

Abstract
Speckle-type POZ protein (SPOP), a novel cancer- associated protein, was previously reported to function as a tumor suppressor or promoter in different malignant tumors. This research aims to investigate the biological functions and underlying molecular mechanisms of SPOP in choriocarcinoma. Our analysis of patient tissues and cell lines showed significantly decreased SPOP expression and highly expressed Nuclear DNA helicase II and RNA helicase A (DHX9), both of them are mainly located into the nucleus. Induction or depletion of endogenous SPOP with a lentivirus-based system correspondingly suppressed or promoted migration and invasion of choriocarcinoma cells. Mechanistically, we found that SPOP bound to DHX9 and induced the ubiquitination and degradation of DHX9 by recognizing a typical SPOP-binding motif in DHX9. SPOP-DHX9 interaction was demonstrated to play a critical role in regulating migration and invasion abilities of choriocarcinoma cells, the promotion of mobility ability in knocking down SPOP was partly counteracted by transfection with siRNA against DHX9. Taken together, our results suggest that SPOP suppresses migration and invasion of choriocarcinoma by promoting the ubiquitination and subsequent degradation of DHX9, which identifies the SPOP-DHX9 interaction may serve as a potential therapeutic target against choriocarcinoma.
Keywords: SPOP, DHX9, choriocarcinoma, ubiquitination, EMT
Introduction
Choriocarcinoma primarily occurs at the chorionic epithelium among females of reproductive ages and exhibits highly aggressive features, including abnormal proliferation of trophoblastic cells and rapid hematogenous dissemination in lung, spleen, brain, and liver [1]. Although many chemotherapeutic agents including etoposide and methotrexate have been used for effective treatment choriocarcinoma with an overall survival rate of > 90% [2-4], a small proportion of patients exhibit drug-resistantance and succumb to this disease due to relapse or solitary metastasis [2]. Thus, great challenges lie ahead in understanding underlying molecular mechanisms of tumorigenesis and metastasis and identifying novel molecular targets to improve patient prognosis.
Cullin-RING Ligase complex is the largest family in the ubiquitin-proteasome system (UPS) which is involved in a variety of physiological processes and pathological conditions, such as apoptosis, cell cycle and cancer. SPOP, an adaptor of the Cul3-Rbx1 E3 ubiquitin ligase complex (CRL3), has been proved that it functions as tumor suppressor or promoter in multifarious cancers via ubiquitination and subsequent degradation of substrate proteins by binding to the SPOP-binding consensus (SBC) motif on the substrate [5-8]. Some of these substrates include the death-associated protein (Daxx), cell division cycle 20 (CDC20), c-MYC, NANOG and other important transcription factors, coactivators and hormone signaling effectors [8-13]. Wild type SPOP has been found decreased expression in great majority human cancers including prostate cancer, breast cancer, gastric cancer, hepatocellular carcinoma and endometrial carcinoma,so it is considered as a tumor suppressor [14]. High expression level of SPOP has been shown to act as a tumor promoter in other tumors such as renal cell carcinoma (ccRCC) [15]. Despite the fact that various roles of SPOP in cancer pathogenesis and progression have been described, little data exist on the expression status and the functions of SPOP in choriocarcinoma.
Nuclear DNA helicase II and RNA helicase A (DHX9) is a member of the DExH/D-box helicase family which can unwind both DNA and RNA [16]. DHX9 consists of a conserved helicase central domain, an N-terminal double-stranded RNA domain, a C-terminal DNA domain and a nuclear localization signal [17]. DHX9 acts as a multifunctional protein, which can interact with a variety of molecular chaperones, participate in regulating multiple biological processes, such as DNA replication, transcription, translation, RNAs processing and transport, repairing DNA damage and maintaining gene stability [18-23]. Recent studies have shown that DHX9 mutations and abnormal expression exist in many malignant tumors. DHX9 had recurrent mutation and this mutation was highly correlated with pathway deregulation and patient survival in nonsmoking patients with lung adenocarcinoma [24]. Previous studies have demonstrated that abnormally high expression of DHX9 often acts as a cancer promoting factor, and inhibiting the expression of DHX9 can cause fatal damage to tumor cells, suggesting that DHX9 may become a potential target for cancer treatment [25,26]. However, other studies have found that DHX9 shows low expression in some tumors, which may have a certain inhibitory effect on tumors [27]. At present, the role of DHX9 in choriocarcinoma has not been explored.
In this study, we sought to identify novel substrate of SPOP to further explore its biological functions and molecular mechanisms in choriocarcinoma. Firstly, we compared SPOP expression in the first trimester villi and choriocarcinoma, and decreased SPOP promoted the proliferation, migration and invasion of cells by facilitating epithelial-mesenchymal transition (EMT) process. Moreover, we found that SPOP acted as a negative regulator of choriocarcinoma processes by binding to and inducing the degradative ubiquitination of DHX9. Our results show that the disorder of SPOP-DHX9 axis promotes the development of choriocarcinoma.
Materials and methods
Patients and clinical specimens
A total of 21 paraffin-embedded choriocarcinoma and 20 normal chorionic specimens were obtained from the Department of Pathology of Chongqing Medical University. Tissue samples were diagnosed as choriocarcinoma by the Department of Pathology; prior written informed consents were obtained from all the patients, and this research was approved by the Ethics Committees of Chongqing Medical University.
Cell culture, treatment, plasmids, transfection and lentiviral transduction
A first-trimester extravillous cytotrophoblast cell HTR8-SVneo was obtained from ATCC (USA), and the human choriocarcinoma cell lines JAR and JEG3 cells were purchased from the Chinese Academy of Sciences (Shanghai, China). All cell lines were identified by the short tandem repeat (STR) genotyping. HTR8 and JAR cells were maintained in RPMI 1640 and JEG3 cells was cultured in DMEM/F12 supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 ng/mL). All cell lines were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Plasmid transfections were performed using EndoFectin TM-Max (GeneCopoeia, EF003). The vectors used for HA-DHX9 and sh-DHX9 were GV366 and GV248, respectively. These vectors were purchased from Jikai Gene Chemical Technology Co., Ltd (Shanghai, China). The vector of HA-DHX9-ΔSBC was synthesized by Wuhan Genecreate Biological Engineering Co., Ltd (Wuhan, China). The Flag-SPOP lentiviral plasmid vector PSE3255 was purchased from Shanghai Shengbo Biomedical Technology Co., Ltd. (Shanghai, China). The DHX9 sequences of shRNAs used in this study are summarized in Table 1.
Table 1.
Sequence of sh-DHX9
| ID | 5’ | stem | loop | stem | 3’ |
|---|---|---|---|---|---|
| DHX9-RNAi (33759-1)-a | Ccgg | gaAGGATTACTA | CTCGAG | TTTCTTGAGTAGT | TTTTTg |
| CTCAAGAAA | AATCCTTC | ||||
| DHX9-RNAi (33759-1)-b | Aattcaaaaa | gaAGGATTACTA | CTCGAG | TTTCTTGAGTAGT | |
| CTCAAGAAA | AATCCTTC | ||||
| DHX9-RNAi (33760-1)-a | Ccgg | agACTTAATATG | CTCGAG | TAGTGTAGCCATA | TTTTTg |
| GCTACACTA | TTAAGTCT | ||||
| DHX9-RNAi (33760-1)-b | Aattcaaaaa | agACTTAATATG | CTCGAG | TAGTGTAGCCATA | |
| GCTACACTA | TTAAGTCT | ||||
| DHX9-RNAi (33762-1)-a | Ccgg | aaGCATGGACC | CTCGAG | TCATTCTTGAGGT | TTTTTg |
| TCAAGAATGA | CCATGCTT | ||||
| DHX9-RNAi (33762-1)-b | Aattcaaaaa | aaGCATGGACC | CTCGAG | TCATTCTTGAGGT | |
| TCAAGAATGA | CCATGCTT |
For lentiviral infection, SPOP expression vector, a control vector, the shRNA targeting SPOP and a negative control vector were designed and constructed by Sunbio Medical Biotechnology Co., Ltd (shanghai, China). The sequence of shRNA oligonucleotides against SPOP was as follows: shSPOP: CCGGCACAAGGCTATCTTAGCAGCTCTCGAGAGCTGCTAAGATAGCCTTGTGTTTTTTG; These vectors were transfected into JAR cells when cells fusion reached 50%. After 24 hours, the culture medium was substituted with fresh medium. After 72 hours of transfection, puromycin was added to the culture medium at a concentration of 5 mg/ml for 2 weeks to generate stable cell lines.
RNA isolation, reverse transcription, and qRT-PCR
Total RNA was extracted from cultured cells with Trizol reagent (Takara, Shiga, Japan). For real-time quantitative PCR, 1 ug of total RNA was reverse-transcribed into cDNA by using the Prime Script RT reagent kit (TakaRa). Synthesized cDNA was then amplified with a SYBR® Premix Ex Taq™ II (Perfect Real-Time) kit (Takara). Melting curve for each target gene was generated to ensure PCR specificity. Relative abundance of mRNA was calculated by normalization to GAPDH mRNA expression. All the primers sequences used for amplification were listed in Table 2.
Table 2.
Primer sequences for qRT-PCR
| Gene | Sequence (5’->3’) |
|---|---|
| SPOP | GCCCTCTGCAGTAACCTGTC |
| GTCTCCAAGACATCCGAAGC | |
| HIF-1α | TTACAGCAGCCAGACGATCA |
| GATTGCCCCAGCAGTCTACA | |
| DHX9 | GCCAATTTCTGGCCAAAGCA |
| CGAGGCTCAATGGGGAGTTT | |
| GAPDH | CGACCACTTTGTCAAGCTCA |
| CCCTGTTGCTGTAGCCAAAT |
Immunoblotting (IB) and co-immunoprecipitation (co-IP)
The equivalent amounts of protein was separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Nonspecific binding sites of the membranes were blocked and incubated overnight with specific primary antibodies at 4°C. Then blots were probed with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies for 1.5 hour at room temperature. Immunoreactive bands were measured by enhanced chemiluminescence (ECL) system (Millipore, WBKLS0100). Signals of the immunoblot bands were analyzed with the Image Lab program.
For co-immunoprecipitation assays, cells were transfected with the indicated constructs and treated with the proteasome inhibitor MG132 (APExBIO Technology, A2585) for 10 h before harvesting. Cells were lysed in Lysis Buffer for IB and IP (Beyotime, P0013). Lysates were incubated with indicated antibodies for 2 h at 4°C. Then lysates were added with protein A/G magnetic beads (MedChem Express, HY-K0202) to form immune complex. Immunoprecipitants were subjected to SDS-PAGE and immunoblotted with indicated antibodies.
Immunohistochemistry (IHC)
Briefly, paraffin-embedded tissue specimens were deparaffinize and rehydrated. Endogenous peroxidase activity was inhibited with 3% hydrogen peroxide/methyl alcohol (H2O2) for 10 minutes. Non-specific binding sites were blocked by non-immune animal serum for 10 min and then sections were subjected to SPOP antibody overnight at 4°C, and then incubation with biotinylated secondary antibody. Cell images were captured with a confocal microscope (Nikon). Quantitative analysis was calculated by two experienced pathologists in at least six fields for each specimen based on both percentage of positively stained cells and staining intensity. For SPOP, a staining intex (values, 0-12) was defined by multiplying the score for staining intensity with the score for positive area. The intensity was scored as follows: negative, 0; weak, 1; moderate, 2; strong, 3. The percentage of positively stained cells was determined as follows: less than 5%, 0; 5%-25%, 1; 26%-50%, 2; 51%-75%, 3; greater than 75%, 4.
Cell proliferation assay
Cell proliferation was performed according to the manufacture’s recommendations. JAR cells were plated at 3000 cells/well in 96-well plates with 100 μL culture medium. After 0 h, 24 h, 48 h, 72 h and 96 h incubation, cells were treated with 10 uL Cell Counting Kit-8 (CCK-8) (Beyotime) solution for 1 h at 37°C. Finally, the optical density (OD) was measured at an absorbance of 450 nm using Microplate Reader.
Cell cycle analysis by flow cytometry
Targeted cells were seeded in 6-well plates at a proper density and grown to about 80% confluence; cells were digested with trypsin to produce a single cell suspension. For a cell cycle detection, suspended cells (1×106 cells/ml) were washed three times with PBS and fixed in ice-cold 75% ethanol for overnight, and then the cells were stained with propidium iodide (PI; BD Biosciences) in the presence of 100 mg/ml RNase A. Cell cycle analysis was analyzed with a flow cytometer.
Transwell assays
The cell invasion and migration assays were conducted in 24-well Transwell plates with or without pre-coated Matrigel. Briefly, 3×104 transfected cells and the corresponding negative control cells were suspended in 200 ul serum free RPMI-1640 medium into the upper chamber. The upper chamber is made with the Boyden chamber containing an 8 um pore size membrane (BD Biosciences, San Jose, CA). In the lower chamber, 500 uL RPMI-1640 medium containing 20% FBS was added. After 48 h of incubation, cells remaining in the upper chamber were softly removed by cotton swab scrubbing. The cells that successfully migrated through the membrane and invaded through the matrigel were fixed in 4% paraformaldehyde for 20 min and stained with crystal violet dye for 15 min. The cell numbers were counted at six randomly selected views, and the average value was calculated.
Wound healing assay
JAR cells were seeded in 6-well plates and cultured to 80% confluence. Scratch wounds across each well were made with a 10-ll pipette tip. The cells were washed 3 times with PBS, removing the cells that had been exfoliated and then fresh medium supplemented with reduced (2%) fetal bovine serum was added. The wound-closing procedure was photographed with an inverted microscope at 0, 24 and 48 h, respectively. Cell migration was quantitated by measuring the width of the distance.
Tumor xenograft experiments
The female nude mice (BALB/c-nu) between 4 and 5 weeks were purchased from Beijing HFK Bioscience Co., Ltd. 2×106 JAR cells infected with SPOP overexpression or control vectors were resuspended in 150 ul of serum-free medium and subcutaneously injected into the axillary fossae. One month after the injection of JAR cells, all mice were sacrificed by dislocation of spin and tumors were carefully removed, photographed and weighed. All in vivo protocols in this study were reviewed and approved by the Animal Ethics Committee of Chongqing Medical University.
Label-free ubiquitination modification quantitative proteomics
JAR cells transfected with Flag-SPOP were treated with the proteasome inhibitor MG132 for 10 h before harvesting. Then cells were added lysis buffer and centrifuged at 12000 g for 10 min to abtain proteins. After cleavage with dithiothreitol, iodoacetamide and trypsin, the proteins were dissolved in IP buffer solution. Then we added the ubiquitinated resin into IP buffer solution to obtain the ubiquitin-binding proteins. Finally, it was analyzed by liquid chromatography-mass spectrometry.
Endogenous and exogeneous ubiquitination assay
JAR and 293T cells were transfected with the indicated constructs. Stable cells were screened by adding antibiotic, and then cells were treated with 10 μg/ml MG132 for 10 h before harvesting. Cells were lysed at 4°C for 20 min and centrifuged to obtain the supernatant. Protein solution were subjected to co-immunoprecipitation with anti-DHX9, and then coupled with the magnetic beads followed by immunoblotting analysis with anti-ubiquitin (proteintech, 10201-2-AP).
Antibodies
Antibodies against SPOP (Absin, abs118000), Flag (Cell Signaling Technology, 2368), DHX9 (proteintech, 17721-1-AP), GAPDH (Beyotime, AF0006), HA (proteintech, 51064-2-AP), ubiquitin (proteintech, 10201-2-AP), N-Cadherin (Beyotime, AF0243), Vimentin (Cell Signaling Technology, 5741), E-Cadherin (Beyotime, AF0138), Cytokeratin 8 (Wanleibio, WL02755). Mouse Anti-Rabbit IgG (Light-Chain Specific) mAb (Cell Signaling Technology, 93702), Peroxidase-Conjugated Goat anti-Rabbit IgG (ZSGB-BIO, ZB-2301), Peroxidase-Conjugated Goat anti-Mouse IgG (ZSGB-BIO, ZB-2305), Alexa Fluor 488-labeled goat anti-rabbit IgG (H + L) (Beyotime, A0423).
Statistical analysis
All statistical analysis was performed with the Graph Pad prism version 8.0 (Graph Pad Software, San Diego, CA, USA). The statistical analysis was evaluated with a two-tailed Student’s t-test and One-way analysis of variance (ANOVA). Data in this study were presented as Mean ± SD. All results were from at least three independent experiments.
Results
The expression and localization of SPOP in choriocarcinoma tissues and cell lines
To determine the expression level of SPOP in choriocarcinoma, we examined the expression of SPOP by immunohistochemistry in selected 21 cases of choriocarcinoma and 20 cases of first trimester villus (FTV). The results showed that SPOP could be labelled in both the nucleus and cytoplasm of tumor cells, but the staining of SPOP in nucleus was more obvious (Figure 1A, indicated by arrows). In contrast to normal villus tissues which consistently displayed moderate or strong staining, the SPOP expression showed significantly reduced levels in choriocarcinoma tissues and this was negatively correlated with tumor stage (Figure 1A-C). To further prove our results, SPOP expression was also determined in choriocarcinoma cell lines. Quantitative RT-PCR (qRT-PCR) (Figure 1D) and immunoblotting analysis (Figure 1E) of two choriocarcinoma cell lines, including JAR and JEG3, exhibited decreased levels of SPOP in comparison to the normal trophoblastic cell line HTR8-SVneo. These data suggested a linkage of SPOP down-regulation and choriocarcinoma progression.
Figure 1.
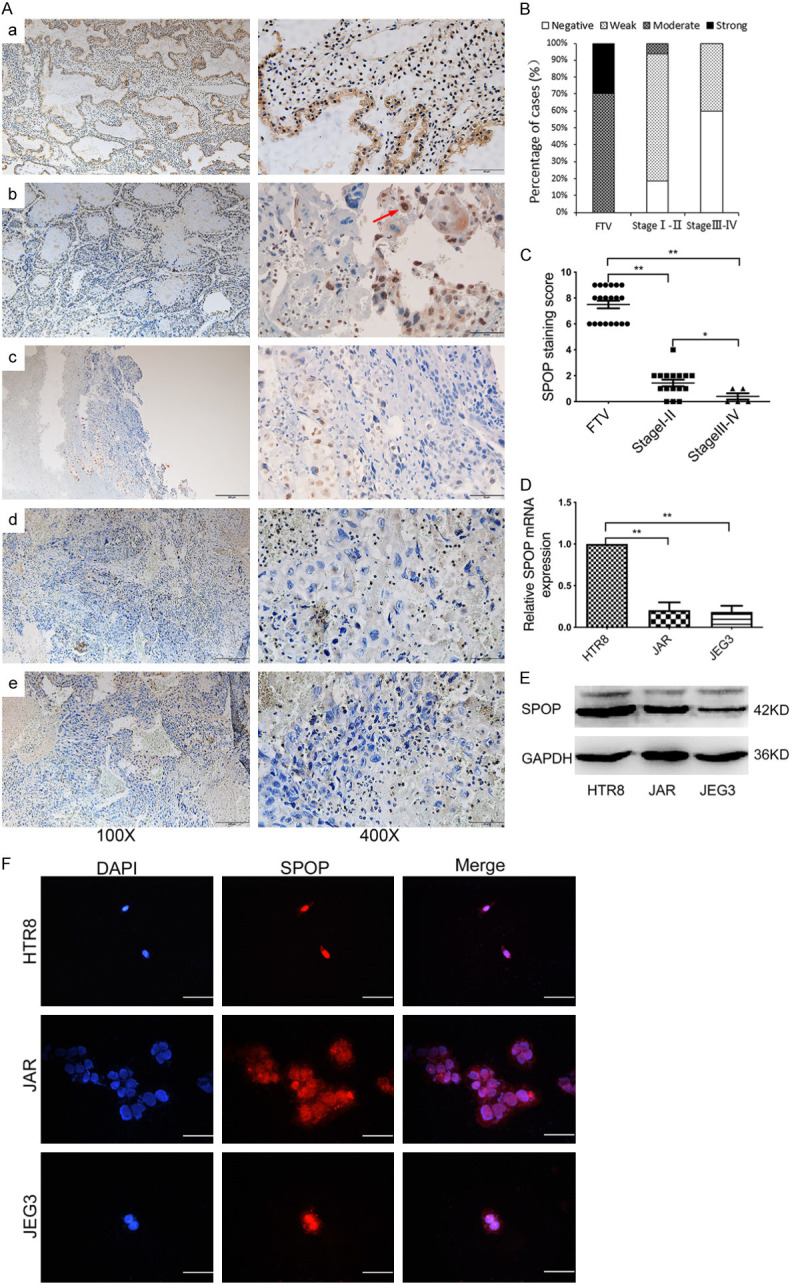
The expression and localization of SPOP in choriocarcinoma tissues and cell lines. (A) The representative images of SPOP expression in FTV (a) and images of TNM stage I (b), stage II (c), stage III (d), stage IV (e) SPOP expression in the tumor tissues, as measured by IHC. Red arrow indicated cytoplasmic and nuclear location of SPOP. FTV=20, TNM stage I=11, stage II=5, stage III =3, stage IV=2. Scale bar, 200 um (left) and 50 um (right), respectively. (B) Percentage analysis of SPOP staining intensity in 21 cases of choriocarcinoma. (C) Comparison of average SPOP staining scores among different TNM stages, showing inversely correlation of SPOP with tumor progression. SPOP mRNA (D) and protein (E) levels in HTR8, JAR and JEG3 cells were analyzed using qRT-PCR and immunoblotting (IB) analysis, respectively. *P < 0.05, **P < 0.01, compared to control groups. (F) Immunofluorescence for HTR8, JAR and JEG3 cells were stained with SPOP antibody (red), and the nuclei were counterstained with DAPI (blue). Scale bar, 50 um.
Moreover, we investigated the subcellular localization of SPOP. Previous studies have shown that SPOP is primarily present in discrete foci scattered in the nucleus, but a small munber of researches have suggested that SPOP is localized in both the cytoplasm and nucleus [28]. The results of our research were similar to those obtained by immunohistochemistry (Figure 1A), SPOP was localized exclusively in the nucleus in the majority of the trophoblast cells by using immunofluorescence analysis (Figure 1F).
Effects of SPOP on proliferation, migration and invasion of choriocarcinoma cells in vitro and vivo
To determine the biological roles of SPOP in the development and progression of choriocarcinoma, we stably depleted or overexpressed SPOP using a lentivirus-based system in choriocarcinoma cell lines JAR and JEG3. Levels of SPOP in these resultant cell lines were confirmed by immunoblotting and qRT-PCR (Figure 2A, 2B). Functionally, stably expressed SPOP significantly attenuated cell proliferation in JAR and JEG3 cells, while depletion of SPOP had the opposite effects (Figure 2C). Cell cycle analysis showed that SPOP overexpression or knockdown significantly affect the cell cycle distribution, especially the S phase. SPOP overexpression reduced the ratio of cells in the S phase whereas SPOP ablation increased S phase populations compared with the control group (Figure 2D). Wound-healing assays revealed that forced expression of SPOP obviously inhibited the wound closure, whereas knockdown of SPOP significantly increased cell motilities (Figure 2E). Besides, similar results were obtained in transwell assays, knockdown of SPOP dramatically enhanced migration and invasion abilities (Figure 2F). It also significantly altered the expression of the EMT associated proteins E-cadherin, cytokeratin 8, N-cadherin and Vimentin (Figure 2G, 2H).
Figure 2.

The expression of SPOP was associated with choriocarcinoma cells proliferation, migration and invasion both in vitro and vivo. (A and B) SPOP knockdown and overexpression in JAR and JEG3 cells were confirmed by immunoblotting (A) and qRT-PCR (B), respectively. (C) Increased or decreased expression of SPOP in JAR and JEG3 cells affected cell proliferation examined by CCK8 assays. (D) Cell cycle analysis of JAR and JEG3 cells after SPOP overexpression and knockdown using flow cytometry. Percentage of cells in G1, S and G2/M phases is shown in the right panel. (E) Wound healing assay showing JAR and JEG3 cells migration abilities at 0, 24 h and 48 h after SPOP overexpression and knockdown, the percentage of wound healing was measured by Image J software. Quantification of the wound closure is presented on the right panel. (F) Transwell assays showing the migration and invasion abilities after SPOP overexpression and knockdown in JAR and JEG3 cells. Results were evaluated as the average number of migrating and invading cells from six random microscope fields. Quantification of the invaded or migrated cells is shown in the right panel. (G) IB analysis of E-cadherin, Cytokeratin 8, N-cadherin and vimentin after up-regulating or down-regulating SPOP in JAR and JEG3 cells. (H) The relative protein abundance of EMT related proteins was quantified by Image Lab software and normalized with control cells. (I) Tumorigenesis ability of JAR cells in a xenograft model after SPOP overexpression. Each experiment was repeated at least three times (mean ± SD), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, compared to control groups.
To further investigate whether the inhibiting growth role of SPOP in choriocarcinoma cells in vitro can be extended in vivo, control and SPOP overexpression JAR cells were injected subcutaneously into 5-week-old nude mice, which were later sacrificed and their tumors weight were measured. As shown in Figure 2I, increased SPOP significantly inhibited tumor growth compared to the control group. Taken together, these in vitro and vivo data demonstrated that SPOP effectively inhibited tumorigenic behaviors of choriocarcinoma cells.
The expression and localization of DHX9 in choriocarcinoma cell lines
In order to obtain the ubiquitination substrate of SPOP, we performed ubiquitination modification quantification proteomics analysis and anti-Flag co-immunoprecipitation coupled with mass spectrometry (IP-MS) analysis in the JAR cells with Flag-SPOP overexpression (Treat/Control > 1.50) (Figure 3A). Then we combined the results of the above two methods with SPOP-binding consensus motif of known substrates [6], and finally screened 4 kinds of proteins, namely: DHX9, KPNB1, RACK1, PSMA6 (Figure 3C) (See Supplementary Materials 1, 2, 3 for detailed differential proteins information). Since DHX9 acts as a multifunctional protein and participates in regulating multiple biological processes; meanwhile, the molecular weight of DHX9 is 140 kD, which is similar to the position of the specific band in the silver staining diagram (Figure 3B). Moreover, we found the mass spectrograms of DHX9 both in the results of intracellular ubiquitination experiment (Figure 3D) and co-immunoprecipitation (Figure 3E). Therefore, DHX9 has attracted our attention and has been further studied. We examined the expression of DHX9 in human trophoblast cells HTR-8/SVneo, JAR and JEG3 cells. These results showed that compared with HTR-8/SVneo, DHX9 was highly expressed in choriocarcinoma cells (Figure 3F, 3G), which is opposite to the expression of SPOP in these three cell lines (Figure 1E). Subsequently, the subcellular localization of DHX9 was detected in HTR-8/SVneo, JAR, and JEG3 cells. We found that DHX9 was localized exclusively in the nucleus of the three trophablast cells (Figure 3H), and our previous research showed that the majority of cells where SPOP was primarily localized in the nucleus, suggesting the co-localization and potential interaction of SPOP and DHX9 in the nucleus.
Figure 3.

DHX9 is low expressed in choriocarcinoma cells and is mainly localized in the nucleus. (A and B) IB analysis (A) and Silver stain assay (B) of JAR whole-cell lysates (WCLs) and anti-Flag immunoprecipitation derived from JAR cells transfected with the Flag-SPOP. The positions of DHX9 and SPOP were indicated by arrows. Rabbit immunoglobulin G (IgG) was used as a negative control for co-immunoprecipitations (co-IPs). Cells were treated with 10 μg/ml proteasome inhibitor MG132 for 10 h before harvesting. Flag, a tag protein. (C) Schematic diagram of three experimental methods. The shaded area indicates the proteins shared by the three experimental results. (D) The mass spectrum of DHX9 in ubiquitination experiment of JAR cells transfected with the indicated lentivirus. (E) The mass spectrum of DHX9 in co-immunoprecipitation assay of JAR cells transfected with the indicated lentivirus. (F and G) IB analysis of WCLs derived from HTR8, JAR and JEG3. Data are shown as mean ± SD of three independent experiments. **P < 0.01, ***P < 0.001, Student’s t-test. (H) Immunofluorescence for trophoblast cells HTR8, JAR and JEG3, cells were stained with DHX9 antibody (green), and the nuclei were counterstained with DAPI (blue). Scale bar, 50 um.
DHX9 interacts with SPOP and depends on the SBC motif
To verify whether DHX9 interacts with SPOP, human embryonic kidney cells 293T were cotransfected with the Flag-SPOP and HA-DHX9 proteins, and stable cells were screened with antibiotics. With the addition of the proteasome inhibitor MG132, we observed that DHX9 can be co-immunoprecipitated by Flag-SPOP (Figure 4A). Under the same conditions, Flag-SPOP was successfully co-immunoprecipitated by HA-DHX9 (Figure 4B), suggesting an interaction between these two exogenously expressed proteins. Next, we investigated whether DHX9 interacted with SPOP in choriocarcinoma cells. Lentivirus with Flag-SPOP was infected into JAR cells and JEG3 cells. Then after 10 hours of adding MG132, Flag antibody successfully immunoprecipitated endogenous DHX9 in both JAR cells and JEG3 cells (Figure 4C). Similarly, only the HA-DHX9 plasmid was transferred into cells, endogenous SPOP were successfully precipitated by HA antibody (Figure 4D). In addition, in order to avoid the impact of exogenous vectors on the interaction between SPOP and DHX9, we used DHX9 antibodies for co-immunoprecipitation and found that endogenous SPOP was successfully precipitated in JAR cells and JEG3 cells (Figure 4E), suggesting an endogenous interaction between SPOP and DHX9. In conclusion, these above exogenous and endogenous results indicate that SPOP interacts with DHX9 in choriocarcinoma cells.
Figure 4.
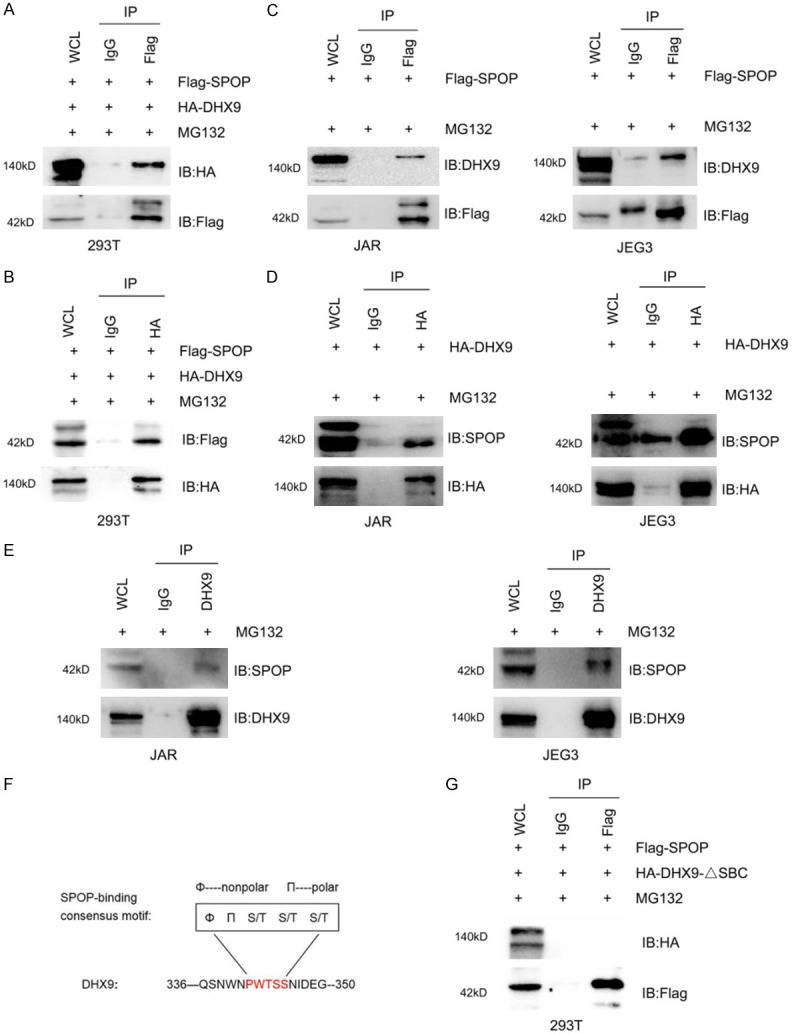
DHX9 Interacts with SPOP and depends on the SBC motif. (A and B) IB analysis of WCLs and anti-Flag co-IPs (A) or anti-HA co-IPs (B) derived from 293T cells transfected with the indicated constructs. (C and D) IB analysis of WCLs and anti-Flag co-IPs (C) or anti-HA co-IPs (D) derived from JAR and JEG3 cells transfected with the indicated constructs. (E) IB analysis of WCLs and anti-DHX9 co-IPs derived from JAR cells and JEG3 cells. (F) Schematic diagram showing the specific amino acid of the SBC motif and the position of the SBC motif contained in DHX9. (G) IB analysis of WCLs and anti-Flag co-IPs derived from 293T cells transfected with the indicated constructs. Rabbit IgG was used as a negative control for co-IPs. Cells were treated with 10 μg/ml MG132 for 10 h before harvesting. HA-DHX9-ΔSBC, HA-DHX9 with SBC motif deleted.
It has been previously reported that SPOP ubiquitinated substrates all contain the SBC motif which is particularly important for the interaction between SPOP and the substrates. We next performed a protein motif search on DHX9 sequence and discovered one perfectly matched SBC motif (336-PWTSS-350 aa) (Figure 4F). To investigate whether this potential motif is required for SPOP-DHX9 interaction, we constructed a HA-DHX9 plasmid with the SBC motif deleted (abbreviated as HA-DHX9-ΔSBC), followed by the lentivirus with Flag-SPOP and HA-DHX9-ΔSBC plasmid co-transfected into 293T cells. Expectedly, co-immunoprecipitation assay showed that HA-DHX9-ΔSBC cannot bind to Flag-SPOP at levels comparable with those of DHX9-WT (Figure 4G), indicating that the SBC motif plays an important role in the interaction between DHX9 and SPOP.
SPOP promotes the ubiquitination and degradation of DHX9
In order to investigate whether SPOP could promote the ubiquitination and degradation of DHX9. We constructed models of over-expressing SPOP and deleting endogenous SPOP in JAR and JEG3 cells. Expectedly, stable overexpression of SPOP decreased the protein level of DHX9 in JAR cells and JEG3 cells, the depletion of endogenous SPOP increased DHX9 protein abundance (Figure 5A, 5B). However, after successfully knocking down SPOP, the mRNA levels of DHX9 both in these cells did not change significantly (Figure 5C, 5D). Thus, these results suggest that SPOP may negatively regulate DHX9 expression at the protein levels.
Figure 5.

SPOP promotes the degradation and ubiquitination of DHX9. (A and B) IB analysis of SPOP and DHX9 protein levels derived from JAR cells (A) or JEG3 cells (B) transfected with the indicated constructs. (C and D) Detection of SPOP and DHX9 mRNA levels by real-time PCR analysis after SPOP depletion in JAR cells (C) or JEG3 cells (D). (E and F) IB analysis of DHX9 and SPOP levels before and after adding MG132 to JAR cells (E) or JEG3 cells (F) transfected with the indicated constructs. (G) IB analysis of WCLs and anti-DHX9 co-IPs derived from JAR cells and 293T cells transfected with indicated constructs. Cells were treated with 10 μg/ml MG132 for 10 h before harvesting. Data are shown as mean ± SD of three independent experiments. NS, P>0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test.
We then examined whether the degradation of DHX9 by SPOP was associated with DHX9 ubiquitination. The results of western blot showed that DHX9 degradation was reduced when MG132 was added into choriocarcinoma cells with SPOP overexpression to inhibit proteasome-dependent protein degradation (Figure 5E, 5F). Furthermore, we also found that DHX9 ubiquitination was increased both in JAR cells overexpressing SPOP and 293T cells overexpressing SPOP and DHX9 simultaneously (Figure 5G). Consequently, these results indicated that SPOP promotes the ubiquitination and degradation of DHX9.
SPOP controls choriocarcinoma cells invasion and migration processes partly through DHX9
We investigated whether SPOP played a tumor suppressor role in choriocarcinoma through modulating DHX9 expression. Firstly, we selected the small hairpin RNA that has the highest knockdown efficiency for DHX9 (Figure 6A-C). We next examined the effects of DHX9 depletion on cell migration and invasion. Compared with the control group, DHX9 knockdown significantly reduced the number of invading and migrating cells (Figure 6D, 6E), which was opposite to the effects of SPOP knockdown on cell migration and invasion abilities. To investigate whether the effect of DHX9 on the invasion and migration of JAR cells was regulated by SPOP, a rescue assay was performed. The results showed that compared with SPOP knockdown, simultaneous depletion of SPOP and DHX9 reduced cell migration and invasion (Figure 6D, 6E), indicating that SPOP modulates cell migration and invasion processes mainly through regulating the abundance of DHX9 protein.
Figure 6.
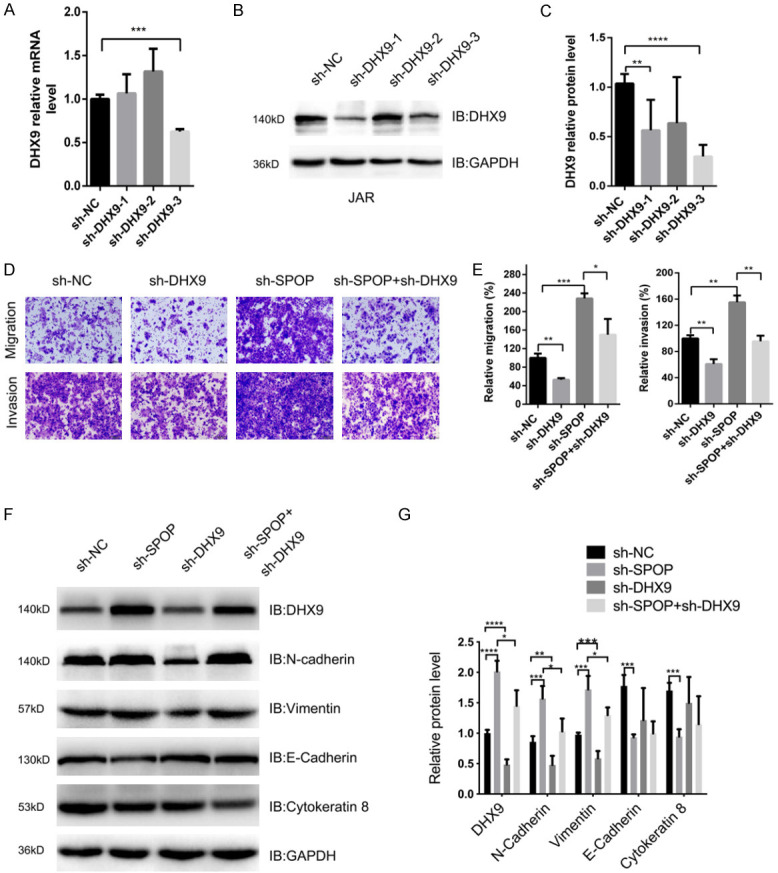
SPOP negatively regulates DHX9-mediated invasion and migration of JAR cells. A. Real-time PCR analysis of DHX9 mRNA levels in JAR cells transfected with sh-RNA plasmid. B and C. IB analysis of DHX9 protein levels derived in JAR cells transfected with the indicated constructs. D. Representative images of migrated and invaded JAR cells transfected with the indicated constructs in migration assay and invasion assay. E. Quantification analysis of migrated JAR cells and invaded JAR cells. F and G. IB analysis of DHX9, E-cadherin, Cytokeratin 8, N-cadherin and vimentin levels derived from JAR cells transfected with the indicated constructs. Data are shown as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test.
More importantly, we observed the changes in EMT markers and found that compared with the control group, the depletion of DHX9 attenuated the expression of N-Cadherin and Vimentin. Simultaneous depletion of SPOP and DHX9 partly counteracted N-Cadherin and Vimentin expression compared with SPOP single knockdown (Figure 6F, 6G). However, the epithelial markers E-Cadherin and cytokeratin 8 did not change significantly between sh-NC group, sh-DHX9 group and the sh-DHX9 + sh-SPOP group (Figure 6F, 6G). In summary, these results demonstrated that SPOP-DHX9 may affect cell invasion and migration by regulating the expression of EMT-associated proteins.
Discussion
SPOP has been extensively studied as an E3 ubiquitin ligase substrate-binding protein, which plays various roles in cancer pathogenesis and progression via complex mechanism [14]. However, its role in choriocarcinoma hasn’t been to be characterized. In present study, we found for the first time that SPOP was markedly down-regulated in choriocarcinoma and was negatively correlated with tumor stage, suggesting its involvement in the development of choriocarcinoma. Meanwhile, dramatically decreased SPOP expression indicated its a tumor promoter role via functional assays. Increasingly more studies reveal that epithelial-mesenchymal transition (EMT) is a main cause for choriocarcinoma invasion and metastasis [29,30], and our results demonstrated that SPOP insufficiency significantly promoted cell proliferation, migration and invasion capacity through the promotion of the EMT transformation.
Human proteomics research found that DHX9 is widely expressed in tissue cells, such as: high expression in skin, intestine, stomach, pancreas, kidney, breast cancer, skeletal muscle, bone marrow and reproductive organs, but is moderately expressed in liver, spleen, lung, heart, smooth muscle, adipose tissue and lymph nodes [31,32]. Studies have shown that abnormally expressed DHX9 has effects on many malignant tumors and may function as either tumor promoter or suppressor. DHX9 was considered to be a tumor suppressor by regulating expression of KIF1Bβ (kinesin family member1B, isoform b) [33], STAT3 [34] and p53 [22]. In contrast, DHX9 interacts with EWS-FLI1 [35], BRCA1 and DBC1 [36-39], NF-κB [40], miR-483-5p [41] and thereby acts as an oncogene in multiple malignant tumors. Similar to its role in these tumors, our results with choriocarcinoma studies have found that DHX9 was up-regulated, and this upregulation was associated with SPOP insufficiency. Recent works have shown that, DHX9 can interact with ubiquitin-conjugating enzyme 9 (Ubc9) and the N terminal of DHX9 is ubiquitinated [42]. Considering that SPOP participates in physiological and pathological processes such as organ development and tumorigenesis via it functioning as an E3 ubiquitin ligase adaptor [43-46], we explore whether the regulation of DHX9 is also through ubiquitin-proteasome system (UPS). Our results showed that SPOP promoted the ubiquitination and subsequent UPS-dependent degradation of DHX9, which depended on SPOP-DHX9 interactions involving the SBC motif (φ-π-S-S/T-S/T) of DHX9 (336-PWTSS-350). In previously published studies, SPOP recognizes the SBC degron of multiple substrates to promote its multimeric ubiquitination and degradation. This provides strong evidence that DHX9 is a new substrate for SPOP in choriocarcinoma. Besides, we investigated the subcellular localization in which SPOP-DHX9 interaction occured. SPOP has been reported to be either distributed as speckles exclusively in the nucleus, or in both the cytoplasm and nucleus [28,47]. In choriocarcinoma, our results indicated that DHX9 and SPOP both were principally localized in the nucleus, and the same subcellular localization further increased the possibility of SPOP-DHX9 interaction.
EMT has been recognized as the crucial mechanism promoting tumor initiation, progression and metastasis in human mammary epithelial cells, which was characterized as the absence of epithelial cell junctions and polarity, and acquisition of invasive mesenchymal phenotype [48,49]. We found that both SPOP overexpression and DHX9 suppression were coupled with a significant decrease in mesenchymal markers vimentin and N-cadherin, and SPOP overexpression was accompanied by an increase of epithelial markers E-cadherin and cytokeratin 8. However, DHX9 knockdown had no effect on the expression of epithelial markers. We speculate that the knockdown of DHX9 may have caused changes in other epithelial markers besides E-cadherin and cytokeratin 8, such as β-catenin, claudin-1, ZO-1 and other members of cytokeratin family, but the specific reasons need to be further explored. Loss of E-cadherin and increase of N-cadherin are critical markers of EMT, resulting in the absence of cell polarity and weak cell-cell adhesion which promotes the motility abilities of tumor cells. Due to the complexity of EMT regulated in different types of tumors, the underlying mechanism by which SPOP or DHX9 regulates the EMT process in choriocarcinoma is still unclear. Previous studies reported that DHX9 inhibited EMT progression in human lung adenocarcinoma cells by regulating JAK/STAT3 pathway [34], SPOP regulated EMT and tumor progression via activation of β-catenin/TCF4 complex in clear cell renal cell carcinoma [50]. The research results above suggested that the pathway associated to cell invasion and metastasis may be involved in the EMT process mediated by SPOP or DHX9. In our previous study on choriocarcinoma, we detected the potential signaling pathways that affect the mobility abilities of trophoblast cells, including ERK1/2 and p-ERK1/2, AKT and p-AKT, STAT3 and p-STAT3, Gli1 and Gli2, and identified the significantly changed pathway PI3K/AKT. We have found that SPOP can inactivate the PI3K/Akt signaling pathway and attenuate the migration and invasion of choriocarcinoma cells (Data were not shown), implying that SPOP-DHX9 may have influenced EMT process in choriocarcinoma cells by regulating PI3K/AKT pathway. However, the specific mechanism of EMT transformation needs to be further explored.
In conclusion, these results spread a novel view on molecular mechanisms of SPOP mediated choriocarcinoma growth and progression. In the future, SPOP-DHX9 interaction may be regarded as a novel molecular target for improving choriocarcinoma patient prognosis.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 81100443), Chongqing science and technology committee Project (cstc2019jscx-msxmX0246) and Chongqing Municipal Health Bureau Scientific Research Project (grant no. 20132151).
Disclosure of conflict of interest
None.
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
References
- 1.Yang C, Lim W, Bazer FW, Song G. Myricetin suppresses invasion and promotes cell death in human placental choriocarcinoma cells through induction of oxidative stress. Cancer Lett. 2017;399:10–19. doi: 10.1016/j.canlet.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Tian Q, Xue Y, Zheng W, Sun R, Ji W, Wang X, An R. Overexpression of hypoxia-inducible factor 1alpha induces migration and invasion through Notch signaling. Int J Oncol. 2015;47:728–738. doi: 10.3892/ijo.2015.3056. [DOI] [PubMed] [Google Scholar]
- 3.Park S, Lim W, Bazer FW, Song G. Naringenin suppresses growth of human placental choriocarcinoma via reactive oxygen species-mediated P38 and JNK MAPK pathways. Phytomedicine. 2018;50:238–246. doi: 10.1016/j.phymed.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 4.Lazovic B, Milenkovic V, Delic M, Mazic S, Jeremic K, Hrgovic Z. Hypersensitivity to Etoposide in case of metastatic gestational choriocarcinoma. Case Rep Oncol. 2013;6:490–492. doi: 10.1159/000353627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mani RS. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov Today. 2014;19:1498–1502. doi: 10.1016/j.drudis.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP, Schulman BA. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36:39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O’Malley BW. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene. 2011;30:4350–4364. doi: 10.1038/onc.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J, Foley C, Dong B, Zhang L, Kwon OJ, Shah SS, Bolaki M, Xin L, Ittmann M, O’Malley BW, Coarfa C, Mitsiades N. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017;36:4767–4777. doi: 10.1038/onc.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, Asara JM, Beck AH, Huang J, Pandolfi PP, Wei W. SPOP promotes ubiquitination and degradation of the erg oncoprotein to suppress prostate cancer progression. Mol Cell. 2015;59:917–930. doi: 10.1016/j.molcel.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu F, Dai X, Gan W, Wan L, Li M, Mitsiades N, Wei W, Ding Q, Zhang J. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Lett. 2017;385:207–214. doi: 10.1016/j.canlet.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O’Malley BW. Tumor suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic coactivator SRC-3/AIB1. Oncogene. 2011;30:4350–4364. doi: 10.1038/onc.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng C, Wang Y, Lu Q, Chen J, Zhang J, Liu T, Lv N, Luo S. SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J Exp Clin Cancer Res. 2014;33:75. doi: 10.1186/s13046-014-0075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan P, Xu Y, Du Y, Wu L, Guo B, Huang S, Zhu J, Li B, Lin F, Yao L. SPOP suppresses pancreatic cancer progression by promoting the degradation of NANOG. Cell Death Dis. 2019;10:794. doi: 10.1038/s41419-019-2017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song Y, Xu Y, Pan C, Yan L, Wang ZW, Zhu X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer. 2020;19:2. doi: 10.1186/s12943-019-1124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Ghanim M, Xue L, Brown CD, Iossifov I, Angeletti C, Hua S, Negre N, Ludwig M, Stricker T, Al-Ahmadie HA, Tretiakova M, Camp RL, Perera-Alberto M, Rimm DL, Xu T, Rzhetsky A, White KP. Analysis of Drosophila segmentation network identifies a JNK pathway factor overexpressed in kidney cancer. Science. 2009;323:1218–1222. doi: 10.1126/science.1157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Grosse F. Nuclear DNA helicase II unwinds both DNA and RNA. Biochemistry. 1994;33:3906–3912. doi: 10.1021/bi00179a016. [DOI] [PubMed] [Google Scholar]
- 17.Zhang S, Grosse F. Domain structure of human nuclear DNA helicase II (RNA helicase A) J Biol Chem. 1997;272:11487–11494. doi: 10.1074/jbc.272.17.11487. [DOI] [PubMed] [Google Scholar]
- 18.Shen B, Chen Y, Hu J, Qiao M, Ren J, Hu J, Chen J, Tang N, Huang A, Hu Y. Hepatitis B virus X protein modulates upregulation of DHX9 to promote viral DNA replication. Cell Microbiol. 2020;22:e13148. doi: 10.1111/cmi.13148. [DOI] [PubMed] [Google Scholar]
- 19.Huan W, Zhang J, Li Y, Zhi K. Involvement of DHX9/YB-1 complex induced alternative splicing of Krüppel-like factor 5 mRNA in phenotypic transformation of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2019;317:C262–C269. doi: 10.1152/ajpcell.00067.2019. [DOI] [PubMed] [Google Scholar]
- 20.Jain A, Bacolla A, Chakraborty P, Grosse F, Vasquez KM. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry. 2010;49:6992–6999. doi: 10.1021/bi100795m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain A, Bacolla A, Del Mundo IM, Zhao J, Wang G, Vasquez KM. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013;41:10345–10357. doi: 10.1093/nar/gkt804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee T, Di Paola D, Malina A, Mills JR, Kreps A, Grosse F, Tang H, Zannis-Hadjopoulos M, Larsson O, Pelletier J. Suppression of the DHX9 helicase induces premature senescence in human diploid fibroblasts in a p53-dependent manner. J Biol Chem. 2014;289:22798–22814. doi: 10.1074/jbc.M114.568535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee T, Pelletier J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget. 2016;7:42716–42739. doi: 10.18632/oncotarget.8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Z, Wang L, Eckloff BW, Deng B, Wang Y, Wampfler JA, Jang J, Wieben ED, Jen J, You M, Yang P. Conserved recurrent gene mutations correlate with pathway deregulation and clinical outcomes of lung adenocarcinoma in never-smokers. BMC Med Genomics. 2014;7:32. doi: 10.1186/1755-8794-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding X, Jia X, Wang C, Xu J, Gao SJ, Lu C. A DHX9-lncRNA-MDM2 interaction regulates cell invasion and angiogenesis of cervical cancer. Cell Death Differ. 2019;26:1750–1765. doi: 10.1038/s41418-018-0242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DD, Zhang HZ, Yuan JQ, Li SJ, Yang QC, Fan CY. Minichromosome maintenance protein 2 and 3 promote osteosarcoma progression via DHX9 and predict poor patient prognosis. Oncotarget. 2017;8:26380–26393. doi: 10.18632/oncotarget.15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee T, Paquet M, Larsson O, Pelletier J. Tumor cell survival dependence on the DHX9 DExH-box helicase. Oncogene. 2016;35:5093–5105. doi: 10.1038/onc.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L, Zhuang M, Hu S, Li X, Zhou L, Li X, Calabrese MF, Watson ER, Prasad SM, Rinker-Schaeffer C, Eggener SE, Stricker T, Tian Y, Schulman BA, Liu J, White KP. SPOP promotes tumorigenesis by acting as a key regulatory. Cancer Cell. 2014;25:455–468. doi: 10.1016/j.ccr.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng X, Zhang Y, Xu H, Zhang T, Xue Y, An R. Secreted frizzled related protein 2 modulates epithelial-mesenchymal transition and stemness via Wnt/beta-catenin signaling in choriocarcinoma. Cell Physiol Biochem. 2018;50:1815–1831. doi: 10.1159/000494862. [DOI] [PubMed] [Google Scholar]
- 30.DaSilva-Arnold S, Kuo CY, Davra V, Remache Y, Kim PCW, Fisher JP, Zamudio S, Al-Khan A, Birge RB, Illsley NP. heZEB2, a master regulator of the epithelial-mesenchymal transition, mediates trophoblast differentiation. Mol Hum Reprod. 2018;25:61–75. doi: 10.1093/molehr/gay053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 32.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. Towards a knowledge-based human protein atlas. Nat Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 33.Chen ZX, Wallis K, Fell SM, Sobrado VR, Hemmer MC, Ramskold D, Hellman U, Sandberg R, Kenchappa RS, Martinson T, Johnsen JI, Kogner P, Schlisio S. RNA helicase A is a downstream mediator of KIF1Bbeta tumor-suppressor function in neuroblastoma. Cancer Discov. 2014;4:434–451. doi: 10.1158/2159-8290.CD-13-0362. [DOI] [PubMed] [Google Scholar]
- 34.Yan X, Chang J, Sun R, Meng X, Wang W, Zeng L, Liu B, Li W, Yan X, Huang C, Zhao Y, Li Z, Yang S. DHX9 inhibits epithelial-mesenchymal transition in human lung adenocarcinoma cells by regulating STAT3. Am J Transl Res. 2019;11:4881–4894. [PMC free article] [PubMed] [Google Scholar]
- 35.Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, Rice AM, Lee SB, Uren A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Rsearch. 2006;66:5574–5581. doi: 10.1158/0008-5472.CAN-05-3293. [DOI] [PubMed] [Google Scholar]
- 36.Schlegel BP, Starita LM, Parvin JD. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene. 2003;22:983–991. doi: 10.1038/sj.onc.1206195. [DOI] [PubMed] [Google Scholar]
- 37.Giguere SS, Guise AJ, Jean Beltran PM, Joshi PM, Greco TM, Quach OL, Kong J, Cristea IM. The proteomic profile of deleted in breast cancer 1 (DBC1) interactions points to a multifaceted regulation of gene expression. Mol Cell Proteomics. 2016;15:791–809. doi: 10.1074/mcp.M115.054619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guenard F, Labrie Y, Ouellette G, Beauparlant CJ, Durocher F INHERIT BRCAs. Genetic sequence variations of BRCA1-interacting genes AURKA, BAP1, BARD1 and DHX9 in French Canadian families with high risk of breast cancer. J Hum Genet. 2009;54:152–161. doi: 10.1038/jhg.2009.6. [DOI] [PubMed] [Google Scholar]
- 39.Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc Natl Acad Sci U S A. 2000;97:1020–1025. doi: 10.1073/pnas.97.3.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zucchini C, Rocchi A, Manara MC, De Sanctis P, Capanni C, Bianchini M, Carinci P, Scotlandi K, Valvassori L. Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines. Int J Oncol. 2008;32:17–31. [PubMed] [Google Scholar]
- 41.Liu M, Roth A, Yu M, Morris R, Bersani F, Rivera MN, Lu J, Shioda T, Vasudevan S, Ramaswamy S, Maheswaran S, Diederichs S, Haber DA. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013;27:2543–2548. doi: 10.1101/gad.224170.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Argasinska J, Zhou K, Donnelly RJ, Hay RT, Lee CG. A functional interaction between RHA and Ubc9, an E2-like enzyme specific for Sumo-1. J Mol Biol. 2004;341:15–25. doi: 10.1016/j.jmb.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Cheng J, Guo J, Wang Z, North BJ, Tao K, Dai X, Wei W. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochim Biophys Acta Rev Cancer. 2018;1869:11–28. doi: 10.1016/j.bbcan.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coquenlorge S, Yin WC, Yung T, Pan J, Zhang X, Mo R, Belik J, Hui CC, Kim TH. GLI2 modulated by SUFU and SPOP induces intestinal stem cell niche signals in development and tumorigenesis. Cell Rep. 2019;27:3006–3018. e3004. doi: 10.1016/j.celrep.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 45.Cai H, Liu A. Spop regulates Gli3 activity and Shh signaling in dorsoventral patterning of the mouse spinal cord. Dev Biol. 2017;432:72–85. doi: 10.1016/j.ydbio.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai H, Liu A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc Natl Acad Sci U S A. 2016;113:14751–14756. doi: 10.1073/pnas.1612520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagai Y, Kojima T, Muro Y, Hachiya T, Nishizawa Y, Wakabayashi T, Hagiwara M. Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett. 1997;418:23–26. doi: 10.1016/s0014-5793(97)01340-9. [DOI] [PubMed] [Google Scholar]
- 48.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 50.Zhao W, Zhou J, Deng Z, Gao Y, Cheng Y. SPOP promotes tumor progression via activation of beta-catenin/TCF4 complex in clear cell renal cell carcinoma. Int J Oncol. 2016;49:1001–1008. doi: 10.3892/ijo.2016.3609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.