

Key Points
Blockade of Rap1 binding to talin-1 F0 and F1 domains phenocopies the defects in integrin activation observed in platelets lacking Rap1a/b.
Rap1–talin-1 interaction in platelets is crucial for hemostasis, with minor impact on granule secretion.
Abstract
Ras-related protein 1 (Rap1) is a major convergence point of the platelet-signaling pathways that result in talin-1 binding to the integrin β cytoplasmic domain and consequent integrin activation, platelet aggregation, and effective hemostasis. The nature of the connection between Rap1 and talin-1 in integrin activation is an important remaining gap in our understanding of this process. Previous work identified a low-affinity Rap1-binding site in the talin-1 F0 domain that makes a small contribution to integrin activation in platelets. We recently identified an additional Rap1-binding site in the talin-1 F1 domain that makes a greater contribution than F0 in model systems. Here we generated mice bearing point mutations, which block Rap1 binding without affecting talin-1 expression, in either the talin-1 F1 domain (R118E) alone, which were viable, or in both the F0 and F1 domains (R35E,R118E), which were embryonic lethal. Loss of the Rap1–talin-1 F1 interaction in platelets markedly decreases talin-1–mediated activation of platelet β1- and β3-integrins. Integrin activation and platelet aggregation in mice whose platelets express only talin-1(R35E, R118E) are even more impaired, resembling the defect seen in platelets lacking both Rap1a and Rap1b. Although Rap1 is important in thrombopoiesis, platelet secretion, and surface exposure of phosphatidylserine, loss of the Rap1–talin-1 interaction in talin-1(R35E, R118E) platelets had little effect on these processes. These findings show that talin-1 is the principal direct effector of Rap1 GTPases that regulates platelet integrin activation in hemostasis.
Visual Abstract

Introduction
The mechanism of platelet aggregation has been an enduring question since the 19th-century work of Bizzozero and Virchow. The seminal discovery that platelet agonists cause integrin αIIbβ3 to bind fibrinogen (“activation”) with high affinity provided a crucial clue as to the mechanism of platelet aggregation.1 Studies with antibody2,3 and peptide4,5 inhibitors ultimately resulted in the development of αIIbβ3 antagonists for clinical use; however, these agents have been hampered by the difficulty in striking a balance between antithrombotic effects and bleeding.6 β3 mutations that reduce activation can lessen thrombosis while ameliorating the pathological bleeding caused by complete lack of αIIbβ3 function, leading to the idea that blocking activation might widen the therapeutic window for αIIbβ3 inhibitors.7 Many different signaling pathways have been implicated in αIIbβ3 activation; however, induction of talin-1 binding to the integrin β subunit cytoplasmic domain is a final common step in platelets and megakaryocytes.7-10
Ras-related protein 1 (Rap1) GTPases are important signaling hubs that control platelet adhesion.11-13 They function as a molecular switch and transition between the active guanosine triphosphate (GTP)-bound form and the inactive guanosine diphosphate (GDP)-bound state.14 Murine platelets express high levels of Rap1b, whereas Rap1a accounts for ∼10% of total platelet Rap1 proteins.15 In response to platelet stimulation with agonists, elevation of the calcium concentration in the cytosol triggers activation of Ca2+- and diacylglycerol-regulated guanine nucleotide exchange factor I (CalDAG-GEFI), which functions as a GEF for Rap1.16 Genetic deletion of both Rap1a and Rap1b in the megakaryocyte lineage causes macrothrombocytopenia due to impaired proplatelet formation, profoundly impaired integrin activation in platelets, and marked defects in hemostasis.12 Thus, Rap1 GTPases are main regulators of talin-1–dependent integrin activation in platelets. Elucidating the connection between active Rap1 and talin-1 in integrin activation is an important remaining gap in our understanding of platelet aggregation.
Talin-1 N-terminal head domain is an atypical FERM (4.1-protein/ezrin/radixin/moesin) domain subdivided into F0, F1, F2, and F3 subdomains.17 Talin-1 is autoinhibited in the cytosol due to the interaction of the talin-1 head domain with the rod domain, which prevents its interaction with the integrin β cytoplasmic tail.18 Pioneering studies showed that Rap1b binds directly to the talin-1 F0 domain through a low-affinity interaction.19 Structural studies subsequently revealed the importance of the lipid environment at the membrane to strengthen the Rap1–talin-1 F0 interaction.20 Quantitative proteomic analyses of murine platelets revealed the high abundance of Rap1b and talin-1 at equal molar ratios (>200 000 per platelet).15 The lack of a known Rap1 effector with such a high abundance in platelets suggests that talin-1 acts as a direct effector of Rap1 to activate αIIbβ3 integrins in platelets. However, we and others showed that blocking the Rap1–talin-1 F0 interaction has a relatively minor effect on platelet integrin activation and hemostasis and cannot account for the dramatic effects of loss of Rap1 activity on platelet functions.21,22 Recently, we identified a second Rap1-binding site in the talin-1 F1 domain of similar affinity to that in F0.23 We found that an R118E mutation in the talin-1 F1 domain, which blocks Rap1 binding, abolishes the capacity of Rap1 to potentiate talin-1–induced integrin activation in A5 CHO cells expressing recombinant αIIbβ3. The ability of the talin-1 F1 domain to mediate Rap1-dependent integrin activation requires a unique unstructured loop that transforms into an amphipathic helix upon binding to membrane lipids. Bromberger et al24 have confirmed the Rap1-binding capacity of talin F1 and shown that mutations in F0 and F1 that block Rap1 binding partially impair activation of β1 integrins in recombinant talin-expressing talin-null fibroblasts. Thus, direct binding of Rap1 to both talin-1 F0 and F1 domains can affect integrin activation in model systems; however, the relative role of these 2 binding sites in platelet integrin activation and in hemostasis is unclear.
In the current study, we generated mice bearing point mutations in either the talin-1 F1 domain (R118E) or both the F0 and F1 domains (R35E,R118E) that block binding to Rap1 without affecting protein expression in platelets. We found that, in contrast to the Rap1–talin-1 F0 interaction, disrupting the Rap1–talin-1 F1 interaction in talin-1(R118E) platelets causes a marked defect in the activation of β1- and β3-integrins. Platelets expressing the talin-1(R35E,R118E) mutant are further impaired; indeed, they phenocopy the defects in αIIbβ3 activation observed in platelets lacking both Rap1a and Rap1b. Thus, talin-1 is the major Rap1 effector involved in integrin activation in platelets. The talin-1(R35E,R118E) mutation does not cause the severe defects in thrombopoiesis, secretion, or generation of procoagulant activity observed in Rap1-deficient platelets, indicating the existence of additional platelet/megakaryocyte Rap1 effectors important in these processes. These data show that direct binding of Rap1 to talin-1 is a major link in the pathway from platelet stimulation to integrin activation, a key event in platelet aggregation and hemostatic plug formation.
Methods
Mice
Tln1FLOX/FLOX,8 Tln1R35E/R35E,21 Rap1A/BFLOX/FLOX,25 and PF4-Cre+/−26 mice have been previously described. Eight- to 16-week-old sex-matched wild-type littermates were control animals for all experiments. Tln1WT/R118E and Tln1WT/R35E,R118E knock-in mice were generated by using a CRISPR/Cas9 approach at the University of California Irvine Transgenic Mouse Facility (UCI TMF). Detailed information is provided in the supplemental Methods (available on the Blood Web site). One founder Tln1WT/R118E and one founder Tln1WT/R35E,R118E were obtained and backcrossed to the C57BL/6J strain to obtain heterozygous Tln1WT/R118E and Tln1WT/R35E,R118E mice. Sex-matched Tln1WT/WT littermates were used as controls for Tln1R118E/R118E mutant animals (indicated as Tln1-R118E) in all experiments. Similar to Tln1−/−mice, Tln1R35E,R118E/R35E,R118E mice were not viable. To circumvent the lethality, we crossed Tln1WT/R35E,R118E;PF4-Cre+/− mice with the Tln1FLOX/FLOX strain to obtain megakaryocyte-specific deletion of Tln1-Flox allele in Tln1FLOX/WT;PF4-Cre+/− (control) and Tln1FLOX/R35E,R118E;PF4-Cre+/− (Tln1-mR35E,R118E) littermates for experiments. Similarly, Tln1FLOX/R35E;PF4-Cre+/− mice (Tln1-mR35E) were compared with Tln1FLOX/WT;PF4-Cre+/− (control) littermates for experiments, and Tln1FLOX/R118E;PF4-Cre+/− mice (Tln1-mR118E) with Tln1FLOX/WT;PF4-Cre+/− (control) littermates. Tln1-mKO refers to the Tln1FLOX/FLOX;PF4-Cre+/− strain, and Rap1a/b-mKO refers to Rap1a/bFLOX/FLOX;PF4-Cre+/−. Mice were housed in the animal facilities of the University of California, San Diego. Experimental procedures were approved by the Institutional Care and Use Committee.
Platelet preparation
Blood was drawn with heparin-coated capillaries (VWR) from the retro-orbital plexus into tubes containing low-molecular-weight enoxaparin sodium (Lovenox, sanofi-aventis). Whole blood was diluted with modified Tyrode’s buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 5 mM glucose, pH 7.3) containing 0.35% bovine serum albumin. Platelet-rich plasma was obtained by 2 successive centrifugation steps at 130g for 4 minutes first and then 100g for 5 minutes at room temperature. The platelet-rich plasma was centrifuged at 700g for 5 minutes at room temperature in the presence of 5 μM prostacyclin to pellet platelets. Platelets were resuspended in modified Tyrode’s buffer, and the platelet concentration was adjusted to 5 × 108/mL.
Flow cytometry
For whole blood assays, blood was diluted 1:12.5 in modified Tyrode’s solution containing 1 mM CaCl2, agonists, and JonA/PE (2 μg/mL) or 9EG7/Alexa Fluor 488 (2 μg/mL) plus anti-GPIX antibody labeled with Alexa Fluor 647 (2 μg/mL). Samples were incubated for 10 minutes at room temperature and then diluted 1:400 in phosphate-buffered saline before analysis with a BD Accuri C6 Plus flow cytometer (BD Biosciences). Platelet phosphatidylserine (PS) exposure was measured by using Alexa Fluor 647–conjugated Annexin V (BioLegend) staining following stimulation with 0.1 μg/mL convulxin + 500 μM protease-activated receptor-4–agonist-peptide (PAR4-AP). For analysis of surface receptor expression levels, 2 × 106 platelets in diluted whole blood (modified Tyrode’s buffer) were stained for 10 minutes with 2 μg/mL phycoerythrin (PE)-conjugated antibodies and immediately analyzed by using flow cytometry.
For real-time αIIbβ3 activation assay, washed platelets were diluted in modified Tyrode’s solution containing 1 mM CaCl2. After establishing a baseline with unlabeled platelets for 30 seconds, JonA/PE and agonist (PAR4-AP or convulxin) were added simultaneously in an equal volume of modified Tyrode’s solution. Jon/A-PE binding was recorded continuously for 10 minutes.
Aggregometry
Platelet-rich plasma was diluted to a concentration of 3 × 108 platelets/mL in modified Tyrode’s buffer containing 0.35% bovine serum albumin and 1 mM CaCl2. Experiments were performed at 37°C under stirring conditions (1200 rpm). Platelets were stimulated with various concentrations of PAR4-AP, fibrillar collagen type I, or adenosine 5′-diphosphate (ADP). Light transmission was recorded on a 4-channel optical aggregation system (Chrono-log).
Saphenous vein laser injury
Mice (6-8 weeks of age) were anesthetized with 2.5% isoflurane before the saphenous vein was surgically exposed. Mice were injected with Alexa Fluor 488–conjugated antibodies to GPIX (2.5 μg) to label circulating platelets and Alexa Fluor 647–conjugated antibodies to fibrin (2.0 μg), and the saphenous vein was injured as previously described.27 Bleeding times and platelet plug formation at the site of laser injury were assessed by intravital microscopy using an Examiner Z1 microscope (Zeiss) equipped with a CSU-W confocal scanning unit (Yokogawa Electric Co.) and Orca Flash 4.0 camera (Hamamatsu). Data were analyzed with SLIDEBOOK 6.0 (Intelligent Imaging Innovations).
Statistics
Statistical significance was assayed by using a 2-tailed Student t test for single comparisons. Analysis of variance with a Tukey post hoc test or a Gehan-Breslow-Wilcoxon test with Bonferroni correction was used to assay statistical significance for multiple comparisons. A Shapiro-Wilk normality test was used to assess the Gaussian distribution of all data sets. A value of P < .05 was considered significant.
Additional methods and more detailed descriptions are provided in the supplemental Methods.
Results
Rap1 binding to the talin-1 F1 domain significantly contributes to integrin activation in platelets
The amino acid substitution R118E in the talin-1 F1 domain does not disrupt the folding of F1 and blocks binding to Rap1.23 To evaluate the contribution of Rap1 binding to the talin-1 F1 domain to integrin activation in platelets, CRISPR/Cas9 was used to introduce the mutation R118E into exon 3 of the murine Tln1 gene (Figure 1A-B). Homozygous Tln1R118E/R118E mice (indicated as Tln1-R118E) were viable and fertile, exhibited no gross developmental abnormalities, and appeared healthy, with normal platelet counts (Table 1). Platelet content of talin-1 (Figure 1C) and surface expression of αIIb, β3, α2, α5, and β1 integrins (Figure 1D) in Tln1-R118E platelets were similar to wild-type platelets, indicating that Tln1-R118E mice are suitable for examination of the effects of blocking Rap1 binding to the talin-1 F1 domain on platelet integrin function in vivo and ex vivo.
Figure 1.
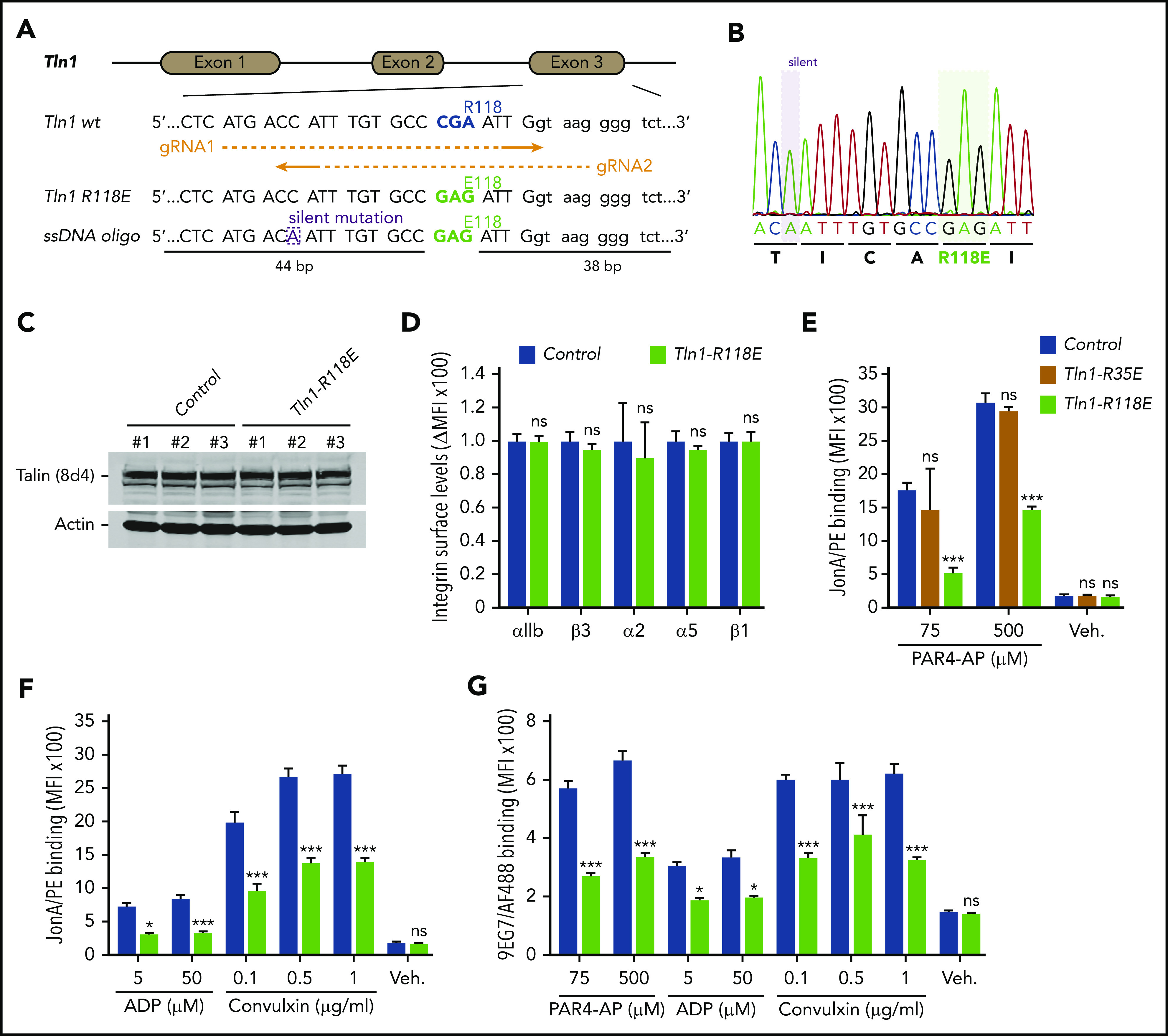
Rap1 binding to talin-1 F1 domain contributes to integrin activation in platelets. (A) Generation of mice harboring Tln1 R118E mutation. Two guide RNAs were used to target Tln1 exon 3. A silent mutation corresponding to the PAM sequence of gRNA2 was inserted into the donor single-stranded DNA oligo to prevent re-editing. (B) Sequencing chromatogram of mutated region of Tln1(R118E) gene. (C) Expression of talin-1 mutant in Tln1-R118E platelets was assayed by using western blotting. Results are representative of 3 independent experiments, n = 3 mice each time. (D) Surface expression of αIIb, β3, α2, α5, and β1 integrins in Tln1-R118E platelets was measured by using flow cytometry. Bar graph represents mean fluorescence intensity (MFI) ± standard error of the mean (n = 6 mice). Two-tailed Student t test. (E-G) Impaired integrin activation in Tln1-R118E platelets. Flow cytometry assay to measure binding of GPIX-labeled platelets in whole blood to the JonA/PE antibody (E-F) or the Alexa Fluor 488–coupled 9EG7 antibody (G) in response to agonist stimulation. Bar graphs represent MFI ± standard error of the mean (n = 6 mice, representative of ≥3 independent experiments). Two-way analysis of variance with Tukey posttest. *P < .05; ***P < .001. ns, not significant.
Table 1.
Peripheral blood cell counts of Tln1-R118E and Tln1-mR35E,R118E mice
| Genotype | PLT (×103/mL) | MPV (fL) | HCT (%) | HB (g/dL) | WBC (×103/mL) |
|---|---|---|---|---|---|
| Tln1WT/WT (n = 13) | 829.2 ± 27.43 | 4.4 ± 0.06 | 44.3 ± 0.69 | 13.7 ± 0.23 | 8.4 ± 0.72 |
| Tln1R118E/R118E (n = 14) | 910.2 ± 34.76* | 4.5 ± 0.09* | 44.8 ± 0.64* | 13.8 ± 0.26* | 15.8 ± 1.29† |
| Tln1FLOX/WT; PF4-Cre+/− (n = 16) | 819.1 ± 29.45 | 4.2 ± 0.05 | 42.7 ± 0.61 | 13.5 ± 0.15 | 10.78 ± 0.65 |
| Tln1FLOX/R35E,R118E; PF4-Cre+/− (n = 16) | 955.8 ± 29.89* | 4.3 ± 0.05* | 43.1 ± 0.56* | 13.5 ± 0.18* | 13.1 ± 0.67† |
Data are presented as the mean ± standard error of the mean. Statistical significance was assayed by a 2-tailed Student t test. No significant differences were observed.
HB, hemoglobin; HCT, hematocrit; MPV, mean platelet volume; PLT, platelet; WBC, white blood cell.
Not significant.
P < .05.
We next measured binding of JonA/PE, an antibody that specifically detects the active conformation of murine αIIbβ3,28 to platelets stimulated with various agonists. Tln1-R118E platelets exhibited a substantial reduction in JonA/PE binding in response to stimulation with various concentrations of PAR4-AP, ADP, and convulxin (Figure 1E-F). Conversely, JonA/PE binding to platelets carrying the amino acid substitution R35E in the talin-1 F0 domain, which blocks its binding to Rap1,21 was similar compared with platelets from wild-type mice (Figure 1E). Binding of 9EG7 antibody, which selectively binds to the active conformation of β1 integrin, was also reduced in Tln1-R118E platelets in response to agonist stimulation (Figure 1G). To verify that the defect was platelet/megakaryocyte cell autonomous, we tested platelets from Tln1FLOX/R118E;PF4-Cre+/− mice (indicated as Tln1-mR118E). In these mice, talin-1(R118E) is the only form of talin expressed in the megakaryocyte lineage, whereas other cells also contain a wild-type allele. Tln1-mR118E platelets exhibited a similar reduction in activation of β1 and β3 as platelets from Tln1R118E/R118E mice (supplemental Figure 1).
Importantly, we checked for off-target mutations that could have been inserted using CRISPR-Cas9 into the genome of Tln1-R118E mice. The top 5 off-target sites were sequenced for each guide RNA, and no detectable mutations were found (supplemental Table 1). Furthermore, the defects in integrin activation were not detected in platelets from Tln1FLOX/R118E;PF4-Cre−/− littermates but were only observed upon loss of the Tln1-Flox allele in Tln1FLOX/R118E;PF4-Cre+/− platelets (supplemental Figure 2A). Thus, the defect in platelet integrin activation in Tln1FLOX/R118E;PF4-Cre+/− mice can be ascribed to the Tln1-R118E mutation. Altogether, our results show that blockade of Rap1 binding to the talin-1 F1 domain inhibits agonist-induced activation of both β1 and β3 integrins, whereas blocking Rap1 binding to F0 has a much weaker effect.
Mutation of the 2 Rap1 binding sites in the talin-1 F0 and F1 domains further impairs platelet integrin activation
Although both the talin-1 F0 and F1 domains contain a Rap1-binding site,21,23 only the R118E mutation in the F1 domain leads to a major reduction in talin-1–mediated integrin activation in platelets. We next generated a mouse strain carrying both amino acid substitutions R35E and R118E in talin-1 F0 and F1 domains, respectively, to block both Rap1-binding sites in talin-1. We used the same CRISPR-Cas9 approach as for Tln1-R118E mice to introduce the R118E mutation into the Tln1-R35E allele (Figure 1A). Intercrosses of heterozygous Tln1WT/R35E,R118E mice only resulted in wild-type and heterozygous pups at a 1:2 ratio. We ruled out the presence of off-target mutations that could have been inserted by CRISPR-Cas9 into the genome of Tln1WT/R35E,R118E mice (supplemental Table 1). We also sequenced the whole coding sequence of the Tln1 gene in Tln1WT/R35E,R118E mice and found no additional mutations. Thus, the absence of homozygous animals indicates that the combination of R35E and R118E mutations leads to a profound loss of function in talin-1 that is incompatible with postnatal life.
To circumvent the embryonic lethality of the Tln1-R35E,R118E allele, we generated Tln1FLOX/R35E,R118E;PF4-Cre+/− mice selectively expressing talin-1(R35E,R118E) mutant protein (indicated as Tln1-mR35E,R118E) in platelets. These mutant mice were viable and fertile, with normal platelet counts, no gross developmental abnormalities, and no spontaneous bleeding (Table 1); the viability of these mice and lethality of the homozygous Tln1R35E,R118E/R35E,R118E mice indicate that Rap1 binding to talin-1 is important in cells other than platelets, as also suggested by the leukocytosis in Tln1R118E/R118Emice (Table 1), the defect in leukocyte migration in mice bearing a combination of 3 mutations in talin-1 F0 domain that disrupts Rap1 binding,22 and defective focal adhesion targeting of Rap1-binding defective talins.20,23,24 Expression of talin-1(R35E,R118E) and kindlin-3 was unaffected in Tln1-mR35E,R118E platelets (Figure 2A), and surface levels of αIIb, β3, α2, α5, and β1 integrins were similar to integrin contents in platelets from littermate controls (Figure 2B). Binding of JonA/PE (Figure 2C) or 9EG7 (Figure 2D) antibody to agonist-stimulated Tln1-mR35E,R118E platelets was drastically reduced compared with controls. Importantly, the defects in integrin activation were not detected in Tln1FLOX/R35E,R118E;PF4-Cre−/− platelets but only observed upon loss of the Tln1-Flox allele in Tln1FLOX/R35E,R118E;PF4-Cre+/− platelets (supplemental Figure 2B); this indicates that defects in integrin activation in Tln1-mR35E,R118E platelets are not due to off-target mutations but rather ascribable to the loss of Rap1–talin-1 interaction.
Figure 2.
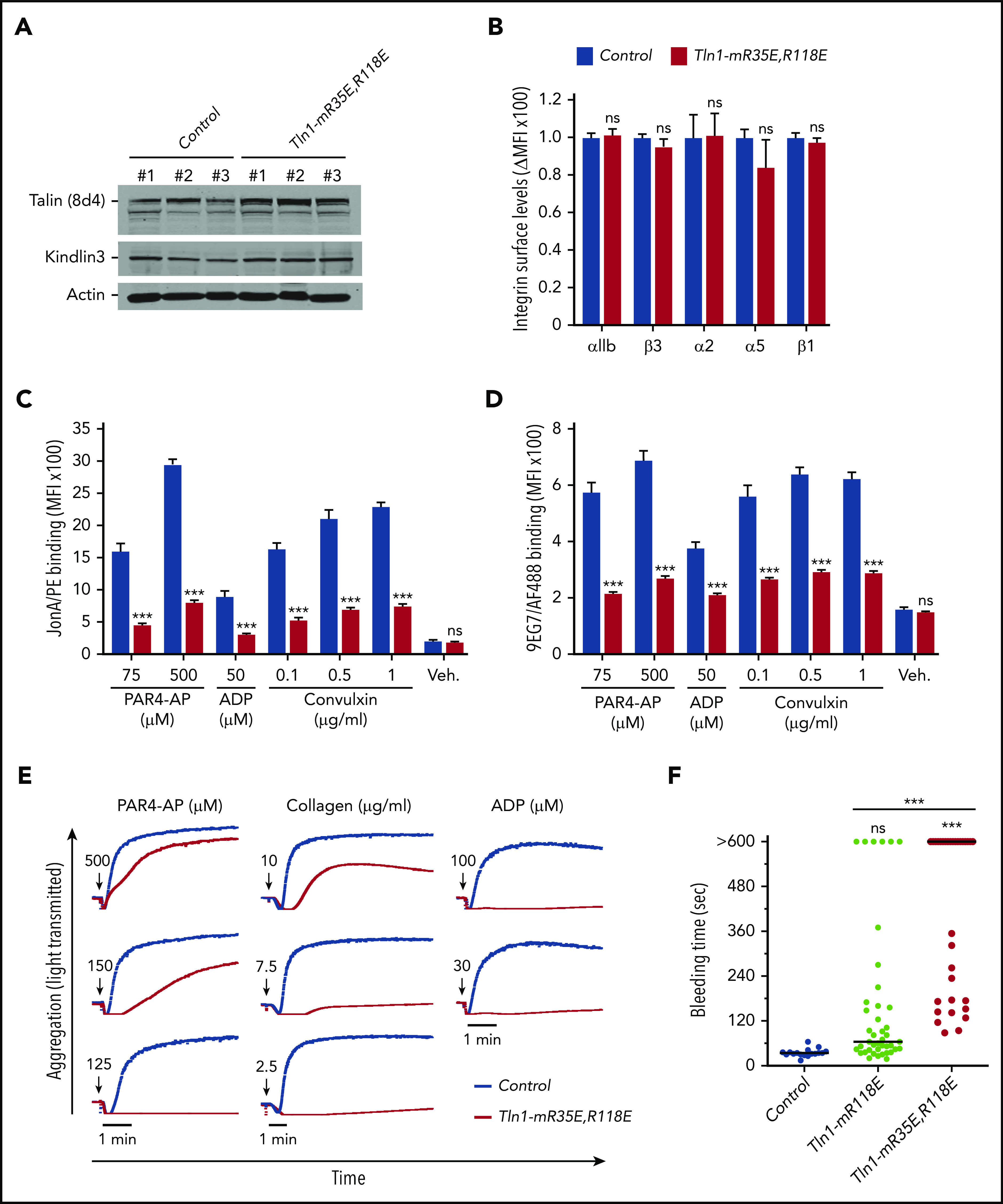
Blockade of Rap1 binding to both F0 and F1 domains in talin-1 prevents integrin activation in platelets in Tln1-mR35E,R118E mice. (A) Expression of talin-1 mutant in Tln1-mR35E,R118E platelets was assayed by using western blotting. Results are representative of 3 independent experiments, n = 3 mice each time. (B) Surface expression of αIIb, β3, α2, α5, and β1 integrins in Tln1-mR35E,R118E platelets was measured by using flow cytometry. Bar graph represents mean fluorescence intensity (MFI) ± standard error of the mean (n = 6 mice). Two-tailed Student t test. (C-D) Impaired integrin activation in Tln1-mR35E,R118E. Flow cytometry assay to measure binding of GPIX-labeled platelets in whole blood to JonA/PE antibody (C) or Alexa Fluor 488–coupled 9EG7 antibody (D) in response to agonist stimulation. Bar graphs represent MFI ± standard error of the mean (n = 6 mice, representative of ≥3 independent experiments). Two-way analysis of variance with Tukey posttest. (E) Representative aggregation responses of Tln1-mR35E,R118E platelets stimulated with various concentrations of agonists (indicated by arrows). (F) Intravital microscopy studies to monitor hemostatic plug formation after laser injury to the saphenous vein in Tln1-mR118E and Tln1-mR35E,R118E mice. The experiment was terminated at the end of 10 minutes to avoid excessive loss of blood. Individual bleeding times with median were plotted for 3 experimental groups. Data depicted are determinations in 3 control mice (17 injury sites), 4 Tln1-mR118E mice (42 injury sites), and 5 Tln1-mR35E,R118E mice (39 injury sites). Statistical significance was assayed by a one-way analysis of variance with Tukey posttest. ***P < .001. ns, not significant.
We next examined the ability of Tln1-mR35E,R118E platelets to form aggregates in response to stimulation with various doses of PAR4-AP, collagen, or ADP. A dramatic reduction was observed in the aggregation response of Tln1-mR35E,R118E platelets, whereas shape change remained intact, indicating an intact Ca++ response to stimulation (Figure 2E). These results highlight the strong contribution of the 2 Rap1-binding sites in talin-1 to integrin activation and platelet aggregation.
To assess the contribution of the Rap1–talin-1 interaction to hemostatic plug formation in vivo, the Tln1-mR35E,R118E mice were then challenged in a laser injury–induced saphenous vein hemostasis model as previously described.27 In this assay, a laser ablation–induced injury with a diameter of ∼50 μm is generated in the saphenous vein endothelium of a mouse, and the time to cessation of bleeding is quantified. About half of the lesions in Tln1-mR35E,R118E mice bled, on average, ∼5 times longer than those in control littermates, whereas the other half of the Tln1-mR35E,R118E lesions bled continuously for the 10-minute observation period (Figure 2F). Notably, hemostasis was less impaired in Tln1-mR118E mice, indicating a detectable contribution of Rap1 binding to talin-1 F0. Thus, disruption of the Rap1–talin-1 interaction profoundly impairs β3 integrin–mediated platelet aggregation and hemostatic plug formation.
Mutation of one or both Rap1 binding sites in talin-1 produces a graded series of defects in platelet integrin activation
To directly determine the relative contributions of the talin-1 F0 and F1 Rap1-binding sites to integrin activation compared with loss of both sites, we compared JonA/PE binding in Tln1-mR35E (Rap1-F0 mutant), Tln1-mR118E (Rap1-F1 mutant), Tln1-mR35E,R118E (both F0 and F1 mutated), and Tln1-mKO (complete loss of talin-1) platelets in response to stimulation with PAR4-AP. A graduated response was observed in talin-1–mediated integrin activation, in which the Rap1–F0 interaction makes a minimal contribution, whereas Rap1 binding to F1 has a strong impact on integrin activation (Figure 3A). Activation of αIIbβ3 was reduced even more when both the Rap1–F0 and Rap1–F1 interactions were hindered in Tln1-mR35E,R118E platelets to reach a level nearly to that of the Tln1-mKO platelets. A similar observation was made when we studied integrin activation kinetics using a flow cytometry–based assay to monitor real-time binding of JonA to Tln1-mR35E,R118E platelets in response to PAR4-AP and convulxin stimulation (Figure 3B-C). Accordingly, agonist-induced aggregation responses of Tln1-mR35E,R118E platelets were affected substantially more compared with Tln1-mR118E platelets (Figure 3D). Together, our findings show the cooperation of the 2 Rap1 interactions with talin-1 F0 and F1 domains to enable optimal integrin activation in platelets and how these novel mutant mouse strains exhibit a graded series of platelet integrin activation defects.
Figure 3.

Graduated αIIbβ3 activation response in Tln1-mR35E, Tln1-mR118E, Tln1-mR35E,R118E, and Tln1-mKO platelets. (A) Flow cytometry assay to measure binding of GPIX-labeled platelets in whole blood to Jon/A-PE antibody in response to PAR4-AP stimulation. Bar graph represents mean fluorescence intensity (MFI) ± standard error of the mean (n = 6 mice, representative of ≥3 independent experiments). Two-way analysis of variance with Tukey posttest. (B-C) Real-time αIIbβ3 activation assay. JonA/PE binding to washed platelets was recorded continuously for 9 minutes by using flow cytometry in response to PAR4-AP (B) or convulxin (C) stimulation. Arrows indicate addition of agonists. (D) Representative aggregation responses of Tln1-mR118E and Tln1-mR35E,R118E platelets stimulated with various concentrations of agonists. Arrows indicate addition of agonists. *P < .05; ***P < .001. ns, not significant.
Tln1-mR35E,R118E platelets phenocopy the defects in αIIbβ3 activation observed in Rap1a/b-deficient platelets
To assess the relative role of the Rap1–talin-1 interaction in αIIbβ3 activation, we compared αIIbβ3 activation in platelets from Tln1-mR35E,R118E mice with platelets deficient for the 2 Rap1a and Rap1b isoforms (indicated as Rap1a/b-mKO). Complete blockade of the Rap1–talin-1 interaction in Tln1-mR35E,R118E platelets inhibited αIIbβ3 activation to a similar extent as in Rap1a/b-mKO platelets upon PAR4-AP stimulation (Figure 4A). However, Rap1a/b-mKO platelets exhibited a slightly more pronounced reduction in αIIbβ3 activation in response to convulxin stimulation (Figure 4B). Agonist-induced aggregation of Tln1-mR35E,R118E and Rap1a/b-mKO platelets were similarly impaired (Figure 4C). Thus, the integrin activation phenotype in Tln1-mR35E,R118E platelets recapitulates the defects observed in combined deficiency of both Rap1a and Rap1b isoforms, thereby showing that other effectors cannot substantially compensate for the loss of the talin-1–Rap1 interaction in αIIbβ3 activation.
Figure 4.

Tln1-mR35E,R118E platelets exhibit impaired αIIbβ3 activation to a similar extent as Rap1a/b-mKO platelets. (A-B) Flow cytometry assay to measure binding of GPIX-labeled platelets in whole blood to JonA/PE antibody in response to PAR4-AP (A) or convulxin (B) stimulation. Bar graphs represent mean fluorescence intensity (MFI) ± standard error of the mean (n = 6 mice, representative of ≥3 independent experiments). (C) Representative aggregation responses of Tln1-mR35E,R118E and Rap1a/b-mKO platelets stimulated with various concentrations of agonists. Curves corresponding to control and Tln1-mR35E,R118E platelets stimulated with PAR4-AP were from the same experiment as those depicted in Figure 3D. Arrows indicate addition of agonists. *P < .05;**P < .01; ***P < .001. ns, not significant.
To compare the importance of the talin-1–Rap1 interaction to Rap1 in platelets to support hemostasis, we challenged Tln1-mR35E,R118 mice and Rap1a/b-mKO mice in the laser injury–induced saphenous vein hemostasis model. Accumulation of Tln1-mR35E,R118E platelets at the site of vascular injury was reduced to a similar extent to that observed with Rap1a/b-mKO mice (Figure 5A). Accordingly, Tln1-mR35E,R118E mice exhibited a significant prolongation in the bleeding time that persisted for up to 10 minutes in half of the tested lesions (Figure 5B). The defect in hemostasis was more severe in Rap1a/b-mKO mice, however, as all lesions were unable to cease bleeding within the 10-minute duration of the assay.
Figure 5.
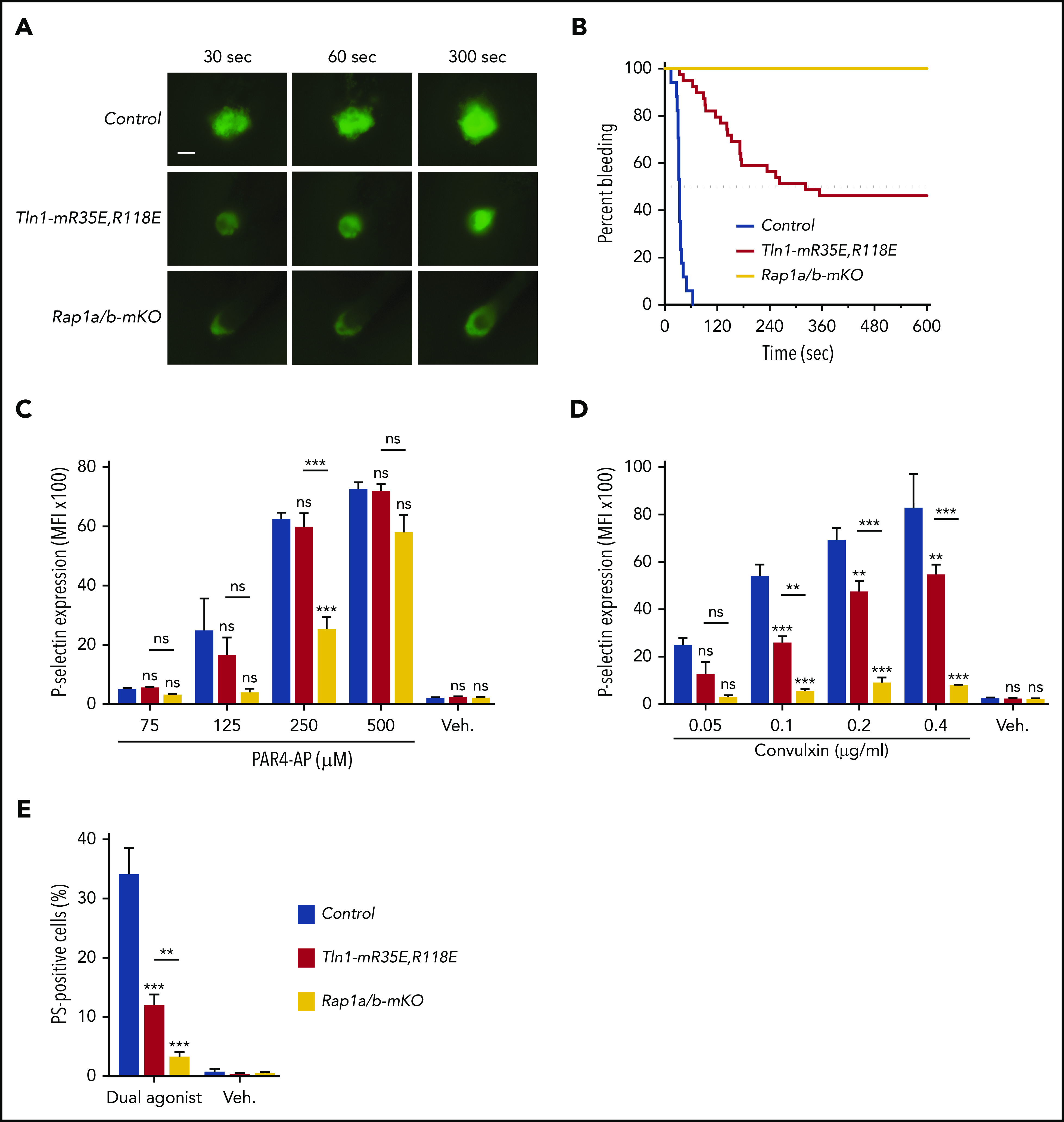
Tln1-mR35E,R118E mice exhibit milder defects in hemostasis compared with Rap1a/b-mKO mice, along with preserved capacity of platelets to secrete α-granules and expose surface PS. (A-B) Intravital microscopy studies to monitor hemostatic plug formation after laser injury to the saphenous vein in Tln1-mR35E,R118E and Rap1a/b-mKO mice. Before laser injury, animals were injected with Alexa Fluor 488–conjugated antibodies to GPIX to label circulating platelets and Alexa Fluor 647–conjugated antibodies to fibrin. The experiment was terminated at the end of 10 minutes to avoid excessive loss of blood. (A) Representative images taken 30, 60, and 300 seconds after laser injury. Scale bar, 50 μm. (B) Percentage of bleeding injury sites was plotted over time for each experimental group. Seventeen injury sites in 3 control mice, 39 injury sites in 5 Tln1-mR35E,R118E mice, and 8 injury sites in 2 Rap1a/b-mKO mice were tested. Statistical significance was assayed by using the Gehan-Breslow-Wilcoxon test with Bonferroni correction. (C-D) Comparisons of the 3 pairs of groups were all statistically significant using a family-wise significance level of 5%. Flow cytometry analysis of P-selectin (CD62P) surface expression onto GPIX-labeled platelets in whole blood in response to PAR4-AP (C) or convulxin (D) stimulation. Bar graphs represent Δ mean fluorescence intensity (MFI) ± standard error of the mean (n = 6 mice). Two-way analysis of variance with Tukey post-test. (E) Determination according to flow cytometry of PS exposure onto GPIX-labeled platelets in whole blood in response to dual stimulation with 100 ng/mL of convulxin and 500 μM of PAR4-AP. Bar graph represents the percent mean ± standard error of the mean (n = 6 mice). Two-way analysis of variance with Tukey posttest. **P < .01; ***P < .001. ns, not significant.
In addition to platelet aggregation, secretion of platelet granule contents and generation of procoagulant activity contribute to hemostasis. Deficiency of Rap1 in platelets impairs α-granule secretion.12,16 Therefore, we next investigated the contribution of the Rap1–talin-1 interaction in platelets to granule secretion. P-selectin exposure was largely intact in Tln1-mR35E,R118E platelets in response to PAR4-AP and partially impaired in response to convulxin stimulation (Figure 5C-D). Similar preservation of P-selectin expression was reported in talin-1–null platelets.8,9 In sharp contrast, Rap1a/b-mKO platelets showed a much more profound secretion defect in response to both agonists, indicating that Rap1 activation is intact in Tln1-mR35E,R118E platelets (Figure 5C-D). These results are consistent with previous reports that Rap1b is more important for platelet secretion in response to GPVI than PAR4 stimulation.29 Agonist-induced exposure of PS on the outer leaflet of the plasma membrane in platelets confers procoagulant activity by increasing the assembly of coagulation protease/cofactor complexes, which enhances thrombin generation. PS exposure in platelets depends on signaling by Rap1,30 which led us to investigate the PS exposure in Tln1-mR35E,R118E or Rap1a/b-mKO platelets that were stimulated with convulxin combined with PAR4-AP to induce PS exposure. Remarkably, PS exposure was reduced in Tln1-mR35E,R118E platelets compared with control platelets (Figure 5E). However, Rap1a/b-mKO platelets exhibited more severe inhibition in PS exposure compared with Tln1-mR35E,R118E platelets. Thus, our findings reveal that the Rap1–talin-1 interaction in platelets is essential for integrin activation but cannot fully account for impaired thrombocytopoiesis, granule secretion, and PS exposure seen in combined deficiency of the 2 Rap1 isoforms.
Discussion
Many different signaling pathways have been implicated in the capacity of agonists such as ADP, thrombin, and collagen to trigger platelet integrin activation required for platelet aggregation and normal hemostasis. All of these signaling pathways converge on activation of Rap1, which in turn results in talin-1 binding to the integrin β cytoplasmic domain, a final step in integrin activation. Here we analyzed mice bearing mutations in one or both of the Rap1-binding sites of talin-1 and used these mice to define the connection between Rap1 and talin-1 in platelets. Compared with the talin-1 F1 domain, the talin-1 F0 domain makes a relatively small contribution to integrin activation in platelets. Disabling both Rap1-binding sites had a greater effect than disabling F1 alone and recapitulated the defect in integrin activation seen in platelets lacking both Rap1a and Rap1b. Thus, talin-1 is the principal and perhaps only platelet Rap1 effector for integrin activation; however, loss of Rap1–talin-1 did not phenocopy defects in thrombocytopoiesis, secretion, and surface exposure of PS observed in Rap1-deficient platelets. These results combined with recent structural studies suggest a mechanism whereby Rap1 mediates initial recruitment of talin-1 to the platelet membrane, resulting in unmasking of the talin-1 integrin-binding site and integrin activation in hemostasis.
Disabling the Rap1-binding sites in talin phenocopies lack of Rap1a and Rap1b with regard to platelet integrin activation and aggregation, indicating that talin-1 is a major Rap1 effector involved in these processes. Deletion of talin-1 itself had a slightly stronger effect, suggesting additional mechanisms, such as disruption of talin-1 autoinhibition by Gα13,31 in talin-1–dependent integrin activation. The fact that convulxin, which binds GPVI and signals via Syk tyrosine kinase, was slightly less affected suggests that tyrosine kinase signaling may provide another such alternative pathway. Interestingly, Rap1a/b-mKO mice exhibited a more profound hemostatic defect than Tln1-mR35E,R118E mice, even though integrin activation in platelets from these mice was impaired to a similar extent. This result can be ascribed to the mild thrombocytopenia and additional platelet function defects, such as impaired granule release and procoagulant response, in mRap1a/b-KO mice. These added defects also highlight the potential importance of one of the many other known Rap1 effectors.32,33 The defects in thrombocytopoiesis and platelet morphology in mRap1a/b-KO mice raised the possibility that the defective integrin activation, in part, could be due to a developmental defect in megakaryocytes rather than lack of Rap1 signaling in platelets. The presence of normal platelet counts, intact gross morphology, and secretion in Tln1-mR35E,R118E platelets further indicates that Rap1 is the major final signaling element in platelet integrin activation.
In combination with recent structural studies,18 the data reported here suggest a model for the main final steps in platelet integrin activation. Earlier work with talin-1 fragments in model systems showed that the talin-1 F3 domain alone is sufficient for activation.34 Even in the absence of Rap1 signaling, the presence of the lipid-binding sites in the F2 domain markedly increases the capacity of talin-1 to disrupt the integrin α and β subunit transmembrane domain interaction, resulting in integrin activation.35-38 In contrast to these talin-1 fragments, full-length talin-1 is autoinhibited,39 and this autoinhibition can be relieved by binding to phosphatidylinositol (4,5)-bisphosphate (PIP2).40 In full-length talin-1, the integrin-binding site in F3 is obscured by its interaction with the R9 helical bundle of the rod domain,41-43 and a recent cryo-electron microscopic structure of talin-1 revealed that the critical PIP2-binding site in F2 is masked by the R12 helical bundle18 (Figure 6). The talin-1 F0 and F1 domains were not part of the autoinhibited structure; mutations in them are therefore unlikely to perturb autoinhibition, and F0 and F1 are thus available to bind to Rap1-GTP in the plasma membrane.18 The lipid-binding loop in F119 enables F1 to make a greater contribution than F0 to this interaction with the plasma membrane. At the membrane, PIP2 can disrupt the F2–R12 and F3–R9 interactions, thus unveiling the integrin-binding site accounting for the critical role of PIP2 in integrin activation.43,44 The binding of the F3 domain to the integrin β tail provides a final cooperative link of the talin-1 head domain to the plasma membrane and induces activation of αIIbβ3 and β1 integrins in platelets.7,45
Figure 6.

Model for the main final steps in talin-1–dependent platelet integrin activation. (A) Talin-1 is autoinhibited where the F3 integrin–binding site (red disc) is obscured by its interaction with the R9 helical bundle, and the PI(4,5)P2 binding site in F2 and F3 are masked by the R12 helical bundle. (B) The Rap1-binding sites in F0 and F1 were not part of the autoinhibited structure and therefore available to bind Rap1-GTP.18 (C) At the membrane, PI(4,5)P2 can disrupt the F3–R9 and F2–R12 interactions to unveil the talin-1 F3 integrin–binding site (red disc). (D) The binding of talin-1 F3 domain to the integrin β tail induces activation of αIIbβ3 and β1 integrins.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the University of California Irvine Transgenic Mouse Facility (UCI TMF) for design help and production of CRISPR-modified mice. The UCI TMF is a shared resource funded in part by the Chao Family Comprehensive Cancer Center Support Grant (P30CA062203) from the National Institutes of Health, National Cancer Institute. The authors also thank Marianna Plozza for her technical help in analyzing the potential CRISPR-Cas9 off-target sites.
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under grant agreement No 841428 (F.L.); the American Heart Association Career Development Award 18CDA34110228 (F.L.), postdoctoral fellowship 17POST33660181 (H.S.), and Grant-In-Aid 16GRNT29650005 (A.R.G.); and National Institutes of Health, National Heart, Lung and Blood Institute grants KO1 HL133530 (M.A.L.-R.), HL078784 and HL139947 (M.H.G.), and R35 HL144976 (W.B.).
Footnotes
Contact the corresponding author for original data.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: F.L. and M.H.G. conceived the study; F.L., D.S.P., W.B., and M.H.G. designed experiments, interpreted data, and wrote the manuscript; F.L., D.S.P., A.J.V., H.S., J.L., and M.N.C. performed and analyzed experiments; F.L. designed the CRISPR/Cas9 approach to generate Tln1-R118E and Tln1-R35E,R118E mutant mice; and A.R.G., H.S., J.N.A., and M.A.L.-R. provided vital reagents and critical expertise.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mark H. Ginsberg, University of California San Diego, 9500 Gilman Dr, MC 0726, La Jolla, CA 92093; e-mail: mhginsberg@ucsd.edu.
REFERENCES
- 1.Bennett JS, Vilaire G. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J Clin Invest. 1979;64(5):1393-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coller BS, Peerschke EI, Scudder LE, Sullivan CA. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest. 1983;72(1):325-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEver RP, Bennett EM, Martin MN. Identification of two structurally and functionally distinct sites on human platelet membrane glycoprotein IIb-IIIa using monoclonal antibodies. J Biol Chem. 1983;258(8):5269-5275. [PubMed] [Google Scholar]
- 4.Ginsberg M, Pierschbacher MD, Ruoslahti E, Marguerie G, Plow E. Inhibition of fibronectin binding to platelets by proteolytic fragments and synthetic peptides which support fibroblast adhesion. J Biol Chem. 1985;260(7):3931-3936. [PubMed] [Google Scholar]
- 5.Kloczewiak M, Timmons S, Lukas TJ, Hawiger J. Platelet receptor recognition site on human fibrinogen. Synthesis and structure-function relationship of peptides corresponding to the carboxy-terminal segment of the gamma chain. Biochemistry. 1984;23(8):1767-1774. [DOI] [PubMed] [Google Scholar]
- 6.Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov. 2016;15(3):173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrich BG, Fogelstrand P, Partridge AW, et al. . The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J Clin Invest. 2007;117(8):2250-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrich BG, Marchese P, Ruggeri ZM, et al. . Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204(13):3103-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieswandt B, Moser M, Pleines I, et al. . Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204(13):3113-3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tadokoro S, Shattil SJ, Eto K, et al. . Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103-106. [DOI] [PubMed] [Google Scholar]
- 11.Stefanini L, Bergmeier W. RAP GTPases and platelet integrin signaling. Platelets. 2019;30(1):41-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefanini L, Lee RH, Paul DS, et al. . Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood. 2018;132(18):1951-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC II. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115(3):680-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2(5):369-377. [DOI] [PubMed] [Google Scholar]
- 15.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crittenden JR, Bergmeier W, Zhang Y, et al. . CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation [published correction appears in Nat Med. 2004;10(10):1139]. Nat Med. 2004;10(9):982-986. [DOI] [PubMed] [Google Scholar]
- 17.Klapholz B, Brown NH. Talin—the master of integrin adhesions. J Cell Sci. 2017;130(15):2435-2446. [DOI] [PubMed] [Google Scholar]
- 18.Dedden D, Schumacher S, Kelley CF, et al. . The architecture of Talin1 reveals an autoinhibition mechanism. Cell. 2019;179(1):120-131.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goult BT, Bouaouina M, Elliott PR, et al. . Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 2010;29(6):1069-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu L, Yang J, Bromberger T, et al. . Structure of Rap1b bound to talin reveals a pathway for triggering integrin activation. Nat Commun. 2017;8(1):1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagarrigue F, Gingras AR, Paul DS, et al. . Rap1 binding to the talin 1 F0 domain makes a minimal contribution to murine platelet GPIIb-IIIa activation. Blood Adv. 2018;2(18):2358-2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bromberger T, Klapproth S, Rohwedder I, et al. . Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood. 2018;132(26):2754-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gingras AR, Lagarrigue F, Cuevas MN, et al. . Rap1 binding and a lipid-dependent helix in talin F1 domain promote integrin activation in tandem. J Cell Biol. 2019;218(6):1799-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bromberger T, Zhu L, Klapproth S, Qin J, Moser M. Rap1 and membrane lipids cooperatively recruit talin to trigger integrin activation. J Cell Sci. 2019;132(21):jcs235531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan BX, Vautier F, Ito W, Bolshakov VY, Morozov A. Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J Neurosci. 2008;28(9):2089-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109(4):1503-1506. [DOI] [PubMed] [Google Scholar]
- 27.Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. J Thromb Haemost. 2015;13(3):417-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. 2002;48(2):80-86. [DOI] [PubMed] [Google Scholar]
- 29.Zhang G, Xiang B, Ye S, et al. . Distinct roles for Rap1b protein in platelet secretion and integrin αIIbβ3 outside-in signaling. J Biol Chem. 2011;286(45):39466-39477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmad F, Boulaftali Y, Greene TK, et al. . Relative contributions of stromal interaction molecule 1 and CalDAG-GEFI to calcium-dependent platelet activation and thrombosis. J Thromb Haemost. 2011;9(10):2077-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiemer J, Bohm A, Lin L, et al. . Gα13 switch region 2 relieves talin autoinhibition to activate αIIbβ3 integrin. J Biol Chem. 2016;291(52):26598-26612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boettner B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol. 2009;21(5):684-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gloerich M, Bos JL. Regulating Rap small G-proteins in time and space. Trends Cell Biol. 2011;21(10):615-623. [DOI] [PubMed] [Google Scholar]
- 34.Calderwood DA, Yan B, de Pereda JM, et al. . The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277(24):21749-21758. [DOI] [PubMed] [Google Scholar]
- 35.Han J, Lim CJ, Watanabe N, et al. . Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol. 2006;16(18):1796-1806. [DOI] [PubMed] [Google Scholar]
- 36.Anthis NJ, Wegener KL, Ye F, et al. . The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009;28(22):3623-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim C, Ye F, Hu X, Ginsberg MH. Talin activates integrins by altering the topology of the β transmembrane domain. J Cell Biol. 2012;197(5):605-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim C, Lau TL, Ulmer TS, Ginsberg MH. Interactions of platelet integrin alphaIIb and beta3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood. 2009;113(19):4747-4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan B, Calderwood DA, Yaspan B, Ginsberg MH. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem. 2001;276(30):28164-28170. [DOI] [PubMed] [Google Scholar]
- 40.Martel V, Racaud-Sultan C, Dupe S, et al. . Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem. 2001;276(24):21217-21227. [DOI] [PubMed] [Google Scholar]
- 41.Banno A, Goult BT, Lee H, Bate N, Critchley DR, Ginsberg MH. Subcellular localization of talin is regulated by inter-domain interactions. J Biol Chem. 2012;287(17):13799-13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goult BT, Bate N, Anthis NJ, et al. . The structure of an interdomain complex that regulates talin activity. J Biol Chem. 2009;284(22):15097-15106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song X, Yang J, Hirbawi J, et al. . A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012;22(11):1533-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chinthalapudi K, Rangarajan ES, Izard T. The interaction of talin with the cell membrane is essential for integrin activation and focal adhesion formation. Proc Natl Acad Sci U S A. 2018;115(41):10339-10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haling JR, Monkley SJ, Critchley DR, Petrich BG. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood. 2011;117(5):1719-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.