

SUMMARY
BAX is a pro-apoptotic protein that transforms from a cytosolic monomer into a toxic oligomer that permeabilizes the mitochondrial outer membrane. How BAX monomers assemble into a higher-order conformation, and the structural determinants essential to membrane permeabilization, remain a mechanistic mystery. A key hurdle has been the inability to generate a homogeneous BAX oligomer (BAXO) for analysis. Here, we report the production and characterization of a full-length BAXO that recapitulates physiologic BAX activation. Multidisciplinary studies revealed striking conformational consequences of oligomerization and insight into the macromolecular structure of oligomeric BAX. Importantly, BAXO enabled the assignment of specific roles to particular residues and α-helices that mediate individual steps of the BAX activation pathway, including unexpected functionalities of BAX α6 and α9 in driving membrane disruption. Our results provide the first glimpse of a full-length and functional BAX oligomer, revealing structural requirements for the elusive execution-phase of mitochondrial apoptosis.
Graphical Abstract
eTOC
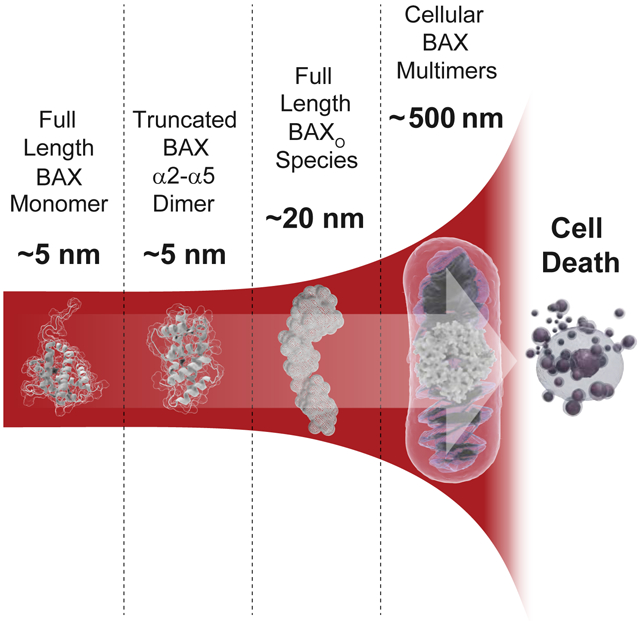
Hauseman, Harvey et al. overcome longstanding barriers in generating a full-length, homogeneous oligomer of BAX for analysis. BAXO is a curvilinear species of 6-8 monomeric units spanning 20 nm. BAXO bridges the gap between monomeric/dimeric BAX and its macromolecular pores, providing mechanistic insights into the execution phase of mitochondrial apoptosis.
INTRODUCTION
Apoptosis is an essential biological process that dictates the balance between new and dying cells, thereby ensuring organism homeostasis. Deregulation of apoptosis gives rise to a host of human diseases characterized by cellular excess or premature cell demise (Czabotar et al., 2014). BCL-2 family proteins control the apoptotic outcome by alternatively guarding or destroying the integrity of the mitochondrial outer membrane (MOM). Thus, defining the structure, function, and druggability of BCL-2 protein interactions remains a high priority goal of basic, translational, and pharmaceutical scientists (Garner et al., 2017). Indeed, the mechanism by which anti-apoptotic BCL-2 proteins deploy their surface groove to trap the BCL-2 homology 3 (BH3) α-helix of pro-apoptotic members has been linked to the development and chemoresistance of human cancer (Hata et al., 2015; Sattler et al., 1997). Small molecule and peptidic inhibitors of the anti-apoptotic groove can reactivate apoptosis in cancer by liberating BAX and BAK, the two essential “executioner” proteins of the BCL-2 family (Oltersdorf et al., 2005; Walensky et al., 2004).
Although anti- and pro-apoptotic BCL-2 family members are similarly globular proteins composed of α-helices, BAX and BAK have the unique capacity to undergo a structural transformation that results in homo-oligomerization at the MOM, leading to its loss of integrity and release of apoptogenic factors (Antonsson et al., 2000; Wei et al., 2000). Whereas structures of the stable complexes between anti-apoptotic proteins and isolated pro-apoptotic BH3 helices have been widely reported, insight into the conformation of functional BAX and BAK homo-oligomers or “death channels” has been elusive, and thus dubbed the “holy grail” of apoptosis research (Youle and Strasser, 2008). The structures of full-length (FL) monomeric BAX (BAXM) (α1-α9) (Suzuki et al., 2000) and a dimeric BAX truncate (α2-α5) (Czabotar et al., 2013) have been determined, both measuring ~5 nm in greatest dimension, and a broad and heterogeneous distribution of membrane-associated BAX multimers or macropores measuring hundreds of nm in size have been visualized in apoptotic cells (Grosse et al., 2016; McArthur et al., 2018; Salvador-Gallego et al., 2016). However, what transpires between these two extremes of the BAX activation continuum represents a critical missing link in our understanding of mitochondrial apoptosis and has awaited the characterization of a homogeneous oligomeric unit of full length BAX, which has thus far been refractory to production and isolation.
BAX- and BAK-mediated apoptosis is initiated by both indirect and direct mechanisms. Activated, BH3-exposed conformers of BAX and BAK are held in check by the BH3-in-groove interaction of anti-apoptotic proteins. In response to cellular stress, anti-apoptotic targeting by the pro-apoptotic “BH3-only” subclass of BCL-2 proteins can release BAX and BAK, resulting in their self-association and permeabilization of the mitochondria (Yang et al., 1995). The question of how BAX or BAK are activated in the first place led to the hypothesis that pro-apoptotic BH3-domain containing proteins could likewise target BAX and BAK directly to turn on these executioner proteins (Cheng et al., 2001; Kuwana et al., 2005; Letai et al., 2002). Structurally-stabilized peptides corresponding to the BH3 domains of select BH3-only proteins, such as BIM and BID, were found to directly interact with BAX at a unique α1/α6 “trigger site” located at its N-terminal surface (Gavathiotis et al., 2008; Walensky et al., 2006) (Figure 1A). This distinctive BH3-in-groove interaction induced a dramatic conformational change in BAX, characterized by displacement of the α1-α2 loop, exposure of the essential BAX BH3 helix, and release of the C-terminal α9 helix for mitochondrial translocation (Gavathiotis et al., 2010). In contrast to BAX, BAK resides within the MOM and thus appears to bypass the N-terminal triggering mechanism required for BAX translocation. Instead, BH3 helices activate BAK by engaging the canonical BH3-binding site at its C-terminal face (Leshchiner et al., 2013; Moldoveanu et al., 2013). A unified model of BAX and BAK activation incorporates the features of indirect and direct mechanisms (Leber et al.,2007; Llambi et al., 2011). Beyond this BAX and BAK activation phase, the structural features and functional determinants of the downstream “execution phase” of BAX- and BAK-mediated mitochondrial apoptosis remain unresolved.
Figure 1. Induction of a stable and homogeneous BAX oligomer by Fos-12.

(A) Structure of full-length, monomeric BAX (PDB:1F16) demonstrating the domains and binding surfaces implicated in BAX activation, including α1/α6 trigger site (teal), BH3 domain (α2, red), and C-terminal translocation helix (α9, orange).
(B) SEC analysis of BAX species generated upon treatment of BAXM with the indicated detergents.
(C) Blue native PAGE of BAXM and Fos-12 induced BAXO. Data shown are representative of two independent biological replicates.
(D) SAXS analysis of BAXO performed at 1, 3, and 5 mg/mL.
(E) Guinier plot of the SAXS data demonstrated a linear dependence of ln(I(Q)) versus Q2, indicating BAXO is a monodispersed oligomeric species.
(F) SAXS calculations indicate the size and spatial characteristics of BAXO.
(G) Plot of P(r) vs particle radius demonstrates a left-skewed distribution, indicating that BAXO is an elongated rather than globular macromolecular assembly.
(H) Curvilinear structure of BAXO based on the 3D envelope generated by SAXS.
See also Figures S1, S2.
The nuclear magnetic resonance (NMR) solution structure of BAXM and heteronuclear single quantum correlation (HSQC) analyses of BH3-triggered BAX have been critical to understanding the initial transformation of inactive BAX into a conformationally-activated monomer (Gavathiotis et al., 2008; Suzuki et al., 2000). Dimeric structures of BAX have also been reported, including a symmetric BH3-in-groove homodimer (e.g. α2-α5) that has been proposed to represent the core unit of full-length oligomeric BAX (Czabotar et al., 2013) and an auto-inhibitory dimer of full-length BAX in which the α9 surface of one monomer engages and thus blocks the α1/α6 trigger site of another (Garner et al., 2016). Structural determination of higher order BAX species beyond dimers has thus far been refractory to modern methods, presumably due to the heterogeneity of recombinant oligomers produced to date (Antonsson et al., 2000; Bleicken et al., 2010; Lai et al., 2019). Therefore, a series of fundamental questions concerning the essential execution phase of mitochondrial apoptosis remain a mechanistic mystery, including: (1) How do full-length activated BAX monomers assemble into an oligomeric species? (2) What structural features of oligomeric BAX confer the critical membrane-disruptive function? (3) How do these structural determinants induce mitochondrial outer membrane permeabilization (MOMP)? What’s more, the inability to generate homogeneous, full-length oligomeric BAX has precluded mechanistic analyses capable of distinguishing between distinct steps of the BAX activation pathway, such as BH3-triggering and oligomeric permeabilization. Here, we sought to overcome the historical challenges associated with generating pure, homogeneous, and full-length BAX homo-oligomers in order to elucidate the conformational and membrane-permeabilizing features of this essential building block of cell death.
RESULTS
Treatment with Fos-12 Yields a Stable and Homogeneous BAX Oligomer
To screen for conditions that could transform full-length BAX into a stable and pure homooligomer, we subjected recombinant BAXM to treatment with a series of detergents (Figure S1A). Analysis of the products by size exclusion chromatography (SEC), performed with detergent in the mobile phase, revealed elution profiles ranging from retained BAX monomer to full transformation to higher molecular weight peaks of variable complexity (Figure 1B). Fos-12 treatment reproducibly generated the most monodispersed peak, which eluted at a volume corresponding to ~150 kDa and was named BAXO. Comparison of the gel migration profiles of BAXM and Fos-12-induced BAX oligomer (BAXO) by blue native PAGE (BNP) confirmed the production of a single oligomeric band at ~146 kDa (Figure 1C). We determined that detergent micelles do not account for the size of BAXO, since BAXO could be generated using concentrations of Fos-12 below its critical micelle concentration (Figure S1B). Once formed, Fos-12-induced BAXO could be purified without detergent in the SEC mobile phase, highlighting its inherent stability (Figure S1C).
To interrogate the homogeneity and macromolecular shape of Fos-12 induced BAXO, we performed small angle X-ray scattering (SAXS) (Figure 1D). The Guinier plot of the SAXS data revealed a linear dependence of ln(I(Q)) versus Q2, indicating that our oligomeric species of BAX was monodispersed and not an aggregate (Figure 1E). The SAXS-predicted average mass of BAXO is 144-148 kDa (Figure 1F), consistent with that observed by SEC and BNP (Figures 1B-C) and suggestive of an oligomeric unit composed of 6-8 monomers. A plot of P(r) versus particle radius demonstrated a left-skewed distribution, revealing that BAXO is an elongated macromolecular assembly (Figure 1G). The 3D reconstruction envelope reproducibly demonstrated a curvilinear structure of ~20 nm in maximum dimension (Figure 1H).
We next prepared grids of BAXO to evaluate its shape by negative stain EM and observed elongated structures resembling the envelope generated by SAXS (Figure 1H, S2). 2D averaging revealed multiple classes that matched the length and shape of the SAXS-based structure, with an exemplary 3D class providing an excellent fit with the SAXS envelope of BAXO in solution (Figure S2A-C). To determine whether the visualized oligomer units generated by Fos-12 induction could be reproduced by an alternative BAX preparation, we repeated the EM analyses on BAXO species generated by exposing BAXM to BIM SAHBA2, a stabilized alpha-helix of BCL-2 domains (SAHB) modeled after the BIM BH3 helix (Gavathiotis et al., 2008), in the presence of liposomes followed by extraction of BAXO, SEC purification, and grid preparation. We again observed a similar curvilinear structure using the BH3-triggering mechanism of BAX activation and self-association (Figure S2D-F). These data indicate that BAXO is a stable and homogeneous BAX homo-oligomer that reproducibly assembles into an elongated curvilinear macromolecule of approximately 20 nm in length, representing a species that is strikingly distinct in uniformity and size from all prior visualizations of BAX.
BAXO Exhibits the Physiologic Activity of BH3-triggered BAX
To determine if BAXO was functional, we compared its activity upon repurification in detergent-free SEC buffer to truncated BID (tBID)-induced BAX in liposomal translocation and release assays. First, we incubated BAXM, BAXO and tBID-triggered BAX with large-unilamellar vesicles (LUVs) bearing the lipid composition of the MOM, and then monitored BAX translocation by western blot of supernatant and liposomal fractions. Whereas BAXM remained in the solution phase, portions of BAXO and tBID-triggered BAX were found in the liposomal fractions, demonstrating the capacity of BAXO to independently undergo membrane translocation (Figure 2A). Further, BAXO induced liposomal release of entrapped fluorophore with the potency and kinetics of tBID-induced BAX-mediated poration (Figure 2B). Whereas anti-apoptotic full-length BCL-w and BFL-1ΔC effectively blocked tBID-induced BAX-mediated poration (Figure 2C), neither protein could inhibit liposomal permeabilization induced by BAXO (Figure 2D), indicating that its BH3 domains are not accessible for entrapment by the anti-apoptotic groove. We performed negative stain EM to visualize the morphology of the treated liposomes (Figure 2E-G). We observed a similar pattern and size of liposomal membrane disruption for BAXO and tBID-triggered BAX, as reflected by focal regions of circular rupture, a pattern similar to what has also been seen on a larger scale in mitochondria upon activation of native BAX (e.g. BAX “rings” measuring hundreds of nm in diameter) (Grosse et al., 2016; Salvador-Gallego et al., 2016). The BAXO findings also mirror those observed upon activation of BAX by BIM SAHBA2 (Figure S3). We next conducted cytochrome c release assays using purified liver mitochondria from AlbCreposBaxfl/flBak−/− mice (Walensky et al., 2006). Consistent with the liposomal release studies, we observed potent, dose-dependent cytochrome c release upon mitochondrial treatment with BAXO (Figure 2G). These data indicate that BAXO can translocate to and permeabilize liposomal and mitochondrial membranes in a manner similar to BH3-triggered BAX poration.
Figure 2. The biochemical activity of BAXO emulates BH3-triggered BAX.

(A) BAXO and tBID-triggered BAXM translocate to liposomes, whereas vehicle-treated BAXM remains in the soluble fraction, as assessed at 1 h by liposomal translocation assay and BAX western analysis. BAXO and BAXM, 500 nM; tBid, 20 nM. Data shown are representative of two independent biological replicates. Fractions: liposomal, 4-6; supernatant, 7-14.
(B) BAXO and tBID-triggered BAXM induce liposomal poration with similar kinetics and potency, as assessed by liposomal release assay. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM, 500 nM; BAXO, 500 nM (equivalent total protein amounts); tBID, 20 nM.
(C-D) Anti-apoptotic proteins effectively blocked liposomal poration induced by BAXM upon tBID triggering (C), but had no inhibitory effect on BAXO (D). Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM and BAXO 500 nM; tBID, 20 nM; anti-apoptotic proteins, 10 μM.
(E-G) Negative stain EM of liposomes incubated with BAXM (E), tBID-triggered BAXM (F), or BAXO (G), showing similar morphology of membrane disruption and pore size induced by BAXO and BH3-triggered BAXM after 15 min. Mean pore diameter (nm) ± s.e.m.: tBID-triggered BAX, 48.9 ± 2.3; BAXo, 56.2 ± 2.6; p value = 0.195.
(H) BAXO induces dose-responsive cytochrome c release from BAX/BAK-deficient mouse liver mitochondria (MLM). tBID-triggered BAX release is shown as a positive control. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM, 200 nM; BAXO, 6.25-200 nM; tBID, 40 nM.
See also Figures S3 and S4.
Biochemical Validation of the Specific Activity of Fos-12-induced BAXO
We sought to validate the specificity of the BAXO species generated by Fos-12 induction even further using a battery of control experiments. Although we repurified BAXo by SEC in the absence of detergent, we tested for any potential effect of Fos-12 in the liposomal release assay and found no evidence of detergent-mediated permeabilization even up to 100 μM dosing (Figure S1D). To evaluate whether BAXO activity was uniquely dependent on Fos-12 induction, we repeated our workflow using an alternative detergent, n-decyl-β-D-maltoside (DM), observed the same kinetics and level of liposomal poration (Figure 2B, S4A-C). In contrast to Fos-12-induced BAXO, DM-induced BAXO was notably heterogeneous, as demonstrated by SEC and BNP analyses (Figure S4A-B). Thus, the functional outcome of inducing BAX oligomerization with Fos-12 or DM is the same (Figures 2B, S4C), but only Fos-12 yielded a homogeneous preparation, underscoring our rationale for selecting Fos-12 to generate BAXO for this study.
We next investigated whether Fos-12 treatment preserved the biochemical and functional fidelity of BAX. That is, would exposure to Fos-12 induce any BCL-2 family protein to oligomerize and porate membranes? We applied the BAXO workflow to anti-apoptotic BCL-w and BFL-1ΔC, and observed no BAXO-sized oligomers upon SEC analysis, no higher-order species of BCL-w or BFL-1ΔC by BNP, and no liposomal poration activity by Fos-12-treated anti-apoptotic proteins (Figure S4D-I). We further tested whether the disruptive effect of an established BAX mutation, G108E, which impairs oligomerization and BAX-mediated apoptosis in vitro and in cells (Kim et al., 2009), would retain physiologic impairment in the context of our Fos-12 workflow. Indeed, Fos-12-treated BAX G108E manifests deficient oligomerization and membrane permeabilization activity by SEC, BNP, and liposomal release assays (Figure S4J-L). These data emphasize that the distinctive, natural functionalities of pro- and anti-apoptotic proteins, and an exemplary BAX mutant, are not altered by Fos-12 treatment, further reinforcing the fidelity of BAXO.
Conformational Features of the Transformation from BAXM to BAXO
Having documented the functional activity of BAXO, we next sought to characterize its conformational features by hydrogen-deuterium exchange mass spectrometry (HXMS) under the same experimental conditions. We have applied HXMS to evaluate the influence of ligand interaction and the membrane environment on the conformational dynamics of BCL-2 family proteins (Barclay et al., 2015; Lee et al., 2016; Pritz et al., 2017). The relative fractional deuterium uptake of BAXM over time in the presence of liposomes demonstrates a range of deuterium uptake across the protein, reflecting the differences in flexibility and solvent exposure of the discrete regions of BAXM. Consistent with the NMR structure of BAXM (PDB: 1F16), the unstructured N-terminus, portions of the α1-α2 loop, exposed loop regions between α-helices, and the extreme C-terminus demonstrate the most rapid exchange, whereas partially or completely buried regions in α1, α2, α5, α6, and α9 exhibit both lower level and slower time-dependent exchange (Figure 3A). In contrast, BAXO has a remarkably distinct deuterium exchange profile (89.1% sequence coverage, e.g., α5-α6 junction not detected) compared to BAXM, indicating structural reorganization of the oligomeric species (Figure 3B). The N-terminal region including α1, and the junction of α-helices 7 and 8, are more readily deuterated, whereas the BH3 domain (α2), junction of α-helices 4 and 5, proximal portion of α5, distal portion of α8, and the C-terminal helix (α9) are substantially more protected from exchange (Figures 3C-D, S5A). The deuterium exchange profile of BAXO is relatively unchanged over time compared to BAXM (Figure 3A-B), highlighting the conformational stability of BAXO. Further, the deuterium exchange profile of BAXO in the presence and absence of liposomes is essentially unchanged (Figure S5A-B). Although regions of conformational change that transition from one buried disposition in solution to another in the membrane would not be observed by HXMS, these data indicate that the pre-formed homo-oligomeric structure does not undergo major conformational reorganization upon membrane interaction.
Figure 3. HXMS identifies regiospecific conformational changes upon conversion of BAXM to BAXO.

(A-B) HXMS profiles of BAXM and BAXO as measured over time in the presence of liposomes. Data are representative of two independent biological replicates.
(C) Deuterium difference plot showing the relative deuterium incorporation of BAXO minus that of BAXM, as measured over time with liposomes. Orange and green color scales indicate extent of conformational deprotection (increased exchange) and protection (decreased exchange), respectively. Data are representative of two independent biological replicates.
(D) Prominent regions of deprotection (N-terminus, α1, α7-α8 junction) and protection (α2, α4-α5 loop, proximal α5, distal α8, and α9) upon conversion of BAXM to BAXO are mapped onto BAXM (PDB: 1F16) according to the orange and green color scales (significance threshold, ± 1.0 Da).
Placed in the context of prior work, the HXMS profile of BAXO reveals several structural insights. First, the very regions deprotected upon transformation of BAXM into BAXO are protected upon incubation of BAXM with a stapled BCL-2 BH4 helix, consistent with our previous finding that BAX-inhibitory BH4 domains preserve the inactive form of BAXM by imposing highly-specific and functionally-relevant structural constraints (Barclay et al., 2015), which are strikingly inverse to those observed in BAXO (Figure S5C-D). Whereas we previously characterized ligand-induced exposure of the N-terminal region and BH3 domain as reflecting the initiating steps of BAX activation (Barclay et al., 2015; Gavathiotis et al., 2010), here we observe for the first time in the context of BAXO the persistence of these N-terminal changes along with marked protection of other functional regions, most notably the pro-apoptotic BAX BH3 and α9 membrane-translocation helices. The defined structural changes in BAXO (Figure 3A-D), and their stability both in solution and upon exposure to membranes (Figure S5A-B), underscores that BAXO achieves a conformationally-distinct and energetically-favorable state required for membrane disruption.
Comparative Chemical Crosslinking Analyses Reveal Key Structural Differences between BAXM and BAXO
To compare the proximity of structural regions in BAX before and after oligomerization, we subjected BAXM and BAXO to a series of established and newly-reported chemical reagents (Mintseris and Gygi, 2020) that either crosslink amine (K) or acidic (D, E) residues within a distance of 33 Å (Figure 4A). Mass spectrometry (MS) analyses were performed to detect the crosslinks generated in independent preparations of BAXM and BAXO, and then compared to identify crosslinks that were at least 10-fold enriched in either BAXM or BAXO. To validate our method, we mapped the list of BAXM-enriched crosslinks onto the BAXM NMR structure (PDB: 1F16). After accounting for the conformational flexibility of loop regions, 100% of the identified crosslinks were compatible with the NMR structure of BAXM, providing robust validation of our methodology (Figure 4B-F, Data File S1). Because this roster of BAXM-specific crosslinks corresponds to structural proximities that are absent in the oligomer, we can conclude that upon BAXM activation and formation of BAXO, the α1-α2 loop is separated from α1 and α6, α1 loses proximity to α2, and the α3-α4 loop and α4 move away from the core α5-α6 hairpin and α9 (Figure 4C-F). These findings are consistent with a major structural reorganization of BAX and the mechanistic framework for BAX activation that includes initiation by BH3-only protein displacement of the α1-α2 loop, followed by conformational release and transient exposure of α2 (BAX BH3 domain) and α9 (C-terminal translocation helix) (Gavathiotis et al., 2010).
Figure 4. BAXM-enriched chemical crosslinks highlight the structural proximities lost upon formation of BAXO.

(A) Molecular structures, crosslinking reactions, and crosslink distance ranges (disuccinimidyl sulfoxide [DSSO], experimental; 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide [EDC], extended [E]-EDC, theoretical) of the chemical crosslinkers.
(B) BAXM-enriched (10-fold) crosslinks as determined by crosslinking acidic and lysine residues of BAXM vs. BAXO and mass spectrometry analyses. Crosslinked residues are colored according to their location on the BAXM structures below.
(C-F) Each of the identified crosslinks are compatible with the structure of BAXM (PDB: 1F16) and reveal the structural proximities in BAXM lost upon BAX oligomerization, including the adjacencies of (C) the α1-α2 loop with α1 and α6 (red), (D) α1 with α2 (orange), (E) α3/α4 with α5/α6 (cyan), and (F) α4 with α5/α6 and α9 (purple).
See also Data File S1.
We next turned to evaluating the BAXO-enriched crosslinks (Figure 5A). We first tested the compatibility of our BAXO crosslinks with the crystal structure of a dimer derived from a BAX construct lacking the C-terminal α9 helix (BAXΔC) (PDB: 4BD2) (Czabotar et al., 2013). Because this domain-swapped dimer was determined to be an off-pathway structure (Czabotar et al., 2013), we wanted to confirm that the structural organization of BAXO was distinct from this non-physiologic species. Indeed, we found that the distance constraints of the majority of crosslinks, whether mapped as intra- or inter-monomeric crosslinks, are violated by the core/latch domain-swapped dimer of BAXΔC (Figure 5B-C, Data File S1). Focusing next on BAXO crosslinks between identical residues, which exclusively report on inter-monomeric proximity, we detected crosslinks between distinct peptides containing (1) E17 and E17/K21 at the proximal to mid-portion of α1, (2) K58 and K57/K58 at the proximal portion of α2, and (3) D69 and D69/D71 at the distal portion of α2 (Figure 5D, Data File S1). These data suggest that the unstructured N-terminus and newly exposed α1 of BAX in the oligomeric state are adjacent to one another and that the hydrophilic faces of the buried α2 helices are in close proximity. We tested the compatibility of these crosslinks with the dimeric structure of a GFP-BAX truncate comprised of α2-α5 only (PDB: 4BDU), which is believed to be the core unit of a proposed BAX oligomer of dimers (Czabotar et al., 2013). We found that one α2-α3 and two α2-α2 intermolecular crosslinks are compatible with the reported α2-α5 dimer, whereas two distinct α2-α2 intermolecular crosslinks (K57-K58 and K58-K58) are incompatible, positioning these residues 37.0 and 35.1 Å apart, respectively 4 Å and 2 Å longer than the maximum experimentally-determined disuccinimidyl sulfoxide (DSSO) crosslinking distance (Gonzalez-Lozano et al., 2020) (Figure 5E, Data File S1). These data suggest that either the physiologic proximities between α2-α5 subunits in a full-length and functional BAX oligomer are different than what occurs upon dimerization of an α2-α5 truncate in solution, or that the incompatible crosslinks reflect our identification of inter-dimeric proximities within the higher-order BAXO species – two distinct explanations that are equally intriguing.
Figure 5. BAXO-enriched chemical crosslinks reveal structural proximity of the N-terminal and BH3 domain regions of oligomerized BAX.

(A) BAXO-enriched (10-fold) chemical crosslinks. Obligate intermolecular crosslinks between the indicated residues at the proximal portion of α1 and the proximal, middle, and distal portions of α2 highlights the adjacency of these structural regions in BAXO. Tabulated intra- and inter-BAX residue distances are based on the dimeric structure of BAXΔC (PDB: 4BD2).
(B-C) Mapping the BAXO-enriched crosslinks onto the BAXΔC dimer (PDB: 4BD2) as inter- (B) or intra- (C) molecular amino acid proximities (orange lines) demonstrates that the structure of BAXO is distinct from the off-pathway dimer, which violates the distance constraints of BAXO crosslinks.
(D) BAXO-enriched (10-fold) intermolecular crosslinks highlight the proximity of BAX BH3 domains in oligomerized BAX. Tabulated inter-BAX residue distances are based on the GFP-BAX α2-α5 dimer (PDB: 4BDU).
(E) Mapping BAXO-enriched intermolecular crosslinks onto the GFP-BAX α2-α5 dimer (PDB: 4BDU) reveals inter-residue distances (orange lines) that are compatible with (K58-E75, E69-E69, D71-E69) and violate (K57-K58, K58-K58) the distance constraints of BAXO crosslinks.
See also Data File S1.
BAXO Distinguishes between the Sequence Determinants of BH3-triggering and Oligomeric Membrane Permeabilization
The observed differences between the structural organization of BAXO as compared to the dimeric units formed using BAX truncates highlight the potential importance of employing full-length protein in structure-function analyses of the BAX activation pathway. Indeed, the interaction between the C-terminal groove and BAX α9, which is absent in the truncate structures, blocks the canonical BH3-binding pocket in inactive BAXM, unmasking the BH3-trigger site on the opposite, N-terminal face of the protein (Edwards et al., 2013; Gavathiotis et al., 2008) (Figure 1A). Point mutations at the α1/α6 trigger site that produce binding incompatibilities at this BH3 interface impair BAX activation, highlighting the physiologic relevance of the direct activation mechanism (Dengler et al., 2019; Gavathiotis et al., 2010; Gavathiotis et al., 2008). Whether such mutations also impair the oligomerization process is difficult to assess without the capacity to bypass this critical initiation step. Now, with BAXO in hand, we sought to deploy this unique “execution-phase” reagent to distinguish between the sequence determinants for two critical steps of BAX activation, BH3-triggering and oligomer-driven membrane permeabilization. For example, I133 is an α6 residue that undergoes chemical shift perturbation by HSQC NMR upon titration of a stapled BIM BH3 helix and is a component of the α1/α6 trigger site predicted to engage in compatible hydrophobic contacts with direct activator helices (Gavathiotis et al., 2008). A recent study confirmed that mutagenesis of trigger site residues, such as I133, impair BAX activation and thus BAX-mediated apoptosis (Dengler et al., 2019). However, beyond the potential effect of such a mutation on triggering, its involvement in critical downstream steps, such as oligomerization and membrane permeabilization, are difficult to distinguish and thus rigorously address.
We compared the influence of I133A mutagenesis in the context of triggering using tBID-induced BAXM I133A activation, with the capacity to oligomerize and permeabilize using BAXO I133A. We observed that I133A mutagenesis markedly impaired the ability of BIM SAHBA2 or tBID to trigger the activation and membrane translocation of BAXM, as assessed by liposomal SEC and BAX western analysis (Figure 6A). Consistent with these data, I133A mutagenesis reduced tBID-triggered, BAX-mediated liposomal release by more than 50% compared to wild-type (WT) BAX (Figure 6B). However, in striking contrast, BAXO I133A retained the capacity to auto-translocate to liposomal membranes (Figure 6A) and was fully competent in inducing liposomal release (Figure 6C). Thus, BAXO can be an effective tool to distinguish between key steps of the BAX activation pathway, as demonstrated here for a mutation that impairs BH3 triggering but has little to no effect on oligomeric permeabilization.
Figure 6. BAXO is an execution-phase reagent that distinguishes between key steps of the BAX activation pathway and reveals dual roles of BAX α9.

(A) I133A mutagenesis impairs BH3-triggered membrane translocation of BAXM, whereas BAXO I133A is essentially unaffected, as assessed at 1 h by liposomal translocation assay and BAX western analysis. BAXM proteins, 500 nM; BAXO proteins, 500 nM; BIM SAHBA2, 500 nM; tBID, 20 nM. Data shown are representative of two independent biological replicates. Fractions: liposomal, 4-6; supernatant, 7-14.
(B-C) tBID-triggered BAXM I133A exhibits decreased kinetics and 50% overall reduction of liposomal release compared to tBID-triggered BAXM (B), whereas I133A mutagenesis has no adverse effect in the context of BAXO (C). Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM proteins, 500 nM; BAXO proteins, 500 nM; tBID, 20 nM.
(D) Membrane translocation kinetics of BAXOΔC vs. BAXO, as monitored by liposomal translocation assay and BAX western analysis. BAXO proteins, 500 nM. Data shown are representative of two independent biological replicates. Fractions: liposomal, 4-6; supernatant, 7-14.
(E-F) Decreased kinetics and extent of membrane permeabilization of tBID-triggered BAXMΔC as compared to tBID-triggered BAXM (E) and of BAXOΔC as compared to BAXO (F), as assessed by liposomal release assay. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM, BAXMΔC, 500 nM; BAXO, BAXOΔC, 500 nM; tBID, 20 nM.
(G) Negative stain EM of liposomes incubated with BAXOΔC for 15 min. Black-squared region enlarged below.
(H) Comparative dose-responsive cytochrome c release upon treatment of BAX/BAK-deficient MLM with BAXOΔC or BAXO. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM and BAXMΔC, 200 nM; BAXO and BAXOΔC, 6.25-200 nM.
See also Figure S6.
The C-terminal Helix of BAX Enhances Both the Kinetics of Translocation and Efficacy of Membrane Permeabilization
Another quandary of the BAX activation mechanism is the role of the α9 helix, which is believed to be the mitochondrial targeting and membrane insertion helix for BAX, but its influence on oligomerization and permeabilization have been otherwise difficult to assess. We harnessed our capacity to generate homogeneous BAX oligomers to evaluate the role of α9 by comparing the conformations and functionalities of BAXO vs. BAXOΔC. We demonstrated that applying the identical Fos-12 treatment and work-up for BAXM to BAXMΔC enabled us to isolate a homogenous BAXOΔC of similar size (minus the mass of α9) to BAXO (Figure S6A). Aside from the absence of α9, the comparative HXMS profile of BAXOΔC and BAXMΔC is also similar to that of BAXO and BAXM (Figures 3C-D, S5A, S6B-C). However, in liposomal translocation assays, we found that despite ultimately achieving a similar extent of membrane accumulation, the kinetics of translocation for BAXOΔC was impaired compared to BAXO (Figure 6D), as was the kinetics of liposomal release for both tBID-triggered BAXΔC (Figure 6E) and BAXOΔC (Figure 6F), consistent with a role for BAX α9 in enhancing the efficiency of membrane targeting. Despite ultimately reaching the liposomal membranes, the absolute quantity of endpoint liposomal release was reduced both for tBID-triggered BAXΔC (Figure 6E) and BAXOΔC (Figure 6F), implicating α9 in the mechanism of membrane permeabilization as well. Compared to the findings with BAXO (Figure 2G), negative stain EM demonstrated that BAXOΔC-treated liposomes were predominantly intact, with both a reduced number and caliber of circular ruptures when evaluated at the same time point (Figure 6G, S6D). Dose-responsive mitochondrial cytochrome c release was likewise reduced for BAXOΔC compared to BAXO (Figure 6H). These data indicate that BAX α9 not only enhances the efficiency of membrane translocation but also plays a role in the effectiveness of membrane permeabilization itself.
BAX α6 is an Arginine-rich Amphipathic Helix with Membrane-lytic Features of Antimicrobial Peptides
Although BAXOΔC showed impaired kinetics of membrane association, key functionalities such as oligomer formation (Figure S6A), translocation (Figure 6D), and at least partial membrane permeabilization (Figure 6F, 6H, S6D) were maintained without α9, raising the question: what other structural determinants of BAXO drive membrane interaction and disruption? Quantifying the hydrophobic landscape along the length of the BAX protein revealed that α6 is second only to α9 in membrane-lytic potential, as calculated by an algorithm developed to predict the “lyticity” of amphipathic α-helices (Mourtada et al., 2019) (Figure 7A). Sequence analyses also revealed relative arginine enrichment in BAX α6 (Figure 7B), with R134 and R145 projecting outward from the protein surface (Figure 7C). We recently determined that the combination of positive charge and high hydrophobic content is a risk factor for generating stapled peptide helices with membrane-disruptive features (Bird et al., 2016). In addition, cation-rich amphipathicity is a hallmark feature of antimicrobial peptide (AMP) helices, which kill bacteria by membrane lysis (Mourtada et al., 2019; Zasloff, 1987). As mitochondria and bacteria share the common feature of negatively-charged membranes, electrostatic engagement by cationic residues can facilitate hydrophobic “carpeting” or direct insertion to lyse the target membrane by barrel stave or toroidal poration (Mechler et al., 2007; Mourtada et al., 2019; Murray et al., 2016; Sengupta et al., 2008). Indeed, a stapled BAX α6 peptide induced dose-responsive mitochondrial membrane disruption, whereas the analogous construct based on α6 of anti-apoptotic BFL-1 had no such effect (Figure 7D-E). Relative to BAX α6, BFL-1 α6 has a significantly lower lyticity index (LI: BAX α6, 538; BFL-1 α6, 390), reflecting decreased contiguous hydrophobic surface area and notably fewer cationic residues (Figure 7D). Thus, we examined the hypothesis that the potential AMP-like features of BAX α6 could contribute to the membrane permeabilization functionality of oligomerized BAX (Garcia-Saez et al., 2006; Mourtada et al., 2019; Valero et al., 2011), a mechanistic study now doable using BAXO constructs.
Figure 7. The lytic properties of BAX α6 contribute to the membrane permeabilization activity of BAXO.

(A) Plot of calculated membrane lyticity indices for consecutive 16-mer peptides along the BAX sequence.
(B) Plot of the arginine content for consecutive 16-mer peptides along the BAX sequence.
(C) Conserved R134 and R145 residues (79% and 85% conservation across 295 BAX sequences) project from the surface of BAX α6 (PDB: 1F16).
(D) Surface features of BAX α6 and BFL-1 α6, highlighting the increased hydrophobicity (yellow) and arginine content (blue) of BAX α6.
(E) A stapled BAX α6 peptide (ELIRTIMGWXLDFXRER) induces dose-responsive cytochrome c release from BAX/BAK-deficient MLM, whereas the corresponding stapled BFL-1 α6 construct (DVDTYKEISYXVAEXIMN) has no such effect. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. X, stapling amino acid; SAHB dosing, 20 nM-2.5 μM.
(F-H) Comparative kinetics and extent of membrane permeabilization of BAXO R134E and BAXO (F), BAXO R134E/R145E and BAXO R134E (G), and BAXOΔC R134E and BAXOΔC (H), as assessed by liposomal release assay. Error bars are mean ± s.e.m. for experiments performed in technical triplicate, with data representative of two independent experiments. BAXM and BAXO proteins, 500 nM.
(I) Western analysis of lysates from Bax−/−Bak−/− MEFs reconstituted with WT or R134E/R145E-mutant BAX.
(J) Cell viability of Bax−/−Bak−/− MEFs and those reconstituted with WT or R134E/R145E-mutant BAX in response to treatment with the indicated doses of S63845 and ABT-737. Data are mean ± s.d. for experiments performed in technical triplicate and repeated twice with similar results using independent cell cultures and treatments.
See also Figure S7.
Given the importance of surface arginine residues in coordinating phospholipid head groups during peptide-membrane interactions (Rice and Wereszczynski, 2017), and in targeting mitochondrial membranes in particular (Horton et al., 2008; Kelley et al., 2011), we mutated the highly evolutionarily-conserved R134 residue of BAX α6 to glutamate in the context of BAXO. BAXO R134E (Figure S7A) was then subjected to comparative testing in liposomal and mitochondrial assays. While liposomal translocation was largely preserved, both liposomal and mitochondrial membrane permeabilization were impaired relative to WT BAXO (Figure 7F, S7B-C). Decreasing positive charge even further by double R134E, R145E mutagenesis (Figure S7D) reduced liposomal translocation and compounded the loss of membrane-disruptive activity in liposomal and mitochondrial release assays (Figure 7G, S7E-F). The adverse impact of R134E mutagenesis on the membrane permeabilization activity of BAXO was equivalent to the detrimental effect of deleting the entire α9 helix in the context of BAXOΔC (Figure 6F, 7F). Strikingly, eliminating both compositional features in BAXOΔC R134E reduced permeabilization activity to background levels observed for BAXM alone (Figure 7H). To examine the implications of BAX α6’s AMP-like features in a cellular context, we reconstituted Bax−/−Bak−/− mouse embryonic fibroblasts (MEFs) with WT and R134E/R145E-mutant BAX (Figure 7I) and then monitored cell viability in response to targeted inhibition of anti-apoptotic MCL-1 and BCL-2/BCL-XL by the selective small molecules S63845 (Kotschy et al., 2016) and ABT-737 (Oltersdorf et al., 2005). Consistent with our functional data using the corresponding BAXo constructs, we observed that cells bearing BAX R134E/R145E were less responsive to anti-apoptotic inhibition compared to those reconstituted with WT BAX (Figure 7J). These results both corroborate the mechanistic insights gleaned from in vitro BAXO studies in a cellular context and validate the utility and fidelity of deploying BAXO to interrogate the critical execution-phase of mitochondrial apoptosis.
DISCUSSION
Since the discovery of BAX (Oltvai et al., 1993), a myriad of studies have advanced our understanding of its biochemistry, structure, and biology. As one of nature’s key mechanisms for disrupting the MOM to induce programmed cell death, BAX activation and oligomerization has been the subject of intensive investigation. An enduring mystery of BAX physiology is the molecular basis for its self-association and poration activity. In addition to its fundamental biological importance, the process of oligomerization and the ultimate porating species could be ideal drug targets for promoting or blocking BAX-mediated cell death in innumerable diseases.
By all accounts, BAX is a difficult protein to work with: it can be lethal upon expression in model systems, unstable to prolonged in vitro study, and extremely heterogeneous upon aggregation or oligomerization. Careful iteration of experimental conditions has led to the production of sufficiently-stable, monomeric, full-length BAX that is amenable to biochemical and structural analyses (Gavathiotis et al., 2008; Suzuki et al., 2000), and even traditional small molecule screening (Pritz et al., 2017). The transition from monomer to oligomer in vivo is – by design – rapid, irreversible, and disruptive. Thus, the dynamic transformation from latent monomer to membrane-toxic oligomer bespeaks protein instability, rendering the capture of stable intermediates somewhat of an oxymoron. Despite an avalanche of cutting-edge methodologies to study higher order, membrane-embedded species, oligomeric BAX has continued to be refractory to mechanistic dissection and definitive structural determination, with the major limitation being heterogeneity of oligomeric species. Here, we sought to overcome this challenge by identifying conditions that could yield more uniform BAX oligomers for conformational and mechanistic studies. We found that transient exposure to the detergent Fos-12 is one such solution, and yields a stable, homogeneous, and functional oligomeric species that persists even after detergent is removed. BAXO is a functional unit of 6-8 monomers, curvilinear in shape, and capable of auto-translocating to liposomal and mitochondrial membranes to induce permeabilization that mirrors the morphology, kinetics, and potency of physiologic BH3-induced BAX activation. Our characterization of BAXO, having a maximum dimension of ~20 nm, provides a missing link along the continuum between the reported structures of monomeric (Suzuki et al., 2000) and truncated dimeric (Czabotar et al., 2013) BAX (~5 nm), and the heterogeneous, macro-oligomeric assemblies (hundreds of nm) observed by high-resolution microscopy in apoptotic mitochondrial membranes (Grosse et al., 2016; McArthur et al., 2018; Salvador-Gallego et al., 2016).
Based on our comparative crosslinking analyses of BAXM and BAXO, characteristic structural features of the BAX monomer, such as proximity of (1) the α1-α2 loop to α1 and α6, (2) α1 to α2, and (3) the α3-α4 loop and α4 to the core α5-α6 hairpin and α9, are lost in BAXO, consistent with a major structural reorganization. Integration of our HXMS and crosslinking analyses revealed that (1) the N-terminal regions, including α1 and the α1-α2 loop, become permanently exposed and accessible to each other in BAXO, (2) α7 and α8 are newly exposed, (3) the essential BH3 domain α2 helix of BAX becomes buried after transient exposure, and lies adjacent to the BH3 domain of its nearest neighbor, and (4) the auto-inhibitory C-terminal α9 becomes transiently exposed, followed by burial in the oligomeric structure. To contextualize these results, the “opening” of the α1-α2 loop to expose α1 is consistent with the initiating structural changes in BAX observed by NMR upon BH3-activation (Gavathiotis et al., 2010) and reactivity of the conformation-specific 6A7 antibody with residues 12-24 of activated BAX (Hsu and Youle, 1997). The very regions that become most exposed in BAXO, namely α1, α7, and α8, are among those regions that are most protected, along with the critical BAX BH3 domain, upon BCL-2 BH4 domain inhibition of the BAX monomer (Barclay et al., 2015), revealing a mechanism to inhibit BAX oligomerization by suppressing exposure of these critical structures. Transient release of the BAX BH3 domain, which exposes its hydrophobic face for protein interaction, is consistent with both the critical role of the BAX BH3 domain in BAX physiology (Wang et al., 1998) and detection of this structural change by NMR upon BH3-triggered BAX activation (Gavathiotis et al., 2010). Prompt sequestration of the BAX BH3 (α2) matches the predicted role of its hydrophobic face in oligomerization interactions, and the proximity of BH3 domains has been observed by X-ray crystallography (Czabotar et al., 2013), double electron-electron spectroscopy (DEER) (Bleicken et al., 2014; Sung et al., 2015), and crosslinking of installed cysteines in the context of α2-α5 dimers and other BAX species (Dewson et al., 2012).
Dissecting the individual dynamic steps along the BAX activation pathway, and especially the “execution-phase” mechanism of self-association and MOMP, has been limited by the lack of full-length species that reflect the conformational intermediates and functional oligomeric units. By generating BAXO, not only have we gained fresh insight into the conformational features of a functional homo-oligomeric species, but now can also distinguish between the structural determinants of the activation and execution steps, including the helices and even particular residues that drive oligomeric permeabilization of model and mitochondrial membranes. For example, we determined that BH3 triggering has specific determinants for initiating the direct conformational activation of BAX that are distinct and isolable from the requirements for oligomeric permeabilization. Indeed, this system can explicitly distinguish between the functional roles of immediately adjacent residues based on the conformational contexts of distinct stages of BAX activation, namely I133 during triggering of BAXM and R134 upon membrane engagement and poration by BAXO. In addition, α9 plays critical roles in ensuring membrane translocation efficiency and in maximizing both the kinetics and extent of membrane permeabilization. Nevertheless, functional oligomers can still form in the absence of α9, implicating additional sequence or structural determinants of membrane disruption. We found that the distinctive amphipathic properties of α6, including conserved cationic residues and a robust hydrophobic interaction surface – key features of membrane-lytic peptides such as AMPs – endow BAXO with mitochondrial membrane tropism and permeabilization activity. Interestingly, a recent model based on live-cell lattice light-sheet microscopy (LLSM) suggests that only a small amount of BAX, not resolved by LLSM, is required for MOMP, with the majority of BAX recruited after cytochrome c release to generate visible macropores (McArthur et al., 2018). It is therefore plausible that the critical initial stage of MOMP that releases cytochrome c derives from outer membrane disruption by a direct lytic mechanism rather than organized pore formation per se. Thus, in addition to providing newfound opportunities for mechanistic inquiry, the production of BAXO represents a critical step forward in the longstanding pursuit of structural insight into one of the cell’s cardinal death channels.
LIMITATIONS OF THE STUDY
BAX is a unique protein that exists in primarily two distinct functional states, a latent cytosolic form and an activated oligomeric species whose function is to disrupt the MOM in response to stress signaling. As such, BAX has at least two energy minima, with a clear propensity to self-associate in response to a diversity of physiologic and non-physiologic triggers, including detergents. One of the inherent paradoxes in studying BAX is that in order to capture an oligomeric species it must be sufficiently stable, yet we know that BAX conformational flexibility, both in its latent and oligomeric states, is the physiologic norm. Without conformational flexibility, latent BAX would be unable to transform into an activated species and without oligomeric flexibility BAX would be unable to embed in membranes and expand into growing macropores. Whereas the BAXO species we report here is sufficiently homogeneous to study its conformation by SAXS, HXMS, and chemical crosslinking, Fos-12 pre-treatment followed by Fos-12 removal preserves the requisite conformationally flexibility that also allows us to study the membrane-permeabilization behavior of BAX, as demonstrated and validated across a battery of biochemical, mitochondrial and cellular assays.
Fos-12 has been reported in some cases to destabilize membrane protein structure (Crichton et al., 2015; Zoonens et al., 2013) and inactivate function (Casiraghi et al., 2016; Matar-Merheb et al., 2011; Telbisz et al., 2013). However, in other studies, Fos-choline detergents like Fos-12 have been successfully used for both functional analyses and structural determination across a variety of methods (Asmar-Rovira et al., 2008; Chipot et al., 2018; Kefala et al., 2010; Liu et al., 2020; Lu and Fu, 2007; Wang et al., 2008). Importantly, we observe that after initial Fos-12 treatment, the BAXO species we generate demonstrates a distinct peak by SEC whether Fos-12 is included in the purification buffer or not. What’s more, in the absence of Fos-12, BAXO continues to migrate as a singlet band by BNP and functions similarly to ligand-triggered BAXM across a battery of gold standard assays, with our mechanistic insights validated both by prior and current work in vitro and in cells (e.g. BAX G108E, BAX I133A, BAX R134E/R145E).
As the field continues to pursue the “holy grail” of determining the structure of oligomeric BAX, it is important to underscore that the level of homogeneity required to achieve a definitive structure is likely to be different than the level of homogeneity that allows for structure-function mechanistic studies of BAX oligomerization and membrane disruption. Indeed, the requisite manipulations to ultimately obtain a definitive structure could alter the natural function of BAX by restraining its inherently dynamic nature, which we aimed to study here. Taken together, we hope that this current body of work brings the apoptosis field ever closer to achieving the “holy grail” oligomeric structure, in addition to providing an important tool for dissecting the structure and function of full-length BAX across each dynamic stage of its fascinating and exquisitely-regulated transformation into an executioner of cell death.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Loren Walensky (loren_walensky@dfci.harvard.edu).
Materials Availability
Plasmids, stapled peptides, and mouse cell lines generated in this study are available upon request to the lead contact.
Data and Code Availability
The data supporting the findings of this study are available within the article and its supplementary materials. This study did not generate deposited datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
Recombinant proteins were expressed in E. coli BL21(DE3) or LOBSTR BL21(DE3) bacteria, which were grown in Luria Broth (LB) at either 30°C or 16°C with shaking at 220 rpm.
METHOD DETAILS
Peptide Synthesis
N-terminally acetylated stapled peptides, including BIM SAHBA2, BAX SAHB α6, and BFL-1 SAHB α6, were synthesized, purified, and quantified as described in detail (Edwards et al., 2013; Gavathiotis et al., 2008). Briefly, all hydrocarbon stapled peptides were generated using solid phase Fmoc chemistry, with amino acids sequentially added to rink amide AM resin (EMD Millipore). Two naturally-occurring amino acids were replaced with the non-natural amino acid S-pentenyl alanine at the i, i+4 positions. Stapling was accomplished by olefin metathesis using Grubb’s catalyst (Sigma Aldrich), followed by peptide deprotection and cleavage from the resin. SAHBs were purified by reverse phase high performance liquid chromatography/mass spectrometry (LC/MS) and quantified by amino acid analysis.
Recombinant Protein Expression and Purification
Recombinant, wild-type full-length BAX (aa 1-192), BAX point mutants G108E, I133A, R134E, R145E, and R134E/R145E, BAXΔC (aa 1-168), and BAXΔC R134E were cloned into pTYB1 (NEB) and expressed in Escherichia coli BL21(DE3) (Edwards et al., 2013; Gavathiotis et al., 2008). BAX protein expression was induced using 1 mM isopropyl β-D-1–thiogalactopyranoside (IPTG) for five h at 30°C, and bacterial pellets were resuspended in lysis buffer (20 mM Tris pH 7.2, 250 mM NaCl) containing complete protease inhibitor cocktail tablets (Roch) and then microfluidized (M-110L, Microfluidics) and centrifuged at 20,000 x RPM for 45 min. The supernatant was passed over a chitin column (NEB) equilibrated with lysis buffer and the column washed twice with lysis buffer. BAX was cleaved from the chitin beads by overnight incubation with elution buffer (20 mM Tris pH 7.2, 250 mM NaCl, 50 mM dithiothreitol). BAX containing eluate was concentrated and loaded onto a Superdex S-75 (GE Healthcare) gel filtration system equilibrated with FPLC buffer (20 mM HEPES pH 7.2, 150 mM KCl). Protein purity and identity was confirmed by western blot analysis using a mouse monoclonal anti-BAX (2D2) antibody (Santa Cruz Biotechnology Cat# sc-20067; RRID: AB_626726).
Recombinant BCL-w was generated as described above for BAX and BFL-1ΔC was produced as described in detail (Guerra et al., 2018; Harvey et al., 2018). Briefly, recombinant, N-terminal hexahistidine-tagged BFL-1ΔC (aa 1-151) was cloned into pET17b (Novagen), expressed in E. coli LOBSTR BL21(DE3) cells (Kerafast) overnight at 16°C, and purified by sequential Ni-affinity and size-exclusion chromatography. Protein expression was induced using 0.5 mM IPTG. Bacterial pellets were resuspended in lysis buffer (20 mM Tris pH 7.5, 250 mM NaCl) containing two complete protease inhibitor cocktail tablets (Roch). Bacteria were lysed using a microfluidizer (M-110L, Microfluidics) and centrifuged at 20,000 x RPM for 45 min to remove insoluble debris. Clarified lysate was passed over a Ni-NTA (Qiagen) column equilibrated with lysis buffer. The column was sequentially washed with lysis buffer and then lysis buffer containing 10 mM, 20 mM, 35 mM, and 50 mM imidazole. His-BFL-1ΔC was eluted in buffer containing 150 mM imidazole. The BFL-1 containing fraction was then dialyzed overnight in lysis buffer, concentrated, and purified by size exclusion chromatography using a Superdex S-75 (GE Healthcare) gel filtration system.
Preparation of BAX Oligomers
Purified BAX monomer (20 μM) was combined with a series of detergents at 3 mM (Figure 1) and incubated overnight at 4°C. Detergent-treated BAX was then concentrated (10 kD spin concentrator, Millipore) and loaded onto a Superose 6 Increase size exclusion column (GE Healthcare) equilibrated with FPLC buffer containing an equivalent concentration of each respective detergent. For activation below the critical micellar concentration of Fos-12, BAX (20 μM) was combined with 1 mM Fos-12 for 7 days at 4°C, concentrated, and loaded onto a Superose 6 column equilibrated with BAX FPLC buffer. For SAXS preparation, BAX (20 μM) was combined with 3 mM Fos-12 overnight at 4°C and consecutively diluted three-fold in BAX FPLC buffer to reduce the detergent concentration to 1 mM. For biochemical studies, BAX (20 μM) was combined with 3 mM Fos-12 overnight at 4°C, concentrated, and loaded onto a Superose 6 column equilibrated with detergent-free BAX FPLC buffer. The fractions containing the highest concentration of protein (peak center) were used.
Blue Native PAGE
BAXM, BAXO, BAXΔC, BAXOΔC, BCL-w, BFL-1ΔC, and BAX G108E were visualized using the Invitrogen NativePAGE Bis-Tris Gel system. Briefly, protein samples were prepared as described and then loaded onto 4-16% Bis-Tris gels in 1x native loading buffer and 0.5% Coomassie G-250. Gel electrophoresis with Coomassie G-250 included in the cathode buffer was performed according to the manufacturer’s instructions at 150 V (Invitrogen). Native PAGE gels were fixed in 40% methanol and 10% acetic acid, and destained in 8% acetic acid.
Small Angle X-ray Scattering
Solutions of BAXO were prepared in aqueous buffer containing 20 mM HEPES, 150 mM KCl, and 1 mM Fos-12, pH 7.2. SAXS measurements were performed at the 12ID-B beamline of the Advanced Photon Source (APS), Argonne National Laboratory. Photon energy was 13.3-KeV and sample-to-detector distance was 2 m to achieve a usable q range of 0.005 < q < 0.90 Å−1, where q = (4π/λ)sinθ, and 2θ is the scattering angle. Concentration series measurements for the same sample were carried out to remove the scattering contribution due to inter-particle interactions and to extrapolate the data to infinite dilution. Sample concentrations were 1, 3 and 5 mg/mL. Thirty two-dimensional (2D) images were recorded for each matching buffer or sample solution using a flow cell, with an exposure time of 0.5-2 sec to minimize radiation damage and to yield optimal signal-to-noise ratio. The 2D images were reduced to 1D scattering profiles, and outliers were eliminated before averaging using the MatLab software package at the beamlines. The buffer background subtraction and intensity extrapolation to infinite dilution were carried out using National Cancer Institute (NCI) in-house developed MatLab script NCI-SAXS. The radius of gyration (Rg) was generated from the Guinier plot in the range of qRg < 1.3. For comparison, Rg was also calculated in real and reciprocal spaces using program GNOM (Svergun, 1992). The pair-distance distribution function P(r) and maximum dimension (Dmax) were also calculated using GNOM. The molecular weights were estimated using three methods based on Porod volume (Petoukhov et al., 2012), correlation volume (Rambo and Tainer, 2013) in the q range of 0 < q < 0.2 Å−1 and apparent volume (Fischer et al., 2010) in the q range of 0 < q < 0.2 Å−1. Thirty-two ab-initio shape reconstructions (molecular envelopes) were generated independently using DAMMIN (Svergen, 1999), and then averaged and filtered (Volkov and Svergun, 2003).
Liposomal Preparation
Large unilamellar vesicles (LUVs) encapsulating the fluorophore/quencher pair 8-aminonapthalene-1,3,6-trisulfonic acid (ANTS) and p-xylene-bis-pyridinium bromide (DPX) were formed by liposome extrusion and size exclusion chromatography (Leshchiner et al., 2013; Pitter et al., 2008). Briefly, a lipid mixture that mimics the composition of the outer mitochondria membrane was generating by dissolving a 48:28:10:10:4 molar ratio of phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, dioleoyl phosphatidylserine, and tetraoleolyl cardiolipin (Avanti Polar Lipids) in chloroform. To produce lipid films, nitrogen gas was used to evaporate the chloroform and the lipids were dried under high vacuum overnight. For long-term storage, lipid films were maintained in a nitrogen atmosphere at −80°C. Lipid films were resuspended in 1 mL of liposomal release assay buffer (10 mM HEPES pH 7, 200 mM KCl, 1 mM MgCl2) containing the fluorophore/quencher pair ANTS (12.5 mM) and DPX (45 mM). Liposomes were formed by exposing the resuspended lipid mixture to ten freeze/thaw cycles and then extruding the liposomes using a 100 nm polycarbonate membrane eleven times. Liposomes were then purified from unencapsulated ANTS and DPX using a Sepharose CL-2B size exclusion column (GE Healthcare).
Liposomal Translocation Assay
Liposomes (40 μL) were incubated with (1) BAXM (0.5 μM) treated with tBID (20 nM), BIM SAHBA2 (0.5 μM) or vehicle, or (2) BAXO, BAXOΔC, BAXO I133A, BAXO R134E, or BAXO R134E/R145E in liposomal release assay buffer for the indicated time points at room temperature. BAX-containing solutions were then added to a Sepharose CL-2B (GE Healthcare) size exclusion column equilibrated with liposomal release assay buffer and 17 equivalent fractions (250 μL) were collected. Liposome-containing fractions were identified by adding 10% Triton X-100 to the fractions and measuring florescence associated with ANTS/DPX release. To determine which fractions contained BAX, gel electrophoresis and western blot analysis was conducted using the anti-BAX N20 antibody (Santa Cruz Biotechnology Cat# sc-493; RRID: AB_2227995).
Liposomal Release Assay
Recombinant BAX proteins (500 nM), with or without tBID (20 nM) or BIM SAHBA2 (500 nM), or anti-apoptotic proteins (10 μM for BAX blockade, 500 nM for Fos-12 control experiments), were added to liposomes generated as described above. ANTS/DPX release was monitored over time at room temperature in a spectrofluorometer (Tecan Infinite M1000) using an excitation wavelength of 355 nm, emission wavelength of 520 nm, and a bandwidth of 20 nm. Maximal release was determined by the addition of Triton X-100 to a final concentration of 0.625% (v/v). Percent release was calculated as ((F–F0)/(F100–F0)) × 100, where F is the observed fluorescence at a given time, and F0 and F100 represent baseline and maximal fluorescence, respectively.
Negative Stain Electron Microscopy
400 mesh copper carbon film microscopy grids (Electron Microscopy Sciences) were glow discharged in preparation for negative stain. To visualize liposomal morphology, liposomes were treated with BAXM or BAXMΔC alone (500 nM), tBID (20 nM) or BIM SAHBA2 (500 nM) triggered BAXM (500 nM), or BAXO or BAXOΔC (500 nM) in 20 mM HEPES pH 7.0, 150 mM KCl, 1 mM MgCl2. After 15 min incubation, a 10 μL sample from each condition was applied to the carbon side of the grid for 1 min, followed by blotting excess liquid. The preparation was washed three times by placing the grid onto a drop of buffer for 10 sec and then blotted dry. Grids were negatively stained by the addition of 10 μL uranyl formate solution (100 mM) for 30 sec and blotted dry. Negative-stained grids were imaged at the Harvard Medical School Electron Microscopy Facility using a Technai G2 Spirit BioTWIN TEM operated at 80 kV and visualized at 50,000 magnification with 3.37 nm/pixel for all samples. Quantitation of porated liposomes and pore size was accomplished by visual identification of undisrupted, pore-bearing, and grossly disrupted liposomes. Pore diameter was measured by quantifying the length of a line spanning the largest visible diameter of round focal pores in adobe illustrator and subsequent normalization using the scale bar.
For the 2D classification of n-dodecylphosphocholine-induced BAX oligomer, 10,000 particles were manually picked with Relion 2.0 (RELION; RRID: SCR_016274) and classified to 25 classes (Scheres, 2012). The best classes with a total number of ~3,500 particles were used to generate the 3D model. 3D classification was performed using an initial model as reference-generated in Relion 2.0 with C1 symmetry and classified to 3 classes. Class number 2 was used as a model for fitting the SAXS model. For 2D classification of BAX activated by BIM SAHBA2 in the presence of liposomes and subsequent extraction, ~1,000 particles were manually picked with Relion 2.0 for the initial 2D classification, which generated a model for auto-picking ~30,000 particles that were classified to 25 classes. The best classes with a total number of ~11,000 particles were used for 3D classification in Relion 2.0. 3D classification was performed using an initial model as reference-generated in Relion 2.0 with C1 symmetry and classified to 4 classes. Class number 2 was used as a model for fitting the SAXS model. Model fitting and 3D model presentation were generated using Chimera (UCSF Chimera; RRID: SCR_004097) and Pymol 2.1.1 (PyMOL; RRID: SCR_000305).
Mitochondrial Cytochrome c Release Assays
Liver mitochondria from AlbCreposBaxf/fBak−/− mice were isolated and release assays performed as described (Walensky et al., 2006). Briefly, mitochondria (1 mg/mL) were incubated with recombinant BAXM proteins (200 nM) in the presence or absence of tBID (40 nM), BAXO proteins (dose range of 6.25 nM - 200 nM), or BAX α6 or BFL-1 α6 SAHBs (dose range of 20 nM - 2.5 μM) for 45 min at room temperature in experimental buffer (200 mM mannitol, 68 mM sucrose, 10 mM HEPES-KOH pH 7.4, 110 mM KCl, 1 mM EDTA, protease inhibitor). The pellet and supernatant fractions were isolated by centrifugation, and cytochrome c was quantitated using a colorimetric ELISA assay (R&D Systems), per the manufacturer’s protocol. Percent cytochrome c released into the supernatant (%cyto c release) was calculated according to the following equation: %cyto c release = [cyto csup]/ [cyto cmax]*100, where cyto csup and cyto cmax represent the amount of cytochrome c detected in the supernatant upon treatment with the indicated conditions or 1% (v/v) Triton X-100, respectively.
Hydrogen Deuterium Exchange Mass Spectrometry
Hydrogen deuterium exchange experiments were performed as described previously (Barclay et al., 2015). BAXM, BAXMΔC, BAXO, or BAXOΔC (BAXM proteins, 7 μM; BAXO proteins, 4 μM) were equilibrated for 15 min at room temperature in the presence or absence of liposomes (4 μL, prepared as described above). Samples were deuterated for the indicated times in labeling buffer, and labeling was stopped with an equal volume of quench buffer (see Tables S1, S2). Proteolysis of BAX (50 pmoles) was performed by adding 40 μg porcine pepsin and 20 μg of Aspergillus satoi factor XIII protease (Sigma Aldrich) to the samples on ice for 5 min. Digested protein was injected into a Waters UPLC HDX manager system (Wales et al., 2008) maintained at 0°C to minimize back-exchange; peptic peptides underwent a 3 min trap and desalting step using a VanGuard pre-column trap (2.1 x 5 mm, ACQUITY UPLC BEH C18, 1.7 μm) flowing at 100 μL/min. Peptides were then separated using an ACQUITY UPLC HSS T3, 1.8 μm, 1.0 x 50 mm analytical column (Waters Corporation) with a 5-35% gradient of increasing acetonitrile (0.1 % formic acid) over 6 min at a flow rate of 65 μL/min. Mass spectra were acquired using a Synapt G2-Si (Waters) in MSE (data independent acquisition) mode with 0.4 scans/sec over a 50-2000 m/z range with ion mobility engaged. BAX peptides were identified using Protein Lynx Global Server (PLGS 3.0.1, Waters Corporation, RRID: SCR_016664) using undeuterated BAX as a reference (see Table S1-2). Data analysis was performed using DynamX 3.0 (Waters Corporation), which includes determination of centroid masses for each isotopic distribution, generation of deuterium uptake plots, and comparison between experimental states. Relative deuterium uptake for each peptide was determined by subtracting the average mass of the undeuterated peptide from the average mass of the deuterated peptide. Deuterium exchange was not corrected for back-exchange and thus expressed as relative deuterium uptake (Wales and Engen, 2006).
Chemical Crosslinking and Mass Spectrometry Analysis
The chemical crosslinkers used in this study are summarized in Figure 4A and the applied proteomic methodology and its validation recently described in detail (Mintseris and Gygi, 2020). For DSSO-crosslinked BAX, BAXM and BAXO samples were subjected to 20x and 100x DSSO for 1 h at room temperature and subsequently quenched with 10x ammonium bicarbonate before being separated by SDS-PAGE. Dimer, trimer, and higher-order bands corresponding to chemically-crosslinked BAX were selected for BAXO samples and compared to the sole monomeric band of BAXM. Gel slices were reduced with 10 mM DTT for 30 min at 56°C, alkylated for 20 min with 50 mM iodoacetamide in the dark, and subjected to tryptic digest with 12.5 ng/μL sequencing grade trypsin overnight at 37°C. Digested samples were desalted after acidifying with 0.1% TFA using Agilent C18 pOmix tips and then dried under vacuum. BAXM and BAXO samples were resuspended to 1 μg/μL in 5% ACN/2% formic acid, and peptides were loaded onto an in-house pulled C18 column (2.6 μm Accucore, Thermo Scientific) and eluted using a gradient from 100% buffer A (5% ACN, 0.125% formic acid) to 30% buffer B (95% ACN, 0.125% formic acid). Peptides were eluted into an Orbitrap Fusion (Thermo Scientific) mass spectrometer and analyzed using the MS2/MS3 strategy for the identification of DSSO-linked peptides (Kao et al., 2011; Liu et al., 2015). Briefly, survey MS1 scans were acquired at a resolution of 60000 (AGC target: 2.0e5, max injection time: 100 ms). Precursors were selected and further fragmented using low CID energy to cleave only the DSSO linker (CID NCE: 23%, resolution: 30000, isolation window: 0.7, AGC target: 1.0e5, max injection time: 150 ms). Both released peptides were further fragmented based on selection of cleaved peptide precursors from the MS/MS scan (targeted mass difference: 31.9721 m/z at +/− 20 ppm) (Kao et al., 2011). Released peptide fragments were analyzed in the ion trap (isolation window: 1.2 m/z, HCD NCE: 34, AGC target: 1.0e5, maximum injection time: 300 ms). Crosslinked peptides were identified using Proteome Discoverer 2.2 and the XlinkX module (Thermo Scientific) (Liu et al., 2015). Briefly, individual peptides were identified using Sequest HF for peptide spectral matching (2 missed cleavage maximum, 6 amino acid minimum peptide length, all DSSO dynamic modifications (Liu et al., 2015)) and matched into crosslinks using XlinkX. Peptide identifications were filtered to a 1% peptide false discovery rate using the target decoy strategy (Elias and Gygi, 2007) and Percolator (The et al., 2016). Proteins were filtered to a 5% FDR and grouped using the rules of parsimony (Nesvizhskii and Aebersold, 2005). Crosslinked peptides were filtered to have a minimum score threshold of 20 and an FDR less than 1%.
For crosslinking using an Extended EDC approach that leverages two diamine linkers, ethylenediamine and 2,2'-(ethylenedioxy)bis(ethylamine) (SIGMA-Aldrich), BAXM and BAXO crosslinking reactions were carried out for 1 h at room temperature in 20 mM sodium phosphate buffer, pH 6.5, 100 mM NaCl with 100 mM or 150 mM EDC, ~18 mM diamine linker, and 15 mM sulfo-NHS. Reactions were quenched with hydroxylamine to a final concentration of 100 mM. Samples were reduced for 1 h in 5 M urea and 10 mM TCEP in 50 mM HEPBS Buffer, pH 8.3, followed by alkylation with 30 mM IAc in the dark for 1 h and quenching with 50 mM β-mercaptoethanol. Samples were then diluted with 50 mM HEPBS to reduce urea concentration down to 1 M and digested with trypsin (Promega) at 1:25 enzyme:substrate ratio overnight at 37°C. Digested peptides were acidified with 10% formic acid to pH ~2 and desalted using stage tips with Empore C18 SPE Extraction Disks (3M) and dried under vacuum. Colorimetric Peptide Assay (Pierce) was used to verify sample amounts and 0.5 μg of each sample was reconstituted in 5% formic acid (FA)/5% acetonitrile and analyzed in the Orbitrap Lumos Mass Spectrometer (Thermo Fischer Scientific) coupled to an EASY-nLC 1200 (Thermo Fisher Scientific) ultra-high pressure liquid chromatography (UHPLC) pump. Peptides were separated on an in-house packed 100 μM inner diameter column packed with 35 cm of Accucore C18 resin (2.6 μm, 150 Å, ThermoFisher), using a gradient consisting of 5–35% (ACN, 0.125% FA) over 60 min at ~450 nL/min. The instrument was operated in data-dependent mode. FTMS1 spectra were collected at a resolution of 120K, with an automated gain control (AGC) target of 1 × 106, and a max injection time of 50 ms. The most intense ions were selected for MS/MS for 2s in top-speed mode. Precursors were filtered according to charge state (allowed 2 <= z <= 7), and monoisotopic peak assignment was turned on. Previously interrogated precursors were excluded using a dynamic exclusion window (45 s ± 7 ppm). MS2 precursors were isolated with a quadrupole mass filter set to a width of 0.7 m/z. Precursors were analyzed by FTMS2, with the Orbitrap operating at 30K resolution, an AGC target of 50K, and a maximum injection time of 150 ms. Precursors were then fragmented by high-energy collision dissociation (HCD) at a 33% normalized collision energy. Mass spectra were processed using a sequence database containing contaminating E. coli proteins and common contaminants in addition to BAX. Concatenated reversed sequences were searched for Target/Decoy-based FDR estimation (Elias & Gygi, 2017). Precursor tolerance was set to 15 ppm and fragment ion tolerance to 10 ppm. Methionine oxidation was set as a variable modification in addition to mono-linked masses of +42.058183 and +130.110613 for the two Extended-EDC diamine crosslinkers. Crosslinked peptides were searched assuming zero-length (−18.010565) and the two Extended-EDC crosslinker masses of +24.047618 and +112.100048, respectively. Crosslinked searches included 50 protein sequences to ensure sufficient statistics for FDR estimation. Matches were filtered to 1% FDR on the unique peptide level using linear discriminant features.
Peptide Lyticity and Arginine Content Analyses
The amino acid sequence of BAX was analyzed in consecutive peptide increments of 16 amino acids along the length of the protein (e.g. aa 1-16, 2-17, 3-18, etc.). The membrane lyticity index of each peptide was calculated as described (Mourtada et al., 2019). Briefly, the lyticity index (LI) was calculated according to the formula below, in which the summation is computed over all hydrophobic amino acids:
Built-in assumptions include (1) a peptide of α-helical structure, (2) hydrophobic amino acids within this structure interact to form a network, (3) each vertex is a hydrophobic amino acid (Hi) and the edges are formed by the interactions with the structurally closest hydrophobic amino acids (i, i+3 and i, i+4 relationships), (4) each edge is weighted by summing the hydrophobicity of the amino acid vertices that form the edge. The hydrophobicity values (Hi) employed are based on Kovacs et al. (Table III, Column 4) (Kovacs et al., 2006). A user interface for this algorithm is available as a web application at http://www.walenskylab.org/HNM/. The arginine content of each 16 amino acid peptide along the BAX sequence was determined by summing the number of arginine residues in each consecutive fragment.
Retroviral Transduction and Cell Culture
Human BAX constructs (wild-type, R134E/R145E) were cloned into the pMIG (MSCV-IRES-GFP) vector, and the presence of insert and the indicated mutations were confirmed by DNA sequencing. Transfection of the packaging cell line GPG-293 yielded amphotropic retroviral particles, which were collected by filtration and ultracentrifugation, and then used to reconstitute Bax−/−Bak−/− MEFs with the indicated BAX proteins, as described (Gavathiotis et al., 2008; Kim et al., 2006). The reconstituted MEFs were sorted for GFP-positivity over two rounds of flow cytometry to ensure comparable levels of expression. Transduction was verified by anti-BAX western analysis using the 2D2 antibody. Cells were maintained in Dulbecco’s Modified Eagle Medium (Gibco) with 10% FBS, 100 U\mL penicillin and streptomycin, and 2 mM glutamine. Cells were verified as mycoplasma-negative using the MycoAlert mycoplasma detection kit (Lonza Biologics).
Cell Viability
Bax−/−Bak−/− MEFs reconstituted with wild-type or R134E/R145E-mutant BAX were seeded at 5,000 cells per well in white, flat-bottomed 96-well plates (Costar), grown overnight, and then treated with the indicated doses of ABT-737 (Cayman Chemical) and S63845 (MedChemExpress), or vehicle (0.1% or 0.05% DMSO, respectively). Viability was measured after 24 h using Cell-TiterGlo in accordance with the manufacturer’s protocol and expressed as a percentage of the corresponding vehicle treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of technical and biological replicates for each experiment are indicated in the corresponding figure legend. Mean ± s.d. or s.e.m. values were calculated using Prism software (Graphpad).
Supplementary Material
Data File S1 Related to Figures 4 and 5. Source data for mass spectrometry analyses of BAXM and BAXO chemical crosslinks.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-BAX 2D2 | Santa Cruz | Cat# SC-20067; RRID: AB-626726 |
| Rabbit polyclonal anti-BAX N20 | Santa Cruz | Cat# sc-493; RRID: AB_2227995 |
| Sheep anti-mouse HRP | Bio-Rad | Cat# AAC10P; RRID: AB-321929 |
| Bacterial and Virus Strains | ||
| One Shot BL21(DE3) Competent Cells | Invitrogen | Cat# C600003 |
| LOBSTR BL21(DE3) Competent Cells | Kerafast | Cat# EC1001 |
| One Shot STBL3 Competent Cells | Invitrogen | Cat# C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rink Amide AM Resin LL 100-200 mesh | Millipore | Cat# 855120 |
| (S)-N-Fmoc-α-(4-pentenyl) alanine | Nagase & Co | Cat# 365023 |
| Grubbs Catalyst 1st Generation | Sigma-Aldrich | Cat# 579726 |
| 8-aminonapthalene-1,3,6-trisulfonic acid (ANTS) | Thermo Fisher Scientific | Cat# A350 |
| p-xylene-bis-pyridinium bromide (DPX) | Thermo Fisher Scientific | Cat# X1525 |
| Complete Protease Inhibitor Cocktail Tablets | Sigma-Aldrich | Cat# 16829800 |
| SimplyBlue SafeStain | Thermo Fisher Scientific | Cat# LC6065 |
| NativePAGE Sample Buffer (4X) | Thermo Fisher Scientific | Cat# BN2003 |
| NativePAGE Cathode Buffer Additive (20X) | Thermo Fisher Scientific | Cat# BN2002 |
| NativePAGE 5% G-250 Sample Additive | Thermo Fisher Scientific | Cat# BN2004 |
| NativePAGE Running Buffer (20X) | Thermo Fisher Scientific | Cat# BN2001 |
| Native Mark Unstained Protein Standard | Thermo Fisher Scientific | Cat# LC0725 |
| NativePAGE 4-16% Bis-Tris Protein Gels, 1.0 mM, 10 well | Thermo Fisher Scientific | Cat# BN1002BOX |
| Chitin bead resin | New England Biolabs | Cat# S6651S |
| SeeBlue Plus2 Pre-stained Protein Standard | Invitrogen | Cat# LC5925 |
| Sepharose CL-2B | GE Healthcare Life Sciences | Cat# 17014001 |
| Agilent C18 pOmix tips | Agilent | Cat# A57003 |
| EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) | ThermoFisher Scientific | Cat# A35391 |
| Ethylenediamine | Sigma Aldrich | Cat# 26266 |
| 2,2’-(ethylenedioxy)bis(ethylamine) | Sigma Aldrich | Cat# 385506 |
| Sulfo-NHS (N-hydroxysulfo Succinimide) | ThermoFisher Scientific | Cat# A39269 |
| DSSO (Disuccinimidyl Sulfoxide) | ThermoFisher Scientific | Cat# A33545 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine | Avanti | Cat# 850457C |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine | Avanti | Cat# 850757C |
| L-α-phosphatidylinositol (Liver, Bovine) (sodium salt) | Avanti | Cat# 840042C |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt) | Avanti | Cat# 840035C |
| Cardiolipin (Heart, Bovine) (sodium salt) | Avanti | Cat# 840012C |
| Fos-choline 12 (Anagrade) | Anatrace | Cat# F308 |
| Detergent Screening Set Classic (n-Octyl-beta-D-glucopyranoside, N,N-Dimethyldodecylamine N-oxide, n-Decyl-beta-maltoside, n-Dodecyl-beta-maltoside, and n-Octyl-1-beta-D-thiglucopyranoside) |
Cube Biotechnology | Cat# 16092 |
| 3-((3-cholamidopropyl)dimethyl ammonio-1-propanesulfonate (CHAPS) | Thermo Fisher Scientific | Cat# 28300 |
| Factor XIII Protease from Aspergillus saitoi | Sigma Aldrich | Cat# P2143 |
| Pepsin from porcine gastric mucosa | Sigma Aldrich | Cat# P6887 |
| S63845 | MedChemExpress | Cat# HY-100741 |
| ABT-737 | Cayman Chemical | Cat# 11501 |
| Lipofectamine 3000 | ThermoFisher Scienfici | Cat# L3000008 |
| BIM SAHBA2 | Walensky Lab | N/A |
| BAX α6 SAHB | Walensky Lab | N/A |
| BFL-1 α6 SAHB | Walensky Lab | N/A |
| Recombinant BAX WT | Walensky Lab | N/A |
| Recombinant BAX I133A | Walensky Lab | N/A |
| Recombinant BAX G108E | Walensky Lab | N/A |
| Recombinant BAX R134E | Walensky Lab | N/A |
| Recombinant BAX R134E/R145E | Walensky Lab | N/A |
| Recombinant BAXΔC (aa 1-168) | Walensky Lab | N/A |
| Recombinant BAXΔC (aa 1-168) R134E | Walensky Lab | N/A |
| Recombinant BCL-w WT | Walensky Lab | N/A |
| Recombinant BFL-1ΔC (1-151) | Walensky Lab | N/A |
| Critical Commercial Assays | ||
| Q5 Site Directed Mutagenesis Kit | New England Biolabs | Cat# E0554S |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat# 23225 |
| Rat/Mouse Cytochrome c Quantikine ELISA Kit | R&D Systems | Cat# MCTC0 |
| Quantitative Colorimetric Peptide Assay | Pierce | Cat# 23275 |
| Experimental Models: Cell Lines | ||
| Bax−/−Bak−/− SV40 MEF | ATCC | Cat# CRL-2913 |
| Bax−/−Bak−/− SV40 MEF + BAX WT | Walensky Lab | N/A |
| Bax−/−Bak−/− SV40 MEF + BAX R134E/R145E | Walensky Lab | N/A |
| 293GPG Retroviral Packaging Line | Cantor Lab | N/A |
| Recombinant DNA | ||
| Plasmid PTYB1 | New England Biolabs | Cat# E6901 |
| Plasmid PTYB1_BAX_WT | Walensky Lab | N/A |
| Plasmid PTYB1_BAX_R134E | Walensky Lab | N/A |
| Plasmid PTYB1_BAX_R134E_R145E | Walensky Lab | N/A |
| Plasmid PTYB1_BAX_I133A | Walensky Lab | N/A |
| Plasmid PTYB1_BAX_G108E | Walensky Lab | N/A |
| Plasmid PTYB1_BAXΔC (aa 1-168) | Walensky Lab | N/A |
| Plasmid PTYB1_BAXΔC (aa 1-168) R134E | Walensky Lab | N/A |
| Plasmid PTYB1_BCL-w | Walensky Lab | N/A |
| Plasmid pet17b_BFL-1ΔC (aa 1-151) | Walensky Lab | N/A |
| Plasmid pMIG_BAX_WT | Genewiz | N/A |
| Plasmid pMIG | Addgene | Cat# 52107 |
| Plasmid pMIG_BAX_R134E_R145E | Walensky Lab | N/A |
| Software and Algorithms | ||
| Prism | Graphpad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Pymol | The PyMol Molecular Graphics System, Version 1.7.4.0 | Schrodinger, LLC |
| Chimera | UCSF | https://www.cgl.ucsf.edu/chimera/ |
| ProteinLynx Global Server (PLGS) 3.0.1 | Waters Corporation | www.waters.com/waters/en_US/ProteinLynx-Global-SERVER-(PLGS)/nav.htm?cid=513821&locale=en_US |
| DynamX 3.0 | Waters Corporation | www.waters.com/waters/library.htm?cid=511436&lid=134832928&locale=en_US |
| Proteome Discoverer 2.2 | Thermo Scientific | Thermo Scientific |
| XlinkX | Liu et al, 2015 | https://www.hecklab.com/software/xlinkx/ |
| Perculator | The et al, 2016 | http://percolator.ms/ |
| MatLab 2018 | Math Works | https://www.mathworks.com/products/matlab.html |
| GNOM | Svergun, 1992 | https://www.embl-hamburg.de/biosaxs/gnom.html |
| DAMMIN | Svergun, 1999 | https://www.embl-hamburg.de/biosaxs/dammin.html |
| Relion 2.0 | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php?title=Main_Page |
| Mathematica 12.0 | Wolfram | https://www.wolfram.com/mathematica/ |
| Other | ||
| Superdex 75 10/300 GL size exclusion column | GE Healthcare Life Sciences | Cat# 29148721 |
| Superose 6 Increase 10/300 GL size exclusion column | GE Healthcare Life Sciences | Cat# 29091596 |
HIGHLIGHTS.
Fos-12 induces homogeneous BAX oligomers that recapitulate physiologic activation.
SAXS, HXMS, and crosslinking analyses reveal conformational features of BAXO.
BAXO distinguishes between the structural determinants of activation and poration.
BAXO uncovered roles for α6 and α9 in the execution phase of mitochondrial apoptosis.
ACKNOWLEDGMENTS
We thank M. Ericsson, K. Korshavn, S. Lee, M. Godes and A. Dates for technical assistance, E. Smith and J. Zar for graphics support, and A. Cantor for 293-CPG cells. This research was supported by NIH grants R35CA197583 to L.D.W., R01GM101135 to J.R.E., R01GM67945 to S.P.G., 5F31CA210590 to Z.J.H., 5F31CA210592 to E.P.H., and 5T32GM7753 to C.E.N., and Chleck Family and Landry Cancer Biology Fellowships to D.T.C., Lauri Strauss Leukemia Foundation and David Abraham Fellowships to H.D.H., Leukemia and Lymphoma Society Fellowship to Y.B.N., NSF Predoctoral Fellowship to N.B.B., and a collaboration between J.R.E. and the Waters Corporation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Antonsson B, Montessuit S, Lauper S, Eskes R, and Martinou JC (2000). Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345 Pt 2, 271–278. [PMC free article] [PubMed] [Google Scholar]
- Asmar-Rovira GA, Asseo-Garcia AM, Quesada O, Hanson MA, Cheng A, Nogueras C, Lasalde-Dominicci JA, and Stevens RC (2008). Biophysical and ion channel functional characterization of the Torpedo californica nicotinic acetylcholine receptor in varying detergent-lipid environments. J Membr Biol 223, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, Stewart ML, Braun CR, Bird GH, Gavathiotis E, et al. (2015). Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell 57, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, and Walensky LD (2016). Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff HJ, and Bordignon E (2010). Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem 285, 6636–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, Garcia-Saez AJ, and Bordignon E (2014). Structural model of active Bax at the membrane. Mol Cell 56, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M, Damian M, Lescop E, Point E, Moncoq K, Morellet N, Levy D, Marie J, Guittet E, Baneres JL, et al. (2016). Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J Am Chem Soc 138, 11170–11175. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, and Korsmeyer SJ (2001). BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8, 705–711. [DOI] [PubMed] [Google Scholar]
- Chipot C, Dehez F, Schnell JR, Zitzmann N, Pebay-Peyroula E, Catoire LJ, Miroux B, Kunji ERS, Veglia G, Cross TA, et al. (2018). Perturbations of Native Membrane Protein Structure in Alkyl Phosphocholine Detergents: A Critical Assessment of NMR and Biophysical Studies. Chem Rev 118, 3559–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton PG, Lee Y, Ruprecht JJ, Cerson E, Thangaratnarajah C, King MS, and Kunji ER (2015). Trends in thermostability provide information on the nature of substrate, inhibitor, and lipid interactions with mitochondrial carriers. J Biol Chem 290, 8206–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, and Adams JM (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15, 49–63. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. (2013). Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531. [DOI] [PubMed] [Google Scholar]
- Dengler MA, Robin AY, Gibson L, Li MX, Sandow JJ, Iyer S, Webb AI, Westphal D, Dewson G, and Adams JM (2019). BAX Activation: Mutations Near Its Proposed Non-canonical BH3 Binding Site Reveal Allosteric Changes Controlling Mitochondrial Association. Cell Rep 27, 359–373 e356. [DOI] [PubMed] [Google Scholar]
- Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, and Kluck RM (2012). Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ 19, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, and Walensky LD (2013). Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol 20, 888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Fischer H, De Oliveira Neto M, Napolitano HB, Polikarpov I, and Craievich AF (2010). Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Crystallogr 43, 101–109. [Google Scholar]
- Garcia-Saez AJ, Coraiola M, Serra MD, Mingarro I, Muller P, and Salgado J (2006). Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. FEBS J 273, 971–981. [DOI] [PubMed] [Google Scholar]
- Garner TP, Lopez A, Reyna DE, Spitz AZ, and Gavathiotis E (2017). Progress in targeting the BCL-2 family of proteins. Curr Opin Chem Biol 39, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner TP, Reyna DE, Priyadarshi A, Chen HC, Li S, Wu Y, Ganesan YT, Malashkevich VN, Cheng EH, and Gavathiotis E (2016). An Autoinhibited Dimeric Form of BAX Regulates the BAX Activation Pathway. Mol Cell 64, 431. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Davis ML, Bird GH, and Walensky LD (2010). BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell 40, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. (2008). BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lozano MA, Koopmans F, Sullivan PF, Protze J, Krause G, Verhage M, Li KW, Liu F, and Smit AB (2020). Stitching the synapse: Cross-linking mass spectrometry into resolving synaptic protein interactions. Sci Adv 6, eaax5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, and Jakobs S (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra RM, Bird GH, Harvey EP, Dharia NV, Korshavn KJ, Prew MS, Stegmaier K, and Walensky LD (2018). Precision Targeting of BFL-1/A1 and an ATM Co-dependency in Human Cancer. Cell Rep 24, 3393–3403 e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey EP, Seo HS, Guerra RM, Bird GH, Dhe-Paganon S, and Walensky LD (2018). Crystal Structures of Anti-apoptotic BFL-1 and Its Complex with a Covalent Stapled Peptide Inhibitor. Structure 26, 153–160 e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Engelman JA, and Faber AC (2015). The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov 5, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton KL, Stewart KM, Fonseca SB, Guo Q, and Kelley SO (2008). Mitochondria-penetrating peptides. Chem Biol 15, 375–382. [DOI] [PubMed] [Google Scholar]
- Hsu YT, and Youle RJ (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272, 13829–13834. [DOI] [PubMed] [Google Scholar]
- Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, and Huang L (2011). Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol Cell Proteomics 10, M110 002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kefala G, Ahn C, Krupa M, Esquivies L, Maslennikov I, Kwiatkowski W, and Choe S (2010). Structures of the OmpF porin crystallized in the presence of foscholine-12. Protein Sci 19, 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley SO, Stewart KM, and Mourtada R (2011). Development of novel peptides for mitochondrial drug delivery: amino acids featuring delocalized lipophilic cations. Pharm Res 28, 2808–2819. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, and Cheng EH (2006). Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8, 1348–1358. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, and Cheng EH (2009). Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. [DOI] [PubMed] [Google Scholar]
- Kovacs JM, Mant CT, and Hodges RS (2006). Determination of intrinsic hydrophilicity/hydrophobicity of amino acid side chains in peptides in the absence of nearest-neighbor or conformational effects. Biopolymers 84, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, and Newmeyer DD (2005). BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17, 525–535. [DOI] [PubMed] [Google Scholar]
- Lai YC, Li CC, Sung TC, Chang CW, Lan YJ, and Chiang YW (2019). The role of cardiolipin in promoting the membrane pore-forming activity of BAX oligomers. Biochim Biophys Acta Biomembr 1861, 268–280. [DOI] [PubMed] [Google Scholar]
- Leber B, Lin J, and Andrews DW (2007). Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wales TE, Escudero S, Cohen DT, Luccarelli J, Gallagher CG, Cohen NA, Huhn AJ, Bird GH, Engen JR, et al. (2016). Allosteric inhibition of antiapoptotic MCL-1. Nat Struct Mol Biol 23, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchiner ES, Braun CR, Bird GH, and Walensky LD (2013). Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A 110, E986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, and Korsmeyer SJ (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. [DOI] [PubMed] [Google Scholar]
- Liu F, Rijkers DT, Post H, and Heck AJ (2015). Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat Methods 12, 1179–1184. [DOI] [PubMed] [Google Scholar]
- Liu J, Kozhaya L, Torres VJ, Unutmaz D, and Lu M (2020). Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence. J Biol Chem 295, 5944–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, and Green DR (2011). A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, and Fu D (2007). Structure of the zinc transporter YiiP. Science 317, 1746–1748. [DOI] [PubMed] [Google Scholar]
- Matar-Merheb R, Rhimi M, Leydier A, Huche F, Galian C, Desuzinges-Mandon E, Ficheux D, Flot D, Aghajari N, Kahn R, et al. (2011). Structuring detergents for extracting and stabilizing functional membrane proteins. PLoS One 6, e18036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H, et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359. [DOI] [PubMed] [Google Scholar]
- Mechler A, Praporski S, Atmuri K, Boland M, Separovic F, and Martin LL (2007). Specific and selective peptide-membrane interactions revealed using quartz crystal microbalance. Biophys J 93, 3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintseris J, and Gygi SP (2020). High-density chemical cross-linking for modeling protein interactions. Proc Natl Acad Sci U S A 117, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, and Green DR (2013). BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol 20, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourtada R, Herce HD, Yin DJ, Moroco JA, Wales TE, Engen JR, and Walensky LD (2019). Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat Biotechnol 37, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B, Pearson CS, Aranjo A, Cherupalla D, and Belfort G (2016). Mechanism of Four de Novo Designed Antimicrobial Peptides. J Biol Chem 291, 25706–25715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, and Aebersold R (2005). Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4, 1419–1440. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, and Korsmeyer SJ (1993). Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609–619. [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HD, Konarev PV, and Svergun DI (2012). New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitter K, Bernal F, Labelle J, and Walensky LD (2008). Dissection of the BCL-2 family signaling network with stabilized alpha-helices of BCL-2 domains. Methods Enzymol 446, 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritz JR, Wachter F, Lee S, Luccarelli J, Wales TE, Cohen DT, Coote P, Heffron GJ, Engen JR, Massefski W, et al. (2017). Allosteric sensitization of proapoptotic BAX. Nat Chem Biol 13, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, and Tainer JA (2013). Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A, and Wereszczynski J (2017). Probing the disparate effects of arginine and lysine residues on antimicrobial peptide/bilayer association. Biochim Biophys Acta Biomembr 1859, 1941–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, and Garcia-Saez AJ (2016). Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. (1997). Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D, Leontiadou H, Mark AE, and Marrink SJ (2008). Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim Biophys Acta 1778, 2308–2317. [DOI] [PubMed] [Google Scholar]
- Sung TC, Li CY, Lai YC, Hung CL, Shih O, Yeh YQ, Jeng US, and Chiang YW (2015). Solution Structure of Apoptotic BAX Oligomer: Oligomerization Likely Precedes Membrane Insertion. Structure 23, 1878–1888. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, and Tjandra N (2000). Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654. [DOI] [PubMed] [Google Scholar]
- Svergun DI (1999). Restoring low resolution solution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J 76, 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI (1992). Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J Appl Cryst 25, 495–503. [Google Scholar]
- Telbisz A, Ozvegy-Laczka C, Hegedus T, Varadi A, and Sarkadi B (2013). Effects of the lipid environment, cholesterol and bile acids on the function of the purified and reconstituted human ABCG2 protein. Biochem J 450, 387–395. [DOI] [PubMed] [Google Scholar]
- The M, MacCoss MJ, Noble WS, and Kall L (2016). Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J Am Soc Mass Spectrom 27, 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero JG, Sancey L, Kucharczak J, Guillemin Y, Gimenez D, Prudent J, Gillet G, Salgado J, Coll JL, and Aouacheria A (2011). Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells. J Cell Sci 124, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V, and Svergun D (2003). Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, and Korsmeyer SJ (2004). Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, and Korsmeyer SJ (2006). A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24, 199–210. [DOI] [PubMed] [Google Scholar]
- Wales TE, and Engen JR (2006). Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Wales TE, Fadgen KE, Gerhardt GC, and Engen JR (2008). High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Gross A, Waksman G, and Korsmeyer SJ (1998). Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol 18, 6083–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Black SS, Edwards MD, Miller S, Morrison EL, Bartlett W, Dong C, Naismith JH, and Booth IR (2008). The structure of an open form of an E. coli mechanosensitive channel at 3.45 A resolution. Science 321, 1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, and Korsmeyer SJ (2000). tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14, 2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, and Korsmeyer SJ (1995). Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80, 285–291. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and Strasser A (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9, 47–59. [DOI] [PubMed] [Google Scholar]
- Zasloff M (1987). Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci U S A 84, 5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoonens M, Comer J, Masscheleyn S, Pebay-Peyroula E, Chipot C, Miroux B, and Dehez F (2013). Dangerous liaisons between detergents and membrane proteins. The case of mitochondrial uncoupling protein 2. J Am Chem Soc 135, 15174–15182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data File S1 Related to Figures 4 and 5. Source data for mass spectrometry analyses of BAXM and BAXO chemical crosslinks.
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials. This study did not generate deposited datasets or code.