

Abstract
Age-related macular degeneration (AMD) is a chronic, multifactorial disorder and a leading cause of blindness in the elderly. Characterized by progressive photoreceptor degeneration in the central retina, disease progression involves epigenetic changes in chromatin accessibility resulting from environmental exposures and chronic stress. Here, we report that a photosensitive mouse model of acute stress-induced photoreceptor degeneration recapitulates the epigenetic hallmarks of human AMD. Global epigenomic profiling was accomplished by employing an Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-Seq), which revealed an association between decreased chromatin accessibility and stress-induced photoreceptor cell death in our mouse model. The epigenomic changes induced by light damage include reduced euchromatin and increased heterochromatin abundance, resulting in transcriptional and translational dysregulation that ultimately drives photoreceptor apoptosis and an inflammatory reactive gliosis in the retina. Of particular interest, pharmacological inhibition of histone deacetylase 11 (HDAC11) and suppressor of variegation 3–9 homolog 2 (SUV39H2), key histone-modifying enzymes involved in promoting reduced chromatin accessibility, ameliorated light damage in our mouse model, supporting a causal link between decreased chromatin accessibility and photoreceptor degeneration, thereby elucidating a potential new therapeutic strategy to combat AMD.
Introduction
In human aging and disease, a variety of genomic and epigenomic changes accumulate over a lifetime, resulting in deleterious effects to overall health (1). Aging is associated with increased transcriptional noise, in which key components of vital cellular signaling pathways are dysregulated as a result of the aberrant production and processing of gene transcripts (2). Unlike DNA mutations, epigenetic alterations are reversible, thereby providing avenues for the development of novel therapies to combat both transcriptional and translational dysregulations in age-associated diseases (3). These epigenetic changes include DNA methylation, histone modifications and chromatin remodeling—all of which could ultimately be targeted therapeutically in efforts to normalize the transcriptome on a global level and improve organismal health.
To date, epigenetic modifications have been implicated in a variety of age-associated disorders, including cancer (4), cardiovascular disease (5) and neurodegeneration (6). In the present study, we focus on epigenetic changes associated with visual dysfunction. Specifically, in age-related macular degeneration (AMD), it has been demonstrated that decreased chromatin accessibility is a hallmark that correlates with disease progression in humans (7). Clinically, AMD is a leading cause of blindness in the elderly, characterized by progressive photoreceptor degeneration in the central retina (8). Up to 200 million individuals worldwide are affected by this disorder (9), yet no effective therapies currently exist for the most common (non-exudative or ‘dry’) form of AMD.
Employing mouse models to study disease offers numerous advantages for accelerating drug development. In particular, for simple monogenic disorders, knock-in/knockout mouse models can quite powerfully recapitulate disease pathogenesis. In the case of complex polygenic disorders such as AMD, mice can be employed as surrogate models that recapitulate key pathological features of disease (8). Since AMD is characterized by photoreceptor degeneration, we reasoned that a light-sensitive mouse strain, in which photoreceptor degeneration is inducible by bright light exposure, could potentially serve as a suitable surrogate model. Enzymatic deficiencies in the visual cycle of Abca4−/−Rdh8−/− double knockout (dKO) mice predispose to accelerated accumulation of cytotoxic by-products (10), a phenomenon similarly observed to occur at a slower rate over a longer time course in normal aging. Therefore, it is conceivable that certain molecular mechanisms of cytotoxicity could be conserved. Indeed, the retinal phenotype of dKO mice exposed to bright light resembles certain clinical features of AMD, including lipofuscin accumulation, drusen deposition and late-onset choroidal neovascularization (11). Here, we utilized an Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-Seq) to investigate whether the chromatin accessibility changes implicated in human AMD are recapitulated in our mouse model of light-induced injury. Through a comprehensive, genome-wide chromatin accessibility analysis, we report that our mouse model exhibits a global decrease in accessibility of open chromatin regions upon induction of photoreceptor degeneration by bright light stress, which closely resembles the epigenetic phenotype characterized in clinical cases of AMD.
Results
Global chromatin accessibility changes in phototoxicity
In this study, we characterized the epigenetic phenotype of our photosensitive dKO mouse model using ATAC-Seq, in order to globally profile chromatin accessibility in the retina and RPE/choroid. Samples were processed in triplicate or as specified for each experimental condition, and in total 63 018 high-confidence peaks representing open chromatin regions were identified in the retina, and 19 950 high-confidence peaks were identified in the RPE/choroid. In dKO mice subjected to bright light exposure, we observed on average a broad decrease in peak intensity with diffusion into intergenic regions of the genome in both the retina and RPE/choroid 1 day after photobleaching (Fig. 1A), resembling the global reduction in accessibility of open chromatin regions previously characterized in clinical cases of AMD (7) (Fig. 1B). Notably, the key photoreceptor genes Rho, Gnat1, Nrl and Nr2e3 exhibited a mixed initial response at 6 h after light exposure, which stabilized into an overall decrease in peak intensity by 1 day after photobleaching (Fig. 1C). In contrast, key inflammatory response genes Ccl4 and Socs3 actually exhibited an increase in peak intensity following photobleaching, consistent with the upregulation of these genes seen in the inflammatory reactive gliosis that follows light-induced photoreceptor degeneration (12).
Figure 1.
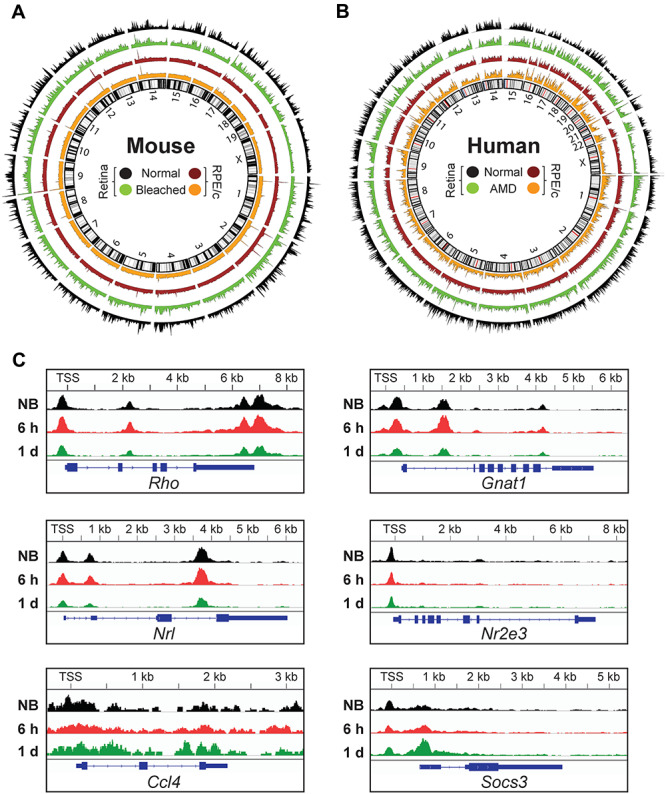
Epigenetic landscape of chromatin accessibility in the retina and RPE/choroid. Circos plots showing genome-wide chromatin accessibility as ATAC-Seq peaks in the retina and RPE/choroid (RPE/c) of (A) photosensitive Abca4−/−Rdh8−/− (dKO) mice reared under normal lighting conditions and 1 day after exposure to bright light stress (bleached), and (B) humans with and without AMD (generated from raw human sequencing data deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE99287). (C) Chromatin accessibility changes induced by photobleaching. Average ATAC-Seq signal in key genes for non-bleached (NB, n = 5) and bleached dKO retina 6 h (n = 3) and 1 day (n = 6) after bright light exposure. TSS, transcriptional start site; kb, kilobases.
In order to visualize the progression of photoreceptor degeneration in mice, we utilized scanning laser ophthalmoscopy (SLO) and optical coherence tomography (OCT) to image the fundus and cross sections of the retina, respectively (Fig. 2A). In photobleached dKO mice, SLO imaging revealed characteristic autofluorescent spots associated with phototoxicity and reactive inflammation, which became increasingly apparent in the days following light damage (12). OCT imaging revealed a concomitant degeneration of the photoreceptor-containing outer nuclear layer (ONL) in bleached dKO mice relative to both wild-type (WT) and non-bleached (NB) controls, with full degeneration of the ONL evident at 7 days after photobleaching (Fig. 2B). At 1 day after light exposure, the majority of ATAC-Seq peaks, 93.7% in the retina and 55.6% in the RPE/choroid of dKO mice, were reduced in signal intensity (Fig. 2C), representative of a global decrease in accessibility of open chromatin regions. This global decrease occurred gradually, with total ATAC signal intensity decreasing in a time-dependent manner following light damage (Fig. 2D). As expected, this analysis showed that the chromatin accessibility changes induced by bright light stress result in distinct and reproducible epigenomic profiles (Fig. 2E).
Figure 2.
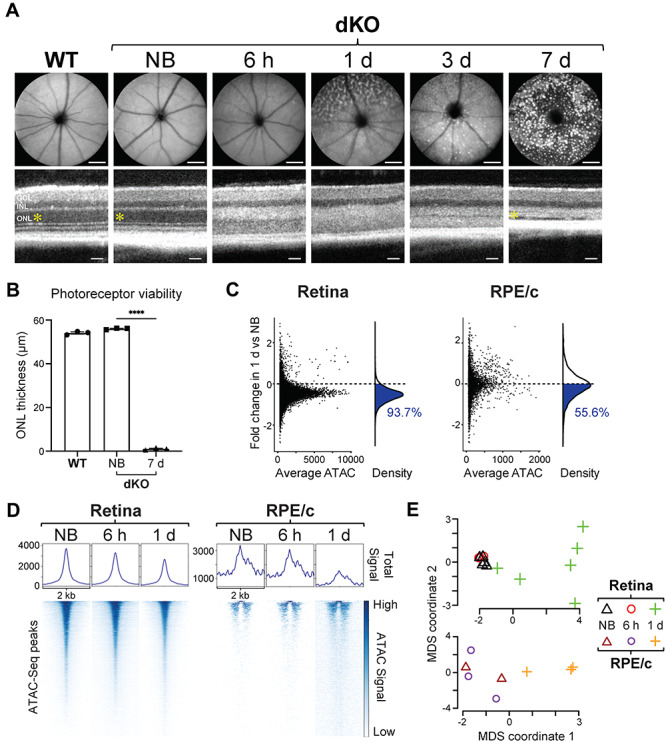
Chromatin accessibility changes in a light-damaged retina. (A) SLO images (top) demonstrate the time course of induction of retinal pathology (autofluorescent spots) in the fundus of photosensitive Abca4−/−Rdh8−/− (dKO) mice 6 h to 1/3/7 days after exposure to bright light stress, as compared to non-bleached (NB) dKO and wild-type (WT) mice. SLO scale bars, 1 mm. OCT images (bottom) were also obtained from these mice, and the thickness of the photoreceptor-containing outer nuclear layer (ONL; yellow asterisk) is quantified in (B), exhibiting complete degeneration in the dKO mice by 7 days post-bleach. OCT scale bars, 50 μm. INL, inner nuclear layer; GCL, ganglion cell layer. n = 3 per group. (C) Global decrease of chromatin accessibility in the retina and RPE/choroid (RPE/c) 1 day after photobleaching relative to non-bleached dKO mice. Each data point (left panel) represents one ATAC-Seq peak, and the population of reduced peaks is highlighted in blue in the density curve (right panel) and quantified as a percentage of all ATAC-Seq peaks. (D) Global heat map of open chromatin regions in non-bleached dKO mice, compared to 6 h and 1 day after bright light exposure. Each row (bottom panel) represents one ATAC-Seq peak, and the degree of chromatin accessibility is represented by color. Peaks are aligned at the center of regions spanning 2 kb. The total ATAC-Seq signal (normalized counts) of all peaks combined is shown in the top panel. (E) Multidimensional scaling (MDS) of all retina and RPE/choroid samples. Non-bleached (n = 5 for retina, n = 2 for RPE/c) and bleached dKO mice 6 h (n = 3 for retina, n = 3 for RPE/c) and 1 day (n = 6 for retina, n = 3 for RPE/c) after light exposure cluster into groups with distinct ATAC-Seq profiles.
Epigenomic changes manifest in the transcriptome
In order to assess how changes at the epigenomic level are reflected in the transcriptome, we utilized next-generation RNA sequencing (RNA-Seq) and performed a correlation analysis. In non-bleached dKO mice, the retina exhibited a higher degree of correlation between ATAC-Seq and RNA-Seq data than the RPE/choroid, with Pearson’s correlation coefficients (R) of 0.33 and 0.21, respectively (Fig. 3A). Taken together, this positive correlation supports a direct relationship between chromatin accessibility and gene transcription. Indeed, MDS analysis of our RNA-Seq data suggests a programmed transcriptional response to bright light stress that results in distinct and reproducible transcriptomic profiles (Fig. 3B). Using differential gene expression analysis, we identified statistically significant differentially expressed (DE) genes in the retina and RPE/choroid of dKO mice at 6 h, 1 day and 3 days after light damage and found that the majority of transcriptomic changes occur in the retina, with the total number of DE genes approximately 10-fold higher than in RPE/choroid (Fig. 3C and D). Moreover, in both the retina and RPE/choroid, the majority of transcriptomic changes occurred 1 day after photobleaching.
Figure 3.
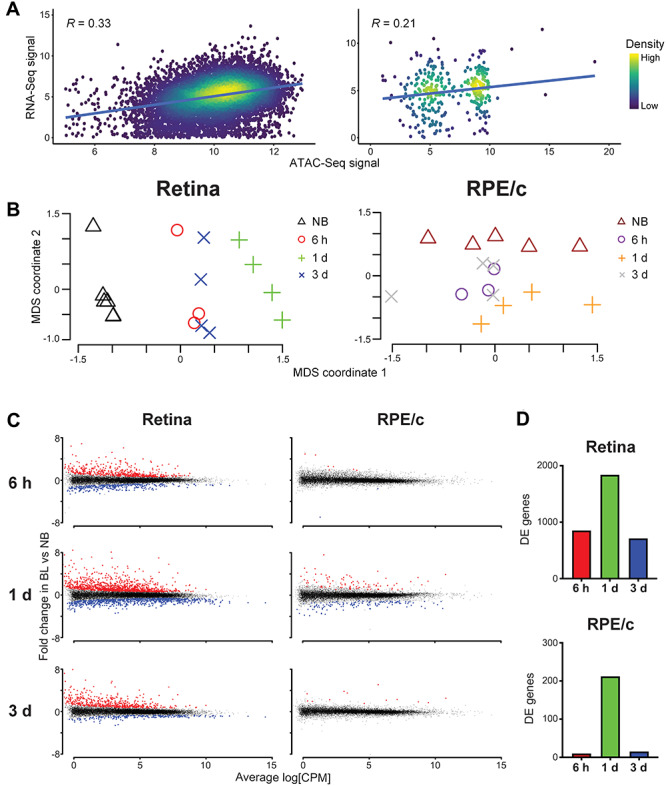
Transcriptome reflects epigenetic changes associated with photoreceptor degeneration. (A) Relationship between chromatin accessibility (ATAC-Seq signal intensity) and gene expression (RNA-Seq signal intensity) in the retina and RPE/choroid (RPE/c) of unbleached dKO mice. Pearson’s product-moment correlation constant (R) is displayed on the graph (P-value < 0.0001). (B) Multidimensional scaling (MDS) of all retina and RPE/choroid samples. Non-bleached (NB) and bleached dKO mice cluster into groups with distinct RNA-Seq profiles. n = 4 per group for all NB, 1-day and 3-day samples. n = 3 per group for all 6-h samples. (C) Differentially expressed (DE) genes in the retina and RPE/choroid 6 h, 1 day and 3 days after photobleaching (BL). Each data point represents one RNA-Seq peak, and the total sum of significantly upregulated (red) and downregulated (blue) DE genes is quantified in (D) at 6 h (red), 1 day (green) and 3 days (blue) post-bleach, respectively; DE genes total 852, 1839 and 712 in the retina and 9, 216 and 13 in RPE/choroid.
To elucidate the pertinent biological pathways involved in the progression of photoreceptor degeneration, we first generated a Venn diagram to quantify the total number of DE genes unique to each post-bleach time point. At 6 h, 1 day and 3 days post-bleach, 317, 980 and 141 unique DE genes were identified in the retina of dKO mice, respectively (Fig. 4A). We then performed gene set enrichment analysis on these unique genes, which revealed early enrichment in transcriptional activation, cell signaling, apoptosis and metabolism pathways, followed by delayed enrichment in phagocytosis and inflammatory response pathways (Fig. 4B). Upon closer inspection of specific DE genes (Supplementary Material, Data 1), we observed early downregulation of key photoreceptor genes Nrl, Nr2e3, Rho and Gnat1 (Supplementary Fig. 1), followed by upregulation of key immune response genes Ccl4, Socs3, Ifi204, Ddx58, Cfi, C3, Nfkb2, Gfap and Cd68 (Supplementary Material, Fig. 2a). Moreover, upregulation of gliosis markers GFAP and CD68 at the protein level was visually confirmed by immunohistochemistry analysis of retinal cross sections (Supplementary Material, Fig. 2b). Taken together, these results suggest an initial transcriptional response that predominates in light-damaged photoreceptors, followed by a delayed inflammatory response mediated by retinal glia. To investigate this possibility, Seurat was used on a generic wild-type murine retina single-cell RNA-Seq (scRNA-Seq) dataset to determine the top 50 marker genes unique to each retinal cell type (Supplementary Material, ata 2), and the DE genes identified from each post-bleach time point were matched to the marker genes and quantified as a percentage of the total number of cell type-specific genes for each time point. Indeed, this analysis revealed the majority of stress-induced transcriptomic changes shift from rod and cone photoreceptors (early) to Müller glia and microglia (late), with a minority of transcriptomic changes occurring in amacrine, horizontal and bipolar cells of the inner retina (Fig. 4C). By integrating Uniform Manifold Approximation and Projection (UMAP) non-linear dimensionality reduction analysis of the scRNA-Seq data with our dKO mouse RNA-Seq data, we employed a novel methodology henceforth known as ‘pseudo-scRNA-Seq’ to map DE genes to individual cells on a UMAP plot for each post-bleach time point (Fig. 4D). As expected from our epigenomic data, this analysis revealed mixed up- and downregulation of transcription at 6 h, followed by a more uniform downregulation in photoreceptors at 1 day post-bleach, and delayed upregulation occurring in activated microglia 3 days after light damage.
Figure 4.
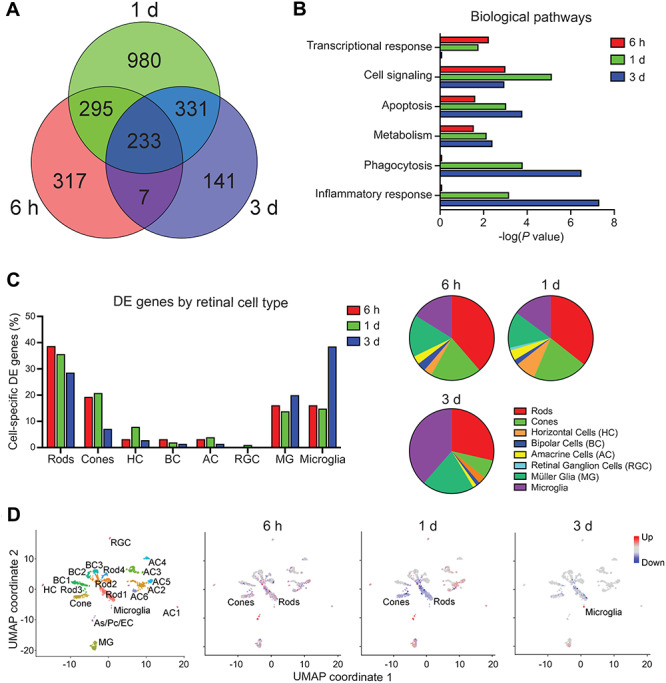
Transcriptomic analysis reveals biological pathways underlying photoreceptor degeneration. (A) Venn diagram of the total numbers of differentially expressed (DE) genes in the retina 6 h, 1 day and 3 days after photobleaching, relative to non-bleached dKO mice. (B) Global transcriptome gene set enrichment analysis identifies top biological pathways associated with bright light-induced damage in the retina of dKO mice at indicated time points after photobleaching. (C) Proportion of cell type-specific DE genes at corresponding time points after photobleaching. Majority of stress-induced transcriptomic changes shift from photoreceptors (early) to glia (late), suggesting a late-onset inflammatory reactive gliosis. DE genes were cross-referenced against a single-cell RNA-Seq (scRNA-Seq) database of the top 50 genes unique to each retinal cell type and are represented as a percentage of the total number of DE genes that are cell type specific at each time point. (D) Pseudo-scRNA-Seq analysis maps of upregulated (red) and downregulated (blue) DE genes in distinct retinal cell types. Uniform Manifold Approximation and Projection (UMAP) non-linear dimensionality reduction was used to cluster individual cells with similar transcriptomic profiles and assign cell types based on the expression of unique marker genes. Cell types exhibiting highest degree of differential gene expression at each time point are labeled. As, astrocyte; Pc, pericyte; EC, endothelial cell.
Chromatin remodeling drives phototoxicity
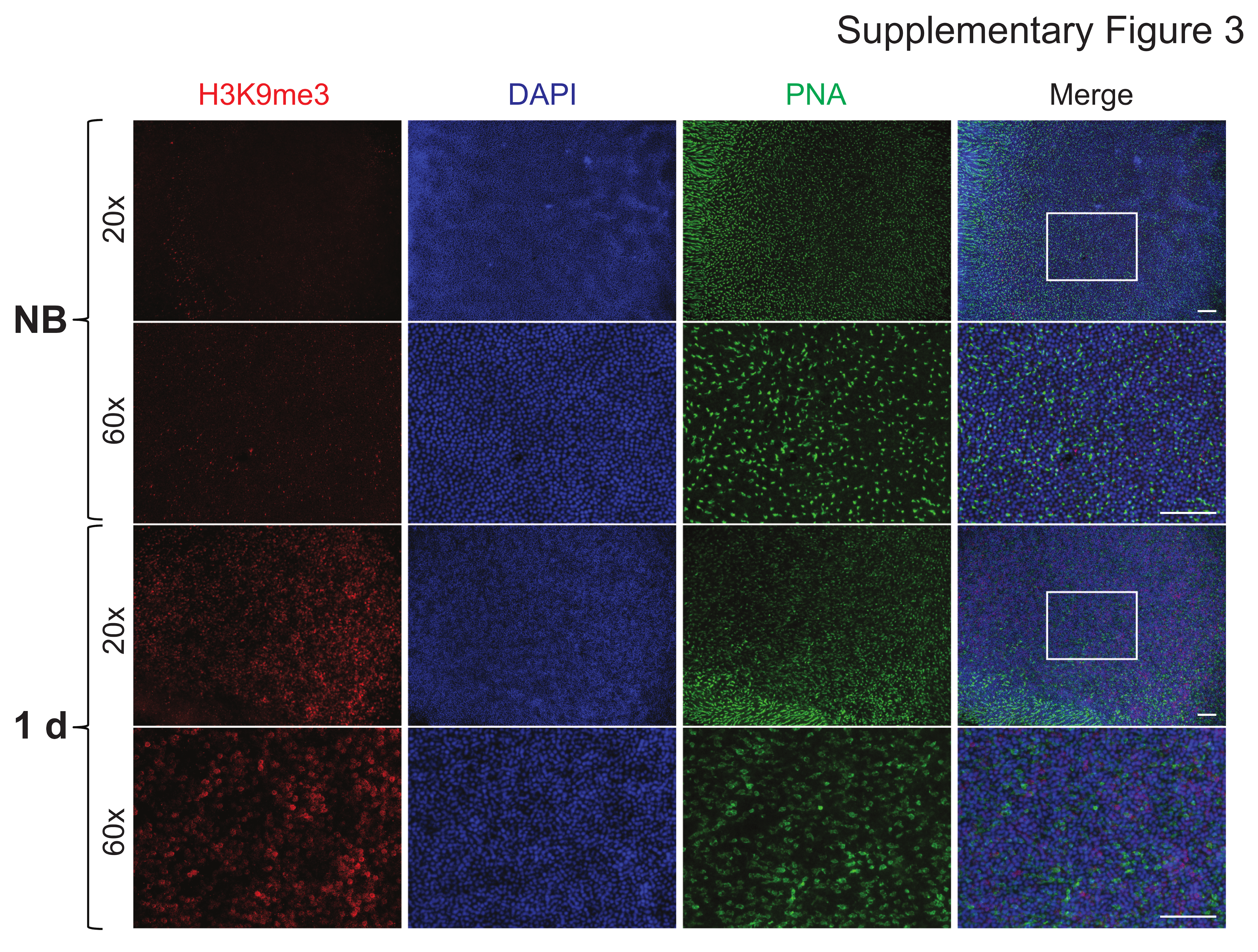
Among the many DE genes identified in our study, we first narrowed our focus on DE genes encoding histone-modifying enzymes that could drive the observed changes in chromatin accessibility (Supplementary Material, Data 3). From this list, we identified HDAC11, which encodes an enzyme that functions to deacetylate histones, thereby promoting reduced chromatin accessibility (13). Notably, upregulation of this gene, along with decreased histone acetylation, has also been observed in clinical cases of dry AMD (7). In bleached dKO mice, we observed statistically significant upregulation of HDAC11 both 1 day and 3 days after light damage in the retina (Fig. 5A), which corresponded with decreased protein abundance of H3K27ac, an acetylated histone marker for open chromatin regions (euchromatin). Likewise, in the RPE/choroid at 1 day post-bleach, we observed statistically significant upregulation of SUV39H2, which encodes a methyltransferase that trimethylates the K9 residue of histone 3, thereby promoting the formation of highly condensed, inaccessible regions of chromatin (heterochromatin) (14). Indeed, by quantitative western blot analysis, we observed a concomitant increase in protein abundance of H3K9me3, a marker for heterochromatin, in the RPE/choroid of bleached dKO mice compared to non-bleached controls (Fig. 5B). In order to visualize the heterochromatin formation contributing to reduced chromatin accessibility, we prepared retina and RPE flat mounts for immunofluorescence microscopy and observed increased H3K9me3 signal in both retina (Supplementary Material, Fig. 3) and RPE (Fig. 5C) of dKO mice at 1 day after photobleaching. Taken together, these results demonstrate that the global reduction of chromatin accessibility induced by bright light stress involves effectors that inhibit euchromatin while promoting heterochromatin formation.
Figure 5.
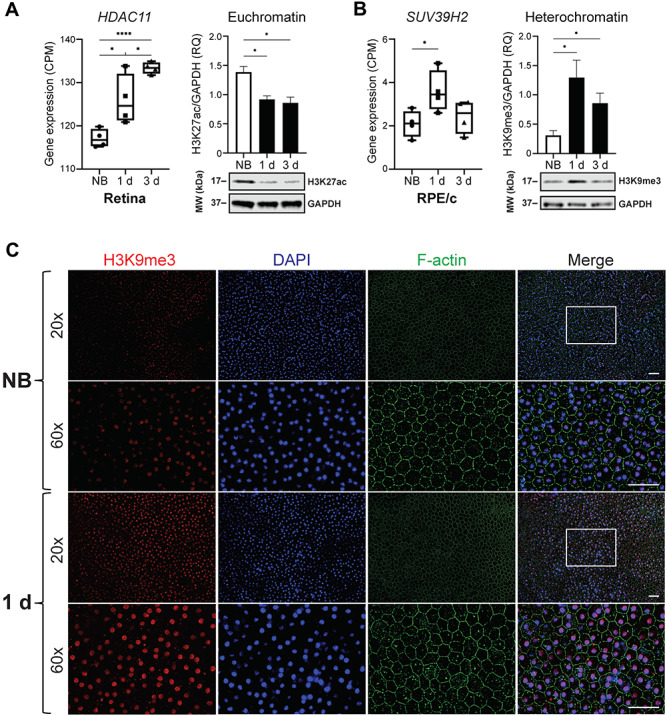
Histone modifications associated with decreased chromatin accessibility. (A) Expression of HDAC11 in the retina (left panel), measured in counts per million (CPM). Increased expression corresponds to histone modifications that decrease chromatin accessibility (n = 4 per group, *P < 0.05, ****P < 0.0001). Euchromatin marker H3K27ac is decreased in dKO mice 1 day and 3 days after photobleaching (right panel). Representative western blot (WB) analysis and quantification of H3K27ac levels, expressed as relative quantity (RQ), in the retina of non-bleached (NB) and bleached dKO mice (n = 3 per group, *P < 0.05). (B) Expression of SUV39H2 in RPE/choroid (RPE/c) (left panel). Heterochromatin marker H3K9me3 is increased in dKO mice 1 day and 3 days after photobleaching (right panel). Representative WB analysis and quantification of H3K9me3 levels in RPE/c of non-bleached and bleached dKO mice. All P-values were calculated by the unpaired t-test. (C) RPE flat mount immunofluorescence microscopy. F-actin labels RPE cell borders, nuclei are labeled by DAPI, and H3K9me3 is a marker for heterochromatin foci. 60× magnification is of boxed region shown in 20× merged window. Increased heterochromatin staining is observed in dKO mice 1 day after photobleaching. Scale bars, 50 μm.
Having identified the correlation between reduced chromatin accessibility and photoreceptor degeneration in our mouse model, we next sought to investigate whether this chromatin remodeling could play a causative role in driving phototoxicity. Knowing that HDAC11 and SUV39H2 are upregulated in photobleached dKO mice, we reasoned that pharmacological inhibition of these enzymes would provide protection from light damage if reduced chromatin accessibility were indeed a driver of phototoxicity. By quantitative western blot analysis, we first confirmed that intraperitoneal administration (60 mg/kg bw) of Mocetinostat (MCT), a pharmacological inhibitor of HDAC11 (15), rescues the stress-induced reduction in euchromatin abundance observed in the retina of photobleached dKO mice (Fig. 6A). Likewise, we confirmed that intraperitoneal administration (60 mg/kg bw) of OTS186935 (OTS), a selective inhibitor of SUV39H2 (16), attenuates the stress-induced increase in heterochromatin abundance observed in the RPE/choroid of bleached dKO mice. In order to visualize the effect of SUV39H2 inhibition on heterochromatin formation throughout the retina of dKO mice, we prepared retinal cross sections for immunohistochemistry analysis and found that the widespread increase in H3K9me3 signal induced by photobleaching was attenuated by OTS therapy, particularly in the photoreceptor-containing outer nuclear layer (Fig. 6B). With confirmation that these pharmacological interventions indeed mitigate the global reduction of chromatin accessibility induced by bright light stress, we performed SLO and OCT imaging and found that either MCT or OTS therapy alone was sufficient to ameliorate stress-induced retinal pathology in photobleached dKO mice (Fig. 6C and D). Altogether, these findings demonstrate proof of concept in support of a causal relationship between decreased chromatin accessibility and photoreceptor degeneration.
Figure 6.
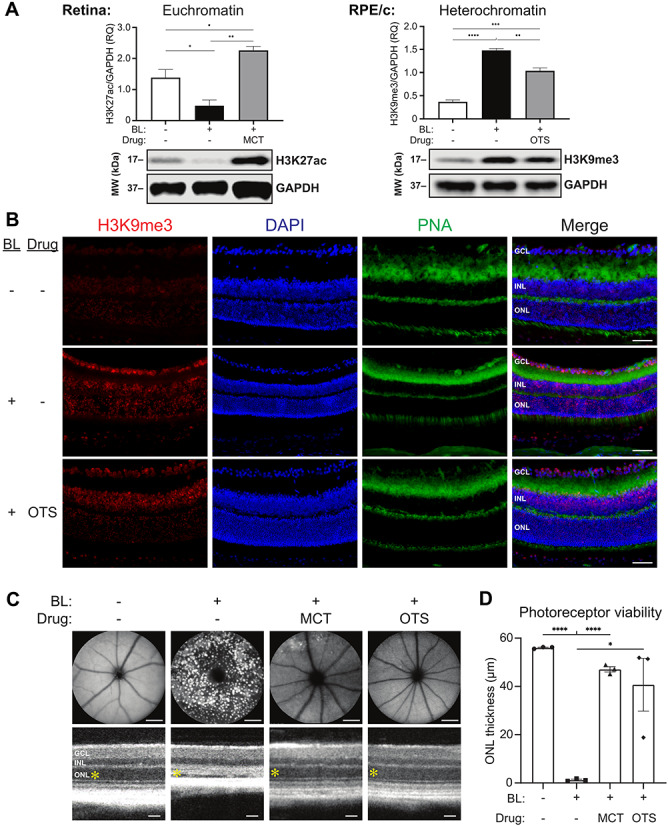
Pharmacological interventions that attenuate stress-induced chromatin remodeling ameliorate photoreceptor degeneration. (A) Representative western blot (WB) analysis and quantification of H3K27ac and H3K9me3 levels, expressed as relative quantity (RQ), in the retina and RPE/choroid (RPE/c) of non-bleached (BL-) and 1 day post-bleach (BL+) dKO mice treated with intraperitoneal injection of DMSO vehicle (drug-), Mocetinostat (MCT) at a dose of 60 mg/kg bw or OTS186935 (OTS) at a dose of 60 mg/kg bw. (B) Immunohistochemistry analysis of retinal cross sections reveals broadly increased H3K9me3 staining throughout the retina of dKO mice 1 day after photobleaching relative to non-bleached controls, which is attenuated in bleached OTS-treated mice relative to DMSO vehicle-treated mice. H3K9me3 is a marker for heterochromatin foci, nuclei are labeled by DAPI, and peanut agglutinin (PNA) labels cone photoreceptors. Scale bars, 50 μm. (C) SLO (top) and OCT (bottom) imaging reveals light-induced retinal pathology in dKO mice is ameliorated by MCT or OTS treatment, and the thickness of the photoreceptor-containing outer nuclear layer (ONL; yellow asterisk) is quantified in (D). SLO and OCT images were acquired from live dKO mice 7 days after photobleaching. SLO scale bars, 1 mm. OCT scale bars, 50 μm. INL, inner nuclear layer; GCL, ganglion cell layer. n = 3 per group, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
The complex pathogenesis of photoreceptor degeneration in AMD has puzzled physicians and biomedical investigators for decades. As a chronic, multifactorial disease, AMD susceptibility, progression and severity are determined by a combination of multiple genetic variants as well as environmental and lifestyle factors (8,17). In this study, we focused on the epigenetic modifications associated with AMD, which accumulate under various environmental conditions over a lifetime. The retinal pathology in AMD involves chronic photoreceptor degeneration, although the underlying mechanisms have remained unclear. To delineate the pathways driving photoreceptor cytotoxicity in a controlled setting, we employed a photosensitive mouse model exhibiting synchronized photoreceptor degeneration following exposure to bright light stress. Using mice as the animal model to study AMD comes with inherent constraints. Given the differing topography between the mouse and human retina, such as the anatomical absence of a macula in mice, the dKO mouse model serves only to approximate the accumulation of stressors that trigger photoreceptor/RPE dysfunction. Retinal photoreceptors and RPE function together as a system, and because of their interdependence, perturbations to this functionally integrated network will lead to cell death and dysfunction, regardless of whether the susceptibility to stress is initially localized in photoreceptors or RPE (18). Whereas human AMD may involve RPE dysfunction preceding photoreceptor degeneration, our dKO mouse model exhibits susceptibility to stress primarily in photoreceptors, with the RPE initially spared. Despite these differences, synchronously induced photoreceptor degeneration in a mouse model confers many advantages over correlative clinical studies on AMD disease progression, namely, with respect to tractability, reproducibility and turnover. Furthermore, as both human and murine cells are derived from a common eukaryotic ancestor, it is conceivable that certain cellular processes governing cytotoxicity could be evolutionarily conserved, and potentially active, in a range of neurodegenerative diseases.
In the current study, we investigated stress-induced retinal pathology in photosensitive mice and ascertained three insights on the pathogenesis of photoreceptor degeneration. The first pertains to how chromatin accessibility changes on a global level in the context of light damage. We found that upon bright light exposure, both the retina and RPE/choroid exhibit a global decrease in accessibility of open chromatin regions, as evidenced by a gradual, time-dependent reduction in total ATAC signal. While our analysis of bulk retina and RPE/choroid samples by ATAC-Seq and RNA-Seq revealed global changes in chromatin accessibility, further studies will be required to determine the nature of chromatin rearrangement occurring at the individual cell level. For instance, single-cell ATAC-Seq (scATAC-Seq) and scRNA-Seq could be employed to investigate the possibility that certain retinal cell types, such as Müller glia and microglia, actually undergo an increase in chromatin accessibility, contrary to the global trend observed in the majority of retinal cells, which might account for the upregulation of glia-specific genes observed in our pseudo-scRNA-Seq analysis. Secondly, we demonstrated the relationship between changes at the epigenomic level and how they manifest in the transcriptome, observing a programmed transcriptional response to bright light stress involving cross-talk between different retinal cell types and a transition from an initial response in photoreceptors to a delayed inflammatory reactive gliosis. Lastly, we identified key histone modifications induced by bright light stress that reduce euchromatin while promoting heterochromatin formation, thereby contributing to the changes in global chromatin accessibility observed in phototoxicity.
In early stages of embryonic development, cellular differentiation results from the interplay between heritable epigenetic modifications and spatiotemporally regulated production of cell type-specific transcription factors (TFs) (19). Cells are able to distinguish their identity through what is known as epigenetic memory (20,21) while maintaining a level of plasticity that enables restoration of tissue homeostasis after injury and exposure to other environmental stimuli over time (22). Rod and cone photoreceptors, which are terminally differentiated cells in the adult murine retina, develop and maintain their cellular identity through a combination of TFs, namely, Nrl (23) and Nr2e3 (24). Upon stress-induced photoreceptor toxicity, we observed decreased ATAC signal for not only Nrl and Nr2e3 but for their respective target genes as well, including Rho and Gnat1. This corresponded to an overall decrease in downstream gene expression, as demonstrated by pseudo-scRNA-Seq, in photoreceptors 1 day after light damage. Taken together, our data suggest that the initial stages of stress-induced photoreceptor degeneration involve globally reduced chromatin accessibility, resulting in decreased TF binding and an altered transcriptome that drives apoptosis (Fig. 7).
Figure 7.
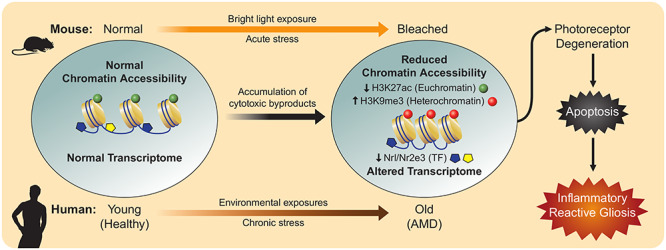
Pathogenesis of photoreceptor degeneration associated with decreased chromatin accessibility. In photosensitive dKO mice and humans that develop AMD, stress-induced cytotoxic by-product accumulation drives global reduction in chromatin accessibility and downregulation of transcription factors (TFs), resulting in an altered transcriptome that culminates in photoreceptor cell death and reactive inflammation.
Following the initial insult to photoreceptors, a delayed immune response emerges, as evidenced by increased ATAC signal in key inflammatory response genes, such as Ccl4 and Socs3. This is accompanied by widespread upregulation of gene expression, as demonstrated by pseudo-scRNA-Seq, in activated microglia 3 days after light damage. Specifically, our analysis revealed significantly increased expression of various pro-inflammatory genes including Ccl4, Ifi204, Ddx58, Cfi, C3, Nfkb2, Gfap and Cd68 following light damage, consistent with inflammasome activation and a reactive gliosis that functions primarily to phagocytose apoptotic photoreceptor debris (12,25–27). Of particular interest, inflammasome activation involving chemokines such as Ccl4 (28), as well as interferon signaling which may involve Ifi204 and Ddx58, has been described in the clinical context of AMD (29,30). Likewise, complement pathway genes Cfi and C3 have also been implicated in genome-wide association studies (GWAS) of AMD (17). Taken together, this evidence supports potentially conserved immune response pathways underlying photoreceptor degeneration in our mouse model and in clinical cases of AMD.
The data presented in this publication demonstrate that our light-sensitive mouse model of stress-induced photoreceptor degeneration recapitulates the epigenetic hallmarks of human AMD. Moreover, the potentially conserved pathways between the two species underlying the pathogenesis of photoreceptor degeneration support the use of our mouse model in subsequent studies to further address causality, whereas human studies have largely been limited to correlative analyses. For instance, in humans, it would be prohibitively difficult to determine whether decreased chromatin accessibility represents a harmful change that compromises photoreceptor viability or an adaptive response that enhances cell survival in disease. In contrast, we were able to tractably approach this question in our mouse model by demonstrating the therapeutic efficacy of pharmacological inhibitors that attenuate chromatin remodeling, thereby identifying HDAC11 and SUV39H2 as potential therapeutic targets in the context of photoreceptor degeneration and AMD. However, for robust target validation, further studies employing conditional Hdac11- and/or Suv39h2-knockout mice crossed onto our light-sensitive dKO genetic background will be required. Nevertheless, our findings from the current study provide the rationale to support such further investigation. Additionally, use of the novel methodology described herein as pseudo-scRNA-Seq, to ascertain cell type-specific insights from bulk RNA-Seq data cross-referenced against a generic scRNA-Seq dataset, establishes a cost-effective framework for numerous potential applications in a variety of experimental contexts. Indeed, this framework may ultimately be leveraged to extend far beyond the context of retinal degenerative disease to provide unique mechanistic insights on disease progression in any organ system of interest.
Materials and Methods
Animals
Male and female Abca4−/−Rdh8−/− mice at 6–8 weeks of age were used for the current study. These mice were maintained on a pigmented C57BL/6 background, and age-matched C57BL/6 mice from the Jackson Laboratory were used as wild-type controls. All mice were housed and maintained in a 12-h light (≤150 lux)/12-h dark cyclic environment in the University Laboratory Animal Resources Center at the University of California, Irvine (UCI) School of Medicine. Bright light-induced retinal damage was generated by exposing photosensitive Abca4−/−Rdh8−/− mice to white light delivered at 10 000 lux (150-W spiral lamp, Commercial Electric, Cleveland, OH) for 30 min. Mice were dark-adapted 24 h prior to photobleaching, and pupils were dilated with 1% ophthalmic tropicamide 30 min prior to light exposure. Mocetinostat (MedChemExpress #HY-12164, 60 mg/kg bw) and OTS186935 (AdooQ BioScience #A18632, 60 mg/kg bw) were dissolved in DMSO and administered in a total volume of 50 μL by intraperitoneal injection 30 min prior to light exposure. All animal handling procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee at the UCI and conformed to recommendations of both the American Veterinary Medical Association (AVMA) Panel on Euthanasia and the Association for Research in Vision and Ophthalmology.
Live in vivo retinal imaging
Mice were anesthetized by intraperitoneal injection of ketamine (20 mg/mL) with xylazine (1.75 mg/mL) at a dose of 5 μL/g bw, and pupils were dilated with 1% tropicamide prior to imaging. Ultrahigh-resolution spectral domain OCT (Bioptigen, Research Triangle Park, NC) was performed for cross-sectional imaging of mouse retinas, as described previously (31). Briefly, five frames of OCT images were acquired in the B-mode and then averaged. For quantitative measurements of photoreceptor viability, ONL thickness was measured in the InVivoVue software at a distance of 0.45 mm from the optic nerve head in the temporal retina, where the most severe damage is found in bright light-exposed Abca4−/−Rdh8−/− mice. SLO (Heidelberg Engineering, Heidelberg, Germany) was also performed for whole fundus imaging of mouse retinas, and images were acquired in the autofluorescence mode, as previously described (12).
ATAC sequencing
Fresh retina and RPE/choroid tissues were harvested from photosensitive Abca4−/−Rdh8−/− mice and dissociated into single cells using the Worthington Papain Dissociation System (Lakewood, NJ). For nuclear extraction, cells (50–75 k) were lysed by adding 50 μL of ice-cold cell lysis buffer (10 mM Tris-Cl pH 7.4, 10 mM NaCl, 3 mM MgCl2) containing 0.03% IGEPAL and protease inhibitors (one tablet per 7 mL of lysis buffer) and mixing three times by pipetting. Cells were then immediately spun down at 500 g for 10 min and washed with 150 μL of ice-cold lysis buffer without IGEPAL and protease inhibitors. For tagmentation, cell nuclei were incubated with 2.5 μL enzyme in 50 μL total volume at 37°C in a thermocycler (Illumina Nextera DNA library prep kit, #FC1211030). DNA was cleaned up using the MinElute PCR purification kit (#28006, Qiagen) and eluted in 10 μL of EB buffer. Tagmented DNA was amplified, and the number of PCR cycles was calculated by following a previously described protocol (32). PCR products (10 μL) were run on a 1.5% agarose gel to confirm expected DNA band pattern. PCR products were then cleaned by double-sized selection using Ampure beads (Agencout AMPure XP, Beckman Coulter, #A63880) to remove large and small DNA fragments. This was performed by using 1:0.5 and 1:1.6 ratios of sample to Ampure beads (v/v). Completed ATAC-Seq libraries were then analyzed by fragment bioanalyzer and sequenced for paired-end 75 cycles using the NextSeq 500 system with ~ 400–500 million reads per run, yielding ~45–50 million reads per sample.
ATAC-Seq differential chromatin accessibility analysis
Adaptors were removed using Trimmomatic (33). ATAC-Seq reads were aligned to the mouse genome (GRCm38) using Bowtie2 with default parameters (34). After filtering the read for mitochondrial DNA, the Y chromosome duplicate reads were removed using the Picard tools MarkDuplicates program (http://broadinstitute.github.io/picard/). ATAC-Seq peak regions of each sample were called using MACS2 with the parameters—nomodel, shift 100 and extsize 200 (35). All peak files were combined together, with overlapping peaks merged into a single peak. We identified 63 018 peaks from retina samples and 19 950 peaks from the RPE/choroid samples. The top 25% of the peaks by signal strength were plotted using R. Integrative genomics viewer was used to visualize the peak intensity for individual genes (36). DeepTools2 was used to create BigWig files. The BigWig files were merged together for each time point to create heat maps for the peak values from the top half of the peaks by signal strength (37). The circos plots were created using circlize (7,38). MDS plots were created from the values of all peaks using edgeR (39).
Retina and RPE extraction for RNA-Seq
Fresh retina and RPE tissues were harvested from photosensitive Abca4−/−Rdh8−/− mice according to published protocols (40). Under a dissecting microscope, spring scissors were used to puncture the eye and remove the cornea, iris and lens. The remaining eyecup had 4 radial incisions made every 90 degrees, resulting in a flat and open eyecup. The retina was then gently removed using curved tweezers and placed in a 1.5-mL microcentrifuge tube containing RNAlater (Qiagen, Hilden, Germany). The RPE-containing eyecup was placed in a 1.5 mL microcentrifuge tube containing RNAprotect (Qiagen). The second eye was processed identically and pooled with the first eye from the same mouse. Total RNA from RPE cells was isolated using the simultaneous RPE cell isolation and RNA stabilization (SRIRS) method (40). Briefly, the tube containing RNAprotect with the two pooled RPE/choroid eyecups was agitated in 10-min intervals for 20 min at RT. After the second agitation, the eyecups were removed to minimize choroid contamination, with dissociated RPE cells remaining in solution. Retina and RPE samples in RNAlater and RNAprotect, respectively, were stored at 4°C for up to 1 week.
RNA sequencing
Retina tissue samples were removed from the RNAlater solution and placed in a fresh 1.5 mL microcentrifuge tube. RPE samples were centrifuged for 5 min at 700 g and the supernatant was then discarded. Total cellular RNA isolation was performed with the miRNAeasy micro kit with an optional DNase step, per the manufacturer’s protocol (Qiagen, Hilden, Germany). RNA samples were sent to the Transcriptomics and Deep Sequencing Core (Johns Hopkins University, Baltimore, MD) for library preparation and sequencing. Briefly, mRNA was polyA-selected from total RNA (100–150 ng per sample) and subjected for library preparation by following the Illumina TruSeq Stranded mRNA Library Prep Kit instructions. Libraries were then pooled and sequenced for paired-end 150 cycles in the Illumina NextSeq 500 system, yielding ~45–50 million raw reads per library.
RNA-Seq differential gene expression analysis
Alignment of sequences to the genome was completed using STAR version 2.5.3, Ensembl GRCm38 was used for STAR mapping (41), and read counts were generated using the featureCounts function of Rsubread (42). Gene transcripts with 1 ≥ CPM in four or more replicates were considered expressed and used in all downstream analyses. Differential gene expression analysis was performed using edgeR. Ggplot2 was used to create the correlation plots. The R package VennDiagram was used to create the Venn diagram (43). Gene set enrichment analysis was performed using the Gene Ontology (GO) functional annotation clustering method of DAVID 6.8 to determine the biological function of differentially expressed genes (44). Seurat was used to determine cell type markers (45) and perform the pseudo-scRNA-Seq analysis by cross-referencing an unpublished wild-type C57BL/6 murine retina scRNA-Seq dataset. For each time point analyzed in the bulk RNA-seq data, the DE genes up- or downregulated relative to non-bleached controls were used to create a ‘meta-gene’ from the scRNA-Seq dataset, which was a collective sum of the values for all the up- or downregulated DE genes. The collective up- and downregulated meta-genes generated were then used through standard Seurat tools to highlight meta-gene expression in individual cells on the UMAP plot.
Preparation of tissue lysates for western blotting analysis
Fresh retina and RPE were harvested from study mice as described previously. Briefly, samples from both eyes of the same mouse were pooled together and homogenized in RIPA buffer supplemented with a protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). Posterior eyecups (sclera–choroid–RPE) were incubated on ice for 20 min with frequent agitation to dissociate the RPE monolayer into solution; then the remainder of the eyecups were removed prior to sonication, vortex and centrifugation at 21 000 g for 15 min at 4°C. Proteins were size-fractionated on 4–12% Bis-Tris Nu-PAGE gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes. The membranes were incubated in Intercept blocking solution (LI-COR, Lincoln, NE) for 1 h at RT, followed by primary antibodies targeting H3K27ac (1:1000, Cell Signaling #8173), H3K9me3 (1:1000, Abcam #8898) and GAPDH (1:1000, Cell Signaling #2118) overnight at 4°C. Membranes were washed with PBS containing 0.1% Tween-20 and incubated with an infrared dye (IR)-labeled goat anti-rabbit secondary antibody (1:5000, LI-COR #926-32 211) for 1 h at RT. The blots were imaged, and IR signals were quantified using a LI-COR Odyssey Fc imaging system.
Immunofluorescence microscopy
Mice were euthanized in a CO2 chamber prior to enucleation. For IF staining of retina and RPE flat mounts, the cornea and lens were first dissected out; then the remaining neural retina was separated from the RPE-containing posterior eyecup, and both were fixed in 4% paraformaldehyde for 30 min. Retina- and RPE-containing eyecups were then flattened by making long radial cuts and mounted on glass slides (Superfrost Plus, Fisher Scientific). For both IHC and flat mount IF staining, slides were incubated in a blocking buffer containing 5% FBS, 1% BSA and 0.2% Triton X-100 in PBS for 2 h at RT. Slides were then incubated with a primary antibody targeting H3K9me3 (1:100, Abcam #8898) overnight at 4°C, followed by a 1-h incubation with a fluorescent goat anti-rabbit secondary antibody (1:250, Invitrogen #A11037). F-actin was labeled by FITC-phalloidin (1:200, Invitrogen #A12379) co-incubated with secondary antibody for 1 h at RT. Fluorescence microscopy images were obtained on a Keyence BZ-X810 fluorescent microscope.
Statistical analyses
Results were collected from at least three mice for each experimental group unless otherwise indicated. Data from at least three independent experiments were presented as mean ± standard error of the mean (SEM). Statistical significance was determined by the Student’s t-test, where differences with P < 0.05 were considered significant. Fold change, false discovery rate (FDR) and Pearson’s correlation coefficient were calculated in the R platform (https://www.R-project.org).
Author Contributions
J.L., L.K. and K.P. designed the research; J.L., L.K., S.B. and K.P. wrote the paper; T.H. performed the tagmentation; L.K. performed data processing and computational analyses; J.L. and D.L. performed the western blotting and immunofluorescence analyses; J.L., L.K., T.H., D.L., Z.D., S.B. and K.P. contributed to methodology; S.B. and K.P. contributed to resources and funding acquisition; J.L., S.B. and K.P. provided project supervision.
Data Availability Statement
All data supporting the findings of this study are available within this paper. Additional data supporting the findings of this manuscript are available from the corresponding author upon request. All ATAC and RNA sequencing data collected for this study have been deposited in the NCBI Gene Expression Omnibus (GEO) data repository under accession numbers GSE153817 and GSE153818.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the members of the Palczewski laboratory for their helpful comments regarding this project. K.P. is the Irving H. Leopold Chair of Ophthalmology. The authors also acknowledge support from an RPB unrestricted grant to the Department of Ophthalmology, University of California, Irvine.
Conflict of Interest Statement.There are no conflict of interests to disclose.
Funding
The National Institutes of Health (NIH) (EY009339, EY027283, EY025451); NIH MSTP training grant T32 GM007250; US Department of Veterans Affairs (IK2BX002683).
References
- 1. Talens R.P., Christensen K., Putter H., Willemsen G., Christiansen L., Kremer D., Suchiman H.E., Slagboom P.E., Boomsma D.I. and Heijmans B.T. (2012) Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell, 11, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bahar R., Hartmann C.H., Rodriguez K.A., Denny A.D., Busuttil R.A., Dolle M.E., Calder R.B., Chisholm G.B., Pollock B.H., Klein C.A. and Vijg J. (2006) Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature, 441, 1011–1014. [DOI] [PubMed] [Google Scholar]
- 3. Rando T.A. and Chang H.Y. (2012) Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell, 148, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones P.A. and Baylin S.B. (2007) The epigenomics of cancer. Cell, 128, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ordovás J.M. and Smith C.E. (2010) Epigenetics and cardiovascular disease. Nat. Rev. Cardiol., 7, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klein H.U., McCabe C., Gjoneska E., Sullivan S.E., Kaskow B.J., Tang A., Smith R.V., Xu J., Pfenning A.R., Bernstein B.E. et al. (2019) Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer's human brains. Nat. Neurosci., 22, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J., Zibetti C., Shang P., Sripathi S.R., Zhang P., Cano M., Hoang T., Xia S., Ji H., Merbs S.L. et al. (2018) ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun., 9, 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luu J. and Palczewski K. (2018) Human aging and disease: lessons from age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A, 115, 2866–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong W.L., Su X., Li X., Cheung C.M., Klein R., Cheng C.Y. and Wong T.Y. (2014) Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health, 2, e106–e116. [DOI] [PubMed] [Google Scholar]
- 10. Maeda A., Maeda T., Golczak M., Chou S., Desai A., Hoppel C.L., Matsuyama S. and Palczewski K. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem., 284, 15173–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maeda A., Maeda T., Golczak M. and Palczewski K. (2008) Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem., 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kohno H., Chen Y., Kevany B.M., Pearlman E., Miyagi M., Maeda T., Palczewski K. and Maeda A. (2013) Photoreceptor proteins initiate microglial activation via toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J. Biol. Chem., 288, 15326–15341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seto E. and Yoshida M. (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol., 6, a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang G., Weng X., Zhao Y., Zhang X., Hu Y., Dai X., Liang P., Wang P., Ma L., Sun X. et al. (2017) The histone H3K9 methyltransferase SUV39H links SIRT1 repression to myocardial infarction. Nat. Commun., 8, 14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fournel M., Bonfils C., Hou Y., Yan P.T., Trachy-Bourget M.C., Kalita A., Liu J., Lu A.H., Zhou N.Z., Robert M.F. et al. (2008) MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther., 7, 759–768. [DOI] [PubMed] [Google Scholar]
- 16. Vougiouklakis T., Saloura V., Park J.H., Takamatsu N., Miyamoto T., Nakamura Y. and Matsuo Y. (2018) Development of novel SUV39H2 inhibitors that exhibit growth suppressive effects in mouse xenograft models and regulate the phosphorylation of H2AX. Oncotarget, 9, 31820–31831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fritsche L.G., Igl W., Bailey J.N., Grassmann F., Sengupta S., Bragg-Gresham J.L., Burdon K.P., Hebbring S.J., Wen C., Gorski M. et al. (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet., 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhutto I. and Lutty G. (2012) Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol. Asp. Med.Mol Aspects. Med., 33, 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atlasi Y. and Stunnenberg H.G. (2017) The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet., 18, 643–658. [DOI] [PubMed] [Google Scholar]
- 20. Rivera C.M. and Ren B. (2013) Mapping human epigenomes. Cell, 155, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell, 125, 315–326. [DOI] [PubMed] [Google Scholar]
- 22. Feinberg A.P. (2007) Phenotypic plasticity and the epigenetics of human disease. Nature, 447, 433–440. [DOI] [PubMed] [Google Scholar]
- 23. Montana C.L., Kolesnikov A.V., Shen S.Q., Myers C.A., Kefalov V.J. and Corbo J.C. (2013) Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc. Natl. Acad. Sci. U. S. A, 110, 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng H., Aleman T.S., Cideciyan A.V., Khanna R., Jacobson S.G. and Swaroop A. (2006) In vivo function of the orphan nuclear receptor NR2E3 in establishing photoreceptor identity during mammalian retinal development. Hum. Mol. Genet., 15, 2588–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis B.K., Wen H. and Ting J.P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol., 29, 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liddelow S.A. and Barres B.A. (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity, 46, 957–967. [DOI] [PubMed] [Google Scholar]
- 27. Wang S.K., Xue Y., Rana P., Hong C.M. and Cepko C.L. (2019) Soluble CX3CL1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A, 116, 10140–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cascella R., Ragazzo M., Strafella C., Missiroli F., Borgiani P., Angelucci F., Marsella L.T., Cusumano A., Novelli G., Ricci F. and Giardina E. (2014) Age-related macular degeneration: insights into inflammatory genes. J. Ophthalmol., 2014, 582842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klettner A., Kauppinen A., Blasiak J., Roider J., Salminen A. and Kaarniranta K. (2013) Cellular and molecular mechanisms of age-related macular degeneration: from impaired autophagy to neovascularization. Int. J. Biochem. Cell Biol., 45, 1457–1467. [DOI] [PubMed] [Google Scholar]
- 30. Jiang K., Cao S., Cui J.Z. and Matsubara J.A. (2013) Immuno-modulatory effect of IFN-gamma in AMD and its role as a possible target for therapy. J Clin Exp Ophthalmol, Suppl, 2, 0071–0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Y., Palczewska G., Masuho I., Gao S., Jin H., Dong Z., Gieser L., Brooks M.J., Kiser P.D., Kern T.S. et al. (2016) Synergistically acting agonists and antagonists of G protein-coupled receptors prevent photoreceptor cell degeneration. Sci. Signal., 9, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buenrostro J.D., Wu B., Chang H.Y. and Greenleaf W.J. (2015) ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol., 109, 21.29.21–21.29.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bolger A.M., Lohse M. and Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langmead B. and Salzberg S.L. (2012) Fast gapped-read alignment with bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. and Liu X.S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G. and Mesirov J.P. (2011) Integrative genomics viewer. Nat. Biotechnol., 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F. and Manke T. (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acid Res., 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gu Z., Gu L., Eils R., Schlesner M. and Brors B. (2014) Circlize implements and enhances circular visualization in R. Bioinformatics, 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- 39. McCarthy D.J., Chen Y. and Smyth G.K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acid Res., 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xin-Zhao Wang C., Zhang K., Aredo B., Lu H. and Ufret-Vincenty R.L. (2012) Novel method for the rapid isolation of RPE cells specifically for RNA extraction and analysis. Exp. Eye Res., 102, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T.R. (2012) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao Y., Smyth G.K. and Shi W. (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acid Res., 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen H. and Boutros P.C. (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics, 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang da W., Sherman B.T. and Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 45. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. 3rd, Hao Y., Stoeckius M., Smibert P. and Satija R. (2019) Comprehensive integration of single-cell data. Cell, 177, e1821, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within this paper. Additional data supporting the findings of this manuscript are available from the corresponding author upon request. All ATAC and RNA sequencing data collected for this study have been deposited in the NCBI Gene Expression Omnibus (GEO) data repository under accession numbers GSE153817 and GSE153818.