw read counts of a species.[24] Note that relative abundance is used when the raw read counts are normalized or converted to percentages.[24]
β-diversity
β-diversity refers to microbiota differences between samples or groups.[15] It is typically used to understand whether differences in the microbiota compositions of two groups are significant.[25] Here, we focus on two commonly used β-diversity indices: Bray-Curtis dissimilarity and UniFrac distance.
The Bray-Curtis dissimilarity is a statistical measure used to quantify the compositional dissimilarity between two samples or groups. Its value ranges from 0 to 1, where 0 means that the two samples or groups share all species, and 1 means that they do not share any.[26] In addition, it gives more weight to common species.[23] Note that Bray-Curtis is not a real distance measure so the term “Bray-Curtis dissimilarity” is more appropriate than “Bray-Curtis distance.”[22]
The UniFrac distance, which can be unweighted or weighted, estimates differences between samples or groups based on phylogenetic distance.[27] The unweighted UniFrac distance takes into account the presence and absence of taxa. It is sensitive for detecting richness changes in rare species but ignores the abundance information in the computation.[28] The weighted UniFrac distance incorporates the abundance information[29] and reduces the contribution of rare species.[25]
Ordination
Ordination is a method to explore data structure in a graph constructed with a reduced set of orthogonal axes. The ordination plot is an effective way to visualize the β-diversity. The ordination can be classified into two types: unconstrained ordination and constrained ordination.[30–32] The ordination is unconstrained if the ordination axes are not constrained by environmental factors (sample metadata); otherwise, it is constrained.[32] The commonly used unconstrained ordinations include principal component analysis (PCA), correspondence analysis (CA), principal coordinate analysis (PCoA), and non-metric multidimensional scaling (NMDS).[30,32] On the other hand, the commonly used constrained ordinations are redundancy analysis (RDA) and canonical correspondence analysis (CCA).[31,32]
The microbiome information corresponds to high-dimensional data. PCA is used to simplify the complexity by geometrically projecting the data onto fewer dimensions called principal components, it uses the Euclidean distance in its computation.[30] In general, it is not suitable for the analysis of microbial abundance data because the underlying structure of the data must be linear.[30] However, it could be used if the data are Hellinger-transformed.[30] In contrast, CA is suitable for the analysis of microbial abundance data without pre-transformation. In CA analysis, all samples are ordinated by using the Pearson χ2 distance.[30] Note, however, that rare species could have an unduly large influence on the CA analysis.[33] If a researcher wishes to ordinate samples or features based on some other dissimilarity measures, then PCoA is a good choice. In microbiome research, the Bray-Curtis dissimilarity and UniFrac distance are most commonly used in PCoA analysis. NMDS is used to represent the relative positions of samples in an ordination plot. Similar to PCoA, any distance or dissimilarity matrix can be used in NMDS analysis. The differences between PCoA and NMDS have been well described in the literature,[30] with the former used in most circumstances.[30]
RDA is a constrained ordination that combines PCA and regression. Its response matrix corresponds to the microbiota data and the explanatory matrix corresponds to clinical indices (sample metadata). It is useful for showing whether the microbiota data are constrained by clinical indices. Note, however, that the dataset may need to be pre-transformed because the underlying structure of the response matrix must be linear due to the PCA procedure.[30] Finally, CCA is a constrained counterpart of CA, which shares the basic properties and drawbacks of CA.[31]
Study Design
Study design schemes
A meticulous study design is important for obtaining accurate and meaningful results. The most popular study designs used in medical microbiome research include cross-sectional studies, case-control studies, longitudinal studies, and randomized controlled trials (RCT). The first three are observational studies that do not apply interventional factors, while the last is a typical experimental study.
The cross-sectional studies are divided into descriptive and analytical cross-sectional studies.[34] The former is purely descriptive and is mainly used to investigate the microbiota composition in one or more populations, while the latter is used to explore the associations between the microbiome and health outcomes. However, the associations between the microbiome and health outcomes may stem from confounding factors such as sex,[35] age,[36] body mass index (BMI),[37] diet,[5,38] season,[39] and medication.[40,41] Moreover, the microbiome and the outcomes are measured simultaneously, making it is difficult to determine the causal relationships between them. Generally, the cross-sectional study is only used for exploring the elementary features of the microbiome, and it could serve as a preliminary experiment for subsequent research.
In most instances, the microbiome is considered as an exposure and a disease is assumed to be an outcome in medical research. Under these assumptions, the conventional case-control study is rarely used in the microbiome research because the previous exposure (the microbiome) is difficult to obtain. However, it works if the exposure and the outcome are reversed.
Similarly, a prospective longitudinal study is also difficult to perform under the above assumptions because it is difficult to know which microbes are the underlying exposures. Moreover, the specific microbiome patterns, which could serve as exposed or unexposed factors, cannot be defined easily so it is difficult to define a participant as being an exposed or an unexposed individual. In practice, participants with or without a disease often serve as a study group or a control group, and samples containing the microbiome are prospectively collected at different time points.[17] That is, the subjects involved in a prospective longitudinal study are often grouped according to a clinical outcome rather than according to the specific microbiome patterns.
Finally, the purpose of an RCT or other experimental studies is to evaluate the effectiveness of an intervention. The intervention could be a medication or the microbiome. For example, the intervention in a fecal transplantation study is the microbiome.[42,43]
It is worth noting that the control group should be selected appropriately. Some confounding factors should be matched in these studies, which will be discussed below. The control selection is sometimes difficult, especially when the intervention is the microbiome itself in a clinical study. In this scenario, a controlled before-after trial or historical controlled trial would be a good option if other study designs may be inappropriate.[44]
Defining the inclusion and exclusion criteria
Defining the exact inclusion and exclusion criteria enables better matching of different groups and limits confounding factors such as age,[36,45] sex,[35] BMI,[46] diet,[47] season,[39] medication,[40,41] ethnicity,[48] geographic region,[45] and comorbidities.[7] Age significantly influences on the microbiome, especially in those younger than 16 years old.[36,45] Thus, age should be well matched in a microbiome research involving children. Diet is another factor contributing to microbiome alterations so it needs to be matched.[47] To improve the comparability between groups, geographic regions where the participants live should also be taken into account when designing a microbiome study.[45] On the other hand, individuals who underwent medications in the preceding several months should be excluded from a microbiome study.[41,49] For example, a patient treated with antimicrobial drugs 3 to 6 months before a microbiome study should be excluded.[49]
Sample size and power calculations for microbiome research
When a researcher designs an experiment, it is important to estimate the sample size. An appropriate sample size enables a microbiome research to discern the differences between groups and to save resources and time. However, sample size and power calculations remain a challenge.[50] The most commonly used methods for sample size and power calculations in microbiome research are based on t-test, analysis of variance, χ2 test, and the Dirichlet multinomial model.[51] Using the t-test as an example, the sample size and power calculations are determined in three steps. First, a small number of amplicon data is acquired through a preliminary experiment. Second, the Shannon-Wiener values of every sample are obtained using the R package vegan.[52] The last step is the calculations of the sample size and power using the power.t.test() function in the R package pwr. The t-test is used to calculate the sample size and power when a researcher only focuses on the differences in species diversity between two groups. Other methods for calculating sample size and power calculations are described well in the reference.[51]
Importance of negative and positive controls
The results of microbiome research could be affected by several factors, such as DNA extraction kits, sampling methods, contaminations, and sequencing methods,[53] which could be reduced by using negative and positive controls. Unfortunately, only 30% of the previous studies reported using negative controls, and only 10% reported using positive controls.[53] Using the controls is important for characterizing the microbiome especially when the samples have low microbial biomass.[54] Previous studies found that the specimens, such as placenta and synovial fluid, which were recognized to be sterile in the past, maybe colonized with microbiome.[55] However, these positive results may be caused by other factors such as contaminations. Interestingly, these low-biomass specimens have been demonstrated to be sterile in recent studies that employed the negative and/or positive controls.[56] Thus, we recommend that negative and positive controls should be considered when the samples are low-biomass specimens such as blood, amniotic fluid, cerebrospinal fluid, synovial fluid, and placenta. It is worth noting that the negative and positive controls are also important in virome research because the virome and microbes are usually explored simultaneously.[16] In addition, R packages decontam can be used to identify and remove contaminant sequences in marker gene and metagenomic data.[57]
Selection of sequencing methods
The sequencing methods used in microbiome research include amplicon, metagenomic and metatranscriptomic sequencing. The amplicon sequencing incorporates the 16S rDNA sequencing for bacteria and archaea and the internal transcribed spacer sequencing for fungi. Every sequencing method has its pros and cons, which were discussed thoroughly in the references.[17,58] In brief, the amplicon sequencing is inexpensive and can be applied to low-biomass specimens contaminated by host DNA, but it is limited to genus level taxonomic resolution and is susceptible to some sources of inherent bias such as the number of polymerase chain reaction (PCR) cycles.[59] The metagenomic sequencing method sequences all DNA present in a sample including bacterial, viral, eukaryotic, and host DNA. It does not only extend its taxonomic resolution to species or strain level but also provides the potential functions.[17] However, both the amplicon and metagenomic sequencing methods cannot discriminate between dead and live microbiota.[17] The metatranscriptomic sequencing only yields active functional information of a community. With the different advantages and disadvantages of these sequencing methods, it is advisable to integrate multi-sequencing methods for optimal study design. Briefly, the selection of the sequencing methods mainly depends on the scientific question of interest, sample types, the quality of samples, and the cost of experiments. Amplicon sequencing is often used to gain an overview of a microbial community,[60] and it is typically applicable to large-scale studies.[6,61] If you have enough project funding, and you would like to gain strain-level resolution and potential functions, or even to recover the whole genomes, the metagenomic sequencing is a preferred method.[62–66]
Multiple measures for improving the reliability of research
Simple cross-sectional studies have limited significance in microbiome research. Hence, in this section, we discuss ways to improve the reliability of research. First, a longitudinal or a RCT is preferred rather than a cross-sectional study or a case-control study.[17,67] Second, the sample size should be calculated.[51] Third, the confounding factors should be matched, and the metadata should be collected carefully. Fourth, the inclusion and exclusion criteria should be defined in detail. For example, the pediatric disease of juvenile idiopathic arthritis has several sub-types, each of which may represent a different disease entity.[68] A researcher should decide whether all the sub-types are included in the patient group. Fifth, it would be better to take negative and/or positive controls into account.[54] Sixth, integrating other omics methods, such as metabolomics, metatranscriptomics, and metaproteomics, is vital for a comprehensive understanding of the structure and function of a microbial community.[17] Thus, plans to acquire the metabolite profiles and/or other multi-omics data of a microbial community should be considered. Currently, research that explores the structure of a microbial community for this sole purpose is no longer considered as a robust study design.[17] Lastly, it is advisable that the preliminary results obtained from a clinical experiment should be verified in an animal model.
Considerations for the design of clinical microbiome research are shown in Table 2, and a typical workflow is presented in Figure 2. Researchers can refer to the considerations for experimental research in the literature.[49]
Table 2.
Checklist for the design of clinical microbiome research.
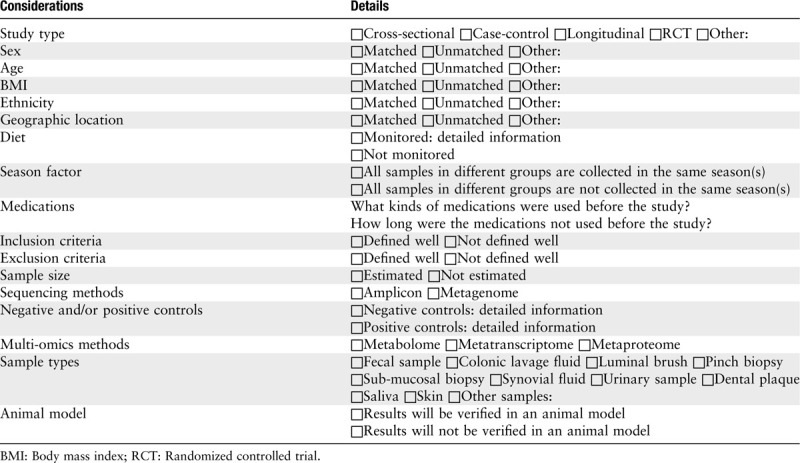
Figure 2.
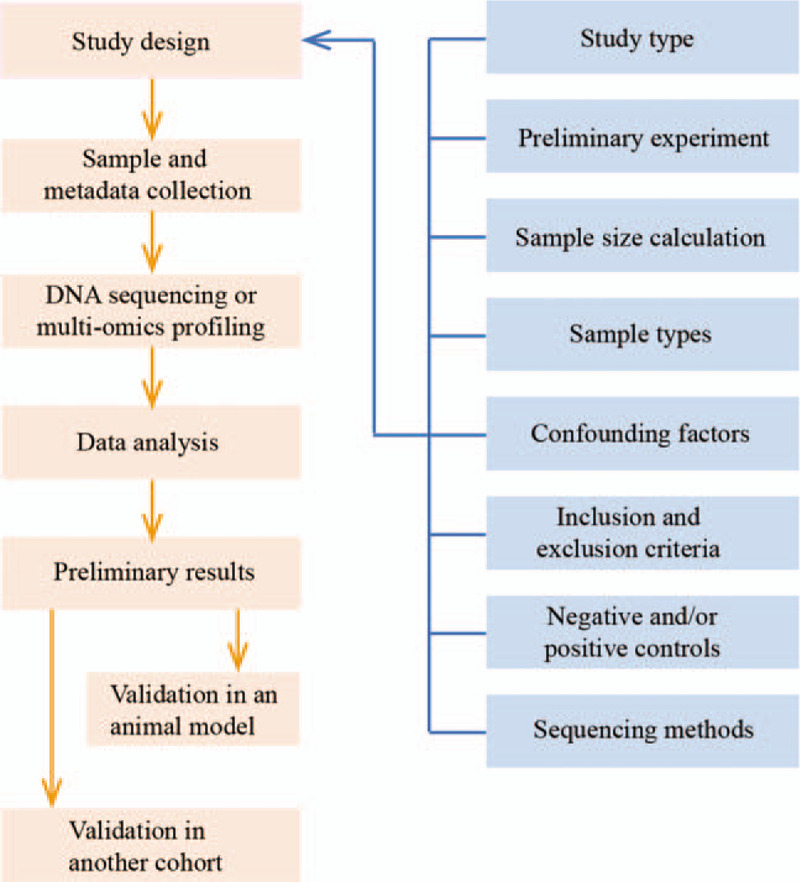
Typical workflow of the human microbiome research.
Sample Types, Preservation, and Storage
Sample types
Sample types in human microbiome research include feces, colonic lavage fluid, luminal brush, pinch biopsy, sub-mucosal biopsy, synovial fluid, urinary sample, dental plaque, saliva, and skin. The choice of a sample type depends on the scientific question of interest. For example, fecal samples are easy to collect and can be used in large-scale and longitudinal studies. On the other hand, biopsy samples are more useful for exploring the interactions between the microbiota and the host.[69] It is important to note that the sampling site should be fixed in one research because different parts of the human body are colonized with different microbiota.[70,71]
Preservation and storage
The methods of sample preservation and storage should be tailored to the experimental method and sample type. The most versatile method is to freeze the samples directly, which can be used in various sequencing and experimental methods such as amplicon, metagenomic, metatranscriptomic sequencing, and metabolomic measurement. It is suggested that the samples should be preserved at −20°C within 15 min after collection,[72,73] and then transferred to a laboratory on dry ice within 24 h of collection and stored at −80°C thereafter. However, samples are commonly collected at home rather than in clinical settings. Under these circumstances, using preservation kits is an alternative. Samples preserved in the kits can be stored at ambient temperature for more than a week.[74] Note that the sample preservation and storage methods should be consistent across all samples to minimize potential confounding variations.
Statistical Analysis in Microbiome Research
Medical researchers are typically familiar with univariate statistical methods, such as t-test, analysis of variance, χ2 test, and the Mann-Whitney U test. Hence, we here discuss problems related to multiple comparisons and other multivariate methods. We first discuss the problems with multiple comparisons and their solutions including P value adjustments and false discovery rates (FDRs). Then, we discuss other multivariate methods such as the permutational multivariate analysis of variance (PERMANOVA) and the Mantel test.
Problems with multiple comparisons and their solutions
Multiple comparisons are commonly used in microbiome research because microbiome data are high-dimensional. For example, the feature table has hundreds or thousands of OTUs or ASVs, and each of them may be compared between groups. Another example often encountered by medical researchers may be more easily understood. Suppose a study has three groups, for example, group A, group B, and group C, and a researcher would like to compare differences between the three groups. In this case, the P value should be adjusted because each group is compared twice, that is, group A vs. group B, group A vs. group C, and group B vs. group C. P value adjustments are needed if each group or variable is compared to limit false-positive discoveries.[75]
The classic method to adjust the P value is to control the family-wise error rate, that is, the type I error or α level. The Bonferroni adjustment is the most commonly used method to control the family-wise error rate. The calculation of an adjusted P value is very easy: the α level for an individual test divided by the number of tests. Thus, in the above example, the adjusted P value is 0.05/3 = 0.017, and only the test results with P < 0.017 are considered to be significant.[75] Note that the Bonferroni adjustment is only applicable to a hypothesis testing with a small number of multiple comparisons, otherwise, it would lead to a high rate of false negatives [Figure 3].[75]
Figure 3.
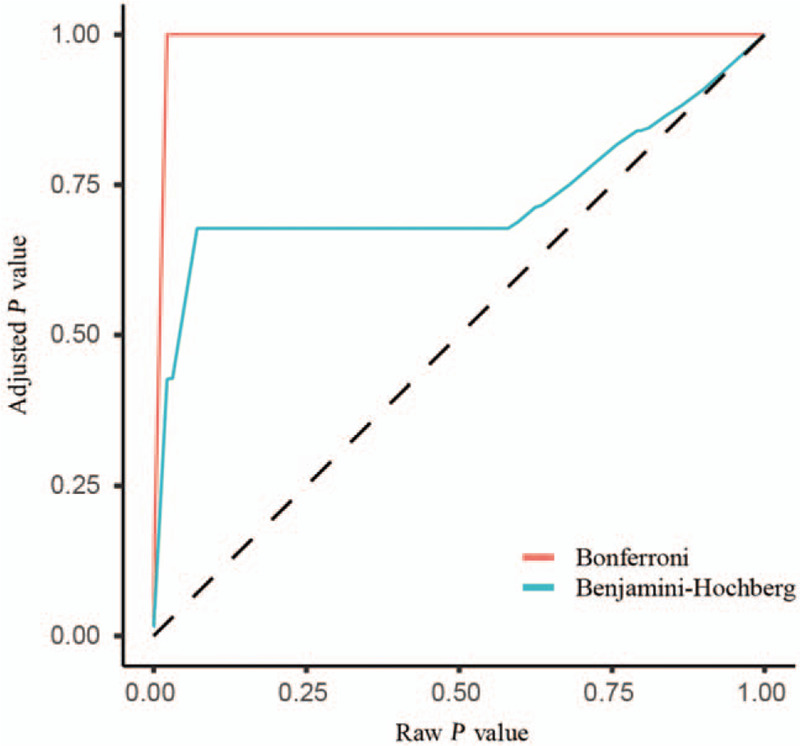
Strength of different P value adjustment methods. The plot shows that the Benjamini-Hochberg method is less conservative than the Bonferroni. The adjusted P values that are generated using the Bonferroni method approach 1.0 sharply as the raw P values increase.
An alternative way to tackle the problems with the multiple comparisons is to control the FDR, which is the expected proportion of type I errors or the number of false positives in all the rejected null hypotheses. For example, if five out of 100 hypothesis tests are false discoveries, then the FDR is 5%. The “Benjamini-Hochberg (BH) adjusted P values” rather than raw P values are often used in microbiome research. The adjusted P = raw P ∗ m/i, where m is the number of tests and i is the rank of each P value.[75] If the adjusted P value is smaller than the chosen FDR you choose, the test is considered to be significant. In contrast to the Bonferroni method, the BH method is less conservative and is often used in multiple comparisons of microbiome features. The Bonferroni and BH are the most commonly used P value adjustment methods.[76] The strength of the two P value adjustment methods is shown in Figure 3.
The PERMANOVA
The β-diversity of different communities can be compared using several statistical methods or models such as the PERMANOVA, the Mantel test, analysis of similarity, and multi-response permutation procedures. The PERMANOVA is the most popular and considered to be the most powerful.[77] It is implemented through the function adonis() in the R package vegan.[52] Four dissimilarity or distance metrics can be processed in the vegan package: the Bray-Curtis dissimilarity, the Jaccard distance, and weighted and unweighted UniFrac distances.[25] If the P value of the permutation test is smaller than 0.05, which indicates that the β-diversity between different communities is statistically significant. Another output of the test is R2, which indicates how much of the total variance can be explained by grouping factor.[25]
The Mantel test
The Mantel test is often used to analyze associations between metadata matrix and community matrix.[77] It is implemented using the function mantel() in the R package vegan.[52,77] The output of the test has at least two main statistics: P value and r. The value of r, similar to other types of correlation coefficients, ranges from − 1 to +1.[25] For example, suppose a researcher would like to know whether the grouping factor (eg, smoking status) of the metadata impacts on the composition of gut microbiome. If the P value is smaller than 0.05 and r is greater than 0, which indicates that the composition of the gut microbiome differs between the smoking group and non-smoking group, then the metadata matrix and community matrix are positively related.
Bioinformatics Analyses
Marker gene analyses: from raw data to taxonomy profile
Several popular software or pipelines, such as QIIME 2,[13] USEARCH,[78] VSEARCH,[79] and mothur,[80] are available for amplicon data analysis. The former two have many advantages and have been recommended by many researchers. The advantages and disadvantages of each software or pipeline have been described in detail in our previous paper.[81] The main steps of amplicon analysis are shown in Figure 4A. We usually start with the raw paired-end Illumina data in fastq format, and the final output is a feature table, which is also known as OTU table. The first step is to recover clean amplicon sequences from the raw data because the raw data include artifacts such as primers and barcodes. It comprises three main procedures: merging paired-end sequences, assigning sample ID by the barcodes, and removing the primers. Due to the raw data having no uniform standard format, we need to design a proper analysis pipeline tailored to the above procedures. Alternatively, we could use the clean amplicon data provided by gene sequencing companies. A typical analysis flowchart for recovering the clean amplicon sequences is shown in Figure 4B. The second step is to filter low-quality reads out to limit the background noise. The third step is to identify non-redundant sequences and their counts. High-quality sequences still have lots of artifacts such as sequence errors and chimera. The counts of the non-redundant sequences are key information to find out credible sequences. The fourth step is to select representative sequences (features). This step is based on unique reads and implemented by clustering the sequences into OTUs or by denoising selected ASVs.[18,82] This step also includes the de novo detection and removal of chimera. The fifth step is a reference-based chimera detection, which is an alternative process to the fourth step.[83] The feature sequences can be further filtered by mapping the sequences into the database such as the comprehensive rRNA gene database, SILVA.[84] It should be noted that the step can reduce false-positive rates and is prone to cause false-negative results. Finally, the feature table is generated by comparing clean amplicon data with feature sequences [Figure 4A]. The feature sequences are then assigned to taxonomic classification using the classifier based on the Ribosomal Database Project[85] or Greengenes[86] database. Additionally, based on the 16S rRNA gene profile, a functional profile can be predicted by PICRUSt,[87,88] FAPROTAX,[87,89] and BugBase.[90]
Figure 4.
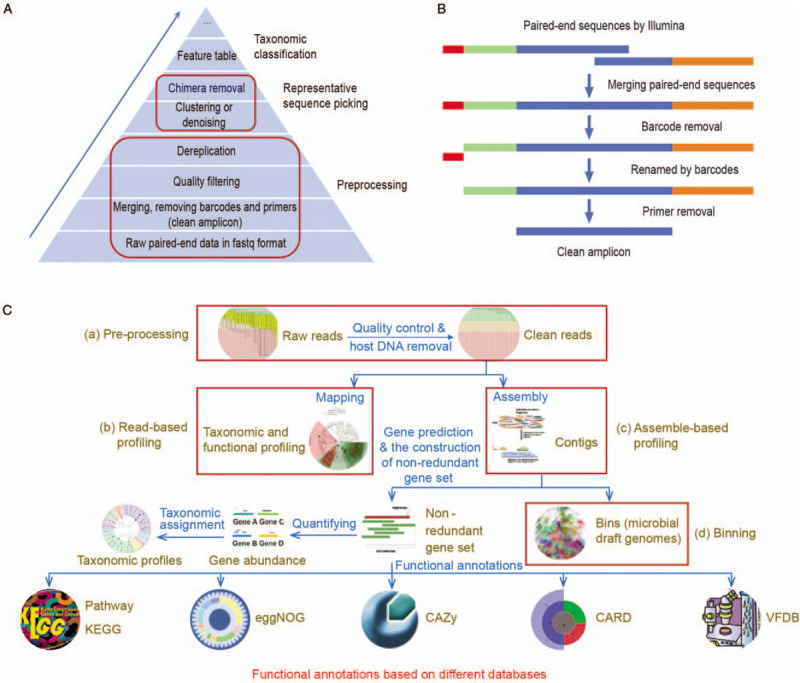
Bioinformatics analysis pipelines for human microbiome research. (A) Main steps for taxonomic profiling of amplicon data. (B) Typical flowchart of pre-processing in amplicon data: from raw paired-end sequences to clean amplicons. (C) Analysis pipeline for metagenomic sequencing data. (a) Pre-processing. It involves removing low-quality, adaptor and host reads. The output corresponds to clean reads. (b) Read-based profiling. It involves that reads map against the databases to infer taxonomic and metabolic profiles. (c) Assemble-based profiling. It involves assembling short reads into contigs, predicting genes, constructing non-redundancy gene catalog, and blasting against the databases to profile taxonomy and functions. (d) Binning. It involves recovering draft genome of uncultured microbe and reconstruction of phylogenetic and metabolic pathways. CARD: Comprehensive antibiotic resistance database; CAZy: Carbohydrate-active enzymes database; eggNOG: Evolutionary genealogy of genes: non-supervised orthologous groups; KEGG: Kyoto encyclopedia of genes and genomes; VFDB: Virulence factor database.
Metagenome analyses: from raw data to taxonomy and functional profiles
Amplicon sequencing only yields taxonomy profile, and the PCR process easily generates bias and chimera.[83] Shotgun metagenomic sequencing provides more detailed genomic information and higher taxonomic resolution than the amplicon sequencing.[67] Compared with the amplicon method, metagenomic analysis is more complex but it provides more accurate taxonomy, multi-dimensional functional profile, and draft genomes of uncultured microbes. The overview of the metagenomic pipeline is shown in Figure 4C. The first step is to pre-process raw sequence data. The raw data contain the contamination of low quality and host-associated reads. We can perform data quality checks using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and then use the KneadData pipeline to perform quality control[91] and to remove host DNA.[92] The KneadData is available at http://huttenhower.sph.harvard.edu/kneaddata. The second step is to profile taxonomy and metabolic potential using the read-based approach. The human microbiome has a high-quality gene catalog and genome,[65,66] so we recommend the read-based method to profile taxonomy and metabolic pathway using the HUMAnN2,[93] which is highly efficient and easy-to-use. However, this method only uses a small part of the sequence information and its use is limited by the known database.[67] If the discovery of new species or gene function is desired, the third step is needed. Several good software tools, such as MEGAHIT[94] and metaSPAdes,[95] were developed for assembling metagenomic clean reads into contigs. The genes are then predicted from contigs by MetaProdigal[96] or Prokka.[97] Additionally, other software tools can also be used for predicting coding genes from metagenomic short reads, such as MetaGeneAnnotator,[98] MetaGeneMark,[99] Glimmer-MG,[100] MetaGUN,[101] FragGeneScan,[102] and Orphelia.[103] To limit duplicated genes, non-redundant gene sets need to be constructed using the CD-HIT when analyzing multiple samples or batches.[104] Gene abundance is calculated by mapping using the Bowtie 2[92] or Salmon.[105] There are at least 20 software tools that can be used to perform taxonomic classification of metagenomic data.[106] We recommend the ultra-fast classifier Kraken 2, which provides fast, accurate, and species-level results.[107] As for functional annotation, DIAMOND, which is a blast-like, fast, and sensitive protein alignment tool, has been recommended by many researchers.[108] Other databases used for functional annotation include Kyoto Encyclopedia of Genes and Genomes,[109] EggNOG (a database of orthology relationships, functional annotation, and gene evolutionary history),[110] Carbohydrate-Active enZYmes Database,[111] Virulence Factors of Pathogenic Bacteria,[112] and Comprehensive Antibiotic Resistance Database.[113] Metagenome usually contains 100 to 1000 species,[65] which is difficult to disentangle from each other. The binning algorithm makes it possible to recover draft genomes from metagenomes and reconstruct phylogenetic and metabolic pathways. The last step is to perform the binning algorithm using the metaWRAP[114] or DASTool [Figure 4C].[115] The software tools have step-by-step tutorials, and there are several sample datasets concerning the human microbiome are available at their websites.[81] Additionally, several integrated pipelines, such as metagenomic analysis toolkit (MOCAT) 2,[116] bioBakery,[98] IMP,[117] and Microbiome Helper,[118] can perform some or all of the above analysis steps. The Chinese tutorials of most popular software can be found in the WeChat subscription account, “meta-genome.”
Now you have owned the taxonomy and functional profiles. It is easy to find out your interesting biomarkers by STAMP or LEfSe.[119,120] All the results can be visualized using R language or ImageGP (http://www.ehbio.com/ImageGP).
Role of Virome in Human Diseases
The role of virome in human diseases has attracted the attention of medical researchers.[121] Many compelling results have been discovered using viral metagenomics in recent years,[122] and some of these technologies have also been used in clinical settings.[123] Viral metagenomics, when integrated with other multi-omics methods, would seem to have a promising application in microbiome research. However, virome research still confronts some challenges. For instance, at least 40% of viral sequences cannot be annotated.[124] Moreover, the sequencing results of the virome are subject to background noises.[17] Lastly, the commercial positive controls, that is, the viral mock communities, used in virome research can hardly be acquired.[16]
Summary and Conclusions
In this review, we discussed the study design, sample collection, statistical methods, and bioinformatics analysis methods for microbiome research. In the “study design” section, we emphasized the importance of the study design, especially the scheme used, the sample size calculation, and the multiple measures used to improve the reliability of research. This is important as a poor study design could yield useless data. In the “statistical analysis” section, we introduced detailed multiple comparison methods. The choice of an appropriate statistical method is important for accurate interpretation of microbiome data. Finally, the “bioinformatics analysis” section illustrated the different bioinformatics methods for analyzing microbiome data. The scripts employed in the figures and examples are available at https://github.com/YongxinLiu/Qian2020CMJ.
In summary, for the microbiome research, the meticulous study design has a pivotal role in obtaining meaningful results, and appropriate statistical methods are important for accurate interpretation of microbiome data. The step-by-step pipelines provide researchers with insights into newly developed bioinformatics analysis methods.
Acknowledgements
The authors would like to thank Shu-Dan Qian and Li-Na Chen for their assistance in managing the project.
Funding
This work was supported by grants from the Project of Young Talent in Medical Field in Zhejiang Province (2015-70) from the Health Commission of Zhejiang Province, and the National Natural Science Foundation of China (No. 31500992).
Conflicts of interest
None.
Footnotes
How to cite this article: Qian XB, Chen T, Xu YP, Chen L, Sun FX, Lu MP, Liu YX. A guide to human microbiome research: study design, sample collection, and bioinformatics analysis. Chin Med J 2020;133:1844–1855. doi: 10.1097/CM9.0000000000000871
Xu-Bo Qian and Tong Chen contributed equally to this work.
References
- 1.Integrative HMP (iHMP) Research Network, Consortium. The integrative human microbiome project. Nature 2019; 569:641–648. doi: 10.1038/s41586-019-1238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.NIH Human Microbiome Portfolio Analysis Team. A review of 10 years of human microbiome research activities at the US National Institutes of Health, fiscal years 2007-2016. Microbiome 2019; 7:31.doi: 10.1186/s40168-019-0620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu Y, Zhao F. Single-cell metagenomics: challenges and applications. Protein Cell 2018; 9:501–510. doi: 10.1007/s13238-018-0544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Vosa U, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet 2019; 51:600–605. doi: 10.1038/s41588-019-0350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018; 359:1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Thingholm LB, Skieceviciene J, Rausch P, Kummen M, Hov JR, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet 2016; 48:1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Xu CM, Liu YX, Wang XQ, Zhang L, Li M, et al. Characteristic dysbiosis of gut microbiota of Chinese patients with diarrhea-predominant irritable bowel syndrome by an insight into the pan-microbiome. Chin Med J 2019; 132:889–904. doi: 10.1097/CM9.0000000000000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong LN, Wang M, Guo J, Wang JP. Role of intestinal microbiota and metabolites in inflammatory bowel disease. Chin Med J 2019; 132:1610–1614. doi: 10.1097/CM9.0000000000000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019; 569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Yu J. The association of diet, gut microbiota and colorectal cancer: what we eat may imply what we get. Protein Cell 2018; 9:474–487. doi: 10.1007/s13238-018-0543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Li HY, Hu XM, Zhang Y, Zhang SY. Current understanding of gut microbiota alterations and related therapeutic intervention strategies in heart failure. Chin Med J 2019; 132:1843–1855. doi: 10.1097/CM9.0000000000000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ 2017; 356:j831.doi: 10.1136/bmj.j831. [DOI] [PubMed] [Google Scholar]
- 13.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019; 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal. Microbiome 2015; 3:31.doi: 10.1186/s40168-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert JA, Lynch SV. Community ecology as a framework for human microbiome research. Nat Med 2019; 25:884–889. doi: 10.1038/s41591-019-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santiago-Rodriguez TM, Hollister EB. Human virome and disease: high-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses 2019; 11:E656.doi: 10.3390/v11070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nat Rev Microbiol 2018; 16:410–422. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 18.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013; 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 19.Westcott SL, Schloss PD. De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 2015; 3:e1487.doi: 10.7717/peerj.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 2017; 11:2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YX, Qin Y, Bai Y. Reductionist synthetic community approaches in root microbiome research. Curr Opin Microbiol 2019; 49:97–102. doi: 10.1016/j.mib.2019.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Xia Y, Sun J, Chen D. Xia Y, Sun J, Chen D. Community diversity measures and calculations. Statistical Analysis of Microbiome Data with R. Singapore:Springer Singapore; 2018. 167–190. [Google Scholar]
- 23.Borcard D, Gillet F, Legendre P. Borcard D, Gillet F, Legendre P. Community diversity. Numerical Ecology with R. Switzerland:Springer International Publishing; 2018. 369–412. [Google Scholar]
- 24.Xia Y, Sun J, Chen D. Xia Y, Sun J, Chen D. Introductory overview of statistical analysis of microbiome data. Statistical Analysis of Microbiome Data with R. Singapore:Springer Singapore; 2018. 43–75. [Google Scholar]
- 25.Xia Y, Sun J, Chen D. Xia Y, Sun J, Chen D. Multivariate community analysis. Statistical Analysis of Microbiome Data with R. Singapore:Springer Singapore; 2018. 285–330. [Google Scholar]
- 26.Bray JR, Curtis JT. An ordination of the upland forest communities of Southern Wisconsin. Ecol Monogr 1957; 27:326–349. doi: 10.2307/1942268. [Google Scholar]
- 27.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, et al. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012; 28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 2007; 73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borcard D, Gillet F, Legendre P. Borcard D, Gillet F, Legendre P. Unconstrained ordination. Numerical Ecology with R. Switzerland:Springer International Publishing; 2018. 151–201. [Google Scholar]
- 31.Borcard D, Gillet F, Legendre P. Borcard D, Gillet F, Legendre P. Canonical ordination. Numerical Ecology with R. Switzerland:Springer International Publishing; 2018. 203–297. [Google Scholar]
- 32.Xia Y, Sun J, Chen D. Xia Y, Sun J, Chen D. Exploratory analysis of microbiome data and beyond. Statistical Analysis of Microbiome Data with R. Singapore:Springer Singapore; 2018. 191–294. [Google Scholar]
- 33.Legendre P, Gallagher ED. Ecologically meaningful transformations for ordination of species data. Oecologia 2001; 129:271–280. doi: 10.1007/s004420100716. [DOI] [PubMed] [Google Scholar]
- 34. Aryal S. Cross-Sectional Study. 2019. Available from: https://microbenotes.com/cross-sectional-study/. [Accessed April 3, 2020] [Google Scholar]
- 35.Rizzetto L, Fava F, Tuohy KM, Selmi C. Connecting the immune system, systemic chronic inflammation and the gut microbiome: The role of sex. J Autoimmun 2018; 92:12–34. doi: 10.1016/j.jaut.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol 2016; 16:90.doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun L, Ma L, Ma Y, Zhang F, Zhao C, Nie Y. Insights into the role of gut microbiota in obesity: pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 2018; 9:397–403. doi: 10.1007/s13238-018-0546-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolodziejczyk AA, Zheng D, Elinav E. Diet-microbiota interactions and personalized nutrition. Nat Rev Microbiol 2019; 17:742–753. doi: 10.1038/s41579-019-0256-8. [DOI] [PubMed] [Google Scholar]
- 39.Davenport ER, Mizrahi-Man O, Michelini K, Barreiro LB, Ober C, Gilad Y. Seasonal variation in human gut microbiome composition. PLoS One 2014; 9:e90731.doi: 10.1371/journal.pone.0090731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willmann M, Vehreschild M, Biehl LM, Vogel W, Dorfel D, Hamprecht A, et al. Distinct impact of antibiotics on the gut microbiome and resistome: a longitudinal multicenter cohort study. BMC Biol 2019; 17:76.doi: 10.1186/s12915-019-0692-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018; 555:623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med 2018; 24:1804–1808. doi: 10.1038/s41591-018-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F, Cui B, He X, Nie Y, Wu K, Fan D, et al. Microbiota transplantation: concept, methodology and strategy for its modernization. Protein Cell 2018; 9:462–473. doi: 10.1007/s13238-018-0541-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sedgwick P. Before and After Study Designs. 2014. Available from: https://www.bmj.com/content/349/bmj.g5074. [Accessed April 3, 2020] [Google Scholar]
- 45.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haro C, Rangel-Zuniga OA, Alcala-Diaz JF, Gomez-Delgado F, Perez-Martinez P, Delgado-Lista J, et al. Intestinal microbiota is influenced by gender and body mass index. PLoS One 2016; 11:e0154090.doi: 10.1371/journal.pone.0154090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014; 505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med 2018; 24:1526–1531. doi: 10.1038/s41591-018-0160-1. [DOI] [PubMed] [Google Scholar]
- 49.Marques FZ, Jama HA, Tsyganov K, Gill PA, Rhys-Jones D, Muralitharan RR, et al. Guidelines for transparency on gut microbiome studies in essential and experimental hypertension. Hypertension 2019; 74:1279–1293. doi: 10.1161/HYPERTENSIONAHA.119.13079. [DOI] [PubMed] [Google Scholar]
- 50.Debelius J, Song SJ, Vazquez-Baeza Y, Xu ZZ, Gonzalez A, Knight R. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol 2016; 17:217.doi: 10.1186/s13059-016-1086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia Y, Sun J, Chen D. Xia Y, Sun J, Chen D. Power and sample size calculations for microbiome data. Statistical Analysis of Microbiome Data with R. Singapore:Springer Singapore; 2018. 129–166. [Google Scholar]
- 52. Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: Community Ecology Package; 2019. Available from: https://cran.r-project.org/web/packages/vegan/index.html. [Accessed April 3, 2020] [Google Scholar]
- 53.Hornung BVH, Zwittink RD, Kuijper EJ. Issues and current standards of controls in microbiome research. FEMS Microbiol Ecol 2019; 95:fiz045.doi: 10.1093/femsec/fiz045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol 2019; 27:105–117. doi: 10.1016/j.tim.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 55.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med 2014; 6:237ra265.doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature 2019; 572:329–334. doi: 10.1038/s41586-019-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018; 6:226.doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rausch P, Ruhlemann M, Hermes BM, Doms S, Dagan T, Dierking K, et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019; 7:133.doi: 10.1186/s40168-019-0743-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sze MA, Schloss PD. The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. mSphere 2019; 4:e00163–e0019. doi: 10.1128/mSphere.00163-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Zheng J, Shi W, Du N, Xu X, Zhang Y, et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut 2018; 67:1614–1625. doi: 10.1136/gutjnl-2018-315988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He Y, Wu W, Zheng HM, Li P, McDonald D, Sheng HF, et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med 2018; 24:1532–1535. doi: 10.1038/s41591-018-0164-x. [DOI] [PubMed] [Google Scholar]
- 62.Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017; 357:802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature 2011; 473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 2014; 32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 66.Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019; 176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 2017; 35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 68.Wu EY, Bryan AR, Rabinovich CE. Kliegman RM, Stanton BF, St Geme JW, III, Schor NF. Juvenile idiopathic arthritis. Nelson Textbook of Pediatrics. The United States:Elsevier; 2015. 1160–1170. [Google Scholar]
- 69.Claesson MJ, Clooney AG, O’Toole PW. A clinician's guide to microbiome analysis. Nat Rev Gastroenterol Hepatol 2017; 14:585–595. doi: 10.1038/nrgastro.2017.97. [DOI] [PubMed] [Google Scholar]
- 70.Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 2016; 14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med 2018; 24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gorzelak MA, Gill SK, Tasnim N, Ahmadi-Vand Z, Jay M, Gibson DL. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PLoS One 2015; 10:e0134802.doi: 10.1371/journal.pone.0134802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choo JM, Leong LE, Rogers GB. Sample storage conditions significantly influence faecal microbiome profiles. Sci Rep 2015; 5:16350.doi: 10.1038/srep16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han M, Hao L, Lin Y, Li F, Wang J, Yang H, et al. A novel affordable reagent for room temperature storage and transport of fecal samples for metagenomic analyses. Microbiome 2018; 6:43.doi: 10.1186/s40168-018-0429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDonald JH. McDonald JH. Multiple tests. Handbook of Biological Statistics. Baltimore, MD, USA:Sparky House Publishing; 2014. 257–263. [Google Scholar]
- 76. Arnold T, Emerson J. The R Stats Package. 2019. Available from: https://www.rdocumentation.org/packages/stats/versions/3.6.1. [Accessed April 3, 2020] [Google Scholar]
- 77.Xia Y, Sun J. Hypothesis testing and statistical analysis of microbiome. Genes Dis 2017; 4:138–148. doi: 10.1016/j.gendis.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 79.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 2016; 4:e2584.doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009; 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu YX, Qin Y, Guo XX, Bai Y. Methods and applications for microbiome data analysis. (In Chinese). Hereditas (Beijing) 2019; 41:845–862. doi: 10.16288/j.yczz.19-222. [DOI] [PubMed] [Google Scholar]
- 82.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016; 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011; 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013; 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 2014; 42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012; 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang J, Liu YX, Zhang N, Hu B, Jin T, Xu H, et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol 2019; 37:676–684. doi: 10.1038/s41587-019-0104-4. [DOI] [PubMed] [Google Scholar]
- 88.Zheng M, Zhou N, Liu S, Dang C, Liu Y-X, He S, et al. N2O and NO emission from a biological aerated filter treating coking wastewater: main source and microbial community. J Clean Prod 2019; 213:365–374. doi: 10.1016/j.jclepro.2018.12.182. [Google Scholar]
- 89.Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016; 353:1272–1277. doi: 10.1126/science.aaf4507. [DOI] [PubMed] [Google Scholar]
- 90.Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, Sidiropoulos D, et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017. 133462.doi: 10.1101/133462. [Google Scholar]
- 91.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 2018; 15:962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015; 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 95.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: a new versatile metagenomic assembler. Genome Res 2017; 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012; 28:2223–2230. doi: 10.1093/bioinformatics/bts429. [DOI] [PubMed] [Google Scholar]
- 97.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 98.Noguchi H, Taniguchi T, Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res 2008; 15:387–396. doi: 10.1093/dnares/dsn027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 2010; 38:e132.doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res 2012; 40:e9.doi: 10.1093/nar/gkr1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y, Guo J, Hu G, Zhu H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinformatics 2013; 14: Suppl 5: S12.doi: 10.1186/1471-2105-14-S5-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rho M, Tang H, Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 2010; 38:e191.doi: 10.1093/nar/gkq747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoff KJ, Lingner T, Meinicke P, Tech M. Orphelia: predicting genes in metagenomic sequencing reads. Nucleic Acids Res 2009; 37:W101–W105. doi: 10.1093/nar/gkp327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012; 28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017; 14:417–149. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ye SH, Siddle KJ, Park DJ, Sabeti PC. Benchmarking metagenomics tools for taxonomic classification. Cell 2019; 178:779–794. doi: 10.1016/j.cell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. bioRxiv 2019. 762302.doi: 10.1101/762302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015; 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 109.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 2019; 47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 2014; 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 2019; 47:D687–D692. doi: 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 2017; 45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018; 6:158.doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 2018; 3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kultima JR, Coelho LP, Forslund K, Huerta-Cepas J, Li SS, Driessen M, et al. MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics 2016; 32:2520–2523. doi: 10.1093/bioinformatics/btw183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Narayanasamy S, Jarosz Y, Muller EE, Heintz-Buschart A, Herold M, Kaysen A, et al. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol 2016; 17:260.doi: 10.1186/s13059-016-1116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Comeau AM, Douglas GM, Langille MG. Microbiome helper: a custom and streamlined workflow for microbiome research. mSystems 2017; 2:e00127-16.doi: 10.1128/mSystems.00127-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 2014; 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60.doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carroll D, Daszak P, Wolfe ND, Gao GF, Morel CM, Morzaria S, et al. The global virome project. Science 2018; 359:872–874. doi: 10.1126/science.aap7463. [DOI] [PubMed] [Google Scholar]
- 122.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet 2019; 20:341–355. doi: 10.1038/s41576-019-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krishnamurthy SR, Wang D. Origins and challenges of viral dark matter. Virus Res 2017; 239:136–142. doi: 10.1016/j.virusres.2017.02.002. [DOI] [PubMed] [Google Scholar]