


Abstract
The ribonucleolytic exosome complex is central for nuclear RNA degradation, primarily targeting non-coding RNAs. Still, the nuclear exosome could have protein-coding (pc) gene-specific regulatory activities. By depleting an exosome core component, or components of exosome adaptor complexes, we identify ∼2900 transcription start sites (TSSs) from within pc genes that produce exosome-sensitive transcripts. At least 1000 of these overlap with annotated mRNA TSSs and a considerable portion of their transcripts share the annotated mRNA 3′ end. We identify two types of pc-genes, both employing a single, annotated TSS across cells, but the first type primarily produces full-length, exosome-sensitive transcripts, whereas the second primarily produces prematurely terminated transcripts. Genes within the former type often belong to immediate early response transcription factors, while genes within the latter are likely transcribed as a consequence of their proximity to upstream TSSs on the opposite strand. Conversely, when genes have multiple active TSSs, alternative TSSs that produce exosome-sensitive transcripts typically do not contribute substantially to overall gene expression, and most such transcripts are prematurely terminated. Our results display a complex landscape of sense transcription within pc-genes and imply a direct role for nuclear RNA turnover in the regulation of a subset of pc-genes.
INTRODUCTION
RNA degradation is essential for maintaining transcript homeostasis in all cells. Together with transcription, it controls steady-state RNA expression levels, which underlie all major cellular transitions in development and disease. While RNA degradation in the cytoplasm is considered to be the main determinant for mRNA half-lives, the extent to which nuclear RNA decay is involved has been less clear. In the nucleus, transcript turnover is often coupled to transcription termination and/or processing of the nascent RNA (1–5). Moreover, it has been suggested that prolonged nuclear residence time correlates with the increased turnover of polyadenylated RNA species (5). Together, this serves to dampen the expression of a large amount of pervasively transcribed RNAs (2), thought to primarily include a multitude of long non-coding RNAs (lncRNAs), which as a group is prone to rapid nuclear degradation (6).
The highly conserved 3′-5′ exo- and endo-nucleolytic RNA exosome complex is a primary caretaker of the decay of capped RNAs in eukaryotic nuclei (3,4). In mammalian nuclei, the exosome is composed of a core unit with associated nucleolytic activities, which in the nucleoplasm may contact one of two exosome adaptor complexes in order to target RNAs for degradation (5). One such adaptor, the nuclear exosome targeting (NEXT) complex targets RNAs, that are primarily short, mono-exonic and non-adenylated RNAs (7–9). These can be lncRNAs, including subsets of enhancer RNAs (eRNAs) and promoter upstream transcripts (PROMPTs)/upstream antisense RNAs (uaRNAs) (10–13). The polyA exosome targeting (PAXT) connection targets similar RNA biotypes as the NEXT complex, but specifically those that are polyadenylated (9). Additionally, PAXT mediates the exosomal degradation of longer and processed nuclear RNAs (14). Of interest, disruption of the nuclear exosome as well as of both the NEXT and PAXT pathways affect stem cell differentiation, suggesting a role for nuclear RNA decay in gene expression regulation (15–17).
Although mRNAs are generally not considered major targets of the nuclear exosome, early reports revealed that annotated mRNA TSSs may produce exosome-sensitive transcripts (12,18,19). Sequencing of capped RNA 5′ ends showed that a subset of alternative mRNA TSSs gives rise to exosome-sensitive RNAs although their exact nature was not established (6). These observations were rationalized in several recent papers, which established that premature termination of transcription (also referred to as ‘attenuation’) can affect the transcriptional output of full-length transcripts from pc-gene TSSs (reviewed in (20)). Such premature transcription termination can be mediated by nascent RNA cleavage by the canonical cleavage and polyadenylation (CPA) machinery or by the Integrator complex, and the resulting short transcripts were shown to be exosome sensitive (21–24). Interestingly, recent work also showed that a substantial number of full-length mRNAs might be nuclear exosome substrates (25). Collectively, these findings demonstrate that a portion of pc-genes emit transcripts that are affected by the nuclear exosome. These can either be prematurely terminated transcripts or full-length RNAs. However, a systematic analysis of such sensitivity, including the nature of the isoforms produced and whether they arise from major and/or cryptic TSSs commonly present in complex genomes (26), has been lacking. This is relevant to assess the impact of such transcription events on the overall output of pc-gene promoters.
Here, we confirm that a substantial number of pc-genes harbor TSSs, producing nuclear exosome-sensitive transcripts. Surprisingly, ∼360 such genes only employ one primary annotated TSS to produce full-length transcripts across diverse cells and tissues. These genes often encode transcription factors and immediate early response genes. Another set of pc-genes also employ a single annotated TSS, but primarily produce prematurely terminated transcripts. We show that this production is likely due to a bystander effect of strong and nearby mRNA initiation on the reverse strand. We also explore multi-TSS genes where at least one TSS produces exosome-sensitive RNAs, and find that such TSSs have a minor contribution to overall gene expression, where the length of exosome-sensitive RNAs produced is correlated to the distance to other TSSs producing exosome-insensitive RNAs. Overall, our work shows that the exosome shapes the expression of several pc genes, many of which are functionally important across cells.
MATERIALS AND METHODS
HeLa cell culture and small interfering RNA (siRNA)-mediated knockdown
HeLa Kyoto cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. siRNA transfections (for SLIC-CAGE and TIF-seq) were carried out using lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were treated with 20 nM siRNA for 4 days, including a re-transfection 2 days after the initial transfection. siRNA sequences: siGFP: GACGUAAACGGCCACAAGUdTdT; siRRP40: CACGCACAGUACUAGGUCAdTdT; siZCCHC8: GGAAUGUACCUCAGGAUAAdTdT; siZFC3H1: GAUUAGAGUCCAUGAUUAAdTdT. RNA was extracted using TRIzol (Invitrogen) and treated with TURBO DNase (Invitrogen) following the manufacturer's instructions.
Western blotting analysis
Cells were lysed with lysis buffer (10 mM Tris–Cl pH 7. 4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% NP-40, 0.5% Triton X-100) on ice for 10 min, then centrifuged at 12 000 rpm for 20 min. The protein concentration in the supernatant was measured using Bradford solution (Bio-Rad). Equal amounts of proteins were loaded onto PAGE gels. After running, proteins were transferred to PVDF membranes, which were blocked with 5% skimmed milk/PBS-T for 1 h at room temperature (RT), and then incubated with primary antibodies diluted in PBS-T at 4°C overnight, followed by washing 3 × 10 min with PBS-T. Membranes were then incubated with HRP-conjugated secondary antibodies diluted in PBS-T for 1 h at RT, followed by washing 3 × 10 min with PBS-T. SuperSignal West Femto HRP substrate (ThermoFisher Scientific) was applied to the membranes and the signal was detected with X-ray film (Konica Minolta). Antibodies: RRP40: ProteinTech, 15062-1-AP, 1:1000; ZFC3H1: Sigma, HPA-007151, 1:1000; ZCCHC8:Novus Biologicals, NB100-94995, 1:1000; Tubulin: Rockland, 200-301-880, 1:2500. Western blotting analysis are shown in Supplementary Figure S11.
SLIC-CAGE library preparation, sequencing
SLIC-CAGE preparation was performed as described in (27) with an input of 2000 ng of total RNA as starting material. Individually prepared SLIC-CAGE libraries with unique barcodes were pooled (8 per lane). The following 8 barcodes were used: # 1 (ACC), #2 (CAC), # 3 (AGT), # 4 (GCG), # 5 (ATG), # 6 (TAC), # 7 (ACG) and #8 (GCT). All used primers and adaptors were purchased from Integrated DNA technologies (IDT). An Illumina NextSeq 500 instrument at the BRIC, University of Copenhagen, was used for sequencing.
SLIC-CAGE data processing, quantification
CAGE reads were trimmed to remove linker sequences at 5′ ends and incorrect ‘G’ calls at 3′ ends using cutadapt (version 1.14) (28) with parameters -u 5 -m 30 –nextseq-trim = 30 -l 70. Trimmed reads were filtered so that only reads with minimum sequence quality of 30 in at least 50% of the bases were kept. rRNAs were further removed using rRNAdust (http://fantom.gsc.riken.jp/5/suppl/rRNAdust/). Remaining reads were mapped to the human genome hg19 using bwa (version 0.7.16a-r1181) (29) with default settings. The number of 5′ ends of CAGE reads were counted at each genomic position to give a unit of CAGE tag start site (CTSS), at single-base resolution. The raw counts were normalized to tags per million mapped reads (TPM) for subsequent quantification.
Public data acquisition, processing and analysis
Public data used in this study were obtained from ENCODE and Gene Expression Omnibus (GEO); the accession numbers were as follows: DNase-seq (ENCODE, ENCSR959ZXU), HeLa S3 H3K4me3, H3K36me3 and H3K27ac ChIPseq (GEO, GSE29611), HeLa S3 nascent RNA-seq (GEO, GSE61332), nuclear RNA-seq of siRRP40 and siEGFP control (GEO, GSE108197), total RNA-seq of siRRP40, siZCCHC8, siZFC3H1 and siEGFP control (GEO, GSE84172), CAGE of siRRP40- and siEGFP control (GEO, GSE62047). For CAGE of siRRP40 and control, the triplicate HeLa siRRP40 and control CAGE libraries were computationally processed as described in (6). In brief, using the FASTX Toolkit (v0.0.13, http://hannonlab.cshl.edu/fastx_toolkit), reads were trimmed from the 5′ end to remove linker sequences, trimmed from the 3′ end to a length of 25 bp and subsequently filtered for a minimum sequencing quality of 30 in 50% of the bases. Trimmed and filtered reads were mapped to the human genome (hg19) using Bowtie (version 0.12.7) (30) with parameters –t –best –strata –v –k 10 –y –p 6 –phred33-quals –chunksmbs 512 –e 120 –q –un. The number of CAGE tag 5′ ends were counted in each genomic position and nearby 5′ ends on the same strand were merged as in (31) to create tag clusters (TCs). The TCs read counts were normalized to tags per million mapped reads (TPM). The CAGE defined TCs were annotated using GENCODE v19 annotation (32) based on a hierarchical ranked classification, where in case of multiple classification overlaps the highest ranked was selected, the hierarchical model is shown in Figure 1C. The categories in priority order and their definitions were as follows: TCs within ±100 bp of the most upstream GENCODE annotated TSS of a gene—primary TSS; TCs within ±100 bp from all other GENCODE annotated TSSs of a gene—alternative TSS; TCs within 5′ UTRs of transcripts with annotated coding regions (CDS)—5′ UTR; TCs within CDS—CDS; TCs within 3′ UTRs of transcripts with annotated CDS—3′ UTR; TCs within exons of transcripts where no CDS is annotated—exon; TCs within introns—intron; TCs within a 10kb window upstream of the most upstream CENCODE annotated TSS of a gene—upstream. For non-CAGE data, replicates were pooled and signals were averaged over replicates for subsequent analysis. For gene level RNA-seq fold change (FC), strand-specific, uniquely mapped and properly paired reads across the GENCODE v19 gene models were counted using featureCounts from the R package Rsubread (1.32.1) (33), to minimize the expression differences between samples for genes with low read counts, a pseudocount of 7 was added when normalizing raw read counts to the library size. FC values were calculated between mean values of the normalized read counts from siRRP40, siZCCHC8 or siZFC3H1 and that from Ctrl libraries. For RNA-seq FC across gene bodies, FC values of siRRP40, siZCCHC8 or siZFC3H1 versus Ctrl were calculated using bigwigCompare from deepTools (34) over a 5 bp window. A pseudocount of 0.05 was added before FC calculation.
Figure 1.

Quantification of sense strand exoTCs within pc-genes. (A) Occurence of exoTCs within pc-genes. The Y-axes of each subpanel show the frequency of sense strand TCs within pc-genes, while the X-axes show exosome sensitivity scores based on CAGE. Each panel corresponds to one TC expression cutoff as indicated on top together with the number of cases satisfying the particular criterion. Vertical dotted lines indicate sensitivity score cutoffs for defining exoTCs (grey-shaded areas), whose overlap of genic features are analyzed in panel B. (B) Overlap between exoTCs and genic features. Y-axes display the percentage of exoTCs from (A) overlapping a given genic annotation feature shown on the X-axes (and as defined in panel C). Expression cutoffs and the number of analyzed exoTCs are indicated on the upper right of each panel. (C) Schematic representation of the gene features used to annotate the exoTCs in panel B. For details see MATERIALS AND METHODS. (D) Co-occurences of exoTCs and non-exoTCs within pc-genes. TCs with expression > 2 TPM in either siRRP40- or Ctrl-samples were analyzed. Matrix cells show the number of genes having a given combination of exo and non-exoTCs and the percentage in parenthesis of total expressed genes (9803). Cell shading indicates genes with only a single TC (grey: analyzed in Figure 2A, Supplementary Figure S1A) and genes with multiple TCs (white: analyzed in Figures 6–8). Total number of genes in each row/column is counted using cells in the box with black solid line.
Sensitivity score calculation
A sensitivity score was designed to quantify the relative amount of expression increase or decrease after depletion of a given factor. It was calculated as:
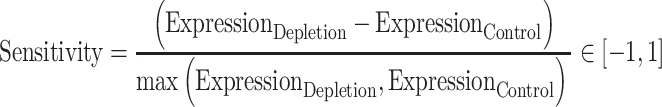 |
where the Expression is the normalized strand-specific CAGE or RNA-seq expression for a given library.
Nascent RNA quantification and directionality calculation
Nascent RNA levels were quantified by data from (35). For quantifying nascent RNAs produced from exoTCs, the strand-specific genomic coverage in −100 to +500 bp regions was computed using computeMatrix reference-point from deepTools; for quantifying nascent RNAs produced from the upstream opposite strand of exoTCs, the strand-specific genomic coverage of NET-seq in -1 to −600 bp regions was computed the same way as exoTCs strand.
A directionality score was designed to measure the biases of transcription or expression levels from opposite strands. It was calculated as follows:
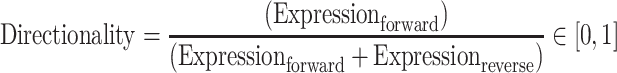 |
where Expressionforward is the transcription or expression levels of the TC on the forward or sense strand and Expressionreverse on the reverse or upstream opposite strand.
Definition of upstream opposite strand TCs
The upstream opposite strand TC of a given TC was defined as the closest CAGE TC, with TPM > 1 in siRRP40, that fell on the upstream opposite strand of the TC within 600 bp.
TIF-seq library preparation and sequencing
TIFseq2 library preparation was performed as described in (36) using 2500 ng of total RNA as starting material. In brief, 5′P RNA was dephosphorylated using Calf Intestinal alkaline phosphatase, purified and decapped using Cap-Clip. Newly exposed 5′P were ligated to chimeric DNA/RNA oligos and reverse transcribed using barcoded oligo dT primers. Full-length cDNA was amplified by PCR and digested with NotI-HF to produce sticky ends. We then circularized the amplified cDNA, removed non- circular fragments and fragmented the purified circles by sonication. Fragments spanning the 5′ and 3′ cDNA ends and containing biotin were bound to streptavidin magnetic beads and then subjected to Illumina library preparation. Samples were sequenced using a NextSeq 500 instrument with the following options: read1 76 bp, read 2 76 bp, index1 6 bp and index2 6 bp.
TIF-seq data processing and analysis
Sequencing reads were converted by using bcf2fastq (v2.20.0) and demultiplexed according to the indexes, allowing two mismatches in index 1 and one mismatch in index 2. TIF-seq2 sequencing primer (AGGTGACCGGCAGGTGT) and Illumina TruSeq adapter (AGATCGGAAG) were removed using cutadapt (v1.16) (28). Then, 8-bp unique molecular identifiers (UMIs) were extracted with UMI-tools [PMID:28100584] (v0.5.4) from the 5′ ends and extra adenine stretches in the 3′ ends were removed with cutadapt (v1.16). STAR (v2.5.3a) (37) were employed for aligning 5′-end reads and 3′-end reads separately to the human reference genome hg38, allowing maximum intron length as 1 Mb. A customized script adapted from UMI-tools was employed to remove PCR duplicates from uniquely mapped read pairs on the same chromosome, allowing 1-bp shifting in the 5′ ends. The hg38 genome coordinates were converted to hg19 using UCSC liftOver tool (38). Paired 5′ end and 3′ end reads located on the same chromosome and opposite strand were used to form 5′-to-3′ end TIF transcripts. To avoid 3′ ends produced by spurious internal poly A priming by the oligo(dT) primer, the sequence immediately downstream of the 3′ end of each TIF transcript was further examined. If the downstream sequence started with five or more contiguous adenines, or had seven or more adenines in the first 10 bp, the corresponding TIF transcript was removed from this analysis. To remove artificially long TIF transcripts, GENCODE v19 genes were merged into transcription units using merge from bedtools (v2.23.0) (39) with parameter -s, TIF transcripts overlapped with more than one transcription units were removed. Replicates were pooled for subsequent analysis. To associate CAGE TCs with TIF transcripts, TIF transcripts whose 5′ ends fell within a ±100 bp window around TC peaks on the same strand were assigned to the corresponding TCs. If a CAGE TC was annotated as primary/alternative TSS of a pc-gene, the associated TIF transcripts were also assigned to the same gene. TIF-seq FC was calculated as the ratio between the library size normalized TIF transcript counts from pooled siRRP40- and Ctrl-libraries, a pseudocount of 1 was added. To annotate 3′ ends of TIF transcripts, a similar hierarchical approach as CAGE TC annotation was used; the 3′ end hierarchical model is shown in Supplementary Figure S1G. The transcription termination site (TES) was defined as the ±200 bp window region around the 3′ end of GENCODE v19 transcripts, the TSS was defined as the ±100 bp window region around the 5′ end of GENCODE v19 transcripts. 3′ UTR, 5′ UTR and CDS, exon regions were defined as in CAGE annotation. First intron was defined as the first intron of GENCODE v19 transcripts of all expressed genes shown in Figure 1D. Full-length TIF transcripts were defined as the TIF-seq reads with a 3′ end annotated as TES or 3′ UTR, premature terminated TIF transcripts were defined as TIF-seq reads with a 3′ end annotated as features within the gene body excluding 3′ UTR and TES.
Classification of exoTCs based on RNA-seq data
We devised a hierarchical decision tree to classify exoTCs that were associated with multi-exonic genes and their cognate transcripts into four classes (shown in Supplementary Figure S2A). This was based on (i) whether the exoTCs produced exosome-sensitive short transcripts, quantified by FC of siRRP40- versus Ctrl in the first intron 1 kb downstream of the first splice site, (ii) whether the TCs produce exosome-sensitive full-length transcripts, quantified by the same ratio but within all exons downstream of the first intron. The raw reads in the defined genomic regions were counted using featureCounts from the R package Rsubread (1.32.1), FC values were calculated between mean values of the library size normalized read counts from siRRP40- and Ctrl-libraries, a pseudocount of 7 was added. If the TCs did not produce exosome-sensitive full-length transcripts according to ii), they were further divided based on whether they produced any full-length transcripts, quantified by the RPKM normalized siRRP40 RNA-seq counts of all exons downstream of the first intron. Mono-exonic genes represented special cases since they have no introns: they were classified as Class 1 if FC of siRRP40 versus Ctrl exceeded the same threshold as in (ii).
Sequence analysis
Sequences were extracted from the reference genome (hg19) using getfasta from bedtools, G/C content was calculated using letterFrequencyInSlidingView function from the Biostrings R package over 5 bp window (version 2.50.2). For pA site and 5′ splice site analysis, the motifs were obtained from (40), ASAP (41) was used to calculate motif prediction scores and a relative score cutoff of 0.9 was used for deciding the occurrence of the motifs.
Evolutionary conservation
Evolutionary conservation of a TC was calculated as the average phastCons score for a ±100 bp window region around the TC. The phastCons score for human genome (hg19) calculated from multiple alignments with other 99 vertebrate species was used (42); data was downloaded from UCSC (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phastCons100way/). As background, random intergenic and intronic regions of length 200 bp were extracted using shuffle from bedtools with default settings. The intergenic regions were randomly chosen from genomic regions that did not overlap with any GENCODE v19 genes. The intronic regions were randomly chosen from regions in the gene body that did not overlap with an exon from any GENCODE v19 annotated transcript isoforms.
FANTOM5 data processing and analysis
We used FANTOM5 CAGE TC expression data from primary cell groups and tissues, taken from SlideBase (43) processed data which in turn is based on CAGE data from (44,45).
Metagene plots
For metagene plots over gene bodies (Figures 2B–D and 4B, Supplementary Figures S1B–F, S4B), the transcript originating from a given TSS was used to represent the gene; for TSSs that have multiple transcript isoforms, the most expressed transcript isoform was used. Salmon (v0.8.2) (46) was used for the isoform expression quantification and the lightweight-alignment (FMD-based) index was used. Strand-specific genomic coverage or log2 FC, was computed using computeMatrix scale-regions from deepTools, where all transcripts were stretched or shrunk to the same length. For metagene plots from a given genomic location, genomic coverage (ChIPseq) and strand-specific genomic coverage or log2 FC (RNA-seq, TIF-seq) was computed using computeMatrix reference-point from deepTools. In TIF-seq coverage plots (Figure 2B, Supplementary Figure S1B, Figure 6C, D), for each TC, the row-normalized relative coverage is calculated as the percentage of TIF-seq read counts at a given position relative to the total number of TIF-seq reads associated with the TC.
Figure 2.

Characterization of transcripts from single exoTC-containing pc-genes. (A) Overlap between single exoTCs and genic annotation. Y-axis shows the percentage of single exoTCs overlapping the respective gene annotation features (X-axis), visualized as in Figure 1B. Gray shading indicates the set of single exoTCs analyzed in Figures 2–5, and referred to as single exoTCs. (B) Coverage of RNAs produced from single exoTCs by TIF-seq. Each row corresponds to one pc-gene body, with added flanking regions (1 kb in both directions), where the ‘TSS’ position corresponds to the exoTC and the ‘TES’ position corresponds to the GENCODE-annotated transcript 3′ end. Blue bars show TIF-seq read coverage from siRRP40 samples, where the 5′- and 3′-end reads are connected by a blue line. Line color intensity shows the row-normalized relative TIF-seq coverage (see MATERIALS AND METHODS), and white color indicates the absence of TIF-seq reads. Blue lines crossing the TES positions are due to transcripts harboring multiple, distinct 3′ ends. Subpanels with callouts show cases where the majority of TIF-seq reads cover the whole gene (top), cases where most RNAs are prematurely terminated (bottom) and cases with a mixture of RNA lengths (middle). (C) TIF-seq-derived exosome sensitivity of RNAs produced from single exoTCs. Heat map representation following the same conventions as in B, but with color intensities showing siRRP40 versus Ctrl TIF-seq log2 FC in 5 bp windows. Genes were sorted in the same order as in B. (D) RNA-seq-derived exosome sensitivity of RNAs produced from single exoTCs. Heat map representation following the same convention as in C, but using RNA-seq data to calculate FC and only analyzing exonic regions within each gene. Genes were sorted in the same order as in B. (E) Classification of single exoTC genes. Left sub-panel shows cartoons of features characterizing each class. Lines beneath gene models depict the RNAs produced. Dotted lines indicate exosome-sensitive RNAs, solid lines indicate exosome-insensitive RNAs. Right sub-panel shows genome-browser examples of each class with RNA-seq tracks from siRRP40- and Ctrl-libraries (average normalized signal per bp across triplicates) at each strand, where blue color indicates the same strand as the exoTCs, while the red color indicates the opposite strand. TSSs on each strand are indicated by arrows. RefSeq gene models (67) are shown on top. (F) Gene Ontology (GO) over-representation analysis of Class 1 and 2 genes. X-axis shows -log10(FDR) of top 5 terms. Numbers on the right of each bar indicate the number of genes from the two classes annotated with the respective GO term.
Figure 4.

Nuclear exosome decay pathways for transcripts from single exoTCs. (A) NEXT- and PAXT sensitivities of single exoTC-derived transcripts. Combined violin-boxplots show the distribution of exosome sensitivity scores based on CAGE (Y-axis) calculated as in Figure 1A, for siZFC3H1 (PAXT, in yellow) and siZCCHC8 (NEXT, in blue) depletion samples versus Ctrl samples and stratified by exoTC class (X-axis). Single non-exoTCs were analyzed for comparison. (B) NEXT- and PAXT sensitivities of single exoTC transcripts across gene bodies. Metagene profile plots show the average FC of the respective depletions based on RNA-seq data (siZFC3H1 (PAXT) and siZCCHC8 (NEXT)) versus Ctrl, stratified by exoTC class as indicated by color. The FC was plotted between the TSS and the annotated gene 3′ end but with introns removed and with 1 kb added both up- and down-stream. Single non-exoTCs are shown for comparison. (C) Nuclear retention of longer RNAs from exoTCs. X-axis shows the RNA-seq signal (TPM) ratio between nuclear and total RNA for transcripts downstream of single exoTCs of classes 1–3 (classes that produce long RNAs) and single non-exoTCs (Y-axis), visualized as boxplots.
Figure 6.

Characterization of exosome-sensitive transcripts from genes with multiple TCs. (A) Genome-browser examples of exoTC:non-exoTC and non-exoTC:exoTC pairs within genes. The CAGE tracks show average normalized signal per bp from siRRP40- and Ctrl-libraries at each strand. Blue color indicates the same strand as the gene with exoTC and non-exoTC pairs. Red color indicates the opposite strand. TSSs on each strand are indicated by arrows. RefSeq gene models (67) are shown on top. (B) Schematic overview of the analyzed combinations of exoTCs and non-exoTCs. The top schematic shows a fictive gene model with five TCs. The bottom schematic shows all pairs of adjacent TCs that were analyzed, and the number of such pairs found across all pc-genes. (C) Length of transcripts originating from non-exoTC:exoTC pairs. Each heat map row shows one non-exoTC:exoTC pair, centered on the non-exoTC position and sorted by increasing TC pair distance. TC positions are shown with vertical dotted lines. X-axis shows the distance from the non-exoTC in bp. Left heat map shows TIF-seq reads starting from the non-exoTC, where blue bars show TIF-seq coverage and the color intensity indicates the relative coverage as in Figure 2B. Horizontal dashed lines indicate specific distances between TCs (indicated on the Y-axis). The number of analyzed TCs per TC pair distance category are indicated on the left side. Right heat map follows the same conventions, but shows TIF-seq reads starting from the downstream exoTC. Schematics on top show the specific TC type analyzed in each pair, highlighted by a red box. Right violin-boxplots show the distribution of median length (log2-scaled) of TIF-seq transcripts from upstream non-exoTCs (turquoise) and downstream exoTCs (red), split by TC pair distances (Y-axis), summarizing TIF-seq data shown in the two heat maps to the left. (D) Length of transcripts originating from exoTC:non-exoTC pairs. Organized as in panel B, but analyzing exoTC:non-exoTC pairs. Heat maps were centered on the exoTC position. (E) Exosome sensitivity of transcripts originating from non-exoTC:exoTC pairs. Heat maps organized as in panel B, but with bar color showing TIF-seq siRRP40 versus Ctrl log2FC. Combined violin-box plots to the right show TIF-seq siRRP40 versus Ctrl log2FC. For each TC, the maximum value of log2FC of positions covered by the associated TIF-seq reads was used to represent the TIF-seq log2FC of that TC. (F) Exosome sensitivity of transcripts originating from exoTC:non-exoTC pairs. Organized as in D, but analyzing exoTC:non-exoTC pairs. Heat maps were centered on the exoTC position. (G) PAXT and NEXT sensitivities of non-exoTC:exoTC pairs. Schematic on top shows the TC types analyzed. The upper violin-boxplot shows the overall distribution of PAXT and NEXT sensitivity scores of TCs based on CAGE data calculated as in Figure 4A. The lower violin-boxplot shows the distribution of PAXT and NEXT sensitivities of the exoTCs in the TC pair, split by the distance to the paired non-exoTCs in bp (X-axis). (H) PAXT and NEXT sensitivities of exoTC:non-exoTC pairs. Organized as in E, but analyzing exoTC:non-exoTC pairs.
Data visualization and statistics
We used R (https://www.r-project.org/) and the ggplot2 R package (47) unless otherwise noted for visualizations.
RESULTS
Many TSSs within pc-genes produce exosome-sensitive transcripts
To assess the prevalence of TSSs within pc-genes, which produce exosome-sensitive RNAs, we measured capped RNA 5′ end abundances by Cap Analysis of Gene Expression (CAGE) data from (6) to compare RRP40/EXOSC3-depleted (siRRP40) HeLa cells with corresponding data from non-depleted control (Ctrl) cells, both in biological triplicates. We first merged nearby nucleotide positions with CAGE tags on the same strand into CAGE tag clusters (TCs) and calculated for each TC the average normalized expression (as TPM) in both the siRRP40- and Ctrl- libraries. For clarity, although many CAGE TCs overlap annotated TSSs, we will refer to them as ‘TCs’ and only use the term ‘TSS’ to indicate an annotated RNA 5′ end. TC expression values were then used to define an exosome sensitivity score, ranging from −1 to 1, where 0 corresponds to equal TPM values between the siRRP40- and Ctrl-libraries, while 1 and −1 correspond to exclusive expression in the siRRP40 and the Ctrl condition, respectively (see MATERIALS AND METHODS). TCs producing exosome-sensitive RNAs (sensitivity score > 0.5) were called ‘exoTCs’, while TCs with values in the range [−0.5, 0.5] were referred to as ‘non-exoTCs’. TCs with sensitivity values < −0.5 were excluded from this study. Because our focus was on transcription initiation events within pc-gene regions, we only analyzed CAGE TCs overlapping GENCODE v19 (32) pc-gene models defined as the gene body and a 10 kb upstream region on the coding strand, thereby omitting annotated antisense- and PROMPT-transcripts from the analysis.
Although the bulk of the analyzed TCs were not exosome-sensitive, regardless of the expression threshold applied, a substantial number of exoTCs could be detected, which declined with increasing expression level threshold (Figure 1A). Using a threshold of 2 TPM, nearly a third of exoTCs overlapped predominantly with annotated transcript 5′ ends (either primary or alternative TSSs, where the most upstream annotated TSS was defined as ‘primary’ and any other annotated TSSs as ‘alternative’); an additional ∼22% of exoTCs were located upstream of the primary TSS (Figure 1B, region definitions in Figure 1C). With increasing thresholds, higher fractions of exoTCs overlapped annotated TSSs. Conversely, lower expressed exoTCs were mostly found within introns. While eRNAs as a group are exosome sensitive, only 18% of these exoTCs overlapped previously defined intronic eRNA-producing loci (45). ExoTCs overlapping 5′- or 3′-UTRs, coding regions or other exons were generally rare, regardless of expression level.
Next, we asked how many pc-genes harbored exoTCs and found that while 59.8% of the 9803 expressed genes (TPM > 2 in either siRRP40- or Ctrl-samples, Supplementary Table S1) exhibited a single non-exoTC, 20.8% contained at least one exoTC (Figure 1D). Of these 2037 genes, 787 exclusively harbored exoTC(s), of which the majority (81.8%) were single exoTC cases. Finally, 12.8% of all expressed genes had combinations of exo- and non-exoTCs. Taking these observations together, we conclude that exoTCs occur within a substantial number of pc-genes. Moreover, it is noteworthy that for many genes having a single expressed TC, that TC was exosome sensitive, at least in HeLa cells. We therefore decided to first characterize such single exoTC cases (analyzed in Figures 2–5), and then later expand our analyses to more complex cases where multiple TCs are present in the same pc-gene (analyzed below in Figures 6–8).
Figure 3.

Class 4 exoTCs are by-products of strong upstream TSSs on the opposite strand. (A) Bidirectional transcription at single exoTC promoters. Left: Boxplots show NET-seq signal distributions (Y-axis) in −100 to +500 bp regions of single exoTCs from each exoTC class (opaque colors), and the −1 to −600 bp region on the opposite strand (pale colors). Genes with a single non-exoTC were analyzed for comparison (red). Right: Boxplots show distributions of corresponding bidirectionality scores for each TC class from the same data, ranging from −1 (only signal upstream of exoTC on the opposite strand) to +1 (only signal on the exoTC strand). (B) Gene annotation overlap of TCs located upstream of, and on the opposite strand of, exoTCs. Bar plots show the percentages of TCs that are upstream and on the opposite strand of single exoTCs and which overlap a given gene annotation feature (GENCODE v19), split by exoTC class as above. (C) Exosome sensitivity of TCs located upstream of, and on the opposite strand of, exoTCs. Boxplots show distributions of exosome sensitivity scores based on CAGE (calculated as in Figure 1A). TCs analyzed as in panel B. (D) Genome-browser examples of Class 4 single exoTCs. The tracks show average normalized RNA-seq signal per bp across triplicates from siRRP40- and Ctrl-libraries at each strand. Blue color indicates the same strand as the exoTCs, while red color indicates the opposite strand. TSSs on each strand are indicated by arrows. RefSeq gene models (67) are shown on top. (E) Evolutionary conservation of exoTCs and their upstream opposite strand TCs. Y-axis shows distributions of evolutionary conservation scores (phastCons 100 vertebrate species, where 0 corresponds to be least conserved and 1 most conserved) in the ±100 bp regions around exoTCs (opaque colors) and upstream opposite strand TCs defined as above (pale colors). X-axis shows TC type. (F) Enrichment of predicted pA sites and 5′ SSs downstream of exoTCs. X-axis shows the distance in bp from exoTCs. Y-axis shows the cumulative fraction of regions having one or more predicted sites at a given bp, moving left to right. ExoTC regions were split by exoTC class as above, as indicated by line color. Regions downstream of PROMPT TSSs and single non-exoTCs are shown for comparison (red and black lines, respectively). (G) G/C sequence content around exoTCs. Heat maps show G/C content centered on exoTCs of different classes. G/C content per base is calculated as the fraction of C or G nucleotides in 5 bp sliding windows. Color intensity indicates average G/C content per base over a 10 bp window.
Figure 5.

Expression of single exoTCs across primary cells. (A) Expression of single exoTCs across primary cell type groups from FANTOM5. Rows show groups of related primary cell types (as defined in (45)). Columns show exoTCs arranged by class as indicated by color-code. Heat map color indicates log2TPM CAGE expression for the respective cell type group. (B) Expression distribution of single exoTCs classes across primary cell types. Combined violin-boxplots show CAGE expression as log2TPM (X-axis) across the same primary cell type groups as in A, split by exoTC class (Y-axis) as indicated by color code. For comparison, the expression distribution of all FANTOM5 TCs is also plotted (gray). (C) Analysis of expression contribution ratio of exoTCs within genes across primary cell types. Boxplots show the distribution of usage ratio of exoTCs across the same primary cell facets as in A, defined as the fraction of cells in which a given exoTC showed the highest CAGE expression within the gene. Y-axis shows exoTC class, single non-exoTCs analysis is shown for comparison.
Figure 7.

Analysis of the relation between TC pair distance, sequence content and transcription outcome. For all plots, the type of TC pairs analyzed is shown as a schematic on top. (A) Heat map representation of predicted pA sites around TCs in non-exo:exoTC pairs (top panel) and exoTC:non-exoTC pairs (bottom panel). The heat maps were organized as in Figure 6C. Positions of TCs are indicated by black lines. Dots indicate predicted pA sites, where the color intensity indicates the motif prediction score (see MATERIALS AND METHODS). Number of analyzed TCs per distance category are indicated on the left side. Dashed lines indicate specific distances between TCs. Red dashed lines (at 200 and 300 bp distances, respectively) and red arrows refer to specific main text discussion points. (B) Heat map representation of predicted 5′SSs around TCs in non-exoTC:exoTC pairs (top panel) and exoTC:non-exoTC pairs (bottom panel). Heat map follows the same convention as in A, but with dots representing predicted 5′ SSs. (C) Heat map representation of G/C content around TCs in non-exoTC:exoTC pairs (top panel) and exoTC:non-exoTC pairs (bottom panel). Heat map follows the same convention as in A, but with color representing G/C content calculated in the same way as in Figure 3G. (D) Exonic overlap of exoTCs in non-exoTC:exoTC pairs. Y-axis shows the percentage of exoTCs, that are within the same exon as their paired non-exoTCs, split by TC pair distance. (E) Distribution of lengths of transcripts starting from exoTCs in non-exoTC:exoTC pair as split by exonic overlap. Y-axis shows the log2-scaled median length of TIF-seq reads starting at the exoTC. X-axis shows whether the exoTC is located within the same exon as the paired non-exoTC. (F) Distribution of lengths of transcripts starting from exoTCs in exoTC:non-exoTC pairs. Combined violin-boxplots show the distribution of log2-scaled median length of TIF-seq reads starting from exoTCs, split by their distance to the first downstream annotated 5′ SS in bp (X-axis). (G) Distribution of ratio of full-length transcripts from exoTCs in exoTC:non-exoTC pairs. For each exoTC, a full-length TIF-seq read ratio was calculated, where 1 corresponds to the case where 100% of the TIF seq reads starting at the exoTC reaches the annotated gene 3′ end. Combined violin-boxplots show the distribution of this ratio for all exoTCs in exoTC/non-exoTC pairs, split by splice site distance as in F (X-axis).
Figure 8.

Expression of exoTCs and non-exoTCs across primary cells. For all plots, the type of TC pairs analyzed are shown as a schematic on top or on the left side. (A) Expression of TCs across primary cells. Combined violin-boxplots show the distribution of CAGE expression of TCs as log2TPM across the same FANTOM5 cell type groups as in Figure 5, split by TC pair type. Color indicates which TC is analyzed (turquoise for the most upstream in the pair, red for the most downstream). Y-axis shows the types of TC pairs and X-axis shows CAGE log2TPM. Median expression values of single exoTCs of different classes and single non-exoTCs across the same FANTOM5 cell facets (from Figure 5B) are plotted as vertical dotted lines. (B) Expression of TCs across primary cells, split by the lengths of the produced transcripts. Combined violin-boxplots show the distribution of CAGE log2TPM of TCs in non-exoTC:exoTC (left panel) and exoTC:non-exoTC (right panel) pairs across cell type groups as in A, but split by the ratio of full-length transcripts (ratio < 0.5: TC produces mostly prematurely terminated transcripts; ratio > 0.5: TC produces mostly full-length transcripts). The reference lines are the same as in A.
Characterization of exoTCs from pc-genes with a single active TSS
As detailed above, we first focused our analysis on the 644 pc-genes harboring a single exoTC with an expression level >2 TPM in either RRP40- or Ctrl-samples. The majority of these single exoTCs (59%, 380/644) overlapped GENCODE-annotated TSSs (Figure 2A, ‘primary and alternative TSS’), as compared to the higher fraction of single non-exoTCs (94%, 5514/5866, Supplementary Figure S1A). Moreover, 15% of single exoTCs were located upstream of the primary TSSs and 16% within introns. Because the majority of TCs overlapped annotated TSSs, we focused our analysis on these 380 single exoTCs, using the set of 5514 genes having a single non-exoTC overlapping with annotated TSSs for comparison.
CAGE reads comprise only the first 30 nt of RNAs and therefore provide limited information about the nature of the RNAs produced from a given TC. We therefore prepared paired end transcript isoform sequencing (TIF-seq) (36,48) libraries from siRRP40- and Ctrl-cells, yielding reads which contain both the capped 5′- and the polyadenylated 3′-end of the same RNA, which can then be used to assess transcript length. We plotted the coverage and fold change (FC) of TIF-seq reads across the single exoTC pc-genes defined above, using a meta-gene heat map representation (Figure 2B and C) anchored at the positions of the TCs and the annotated gene 3′ ends (see MATERIALS AND METHODS). This revealed that many single exoTCs produced exosome-sensitive prematurely terminated RNAs (bottom panels in Figure 2B and C), even though these TCs by selection overlapped an annotated 5′ end of a full-length transcript. Previously produced total RNA-seq data, from the same cell samples (14) showed similar results (Figure 2D, bottom panels of Supplementary Figure S1F), which prompted us to analyze the exact location of these premature 3′ ends. Most (76.68%, Supplementary Figure S1H) were located in the first intron downstream of the exoTC and on average ∼1000 nt from the 5′ splice site (Supplementary Figure S1I). This, and the exosome sensitivity of these transcripts, was further confirmed by RNA-seq FC in the first intron (Supplementary Figure S1J) and is consistent with previous results describing prematurely terminated exosome-sensitive transcripts (22,49). However, at least one third of the analyzed genes showed a substantial TIF-seq coverage across the whole gene (top panel in Figure 2B). Both TIF-seq and RNA-seq data confirmed that a substantial number of these cases represented full-length RNAs, displaying robust exosome sensitivity throughout the gene (Figure 2C and D, top panel). Others contained a mixture of shorter exosome-sensitive transcripts and longer transcripts covering the whole gene (middle panel in Figure 2B−D), where the longer transcripts in some cases were also exosome sensitive (Figure 2C and D, middle panel). In contrast, transcripts produced from single non-exoTCs were predominantly full-length and exosome insensitive (Supplementary Figure S1B−E).
Based on the above observations and in order to facilitate downstream analysis, we devised a hierarchical classification system of single exoTCs with decision rules based on the above properties (see decision tree in Supplementary Figure S2A, and MATERIALS AND METHODS). This comprised four classes with the following properties (visualized in Figure 2E, left; and with specific gene examples shown in Figure 2E, right): (i) Class 1 (N = 49) exoTCs almost exclusively producing full-length exosome-sensitive transcripts, (ii) Class 2 (N = 68) exoTCs producing both full-length and prematurely terminated transcripts, both of which were exosome sensitive, (iii) Class 3 (N = 99) exoTCs producing prematurely terminated exosome-sensitive transcripts, that also give rise to full-length exosome-insensitive transcripts (the exoTC captures only the 5′ ends, and therefore the average sensitivity, of both transcript types) and (iv) Class 4 (N = 64) exoTCs almost exclusively producing prematurely terminated exosome-sensitive transcripts (Supplementary Table S2).
To investigate whether genes in these established classes might share specific functions, we performed Gene Ontology (GO) over-representation analysis. Class 1 and 2 genes were enriched for GO terms related to transcription factor and regulator activities, agreeing with previous results (25), and included well known immediate early response transcription factor genes such as JUN, KLF6, ATF3, MAFF and DDIT3 (Figure 2F). Immediate early response genes often encode short primary transcripts with few exons (50). Consistently, Class 1 and 2 genes produce shorter primary transcripts and with fewer exons than RNAs from genes with single non-exoTCs (Supplementary Figure S2B). Class 1 genes in particular were often mono-exonic and had longer first exons than other classes, and both classes had shorter first introns, consistent with the above (Supplementary Figure S2B). However, Class 1 and 2 genes did not exhibit substantially higher degrees of intron retention than other classes (Supplementary Figure S2B). Overall, this led us to conclude that the exosome likely participates in regulating mRNA levels of such early-response transcription factors. While we found no significantly enriched GO terms for Class 3 and 4 genes, it is interesting to note that Class 3 included the PCF11 gene, which was recently reported to autoregulate its expression levels by transcription attenuation (22).
The establishment of the four classes suggested the possibility that genes within specific classes may utilize different, perhaps exosome-related, mechanisms to regulate their expression. In Class 1, the exosome might control full-length mRNA expression, while the Class 3 genes are likely subjected to partial premature termination of transcription within their first introns, which may influence their final overall gene expression (reviewed in (20)). RNAs deriving from such an attenuation mechanism would then be substrates of the exosome. Class 2 transcripts can be viewed as a hybrid of classes 1 and 3, where both prematurely terminated and full-length transcripts are exosome-sensitive, while Class 4 constitutes cases where premature transcription termination dominates and full-length transcripts are rare.
Prematurely terminated transcripts are often reverse strand byproducts of nearby mRNA TSSs
Next, we investigated the transcription levels of exoTCs of each class using native elongating transcript sequencing (NET-seq) of nascent RNA from HeLa cells (35). Interestingly, exoTCs from all four gene classes exhibited similar transcription levels, which in turn were on average slightly lower than those of single non-exoTCs (Figure 3A, left panel). As discussed in the introduction, the large majority of human gene promoters are bidirectionally transcribed (51,52). Analysis of opposite strand transcription upstream of the respective TCs showed that Class 4 gene promoters were highly balanced with roughly equal amounts of transcription in the forward and reverse directions, while promoters from the other gene classes displayed a higher transcription on the strand from which the exoTC of interest was present (Figure 3A, left and right panels). Notably, for Class 4 exoTCs, the reverse strand TCs were in 60% of cases overlapping an annotated pc-gene TSS within 600 bp (Figure 3B). In other words, more than half of Class 4 exoTCs were components of annotated mRNA-mRNA bidirectional promoters, which was roughly twice as much as that of any other class, despite the fact that Class 4 TCs were required to overlap annotated pc TSSs. We therefore reasoned that Class 4 exoTCs, and their predominantly prematurely terminated RNA products, might be consequences of highly transcribed mRNA TSSs on the other strand, similar to canonical mRNA-PROMPT pairs. Consistently, opposite strand TCs upstream of Class 4 promoters were typically non-exoTCs that are not exosome sensitive, as opposed to other corresponding opposite strand TCs upstream of classes 1-3 exoTCs (Figure 3C, selected examples are shown in Figure 3D). Moreover, the region downstream of such opposite-strand TCs was more evolutionarily conserved than the correspoding region downstream of Class 4 exoTCs, with similar conservation levels as regions downstream of non-exoTCs (Figure 3E).
Chromatin data from the HeLa cell ENCODE project (53) showed enrichment patterns consistent with the above observations; while exoTCs of all classes showed similar chromatin accessibilities and levels of H3K27ac, H3K4me3 histone marks, implicating active transcription. These levels were higher upstream of Class 4 exoTCs. Notably, Class 4 exoTCs themselves also lacked a gene body enrichment of the H3K36me3 histone mark, consistent with their inefficient transcription elongation (Supplementary Figure S3).
Next, we asked whether the properties of each TC class might be related to its surrounding sequence content. As previously reported, polyadenylation (pA) sites and 5′ splice sites (5′ SSs) are over- and under-represented, respectively, downstream of 5′ ends of known exosome-sensitive transcripts (e.g. PROMPTs) compared to their forward strand mRNA counterparts (18,54). All four single exoTC classes fell between these two reference sets in terms of predicted pA site occurrence, where Class 1 exoTCs were the most similar to single non-exoTCs, while Class 4 exoTCs showed a similarly strong pA enrichment as PROMPTs from ∼1200 bp downstream from the exoTC (Figure 3F, left panel). This is roughly consistent with the typical position of prematurely terminated transcript 3′ ends (median Class 4 transcript length by TIF-seq was 1391 bp). Class 2 and 3 genes displayed a similar enrichment of predicted 5′ SSs as non-exoTCs, while Class 4 genes had a similar 5′ SSs enrichment profile as PROMPT regions from ∼1200 bp downstream from the exoTC (Figure 3F, right panel). Class 1 genes fell between these two, possibly due to the fact that many Class 1 transcripts were short and mono-exonic. Related to the above, we have previously shown that pA site depletion, downstream of the TSS of exosome-insensitive transcripts, often coincides with CpG-enriched regions, which in turn are often limited to the first 500 bp (55). Plotting G/C content up- and downstream of TCs, in each class, showed that exoTCs from classes 1–3, which all largely produce full-length transcripts, had a clear G/C enrichment around the exoTC peak, which often extended downstream (Figure 3G). In contrast, Class 4 exoTCs had a much higher G/C enrichment upstream the exoTC than downstream, which likely reflects a G/C enrichment around their commonly occurring upstream mRNA TSSs on the opposite strand, as discussed above.
In summary, several lines of inquiry - transcription initiation bidirectionality, evolutionary conservation and sequence motif enrichment/depletion indicate that Class 4 exoTCs and their associated transcripts are by-products of the initiation of canonical, exosome-insensitive mRNAs upstream and on the opposite strand of the exoTCs. In this sense, Class 4 exoTC regions share properties with canonical PROMPT regions, although Class 4 transcripts are on average 2–3 times longer than typical PROMPTs (1391 versus ∼500 bp (18)). This difference in length is also reflected in sequence content: once the 3′ end is reached for Class 4 transcripts, the pA site and 5′ SS enrichment is similar to those of PROMPTs (Figure 3F).
Exosome-sensitive full-length mRNAs are PAXT targets
Exosome-directed decay of nuclear RNA is mediated by adaptor complexes (56). We therefore investigated which adaptor is implicated in the degradation of transcripts deriving from the four single exoTC classes, focusing on the PAXT connection and the NEXT complex, which primarily target longer polyadenylated and short non-adenylated transcripts, respectively (7,9,14). To enable such analysis, we prepared CAGE libraries from cells subjected to siRNA-depletion of ZFC3H1 (PAXT) or ZCCHC8 (NEXT), and plotted the distribution of CAGE exoTC sensitivity scores of siZFC3H1 versus Ctrl and siZCCHC8 versus Ctrl (Figure 4A). In general, all exoTC classes were to some degree sensitive to both nuclear RNA decay pathways (median sensitivity > 0), however, classes 1 and 2 showed significantly (P = 0.016 and 0.037, respectively; one-sided Mann–Whitney test) higher siZFC3H1 than siZCCHC8 sensitivity, while Class 4 displayed the opposite pattern (Figure 4A). Corresponding analyses using RNA-seq data from the same cell samples, summing all reads across the gene models, gave consistent results (Supplementary Figure S4A). Importantly, these analyses showed the average sensitivity of all transcript isoforms from loci starting at the respective exoTCs. To investigate changes in PAXT- and NEXT-sensitivity across gene bodies, we plotted the average FC of RNA-seq signals for factor depletions versus Ctrl as metagene profiles (Figure 4B). This revealed that for classes 1 and 2, the whole gene body showed only moderately higher RNA-seq signal in ZCCHC8-depleted cells versus Ctrl cells, while RNA-seq signal was strongly increased in ZFC3H1-depleted cells. Conversely, for Class 4, the increase in RNA-seq signal in ZCCHC8- and ZFC3H1-depleted cells compared to Ctrl cells was only visible in the first ∼20% of the gene body, consistent with the premature termination of these transcripts.
For Class 3 genes, no substantial RNA-seq signal increase was observed in ZCCHC8- or ZFC3H1-depleted cells except for a modest increase in ZFC3H1-depleted cells in the first ∼30% of the gene body. Similar trends were observed when plotting log2 FC of TIF-seq data from RRP40 depleted cells (Supplementary Figure S4B). We interpret this pattern as a mixture between production of primarily PAXT-sensitive prematurely terminated RNAs and the production of longer, exosome-insensitive RNA isoforms.
While mRNAs are generally quickly exported to the cytoplasm and therefore are not usually targets of the nuclear exosome, studies have shown that some mRNAs, that are retained in the nucleus, undergo decay (25,57,58). Consistently, exosome-sensitive full-length transcripts produced from exoTCs of classes 1 and 2 were enriched in RNA-seq libraries from nuclear RNA versus total RNA samples from control HeLa cells, compared to full-length transcripts from Class 3 exoTCs or from single non-exoTCs (Figure 4C).
Overall, we conclude that the examined exosome-sensitive transcripts are substrates of both the PAXT and NEXT decay pathways. However, longer exosome-sensitive RNAs (from classes 1 and 2) are primarily targeted by PAXT, consistent with the similarity of these transcripts to canonical polyadenylated mRNAs. In line with NEXT primarily targeting short cryptic transcripts, this decay pathway plays a more prominent role for Class 4 genes.
Long exosome-sensitive RNAs from pc-genes are ubiquitously expressed across cells and tissues
An important question is to what extent exoTCs, and in particular those that primarily produce full-length exosome-sensitive transcripts, are used across normal cells and tissues, and if so, whether these TCs are the main expression contributors of their cognate genes. To investigate this, we employed CAGE data from the FANTOM5 consortium, covering most human primary cells and tissues (45). Heatmap visualization of the expression of Class 1–4 exoTCs showed that classes 1–3 were expressed roughly uniformly across primary FANTOM cell groups; classes 1 and 3 exoTCs were more highly expressed while Class 4 exoTCs, as expected, were lower expressed across all cell facets (Figure 5A, B). Corresponding analysis on FANTOM5 CAGE tissue samples showed similar patterns (Supplementary Figure S5).
We then wondered whether exoTCs might be the main drivers of gene expression across cell types, or if they merely correspond to secondary TSSs with more modest expression contributions in non-HeLa cells. To address this, we calculated, for each exoTC, an expression contribution score, representing the fraction of FANTOM5 primary cell types in which the TC had the highest CAGE expression of all FANTOM5 TCs within the specific gene (Figure 5C). This revealed that Class 1–3 exoTCs were the main contributors in ∼95% of primary cell groups (median contribution score 0.94–0.99), while Class 4 exoTCs showed more variance and contributed less, albeit still with a high median contribution score of 0.9.
Taken together these data imply that single exoTCs identified in HeLa cells correspond to bona fide TSSs used across most cell types, and often corresponding to the most used TSS. Thus, the exoTCs identified here are likely physiologically relevant given that many of the genes with exoTCs are functionally important, e.g. JUN, KLF6, ATF3, MAFF and DDIT3. Moreover, the exosome sensitivity of these transcripts suggest that their gene expression might be regulated post-transcriptionally at the level of nuclear RNA turnover.
The NEXT sensitivity of exoTCs is correlated with their proximity to non-exoTCs
In the above analyses, we have focused on ‘simple’ cases where a given pc-gene was utilizing a single TC. However, as many genes contain multiple active TCs, and thereby the potential to employ combinations of exo- and non-exoTCs (Figure 1D, also exemplified in Figure 6A), we set out to explore such relations in terms of genomic distance, sequence content, expression level and utilization across primary cells and tissues.
For this analysis, we focused on pc-genes harboring at least two TCs on the coding strand, using the same expression cut-offs as employed for our single TC analysis. We considered all adjacent pairs of TCs within 3000 bp of one another and within the same gene, including a 10 kb upstream region of its primary annotated TSS. These pairs were then stratified by whether their TCs were exosome-sensitive or not, which resulted in the four TC combinations shown in Figure 6B. TC pairs consisting of two non-exoTCs did, as expected, account for the large majority of cases (67%, 2611 pairs), while the remaining pairs, involving at least one exoTC, were roughly evenly divided between the remaining three possible pair types. The spacing between TC pairs was typically 300–500 bp, except for exoTC:non-exoTC pairs, which displayed highly varied spacing but on average were further apart (Supplementary Figure S6A, median 876 bp, P < 2.2–16, two-sided Mann-Whitney test). Intersection with gene annotations showed that non-exoTCs were primarily overlapping annotated TSSs, whereas exoTCs were not; in exoTC:non-exoTC pairs the exoTC was primarily located in the unannotated upstream region, while in non-exoTC:exoTC pairs the exoTC was typically located within introns (Supplementary Figure S6B).
We hypothesized that the lengths of transcripts deriving from exoTCs might be influenced by the distance to the closest up- or down-stream non-exoTC. Indeed, while TIF-seq reads from exoTCs were generally short (median 1281 nt in siRRP40), when an exoTC was close to a non-exoTC (<200 bp), transcripts initiating at the exoTC were longer (Figure 6C, D: heat map visualizations to the left, TIF-seq length distributions to the right, P = 8.159e–08 and 2.729e–11 for exoTC:non-exoTC and non-exoTC:exoTC pairs, respectively) regardless of whether the exoTC preceeded the non-exoTC or vice versa. The 3′ ends of these longer transcripts overlapped the annotated TES of the pc-gene in 80% of cases, similar to the TIF-seq reads originating from non-exoTCs (Supplementary Figure S7A).
To inquire whether these longer transcripts originating from exoTCs were exosome sensitive, we assessed the TIF-seq FC between siRRP40- and Ctrl-library data (while corresponding RNA-seq data was available, the overlapping transcripts originating from TC pairs made it difficult to assess the exosome targeting of individual transcripts using RNA-seq data). This revealed that both prematurely terminated as well as full-length transcripts, originating from exoTCs within the pair types analyzed above, were similarly exosome-sensitive (Figure 6E, F: heat map visualizations to the left, distributions of log2FC to the right). This in turn suggested that the CAGE-based depletion sensitivities were reasonable approximations for whole transcript sensitivities.
We hypothesized that exoTCs close to non-exoTCs would give rise to predominantly PAXT-targeted transcripts similar to the classes 1–2 single exoTC cases analyzed above, while transcripts from distal exoTCs might give rise to predominantly NEXT-targeted transcripts similar to those from Class 4. To test this, we analyzed PAXT and NEXT sensitivities of exoTCs from the same TC pairs as above, using our CAGE siZFC3H1 and siZCCHC8 libraries. When averaging over all TC pairs, exoTCs were generally NEXT- but not substantially PAXT-sensitive, regardless of pair type (Figure 6G, H, top panels, and Supplementary Figure S7B). Moreover, the NEXT sensitivity of exoTCs increased with the distance between exoTCs and non-exoTCs, where exoTCs close to non-exoTCs were neither substantially NEXT- nor PAXT-sensitive despite being RRP40 sensitive (Figure 6G, H, bottom panels). While this increase in NEXT sensitivity with increased TC-TC distance was compatible with the initial hypothesis, the putative exosome adaptor responsible for the observed RRP40/EXOSC3 sensitivity of exoTCs close to non-exoTCs is presently unclear.
ExoTC proximity to annotated splice sites is correlated with the generation of long, exosome-sensitive transcripts
Next, we asked whether sequence content around TCs could explain the correlation between transcript length and TC pair distance. By displaying predicted pA sites, 5′SSs, and G/C content for each TC pair analyzed above as a heat map (Figure 7A–C, Supplementary Figure S8A–C), we observed drastic shifts in sequence content at the respective TC positions and its immediate downstream region in pairs with one exoTC and one non-exoTC (Figure 7A–C). This was not observed in other TC pairs (Supplementary Figure S8A–C).
In particular, non-exoTCs displayed a strong occurrence of predicted 5′SSs just after, but not before their TC peak, and a corresponding depletion of predicted pA sites. In the case of non-exoTC:exoTC pairs (Figure 7A–C, top row), the depletion of pA sites extended up until the exoTC location, while the 5′SSs accumulation was strongest just after the non-exoTC. The same pattern was also evident when assessing G/C content. Thus, such TC pairs delineated a G/C (and predicted 5′SSs)-rich boundary in between them. For exoTC:non-exoTC pairs, the properties were expectedly reversed: there was an increase in G/C content before the exoTC and after the non-exoTC, but a depletion in between. The same pattern was reflected in predicted pA sites, which were depleted downstream of the non-exoTC only, and 5′ SSs, which were highly enriched directly downstream of the non-exoTC, but depleted between the TCs.
For both pair types, the exception to the above observations was when TCs were close (< 200–300 bp), where the sequence properties of the non-exoTCs overtook that of the exoTC, most visible in terms of G/C content (Figure 7A–C, indicated with red arrows). These sequence properties of exoTCs close to non-exoTCs coincided with their production of full-length transcripts as analyzed in Figure 6C and D. Based on this observation, we hypothesized that the occurrence of longer exoTC transcripts, when TCs were close, was not due to the TC distance itself but rather because exoTC transcripts might co-opt the first 5′SS used by the non-exoTC transcript. We tested this hypothesis by focusing on the subset of TC pairs where the non-exoTC overlapped with an annotated TSS, or was within the 5′ UTR of a GENCODE gene model (71% of analyzed pairs), so that analyzed non-exoTCs were always associated with an annotated first exon. Consistent with the hypothesis, in non-exoTC:exoTC pairs, exoTCs were rarely located within the first exon originating from the non-exoTC unless the distance between TCs was <200 bp (Figure 7D). ExoTCs that were located within the first exon initiated significantly longer transcripts than exoTCs located downstream of the first exon, as assessed by median TIF-seq lengths (P = 1.731e–09, one-sided Mann–Whitney test, Figure 7E).
For exoTC:non-exoTC pairs, the exoTC can per definition not be within the same annotated exon, originating from the non-exoTC, so the same analysis was not meaningful. However, we observed a clear correlation between the median TIF-seq lengths of transcripts originating from the exoTC and the distance to the next downstream annotated 5′ SS: predominantly full-length transcripts were produced from exoTCs within ∼500 bp of a 5′ SS, and the proportion of prematurely terminated short transcripts increased when exoTCs were further from annotated 5′ SSs, especially when the distance was > 1 kb (Figure 7F, G). This was consistent with our observations for non-exoTC:exoTC pairs discussed above; transcripts become long if the exoTC is proximal to a strong 5′ SS, which either could be specific for the exoTC or shared with the non-exoTC. Otherwise, they would prematurely terminate before the non-exoTC.
In summary, these analyses demonstrated that distances between exoTCs and non-exoTCs correlate strongly with the lengths of the transcripts produced from exoTCs, a property most likely linked to the availability of strong splice donor sites utilized by exosome-insensitive transcripts, that can only be co-opted if the exoTC is proximal to the splice site.
The majority of exoTCs within multi-TC genes are secondary TSSs across cells and tissues
An important parameter for establishing whether exoTCs within multi-TC genes are perhaps functionally relevant is their relative expression levels compared to their paired non-exoTCs. To answer this question, we interrogated FANTOM5 CAGE data across primary cells, as when analyzing single exoTCs above (Figure 5). In order to make results between these two analyses (single versus multi-TC genes) comparable, we focused on cases where both TCs in exoTC:non-exoTC and non-exoTC:exoTC pairs overlapped annotated TSSs (25%, 97/385 and 29%, 148/514 of pairs, respectively). In general, exoTCs paired with non-exoTC were substantially less expressed across FANTOM5 primary cells (median 0.9 TPM) than their paired non-exoTCs (median 3.9 TPM), and at the same level as exoTCs in exoTC:exoTC pairs (Figure 8A, Supplementary Figure S9). Moreover, the expression level of exoTCs of any pair type was significantly (P = 6.321e–13, one-sided Mann–Whitney test) lower than those of single exoTCs for classes 1–3 analyzed above (see dotted lines in Figure 8A). Somewhat surprisingly, there was no substantial expression difference between exoTCs that produced full-length or prematurely terminated transcripts as defined in Figure 6 (Figure 8B). Similar trends were observed when analyzing expression data at the level of tissues (Supplementary Figure S10).
Taken together, these results indicated that most exoTCs are minor contributors to overall gene expression, provided other non-exoTCs are present in the same gene. This is regardless of their position and whether they produce prematurely terminated transcripts or not. In that sense these exoTCs are more similar to single exoTCs of the Class 4 genes analyzed above, and indeed have comparable expression levels (see dotted lines in Figure 8A). However, our observation that Class 4 single exoTCs were often linked to mRNA TSSs on the opposite strand was not mirrored for exoTCs within multi-TC genes (only 19 versus 58% in Class 4 single exoTCs). Overall, these observations indicate that most exoTCs, and their transcripts, within multi TC genes have limited physiological relevance, since they are lowly expressed and their lengths are most likely a side-effect of their proximities to non-exoTCs and their downstream splice sites (Figure 7).
DISCUSSION
In the present study, we have established that TSSs that produce exosome-sensitive transcripts occur within many pc-genes. In many cases, these TSSs, captured as exoTCs using CAGE, overlap annotated mRNA TSSs, established by full-length cDNA sequencing. We found that the properties of exosome-sensitive transcripts can be classified largely based on two parameters: (i) whether the exoTC is the only active TC in the gene region and (ii) whether the produced transcripts are prematurely terminated, or whether they share their 3′ ends with the annotated full-length mRNAs (Figure 9).
Figure 9.
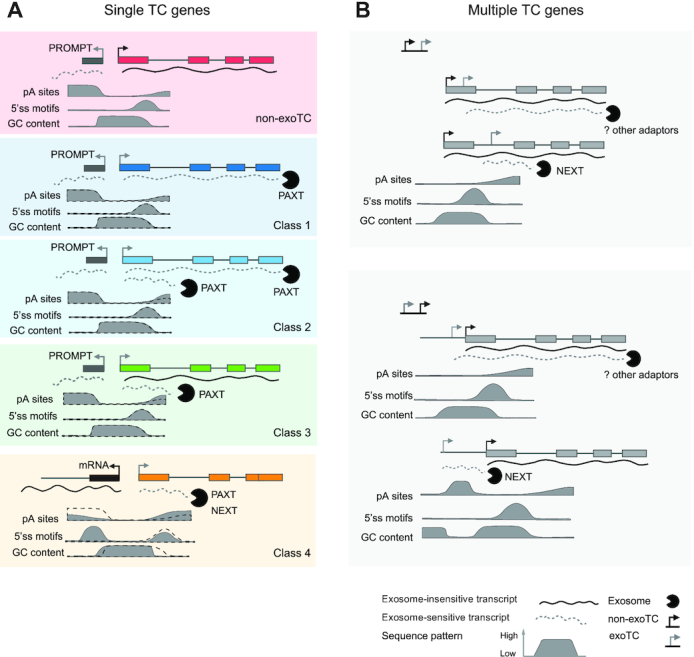
Models for exoTCs positioning and expression within pc-genes. For each gene/TC cartoon model, typical transcripts and their most common targeting fates are shown. Solid lines represent exosome-insensitive RNAs, dotted lines represent exosome-sensitive RNAs, where the primary exosome adaptor responsible for degradation is indicated. Below: typical enrichment/depletion of pA sites, 5′ SSs and G/C content are shown as solid density plots: the dotted lines show corresponding enrichments at non-exoTCs as a reference. Also see DISCUSSION. (A) Genes with a single TC. Cartoon models of single TC genes: non-exoTCs and exoTCs of classes 1–4 are shown from top to bottom. ExoTCs were divided into four classes based on the lengths of transcripts and their exosome sensitivities. Similar to non-exoTCs, exoTCs that are at least partially producing full-length transcripts, have canonical PROMPT regions on their reverse strand, and share similar downstream sequence patterns (pA sites, 5′ SSs, G/C content) as single non-exoTC (in classes 1–4, the black dashed line indicates sequence pattern of single non-exoTCs as a reference), although for classes 2–3, which also produce exosome-sensitive prematurely terminated transcripts, have a slightly higher occurrence of pA sites downstream. Class 4 exoTCs produce predominantly short, prematurely terminated transcripts and most are initiated head-to-head with the TSS of another pc-gene, thus appearing like ‘PROMPTs’ of the other gene with the sequence pattern more similar to PROMPTs, and produce short transcripts, that are redundantly targeted by the NEXT and PAXT pathways. (B) Genes with multiple TCs. A non-exoTC:exoTC pair cartoon is shown in the upper panel, and an exoTC:non-exoTC cartoon in the lower panel. In non-exoTC:exoTC pairs: when exoTCs are close to non-exoTCs, they are mostly within the same exon, and share downstream sequence properties with the non-exoTC due to their proximity. When such exoTCs are positioned further away from non-exoTCs, they are mostly located in introns with sequence patterns similar to PROMPT TSSs and producing short transcripts that are targeted by NEXT. In exoTC:non-exoTC pairs, there is a similar distance effect as non-exoTC:exoTC pairs: when exoTCs are close to non-exoTC, exoTCs are more likely to be annotated TSSs and produce long exosome-sensitive transcripts, that likely use the same splice sites as transcripts from the non-exoTC, due to that the sequence pattern of non-exoTC ‘bleeds into’ that of exoTC. More distal exoTCs are not subject to sequence constraints of the non-exoTC, and produce NEXT-sensitive short transcripts, and are similar to PROMPTs in terms of sequence patterns and products. For both configurations, long exosome-sensitive transcripts appear not to be targeted by NEXT or PAXT, suggesting the possible existence of additional adaptors.
Genes that only use one exoTC and no non-exoTCs (Figure 9A) are arguably interesting, as the abundance of their expressed transcripts will depend on exosome availability. Interestingly, a subset of these exoTCs overlap an annotated TSS and mainly produce full-length exosome-sensitive transcripts, which are primarily PAXT targets (classes 1–2). Those TSSs remain active across many cells and tissue types and in most cases constitute the main TSSs for their respective host genes. Genes in this category include well-known transcription factors as JUN, KLF6, ATF3, MAFF and DDIT3, many of which are so-called immediate early response genes. Single exoTCs of these classes share many properties with canonical non-exoTCs (53,57). It is somewhat surprising that immediate early gene full-length transcripts would benefit from being exosome sensitive, since they by definition need to exhibit strong transcriptional/expression responses within minutes after cells are exposed to external stimuli. A high nuclear exosome sensitivity would have the effect of decreasing the overall expression amplitude, but could be important for the rapid removal of remaining RNA copies once the transcriptional burst has ended. Moreover, constitutive nuclear degradation of full-length transcripts could serve to allow these genes to be constitutively lowly transcribed instead of fully inactive when stimuli are not present, which would allow for a faster response upon stimulation. When cells are not stimulated, the exosome would dampen RNA copy numbers, while during induction, robust production of transcripts would saturate the PAXT/exosome pathway and thereby achieve sufficient expression. A similar model has been proposed for nutrient response in budding yeast (59). Alternatively, the exosome sensitivity of these transcripts may be a necessary tradeoff: gene features that facilitate early gene response (short gene lengths, few or no introns) are also correlated to high exosome sensitivity (6).
Classes 2–4 also produce short, prematurely terminated RNAs which are targeted by both the NEXT and PAXT pathways. ExoTCs initiating such transcription exhibit a higher and lower average occurrence of predicted TSS-proximal pA sites and 5′SS, respectively. This is quite similar to the sequence features present within PROMPT regions, although with enrichments/depletions of lower magnitude. Class 3 genes, which produce long, exosome-insensitive RNAs in combination with short prematurely terminated RNAs, are the most similar to canonical mRNA genes. We speculate that this class may be characterized by less efficient RNAPII elongation, meaning that higher transcription initiation is needed to attain a given mRNA copy number. Possibly related to this, reduced RNAPII elongation has been shown for genes that undergo premature termination by the Integrator complex at sites of paused RNAPII (23), resulting in transcription attenuation. Because the CAGE, TIF and RNA-seq techniques we employ only assess RNAs that are >100 nt, we will in most cases not capture attenuation events close to the TSS, but premature termination further downstream the gene is widespread in pc-genes and could also lead to attenuation (reviewed in (20)). The short prematurely terminated transcripts observed in classes 2–4 could therefore be results of transcription attenuation, as reported for PCF11 (22), a Class 3 gene in our analysis
Surprisingly, our data demonstrate that some genes contained in Class 4 harbor single TCs that almost exclusively produce prematurely terminated and exosome-sensitive transcripts. This is despite the fact that the related exoTCs correspond to annotated mRNA TSSs, which in most cases are the dominant TSSs for these genes across most cell and tissue types. However, in many cases the expression of such TSSs is likely a bystander effect of strongly expressed upstream mRNA TSSs on the other strand. Thereby, these RNAs, which share properties with canonical PROMPTs, are under lower selective pressure.
An outstanding question is which features drive exosome targeting of the sensitive transcripts. A commonly accepted idea is that mRNAs that are slowly or inefficiently processed are subject to inefficient nuclear RNA export (60) and therefore targeted by the PAXT pathway. In line with this, Class 1 genes are often mono-exonic, and Class 1 and 2 transcripts were found to be more enriched in the nucleus than exosome-insensitive transcripts. Hence, this may, at least in part, explain the PAXT sensitivities of these transcripts (56,61). Prematurely terminated transcripts in classes 2–4 may share many of these features, since their 3′ ends predominantly reside in the first intron and they are likely not spliced. However, a clear difference to the above is the high incidence of pA sites, reminiscent of the link between exosome targeting of PROMPTs and TSS-proximal pA sites (18,54). As many of these prematurely terminated transcripts are enriched in both PAXT and NEXT depletions, these transcripts are targeted by the exosome through different mechanisms. As reported previously (9), the recruitment of PAXT could occur through recognition of pA sites and conventional 3′end processing by the CPA machinery. NEXT could be recruited to these transcripts through its interaction with the CBC (62–64). For both full length mRNAs and prematurely terminated transcripts, it is also possible that exosome targeting is further increased by additional RNA-bound proteins. However, since the transcripts are short-lived, it is challenging to comprehensively identify the mechanisms that target them for degradation.
While single exoTCs are probably largely physiologically relevant since they remain the main TSS across cells and tissues, exoTCs co-existing with non-exoTCs (Figure 9B) within the same gene are more common, but likely of lesser physiological importance. This is because they are more lowly expressed than non-exoTCs within the same gene, both in HeLa cells and across the FANTOM5 cells and tissues. Moreover, these exoTCs mostly produce short, prematurely terminated transcripts. Longer transcripts produced from such exoTCs are rare and likely consequences of proximity to stronger non-exoTCs on the same strand, where DNA sequence constraints, in particular splice sites, of the non-exoTC and its transcripts are also imposed on the exoTCs and their products. Conversely, when exoTCs are positioned further away from non-exoTCs, either in introns or in the region upstream of the non-exoTC, the sequence pattern downstream of them will be similar to that of PROMPTs and consequently produce exosome-sensitive short transcripts.
Our analysis leads to two important open questions regarding the metabolism of longer exosome-sensitive transcripts, originating from exoTCs from within multi-TC genes. First, their exosome sensitivity is surprising given that they share 3′ ends with transcripts originating from non-exoTCs, that reside only a few hundred bp away. Hence, the DNA that encodes them is almost identical. This would suggest that either the local sequence downstream of each TC is highly informative for exosome targeting, or that the transcripts are biochemically different in some other way, e.g. with regards to processing and/or nuclear export efficiency. To investigate the latter in detail, it would be necessary to employ long read sequencing, as isoform convolution from standard RNA-seq does not have the resolution to distinguish different RNAs initiating from nearby TSSs. Second, although the longer transcripts appear clearly RRP40/EXOSC3 sensitive in both our CAGE and TIF-seq experiments, they are not enriched in neither NEXT nor PAXT-depleted cells. Perhaps, these transcripts are targeted by alternative exosome adaptors, which remain to be discovered.
Given our results discussed above, there may be several mechanisms underlying the production of exosome-sensitive transcripts within mRNA genes. Exosome-sensitive full-length mRNAs might have evolved to be co-regulated with exosome levels (in particular classes 1–2 of single exoTC genes). Alternatively, their exosome sensitivity may be a ‘necessary evil’ to accommodate other constraints, like mediating burst transcription of immediate early response genes. Such transcripts are typically PAXT sensitive and this might be a contributing reason to why depletion of a factor in the PAXT pathway impairs mouse embryonic stem cell differentiation (16). Other exosome-sensitive transcripts are prematurely terminated and may in many cases be bystander effects of the transcription initiation of other loci. Alternatively, such RNAs may be the results of processes that are not directly linked to the host gene, e.g. transcription of eRNAs or other non-coding RNAs. Close-by TSSs may also affect each other functionally by the act of transcription, even though they are not producing full-length transcripts, e.g. by transcriptional interference by RNAPII elongation through downstream TSSs (65).
More generally, sensitive 5′ sequencing-based methods like CAGE have the ability to discover a wealth of uncharacterized alternative TSSs, but not all of these will produce physiologically relevant RNAs (6,66). Exosome sensitivity and the lengths of the produced RNAs, as presented here, are therefore important features for predicting alternative TSS relevance within complex genomes.
DATA AVAILABILITY
All TIF-seq, SLIC-CAGE datasets are available at the Gene Expression Omnibus (GEO) under accession number: GSE147655. Code for TIF-seq processing and CAGE annotation is available at GitHub (https://github.com/PelechanoLab/TIFseq2 and https://github.com/MengjunWu/Exosome_sensitive_TSS). Supplementary Data are available at NAR online.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jette Bornholdt for helping with the SLIC-CAGE experiment, Hjörleifur Einarsson for sharing SLIC-CAGE processing methods, Kristoffer Vitting Seerup, Manfred Schmid, Alfredo Rago, Signe Schmidt Kjølner Hansen for comments on the manuscript and for constructive criticism.
Author contributions: M.W., E.K. analyzed data. G.W. and M.G. carried out siRNA knockdown experiments in HeLa for SLIC-CAGE and TIF-seq libraries. C.S. made SLIC-CAGE libraries. B.L., J.W. and V.P. made and processed TIF-seq libraries. M.W., E.K., M.L., J.O.R., M.M., T.H.J., A.S. interpreted data. M.W., M.L., E.K, T.H.J., A.S. wrote the paper with input from all other authors.
Contributor Information
Mengjun Wu, The Bioinformatics Centre, Department of Biology and Biotech and Research Innovation Centre, University of Copenhagen, Ole Maaløes Vej 5, DK2200 Copenhagen N, Denmark.
Evdoxia Karadoulama, The Bioinformatics Centre, Department of Biology and Biotech and Research Innovation Centre, University of Copenhagen, Ole Maaløes Vej 5, DK2200 Copenhagen N, Denmark; Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Marta Lloret-Llinares, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark; European Bioinformatics Institute (EMBL-EBI), European Molecular Biology Laboratory, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK.
Jerome Olivier Rouviere, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Christian Skov Vaagensø, The Bioinformatics Centre, Department of Biology and Biotech and Research Innovation Centre, University of Copenhagen, Ole Maaløes Vej 5, DK2200 Copenhagen N, Denmark.
Martin Moravec, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Bingnan Li, SciLifeLab, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Solna 171 65, Sweden.
Jingwen Wang, SciLifeLab, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Solna 171 65, Sweden.
Guifen Wu, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Maria Gockert, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Vicent Pelechano, SciLifeLab, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Solna 171 65, Sweden.
Torben Heick Jensen, Department of Molecular Biology and Genetics, Aarhus University, C.F. Møllers Alle 3, Building 1130, Aarhus 8000, Denmark.
Albin Sandelin, The Bioinformatics Centre, Department of Biology and Biotech and Research Innovation Centre, University of Copenhagen, Ole Maaløes Vej 5, DK2200 Copenhagen N, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Work in the Sandelin laboratory was supported by grants from the Lundbeck Foundation; Danish Cancer Society; Novo Nordisk Foundation; Independent Research Fund Denmark; Carlsberg Foundation; Work in the Jensen laboratory was supported by the Danish Cancer Society; Lundbeck Foundation; Independent Research Fund Denmark; Work in the Pelechano laboratory was supported by the Swedish Research Council; Wallenberg Academy Fellowship; Swedish Foundations’ Starting Grant (Ragnar Söderberg Foundation); Karolinska Institutet. Funding for open access charge: Novo Nordisk Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kilchert C., Wittmann S., Vasiljeva L.. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016; 17:227–239. [DOI] [PubMed] [Google Scholar]
- 2. Jensen T.H., Jacquier A., Libri D.. Dealing with pervasive transcription. Mol. Cell. 2013; 52:473–484. [DOI] [PubMed] [Google Scholar]
- 3. Houseley J., Tollervey D.. The many pathways of RNA degradation. Cell. 2009; 136:763–776. [DOI] [PubMed] [Google Scholar]
- 4. Schmid M., Jensen T.H.. The exosome: a multipurpose RNA-decay machine. Trends Biochem. Sci. 2008; 33:501–510. [DOI] [PubMed] [Google Scholar]
- 5. Schmid M., Jensen T.H.. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018; 19:518–529. [DOI] [PubMed] [Google Scholar]
- 6. Andersson R., Refsing Andersen P., Valen E., Core L.J., Bornholdt J., Boyd M., Heick Jensen T., Sandelin A.. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014; 5:5336. [DOI] [PubMed] [Google Scholar]
- 7. Lubas M., Christensen M.S., Kristiansen M.S., Domanski M., Falkenby L.G., Lykke-Andersen S., Andersen J.S., Dziembowski A., Jensen T.H.. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011; 43:624–637. [DOI] [PubMed] [Google Scholar]
- 8. Lubas M., Andersen P.R., Schein A., Dziembowski A., Kudla G., Jensen T.H.. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep. 2015; 10:178–192. [DOI] [PubMed] [Google Scholar]
- 9. Wu G., Schmid M., Rib L., Polak P., Meola N., Sandelin A., Heick Jensen T.. A two-layered targeting mechanism underlies nuclear RNA sorting by the human exosome. Cell Rep. 2020; 30:2387–2401. [DOI] [PubMed] [Google Scholar]
- 10. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seila A.C., Calabrese J.M., Levine S.S., Yeo G.W., Rahl P.B., Flynn R.A., Young R.A., Sharp P.A.. Divergent transcription from active promoters. Science. 2008; 322:1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Preker P., Nielsen J., Kammler S., Lykke-Andersen S., Christensen M.S., Mapendano C.K., Schierup M.H., Jensen T.H.. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008; 322:1851–1854. [DOI] [PubMed] [Google Scholar]
- 13. Flynn R.A., Almada A.E., Zamudio J.R., Sharp P.A.. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10460–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meola N., Domanski M., Karadoulama E., Chen Y., Gentil C., Pultz D., Vitting-Seerup K., Lykke-Andersen S., Andersen J.S., Sandelin A. et al.. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell. 2016; 64:520–533. [DOI] [PubMed] [Google Scholar]
- 15. Lloret-Llinares M., Karadoulama E., Chen Y., Wojenski L.A., Villafano G.J., Bornholdt J., Andersson R., Core L., Sandelin A., Jensen T.H.. The RNA exosome contributes to gene expression regulation during stem cell differentiation. Nucleic Acids Res. 2018; 46:11502–11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garland W., Comet I., Wu M., Radzisheuskaya A., Rib L., Vitting-Seerup K., Lloret-Llinares M., Sandelin A., Helin K., Jensen T.H.. A functional link between nuclear RNA decay and transcriptional control mediated by the polycomb repressive complex 2. Cell Rep. 2019; 29:1800–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belair C., Sim S., Kim K.-Y., Tanaka Y., Park I.-H., Wolin S.L.. The RNA exosome nuclease complex regulates human embryonic stem cell differentiation. J. Cell Biol. 2019; 218:2564–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ntini E., Järvelin A.I., Bornholdt J., Chen Y., Boyd M., Jørgensen M., Andersson R., Hoof I., Schein A., Andersen P.R. et al.. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat. Struct. Mol. Biol. 2013; 20:923–928. [DOI] [PubMed] [Google Scholar]
- 19. Chiu A.C., Suzuki H.I., Wu X., Mahat D.B., Kriz A.J., Sharp P.A.. Transcriptional pause sites delineate stable nucleosome-associated premature polyadenylation suppressed by U1 snRNP. Mol. Cell. 2018; 69:648–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamieniarz-Gdula K., Proudfoot N.J.. Transcriptional control by premature termination: a forgotten mechanism. Trends Genet. 2019; 35:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iasillo C., Schmid M., Yahia Y., Maqbool M.A., Descostes N., Karadoulama E., Bertrand E., Andrau J.-C., Jensen T.H.. ARS2 is a general suppressor of pervasive transcription. Nucleic Acids Res. 2017; 45:10229–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamieniarz-Gdula K., Gdula M.R., Panser K., Nojima T., Monks J., Wiśniewski J.R., Riepsaame J., Brockdorff N., Pauli A., Proudfoot N.J.. Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol. Cell. 2019; 74:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elrod N.D., Henriques T., Huang K.-L., Tatomer D.C., Wilusz J.E., Wagner E.J., Adelman K.. The integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol. Cell. 2019; 76:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tatomer D.C., Elrod N.D., Liang D., Xiao M.-S., Jiang J.Z., Jonathan M., Huang K.-L., Wagner E.J., Cherry S., Wilusz J.E.. The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 2019; 33:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silla T., Karadoulama E., Mąkosa D., Lubas M., Jensen T.H.. The RNA exosome adaptor ZFC3H1 functionally competes with nuclear export activity to retain target transcripts. Cell Rep. 2018; 23:2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engström P.G., Frith M.C. et al.. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006; 38:626–635. [DOI] [PubMed] [Google Scholar]
- 27. Cvetesic N., Leitch H.G., Borkowska M., Müller F., Carninci P., Hajkova P., Lenhard B.. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Res. 2018; 28:1943–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 2011; 17:10. [Google Scholar]
- 29. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ben Langmead, Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyd M., Coskun M., Lilje B., Andersson R., Hoof I., Bornholdt J., Dahlgaard K., Olsen J., Vitezic M., Bjerrum J.T. et al.. Identification of TNF-α-responsive promoters and enhancers in the intestinal epithelial cell model Caco-2. DNA Res. 2014; 21:569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liao Y., Smyth G.K., Shi W.. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mayer A., di Iulio J., Maleri S., Eser U., Vierstra J., Reynolds A., Sandstrom R., Stamatoyannopoulos J.A., Churchman L.S.. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015; 161:541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J., Li B., Marques S., Steinmetz L.M., Wei W., Pelechano V.. Improved transcriptome annotation and read-through transcript identification with TIF-Seq2. 2019; bioRxiv doi:29 November 2019, preprint: not peer reviewed 10.1101/859488. [DOI]
- 37. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E., Yusuf D., Lenhard B., Wasserman W.W., Sandelin A.. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010; 38:D105–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marstrand T.T., Frellsen J., Moltke I., Thiim M., Valen E., Retelska D., Krogh A.. Asap: a framework for over-representation statistics for transcription factor binding sites. PLoS One. 2008; 3:e1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ienasescu H., Li K., Andersson R., Vitezic M., Rennie S., Chen Y., Vitting-Seerup K., Lagoni E., Boyd M., Bornholdt J. et al.. On-the-fly selection of cell-specific enhancers, genes, miRNAs and proteins across the human body using SlideBase. Database. 2016; 2016:baw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M. et al.. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al.. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wickham H. ggplot2: elegant graphics for data analysis. 2009; NY: Springer. [Google Scholar]
- 48. Pelechano V., Wei W., Steinmetz L.M.. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013; 497:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iasillo C., Schmid M., Yahia Y., Maqbool M.A., Descostes N., Karadoulama E., Bertrand E., Andrau J.-C., Jensen T.H.. ARS2 is a general suppressor of pervasive transcription. Nucleic Acids Res. 2017; 45:10229–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tullai J.W., Schaffer M.E., Mullenbrock S., Sholder G., Kasif S., Cooper G.M.. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J. Biol. Chem. 2007; 282:23981–23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andersson R., Chen Y., Core L., Lis J.T., Sandelin A., Jensen T.H.. Human Gene Promoters Are Intrinsically Bidirectional. Mol. Cell. 2015; 60:346–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andersson R., Sandelin A.. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020; 21:71–87. [DOI] [PubMed] [Google Scholar]
- 53. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Almada A.E., Wu X., Kriz A.J., Burge C.B., Sharp P.A.. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013; 499:360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y., Pai A.A., Herudek J., Lubas M., Meola N., Järvelin A.I., Andersson R., Pelechano V., Steinmetz L.M., Jensen T.H. et al.. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat. Genet. 2016; 48:984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmid M., Jensen T.H.. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018; 19:518–529. [DOI] [PubMed] [Google Scholar]
- 57. Soucek S., Zeng Y., Bellur D.L., Bergkessel M., Morris K.J., Deng Q., Duong D., Seyfried N.T., Guthrie C., Staley J.P. et al.. The evolutionarily-conserved polyadenosine RNA binding protein, Nab2, cooperates with splicing machinery to regulate the fate of pre-mRNA. Mol. Cell. Biol. 2016; 36:2697–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schmid M., Poulsen M.B., Olszewski P., Pelechano V., Saguez C., Gupta I., Steinmetz L.M., Moore C., Jensen T.H.. Rrp6p controls mRNA poly(A) tail length and its decoration with poly(A) binding proteins. Mol. Cell. 2012; 47:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bresson S., Tuck A., Staneva D., Tollervey D.. Nuclear RNA decay pathways aid rapid remodeling of gene expression in yeast. Mol. Cell. 2017; 65:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garland W., Jensen T.H.. Nuclear sorting of RNA. Wiley Interdiscip. Rev. RNA. 2020; 11:e1572. [DOI] [PubMed] [Google Scholar]
- 61. Meola N., Jensen T.H.. Targeting the nuclear RNA exosome: poly(A) binding proteins enter the stage. RNA Biol. 2017; 14:820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andersen P.R., Domanski M., Kristiansen M.S., Storvall H., Ntini E., Verheggen C., Schein A., Bunkenborg J., Poser I., Hallais M. et al.. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat. Struct. Mol. Biol. 2013; 20:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hallais M., Pontvianne F., Andersen P.R., Clerici M., Lener D., Benbahouche N.E.H., Gostan T., Vandermoere F., Robert M.-C., Cusack S. et al.. CBC-ARS2 stimulates 3′-end maturation of multiple RNA families and favors cap-proximal processing. Nat. Struct. Mol. Biol. 2013; 20:1358–1366. [DOI] [PubMed] [Google Scholar]
- 64. Giacometti S., Benbahouche N.E.H., Domanski M., Robert M.-C., Meola N., Lubas M., Bukenborg J., Andersen J.S., Schulze W.M., Verheggen C. et al.. Mutually exclusive CBC-Containing complexes contribute to RNA fate. Cell Rep. 2017; 18:2635–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. duMee D.J.M., Ivanov M., Parker J.P., Buratowski S., Marquardt S.. Efficient termination of nuclear lncRNA transcription promotes mitochondrial genome maintenance. Elife. 2018; 7:e31989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thodberg M., Thieffry A., Bornholdt J., Boyd M., Holmberg C., Azad A., Workman C.T., Chen Y., Ekwall K., Nielsen O. et al.. Comprehensive profiling of the fission yeast transcription start site activity during stress and media response. Nucleic Acids Res. 2019; 47:1671–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All TIF-seq, SLIC-CAGE datasets are available at the Gene Expression Omnibus (GEO) under accession number: GSE147655. Code for TIF-seq processing and CAGE annotation is available at GitHub (https://github.com/PelechanoLab/TIFseq2 and https://github.com/MengjunWu/Exosome_sensitive_TSS). Supplementary Data are available at NAR online.