

Keywords: asthma, endotypes, eosinophils, inflammation, phenotypes, precision medicine, type 2
Abstract
While the term asthma has long been known to describe heterogeneous groupings of patients, only recently have data evolved which enable a molecular understanding of the clinical differences. The evolution of transcriptomics (and other ‘omics platforms) and improved statistical analyses in combination with large clinical cohorts opened the door for molecular characterization of pathobiologic processes associated with a range of asthma patients. When linked with data from animal models and clinical trials of targeted biologic therapies, emerging distinctions arose between patients with and without elevations in type 2 immune and inflammatory pathways, leading to the confirmation of a broad categorization of type 2-Hi asthma. Differences in the ratios, sources, and location of type 2 cytokines and their relation to additional immune pathway activation appear to distinguish several different (sub)molecular phenotypes, and perhaps endotypes of type 2-Hi asthma, which respond differently to broad and targeted anti-inflammatory therapies. Asthma in the absence of type 2 inflammation is much less well defined, without clear biomarkers, but is generally linked with poor responses to corticosteroids. Integration of “big data” from large cohorts, over time, using machine learning approaches, combined with validation and iterative learning in animal (and human) model systems is needed to identify the biomarkers and tightly defined molecular phenotypes/endotypes required to fulfill the promise of precision medicine.
Clinical relevance of molecular phenotyping:
Asthma is a heterogeneous disease, in which identifying phenotypes can lead to better responses to both nonspecific and specific targeted therapies.
Patients who are not responding to broad anti-inflammatory therapies should be evaluated for elevations of currently available biomarkers, including blood eosinophils and fraction exhaled nitric oxide.
Severe asthma patients in whom comorbidity and adherence issues have been addressed should be considered for targeted type 2 (T2)-biologic therapies.
Not all patients will respond equally well to every T2-targeted biologic, and more studies are needed to determine which biologic for which patient.
T2-Lo asthma remains poorly defined and requires rule out of T2 activity/biomarkers over time.
I. INTRODUCTION
Although “asthma” arose from the Greek term for “short of breath, ” it likely was not associated with a broadly recognizable entity, or disease, until the 19th century. Henry Hyde Salter, in his treatise “On asthma and its treatment,” specifically described asthma as “Paroxysmal dyspnoea of a peculiar character with intervals of healthy respiration between attacks.” This definition, with some adaptation, remains recognizable today, as asthma is classically defined as reversible airflow limitation (or airway hyperresponsiveness) in the face of variable symptoms, which include wheeze, dyspnea, and cough. Spirometric measurement of airflow limitation with reversible improvement of at least 12% (and 200 ml) was recognized as increasing the likelihood of diagnosis in the 1980s (1). In the absence of reversible airflow limitation, hyperresponsiveness can be measured by inhalational challenges with various stimulatory agents (or exercise), which, in asthmatic patients, induce bronchoconstriction at a lower threshold than individuals without asthma. Additionally, most guideline-associated definitions include inflammation as a key characteristic, but without specific inflammatory biomarkers. This current definition of asthma includes nonspecific characteristics that allow inclusion of a wide range of patients. In fact, many patients receive a clinical asthma diagnosis without meeting the physiological definitions When asthma is defined by doctor diagnosis and asthma medication use, as in most large epidemiologic and genetic population studies, up to 30% may not in actuality have asthma (2, 3). Thus asthma is ripe for investigations which refine the understanding of the mix of clinical, physiological, pathobiologic, and genetic characteristics to enable identification of molecular phenotypes, their biomarkers, and precision medicine therapies.
II. BACKGROUND
A. Heterogeneity of Clinical Asthma
A phenotype is defined as the observable characteristics of an organism that result from the interaction of its genotype (total genetic inheritance) with the environment (https://www.britannica.com/science/phenotype). While the term phenotype has only recently been popularized, asthma has long been recognized as a heterogeneous disease, with the first clinically defined phenotypes those of intrinsic versus extrinsic asthma, described by Francis Rackemann in the 1940s (226). From the early 20th century, asthma was considered an allergic disease, often treated with allergy immunotherapy (71). However, using his clinician’s recognition of overlapping characteristics, Rackemann (226) identified differences between patients who typically developed their disease in childhood, associated with allergic sensitization (extrinsic asthma), and those who developed their disease in adulthood (age 40 and above) (intrinsic asthma), whose disease was less associated with allergies and more likely associated with infections and lower lung function. Similarly, a clinical phenotype of aspirin exacerbated respiratory disease (AERD) was described in 1922, but it was not until 1968 that the phenotype of aspirin sensitivity, asthma, and nasal polyposis became widely recognized (167, 234). These early clinical observations have now been reproduced in multiple hypothesis-driven and unsupervised approaches (184, 186, 190).
In the ensuing era of broadly effective anti-inflammatory/corticosteroid (CS) treatment (280), it eventually became recognized that not all asthma patients responded optimally to this nonspecific immunosuppression. This recognition reignited the concept of heterogeneity in the late 1990s (28, 282). This clinical recognition of asthma heterogeneity, combined with therapeutic needs unmet by CSs, contributed greatly to the evolution of molecular phenotyping.
B. Breaking Down the Characteristics of Asthma
The definition of asthma encompasses multiple clinical and physiological characteristics, which contribute to the heterogeneity of patients’ presentations (280). In the last several years, efforts have been made to deconvolute asthma into these essential characteristics or “treatable traits” (5, 213). These traits include clinical, physiological, and pathobiologic variables such that patients can be identified as having airway obstruction which normalizes with treatments or by having a specific eosinophil-associated airway process, cough, or repeated infections. Numerous other traits could also be considered (TABLE 1). While this simplifies approaches to some degree, in isolation, it does not account for overlapping or conflicting traits. In fact, “treatable traits” typically exist in relation to other characteristics. Traits also have varying definitions and underlying mechanisms, with eosinophilia and airway obstruction often defined in different ways. Similarly, multiple mechanisms could drive a trait like airway obstruction. These include excess mucus, smooth muscle contraction, fibrotic changes, or even alveolar changes, each of which could be driving obstruction in some patients more than others and all with differing treatment implications. Thus, although deconvoluting a disease into its simplest parts clearly offers guidance (see next section), a more integrated approach may be more beneficial.
Table 1.
“Treatable” traits and their nonspecificity
| Trait | Definition | Related Factors | Overlap with Other Traits | Range of Potential Treatments |
|---|---|---|---|---|
| Eosinophilic inflammation | Defined by blood/sputum eosinophils with variable thresholds | T2-related inflammation, eosinophils in blood/lung | Associated with worse airflow limitation, exacerbations, leaner BMI | Corticosteroids, T2-targeted biologics |
| Airflow limitation | Obstruction: various definitions of low FEV/FVC. Reversibility: 12% and 200-ml improvement in FEV1. Full reversibility: FEV1 improves to normal range. | Obstruction: smooth muscle hypertrophy/hyperplasia and contraction, fibrosis, loss of alveolar attachments, mucoid obstruction, edema. Reversibility: contraction, T2 inflammation. | Reversibility associated with eosinophilia. Airway obstruction associated with infections. | Bronchodilators, corticosteroids, mucus modifying agents, possibly T2 biologics. Comment: smooth muscle targeted bronchodilators only treat contraction, possibly mucus clearance. |
| Airway infection | Positive sputum cultures | Bronchiectasis, bronchiolectasis, microbiome dysbiosis | Antibiotics, possibly macrolides long term | |
| Cough | Cough hypersensitivity to challenges, truncated inspiratory flow volume loops, direct examination of inspiratory vocal cord closure | Neuropeptide abnormalities, gastroesophageal reflux, postnasal drip, vocal cord dysfunction, asthma | Treatment directed to cause | |
| Obesity | BMI, % body fat, CT | High-dose corticosteroids, inactivity from disease, metabolic dysfunction, microbiome, differences by age at onset | Low eosinophils | Reduction in corticosteroid dose, improved asthma, exercise and diet |
| Psychosocial issues | Psychiatric diagnosis? | Poor adherence, access to meds, socioeconomic, cultural, psychological, very severe asthma | Counseling, understanding of social issues, better asthma treatment | |
| Age at onset | Variably defined cut-points from 12 to 40 | Presence of allergic features, sinus disease, eosinophilia | Eosinophilia, airway obstruction, reversibility | Anti-IL-5 may do better in later onset |
BMI, body mass index; CT, computed tomography; FEV, forced expiratory volume; FVC, forced vital capacity; IL, interleukin; T2, type 2.
III. LESSONS FROM CLINICAL TRAIT-TARGETED AND CLUSTERING STUDIES
Renewed interest in identifying new subgroups of asthma patients emerged in the late 1990s. Three factors contributed to this renewed interest, including that 1) responses to CSs were highly variable, 2) pathological heterogeneity was increasingly appreciated (28, 97, 282), and 3) studies of Th2 (now type 2)-pathway targeted biologics were negative across “all-comers” with asthma (83).
A. Targeted, Hypothesis-Driven Approaches and Application of Precision Medicine
Early trait-based studies often targeted single characteristics. Age at onset was consistently identified as a distinguishing trait, confirming and expanding on the concepts of extrinsic and intrinsic asthma (184, 190, 226). Importantly, these studies also shed light on eosinophilic inflammation, measured in blood or sputum. Despite earlier biases, it was not present in all patients with asthma or even severe asthma, but was more common in exacerbation-prone, CS-responsive, and late-onset asthma (28, 101, 184). This recognition was critical to completion of the first successful precision medicine trials utilizing eosinophil targeted biologic therapies only in patients with eosinophilic exacerbation-prone severe asthma (110, 199). Similar hypothesis-driven studies identified interleukin (IL)-6 and related pathways, particularly in blood, in association with more severe obese and exacerbation-prone asthma (218). Finally, fungal-associated asthma has also been suggested as a clinical phenotype, based, among other things, on the biologic association of fungi with extrinsic proteolytic activity (169). It was included on an early list of asthma endotypes as well (174). Clinically, fungal asthma is variably defined based on levels of fungal specific IgE-associated sensitivity and/or fungal presence. Three small double-blind placebo controlled trials of antifungal therapies have been performed. Of the two larger studies, one, which studied voriconazole in patients sensitized to Aspergillus fumigatus, was negative, while the other, which studied itraconazole in those sensitized to any of a number of fungi, was positive (4, 62). Both utilized asthma quality of life as the primary end point, suggesting that molecular approaches may be needed to better define a fungal-driven asthma phenotype. However, these hypothesis-driven studies all identified single pathways (i.e., eosinophils, fungi, IL-5, IL-6) in relation to certain traits (exacerbations/obesity/age at onset, fungal sensitivity), initiating the application of precision medicine to biomarker-identifiable patients.
B. Less Biased/Supervised Clinical Clustering Approaches
As statistical methods evolved integrating increasing amounts of data into single analyses, less biased (or less hypothesis-driven) approaches emerged. One early study used only seven selected/hypothesis-driven variables (including sputum eosinophil percentages, symptoms, and peak flow variability) and a K-means clustering approach (see analytic approaches) on two separate clinic populations (111). Four reproducible asthma patient clusters were identified, on the basis of symptoms, eosinophilic inflammation, and age at onset. While simple in design, these clusters (or phenotypes) are broadly reproduced in more complex studies, including 1) a benign early-onset asthma phenotype; 2) a symptom predominant, minimally obstructed obese phenotype; 3) an early-onset increasingly severe phenotype; and 4) a late-onset more severe eosinophilic phenotype.
The maturation of large asthma networks allowed incorporation of increasing numbers of characteristics into clustering approaches of larger populations. Using only clinical and physiological characteristics collected as part of the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program (SARP) network, Moore et al. (118) clustered 34 asthma characteristics, derived using a random forest approach, to reveal 5 different patient clusters, 3 of which were severe, and differed primarily by lung function variables and age at onset. As described in earlier hypothesis-driven studies, later onset disease was less likely to be allergic, but was also more obese and female, while the most severe disease had the most exacerbations, highest systemic CS use, and the worst airflow limitation which failed to normalize after bronchodilators. This study was followed by an analysis of 378 asthmatic and healthy controls (HC), which incorporated lung-specific inflammatory characteristics [i.e., bronchoalveolar lavage (BAL) cell differentials] from those who had undergone bronchoscopy (186). This study included a greater number (and range) of variables into the clustering solution, as well as HCs. With the use of hierarchical clustering, 10 variable clusters and 6 clinical clusters were identified: 1 healthy cluster, 3 severe, and 2 milder asthmatic clusters. Validating previous studies, a late-onset, nasal polyposis-prone highly eosinophilic cluster was recognizable, as were several early-onset more allergic groups. Similar to the Moore SARP clusters, a very severe cluster was identified with persistent obstruction, despite a large bronchodilator response. Inclusion of inflammatory characteristics showed that this most severe cluster associated with high levels of the asthma biomarker, fraction exhaled nitric oxide (FeNO), as well as a mixed neutrophilic/eosinophilic inflammatory process. These variable associations were validated in separate studies where FeNO levels were identified as the strongest independent predictor of oral CS use and where a mixed granulocytic process was observed in sputum from the most severe patients (191, 291). Finally, a large two-way cluster analysis was performed on over 300 patients in the British Thoracic Society Severe Asthma Registry, using factor analysis to first do initial dimension reduction (203). With the use of 23 variables including allergy, obesity, blood eosinophils, treatment, exacerbations, and lung function, late-onset eosinophilic, late-onset obese, and early-onset allergic clusters again emerged. Although lacking molecular input, consistent clinical patterns were emerging, laying the groundwork for the approaches to molecular phenotyping, endotyping, and precision medicine outlined in FIGURE 1.
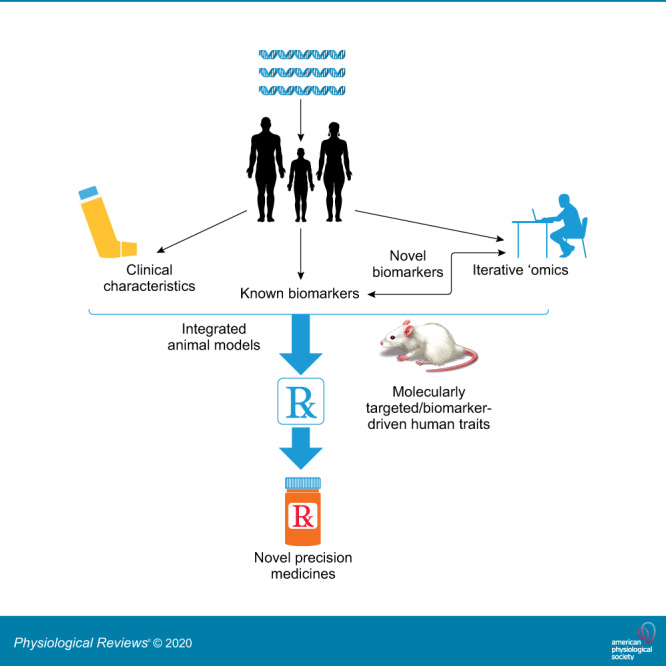
FIGURE 1.

Generalized paradigm for the implementation of precision medicine. Asthma presents in distinctly different ways based on integration of genetics and environmental exposures. Granular clinical and molecular phenotyping can generate novel target pathways that can be validated over time and through the use of animal models. These approaches should lead to development of biomarker targeted therapeutic trials which improve the overall efficacy and safety of drugs, bringing the best drug option to the correct patient.
IV. EVOLUTION OF MOLECULAR PHENOTYPING AND ENDOTYPING
The term molecular phenotyping alludes to identification of specific molecular pathways in relation to clinically distinguishable traits or phenotypes. Molecular phenotyping requires identification of consistent plausible molecular pathways in relation to disease, association with clinical outcomes, and, ideally, the improvement of outcomes when the identified pathways are specifically targeted (TABLE 2, FIGURE 1). Endotyping is the penultimate phenotype, or even disease. It is a condition which is defined by a distinct functional or pathobiologic mechanism (or treatment response) (10). This implies that endotypes as defined above require identification of a specific pathway(s) that controls most aspects of the disease. Application of this definition to asthma is controversial as interpretations of the overall importance of a specific pathway to a molecular phenotype may vary. Some might suggest an endotype includes all T2-Hi phenotypes. However, a more literal interpretation is that a molecular pathway (or treatment) truly defines the disease. While T2 (or associated) inflammation is present and relevant to mild to severe early-onset allergic asthma, the data per se do not yet fully support T2 inflammation as the defining pathway for those molecular phenotypes. In fact, it is increasingly recognized that T2 biologics (treatment responses related to IL-4, -5, and -13 pathways) are not as effective in early-onset disease (23, 29). However, considerable data now support the concept that a T2-Hi, adult-onset eosinophilic asthma endotype is emerging, with the dramatic responses to anti-IL-5/5R and perhaps anti-IL-4Rα as well, supporting involvement of ILC2 (or other) cells. As noted, treatment response could also eventually define an endotype, if, for whatever reason, a patient had a dramatic response to a specific pathway blocker but did not fall into any previously defined clinical phenotype. However, ideally additional clinical characteristics would associate with that response. Thus newly defined and characterized endotypes could emerge either as outshoots of the current molecular phenotypes, or alternatively as distinctly new entities.
Table 2.
Postulates that define molecular phenotype/endotypes
| 1. Identification of plausible biologic pathway |
| 2. Consistency of expression of the pathway over time |
| 3. Correlation of biologic pathway expression level with relevant clinical outcomes |
| 4. Identification of measurable biomarker for the pathway |
| 5. Reduction (molecular phenotype) or resolution (endotype) of clinical manifestations of disease when the pathway is targeted |
The development of multiple/’omic platforms, capable of measuring hundreds to even millions of analytes, typically of a specific type, on a single specimen, opened the flood gates for molecular phenotyping by vastly increasing the complexity and granularity of molecular data (FIGURE 1). These platforms included microarrays, RNA sequencing, two-dimensional gels, multiple-antibody bead-targeted proteomic assays (luminex and others), mass spectroscopy-based multi-antigen cell sorting techniques (CyTOF and others), as well as mass spectroscopy studies of proteins, lipids, and metabolic profiles. Most ‘omic data have been transcriptomic, with newer approaches including single cell sequencing, CITE-sequencing, ribo-sequencing, which identifies mRNA sites undergoing active ribosomal translation and ATAC-seq which identifies open chromosomal regions linked with gene transcriptomics (See TABLE 3 for a short list of current and evolving approaches). In all cases, vast amounts of data are generated on a single subject’s samples, or in some cases thousands of sequences on a single cell from a single individual (single cell RNA sequencing). As of yet, there is no gold standard approach. In all cases, validation is necessary, ideally at the protein, pathway level, with model systems and with additional support through alternate computational approaches.
Table 3.
Emerging ‘omics platforms
| Platform/Array | Description | Reference Nos. |
|---|---|---|
| Single cell RNA sequencing (scRNA-seq) | Manual or FACS isolation of single cells which are then lysed, undergo poly-A selection, cDNA preparation, and amplification and sequencing similar to bulk NGS (see above). Allows single cell resolution, but can be hampered by quality control limiting ability to compare cells. | Stegle et al. (251) |
| Imaging flow cytometry | Combined antibody fluorescence with bright- and dark-field microscopy. This allows protein quantification and subcellular visualization. Limited number of channels can limit the ability to investigate large numbers of markers. | McGrath et al. (181a) |
| Mass cytometry (CyTOF)/HD flow cytometry | High-dimensional flow cytometry techniques to assess large panels of antibodies. CyTOF utilizes metal isotope labels and mass spectroscopy, while HD cytometry uses photomultiplier tubes across multiple channels to allow a large number of fluorophores. | Han et al. (113), Mair and Prlic (178) |
| High-dimensional scRNA-seq | Cell populations can be analyzed and sorted by CyTOF or HD cytometry. Isolated cells are then lysed and undergo single cell RNA-seq. Expense and similar issues associated with scRNA-seq can limit ability to apply in large cohorts. | Stegle et al. (251) |
| Assay of transposase-accessible chromatin sequencing (ATAC-seq) | Tn5 transposase cleaves DNA fragments from accessible regions of the cell’s DNA. These sequences are then read and mapped to the chromosome. Areas with frequent reads show up as peaks, and these peaks are used to determine accessible regions of chromatin. | Chang et al. (44) |
| Chromatin immunoprecipitation sequencing (ChIP-seq) | DNA binding proteins are crosslinked in vivo using formaldehyde. Chromatin is then digested and antibodies against the DNA binding proteins are used to precipitate out these proteins with their associated chromatin. The recovered chromatin is then sequenced and mapped. | Davies et al. (57) |
| Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) | Antibodies with coupled oligonucleotide barcodes are able to bind to cell surface proteins. These cells then undergo scRNA-seq, and the unique barcode allows protein quantification in addition to RNA sequencing in the same readout. | Stoeckius et al. (253) |
| MethylC-seq | Bisulfite treatment converts nonmethylated cytosine to uracil, providing a sequencing marker for methylation status of cytosine residues. This allows assessment of potential epigenetic changes in tissues and cells | Urich et al. (266) |
In almost all ‘omic studies, millions of data points are generated. Broad analysis of lung diseases requires sampling of multiple different organ compartments, including 1) site-directed bronchoscopic approaches (i.e., bronchial brushings, primarily for epithelial cells), 2) BAL fluid and cells (luminal distal airway and alveolar cells), and 3) endobronchial biopsies (large airway structural and inflammatory cells). Sputum (large airway luminal/primarily immune-inflammatory) cells and nasal brushings, in addition to more accessible peripheral compartments, including blood and urine, are also targets for study. Each of these compartments can be collected at the same or at different time points and yield different, sometimes complementary information, from the same or different platforms. Interdigitating multiple compartments, cell types, time elements, as well as the large numbers of data points from each measurement poses enormous statistical challenges. Few studies have satisfactorily combined (or harmonized) these differing data sets, or even more importantly, different data types, such that this remains an ongoing challenge.
A. Analytic Approaches to Phenotyping and Endotyping
While clinical phenotyping often utilizes less than 100 variables, ‘omics studies require approaches capable of integrating thousands if not millions of variables. Not surprisingly, the use of computational pattern detection is integral to making sense of these big data (FIGURE 1). Machine learning, the ability of software to learn from data without human direction, allows patient classification based on underlying similarities in the data set variables in a manner too complex for traditional assessments.
1. Dimension reduction
Clustering approaches using supervised or unsupervised techniques were developed to group large numbers of similar variables. Several steps are required before clustering analysis. Dimension reduction utilizes approaches to decrease variable numbers thereby facilitating analysis. In many cases, input variables are selected based on subjective or objective measures. Less biased approaches include principal component analysis (PCA) and t-stochastic neighbor embedding (tSNE) (171). PCA identifies small numbers of uncorrelated variables from large data sets, is computationally fast, and generally maintains large differences between individuals. However, it performs less well with nonlinear data. tSNE (often used with ‘omics studies of mixed cell types) maintains similarities between data points but is less informative at describing magnitudes of differences (268a). Regardless of method, dimension reduction involves optimizing distance measures among data points but is confounded by increases in variance at the extreme end of observations in big data sets. Algorithms such as rLog transformation of RNA sequencing data can stabilize variance to facilitate meaningful analysis (175).
2. Clustering
Following dimension reduction, popular clustering methods include hierarchical, partitioning (K-means)- and model-based or probabilistic approaches. Both hierarchical and K-means clustering have been used in asthma phenotyping studies (186, 290). Hierarchical clustering can be performed utilizing a bottom-up approach where each data point is iteratively merged with its neighbor until all have been joined to a single cluster (58), or as a top-down approach where objects are recursively divided (105). Hierarchical clustering is dependent on input distance measure and linkage criteria used to merge data points. Distance measures include Euclidean or squared Euclidean, which represent “ordinary” straight line distances in coordinate space (or its squared value). Less often, Manhattan, representing absolute distance between vectors, or Maximum distance between vectors are used.
In single-linkage hierarchical clustering, cluster relationships are determined by the similarity of their most alike members (195). The merge criterion is local, meaning only the area where two clusters come closest is taken into account, while the overall structure is not. This can result in a straggling chain of points representing local relationships without providing information on higher level structure. In contrast, complete-linkage clustering utilizes similarity between the most dissimilar members of a cluster and takes the entire clustering structure into account (37). Perhaps the most popular, Ward’s method, which considers the distance between two clusters as how the sum of square errors increases when they are merged, was developed to minimize error within clusters (272).
K-means clustering algorithms belong to a class of approaches known as partitioning methods (117). K-means is considered the fastest and simplest method for clustering data starting with a user defined number of data points that defines the initial clusters to which all other points are assigned (138). The centroid, or resultant mean from the newly generated cluster, is then updated and cluster assignment processing is repeated until a stopping-criterion such as distance minimization or maximum iterations is reached. K-means clustering is sensitive to outliers and tends to perform poorly on data where attribute variance is unequally distributed. To address these issues, the partitioning around medoids (PAM) and CLustering LARge Applications (CLARA) algorithms utilize the most centrally located point in a cluster, or medoid, for dissimilarity calculation, rather than the mean value of all the data points in the cluster (117, 268).
Model-based clustering, also known as latent class analysis or mixture modeling, assumes that each cluster present in the data set corresponds to a different model (182). Although this allows for nonuniform cluster shapes as well as fuzzy cluster assignments, the process is relatively complex and requires user input based on Bayesian information criteria.
A common feature of these methods is that they require user specified cluster assumptions to define groups. Although hierarchical clustering provides a dendrogram for visual inspection of relationships, the level to cut the tree to define groupings/clusters is subjective. Methods for estimating the number of clusters in a data set include optimization of criterion via the elbow or silhouette approaches, as well as calculation of the gap statistic (263). No approach is perfect, and each requires assumptions regarding what the right solution would represent. One way to estimate veracity is through cross-validation, whereby the rules for assignment from a training set are used to classify a testing set, giving a sense of how well a clustering solution can be generalized. However, in asthma studies, this is often problematic, as parallel data sets may not be available.
3. Pathway analysis
The importance of these clustering approaches to biologic or clinical reality is often unclear. To this end, identifying patterns to provide meaningful biologic context is not only important but also controversial. As examples, differential gene expression analysis from RNA sequencing/microarray data can be queried for meaning using databases such as protein analysis through evolutionary relationships (PANTHER); Database for Annotation, Visualization and Integrated Discovery (DAVID); or Kyoto Encyclopedia of Genes and Genomes (KEGG) for enrichment in functional terms (132, 145a, 196, 293). Patterns of upregulated genes may also be used to infer transcription factor activity by querying data sets such as ENSEMBLE, TRANSFAC, or MotifMap (181, 292, 298a). Patterns have also been identified from previously annotated and targeted mouse or human gene expression studies by gene set variation analyses (GSVA) (121, 122, 231). Gene Set Enrichment Analysis (GSEA) compares gene expression using sets of transcripts more broadly associated with biologic processes or diseases (217, 254). Weighted gene coexpression analysis (WGCNA),unlike GSEA, does not rely on curated or annotated lists, but rather constructs correlated gene modules that are then compared with a clinical trait or annotated through one of the platforms described above (161, 185, 217). WGCNA also facilitates extraction of key “hub” genes that connect networks or summarize the behavior of modules (eigengenes). These approaches all promote development of molecular hypotheses which must be tested in vitro and in vivo before results can be fully validated, a bar which most studies fail to reach.
Although tools available for evaluating complex analyses grow every day, there are still limitations. New and emerging tools improve integration of multiple data sets (i.e., data-harmonization) across cellular compartments or differing time points, including multi-view clustering recently applied to identify corticosteroid response patterns in asthma (289). Expanded analytic approaches are still needed.
B. Molecular Phenotypes: Linking Molecular Pathways to Clinical Traits
Breaking a disease into its components allows for a more simplified analysis linking broad ‘omic profiles with disease or specific characteristics. Three general approaches have been used. The most common uses the trait as the dependent (fixed) characteristic, with transcriptomic (or other ‘omic) data, either individual genes/proteins or gene signatures, evaluated in relation to that characteristic only (16, 17, 121, 122, 156). Others have identified all genes related to a specific trait, then clustered those genes to identify expression patterns in relation to individual patients (156, 186). Finally, unbiased approaches have clustered genes and then related the gene clusters to specific defined characteristics/traits (185, 217).
Multiple transcriptomic studies have evaluated differentially expressed genes between two or more categorical variables (traits), from persistent airflow limitation to inflammatory phenotypes, to clinically defined clusters. The simplest approaches identified differentially expressed genes between categorical variables, such as asthma versus health (287). Others have taken a second step and evaluated the differentially expressed genes for the presence of predefined, typically human in vitro or mouse-model related, gene clusters using GSVA, to give insight into the functional aspects of the differential gene expression patterns (121, 122, 231). Clustering of GSVA gene sets has also been used to identify clinical phenotypes (156). Limitations to these approaches begin with using an arbitrary/fixed definition of the comparator groups or traits, often based on categorical cut-points with limited biologic validation. This biased approach simplifies continuous characteristics into single prespecified categorical variables, which for statistical analysis are now the dependent variable. In some cases, statistically identified clinical clusters have been used as the anchors for the molecular analysis (166). Depending on the accuracy of these clinical clusters, their predictive characteristics may have less value. Additionally, GSVA gene sets can give limited or even misleading information on pathway analyses, especially when mouse comparator gene sets are included. It is likely that deficiencies in both the independent and dependent variables contribute to the modest associations between GSVA and clinical characteristics.
Despite these limitations, these studies have consistently produced relationships of eosinophilic or type 2/IL-4-IL-13 gene sets with traits such as persistent airflow limitation and eosinophilic asthma phenotypes (FIGURE 2) (121, 156). Additional differences have been observed in inflammasome-related pathways, although its presence is complex, with certain elements (IL1RL1, the IL-33 receptor) upregulated in association with eosinophilic inflammation and others (IL-1-related genes) more likely associated with neutrophilic processes (FIGURE 3) (149, 159, 231). Inconsistencies in associations of these pathways are also apparent when comparing multiple compartments and cell types. In cases where mixed cell types are included (sputum or biopsy tissues in particular), controlling for background differences in cell types, each with their own specific gene signatures, is often not done, leading to difficulty in interpretation. However, this approach confirmed an overall association of T2 immune pathways with asthma characteristics or eosinophilic phenotypes and suggested complex relationships to inflammasome-related pathways.
FIGURE 2.

Complex interplay between type 1 and type 2 immune pathways contributes to differing asthma phenotypes. Repeated exposure to allergens in genetically susceptible individuals induces the development of Th2 cells, which produce interleukin (IL)-4, IL-5, and IL-13. These type 2 cytokines can be also produced by alternate mechanisms involving cells of the innate immune system. In this regard, pathogens including bacteria, viruses, and fungi as well as allergens can cause epithelial damage in which proteases encoded by these agents play a prominent role. Epithelial damage leads to increased expression and release of IL-33 and thymic stromal lymphopoietin (TSLP), which stimulate ILC2 cells to produce IL-5 and IL-13. Tissue resident mast cells and basophils recruited from the periphery or generated from in situ differentiation of progenitors can also generate these cytokines. IL-5 released from circulating Th2 cells and other growth factors such as IL-3 and granulocyte-macrophage colony stimulating factor (GM-CSF) stimulate eosinophil differentiation, proliferation in the bone marrow, which are then recruited to the airways under the influence eosinophilic chemokines. Mast cells also produce prostaglandin D2 (PGD2) that binds its cognate receptor CRTH2 on Th2, ILC2 cells, triggering cytokine release. IL-4 and IL-13 produced by these different cell types augment mucus production via expression of mucin genes including MUC5AC. In certain individuals, gene-environment interactions also promote a type 1/interferon (IFN)-γ response with or without IL-17 production from T cells (not shown). IFN-γ in turn stimulates production of the chemokines CXCL9 and CXCL10 from both airway epithelial cells and resident macrophages with the potential to create a positive feedback loop promoting recruitment of Th1 cells and eosinophils via interaction with the receptor CXCR3. Increased expression of the cytokine IL-18 in macrophages can also promote development of Th1 and Th2 cells. Synergism between T1 (IFN-γ) and T2 (IL-13) cytokines augments expression of the enzymes inducible nitric oxide synthase (iNOS) and dual oxidase 2 (DUOX2) in the airway epithelial cells driving production of nitric oxide (NO) and H2O2, respectively, with increases in nitrative and oxidative stress. APC, antigen presenting cell; FeNO, fraction exhaled nitric oxide.
FIGURE 3.

“Inflammasome” linked interleukin (IL)-1β, IL-18, and IL-33 pathways in relation to downstream pathway activation and cellular responses. Both genetic and transcriptomic studies have strongly supported involvement of these cytokines in asthma phenotypes. These pathways are all activated by danger signals/receptors, with responses likely dependent on initiating signal, cell type, and perhaps genetic factors. IL-1β activation, through a classical inflammasome process, leads to broad activation of many cell types, generally linked to neutrophilic inflammation, perhaps through interferon (IFN)-γ or IL-17 pathways. Genetically associated IL-33 activation, arising from epithelial damage and augmented by inflammatory cell protease activity, stimulates ILC2 cells and mast cells/basophils to generate type 2 cytokines increasing type 2 immune responses. IL-18, strongly linked genetically with asthma through IL-18R1, can augment either type 1 or type 2 immune responses depending on the presence of IL-12. Interestingly, all 3 receptor pathways signal through MyD88, NF-κB, and Jun kinase (JNK) pathways. IL18RAP, IL-18 receptor activating protein; IL-18BP, IL-18 binding protein; sST2, soluble ST2/IL1RL1.
1. Identifying new patient clusters based on trait-based molecular profiles (limited molecular phenotypes)
The second approach utilizes gene sets to identify clinically distinguishable clusters exclusively derived from molecular characteristics. Thus “biased” molecular pathways drive clinical outputs. This approach was applied to 154 participants in the SARP with bronchoscopic microarray data from fresh human airway epithelial cells (HAEC) (186). Over 500 genes were identified in the brushings, which correlated strongly with the asthma biomarker FeNO. A K-means clustering approach identified five clinical clusters, three with high FeNO and two with low levels. The three high FeNO clusters included a very severe T2-Hi gene expression group, associated with low epithelial growth and repair expression, an older-onset group, with more complex inflammatory profiles, as well as a younger, milder, more traditionally allergic cluster. The two low FeNO clusters, which contained nearly all the HCs, varied by atopic status, asthma severity, and disease duration. A similar approach was used on ~500 genes which differentiated patients with ≥1.5% eosinophils compared with those with less (156). Hierarchical gene clustering identified three different “transcriptome associated clusters” (TACs). Sputum eosinophils strongly differentiated the TACs, identifying one high, one low, and one sputum eosinophil intermediate. Using GSVA, potential pathways were identified, including T2/IL-13, ILC2, neutrophilic inflammation, inflammasome, and Th17. Pathways linked to ILC1 and aging were also identified. Unfortunately, clinically or statistically significant differences across the three clusters were small. Thus the ability of this approach to identify clinically meaningful clusters may vary by study, depending on biomarker/associated genes and compartment chosen.
2. Molecularly defined phenotypes (FIGURE 1)
The most unbiased approach is to start with expressed genes and allow them to identify molecularly defined clinical phenotypes. The earliest (and biased) example was the original identification of what was then defined as “Th2-High” asthma (now T2-Hi), based around a highly selected signature of three genes upregulated by the addition of IL-13 in cultured HAECs (287, 288). Evaluating a group of mild, CS naive patients and HCs, ~50% of asthmatic patients had a broad increase in expression of these genes in fresh HAECs, establishing a standard for a HAEC T2 gene signature. These T2-Hi patients were more allergic and had higher eosinophils and more bronchial hyperreactivity than patients with low expression. Importantly, this was the first time response to an anti-inflammatory therapy was evaluated by molecular cluster, with Th2/T2-Hi patients having robust improvement in forced expiratory volume in the first second (FEV1) to inhaled CSs, while those with low expression did not. Confirming the CS responsiveness of this gene signature in mild asthma, follow-up bronchoscopies showed a reduction in the T2 gene signature expression in response to the inhaled CS.
This initial identification of the two T2 molecular phenotypes was based on a very small number of biologically identified T2 signature genes. A less biased approach was undertaken on microarray data from sputum samples utilizing KEGG pathways to identify samples, which differed in relation to the presence (numbers and percentages) of genes from these identified pathways (293). Three transcriptomic endotypes of asthma (TEA) clusters were identified which integrated the expression of the enriched KEGG pathways across the samples. Several KEGG pathways distinguished the clusters, including pathways linked to epithelial and neuronal function. These three TEA clusters differed by overall severity and biomarkers of T2 inflammation but, surprisingly, were not enriched for T2-related genes, perhaps because of low T2 genes in sputum cells. There was also little overlap of the differentially expressed genes from this study compared with results from other approaches.
Identifying molecularly defined clinical phenotypes has also been limited by the number of genes evaluated, primarily due to lack of approaches to analyze many more than a thousand variables. Microarrays and more recently RNA-sequencing now provide access to differential expression of thousands of genes. WGCNA first clusters genes in relation to each other and then weights these clustered genes in relation to specific categorical and continuous trait variables (see sect. IVA). With the use of this approach on HAEC microarray data, over 50 different “related gene” modules were identified, with plausible gene relationships by pathway analysis (185). Genes in these related modules identified cohesive functional, immune, or structural cell pathways, including those related to T2 signature genes and epithelial dysfunction (FIGURE 2). This less biased approach also identified new relationships between epithelial growth and repair, neural processes, and mitochondrial gene modules with severity traits, which could also contribute to the pathogenesis of severe asthma.
This approach was recently applied to BAL cell microarray data from 154 SARP subjects, 104 of whom overlapped with previous epithelial brushing data (185, 273). Due to the complex nature of BAL cells, the data were first “deconvoluted” by cell differentials and then by race, age, sex, and body mass index (BMI), all of which influenced gene relationships. All expressed genes were then related to various clinical outcomes, while controlling for cell type, demographics, and medication use. BAL cell genes associated with the cAMP pathway inversely linked to severity outcomes, including amount of β2-agonist use. WGCNA then identified 49 gene networks, only one of which, regulation of cAMP-dependent protein kinase activity, related to disease severity parameters, and in particular to β2-agonist use. Additional genes in this pathway include several growth and repair factors, as well as dendritic cell markers, suggesting β2 agonist treatment, especially in excess, could also contribute to molecular phenotypes, and confirming the potential for large effects of treatments on molecular patterns. Using the hub genes identified by the WGCNA approach to prioritize hypothesis-driven investigations, the authors expanded and validated the β2-agonist hypothesis using in vitro studies. Unlike β2 agonist use, CS-related gene expression patterns did not relate to disease severity.
V. MOLECULAR PHENOTYPES: TYPE 2 AND BEYOND
A. Identification of T2-Hi Molecular Phenotypes: Biologic and Clinical Validity
Type 2 asthma has clearly reached the status of a broad molecular phenotype. The asthmatic inflammatory response was first associated with the Th2 arm of the adaptive immune system in the early 1990s (230), a few years after mouse immune system studies helped establish the Th1/Th2 T-lymphocyte-focused paradigm (192) (FIGURE 4). Subsequent research led to identification of GATA-3 as the master regulator of Th2 cell development (299, 302). GATA-3 was also associated with allergic asthma in an experimental mouse model (300), and increased expression of GATA-3 was described in the airways of asthmatics (201). Connections of Th2 immunity to human asthma were driven by associations with IgE/allergic type 1 hypersensitivity reaction, eosinophils, and CD4 lymphocytes (192, 230, 270). Human studies reported IL-4, -5, and -13 expression in both peripheral blood cells and airways (54, 135, 153). Transgenic animal models of allergic asthma followed in the 1990s, which confirmed the importance of the Th2 cytokine IL-13 to allergic responses in mice (103, 285). Unfortunately, although it appeared obvious that Th2 immunity was critical for asthma, human trials of targeted therapies failed. At the same time, human studies of Th2 cytokine expression and eosinophilic inflammation revealed wide variability across asthma (153, 282, 295). It also became clear that the canonical Th2 cytokines (IL-4, IL-5, and IL-13) were produced by many cells beyond Th2 cells, including ILC2, mast cells/basophils, and even eosinophils (reviewed in Zhu, Ref. 304) (FIGURE 2). This led to a broad acceptance that Th2 immunity, especially in relation to asthma phenotypes, is more properly referred to as type 2 or T2 immunity, encompassing the broader cellular sources and potential immune mechanisms. While these variations were originally believed to be “noise” associated with human studies, nearly all ‘omic studies to date have identified subsets of asthma patients who manifest consistent evidence for T2-related immune processes, without specific relation to Th2 or other immune cells. Simultaneously, new tools led to increasing appreciation of the reach of the immune system beyond Th2 cells to the importance of other arms of the adaptive immune system, including Th1 and Th17 (or less well appreciated T1 and T17) (FIGURE 2). Thus, while T2 immunity was apparent in human asthma, complexities in its levels, cell sources, and accompanying immune pathways likely contribute to differing clinical manifestations and importance.
FIGURE 4.

Timeline in establishment of the Th1/Th2 paradigm, identification of master regulators of T helper cells, and association with asthma. Naive CD4+ T cells differentiate in response to antigens and costimulation to different subsets, each characterized by its expression of specific cytokines, a paradigm established in the mid 1980s. Increased numbers of Th2 cells were identified in the airways of asthmatics. A surge of interest in molecules that are essential for lineage development led to the association of GATA-3 with interleukin (IL)-5 gene expression and as a master regulator of Th2 cells and subsequently of ILC2 cells as well. Likewise, T-bet and RORγt are essential for the development of Th1 and Th17 cells, respectively. Increase in GATA-3 expression was observed in human asthma, and expression of a dominant-negative (DN) mutant of GATA-3 in mice blocked allergic eosinophilic airway inflammation in a mouse model. IFN, interferon; BAL, bronchoalveolar lavage; ConA, concanavalin A; TCR, T-cell receptor; HSC, hematopoietic stem cell; CLP, common lymphoid progenitor.
1. Consistency and complexity of T2-Hi molecular phenotypes (FIGURES 2 and 5)
FIGURE 5.

Schematic of the current understanding of T2-Hi molecular and clinical phenotypes. Type 2 inflammation is characterized by enhanced activity of interleukin (IL)-4, -5, and -13 which commonly leads to eosinophilic inflammation, mast cells, mucus, and elevated fraction exhaled nitric oxide (FeNO), all of which are likely to overlap to varying degrees. However, depending on the mix of cytokines and these accompanying immune processes, at least 4 distinct type 2 molecular phenotypes are recognizable, with varying response to therapy, ranging from simple mild allergic early-onset asthma to very severe, T2-Hi+ asthma. IFN, interferon; CS, corticosteroid.
A T2-molecular phenotype is identifiable in a large percentage of the overall asthma population (280). This phenotype is associated with evidence for the presence or downstream activation of IL-4, -5, and/or -13, in varying lung compartments (tissue, fresh HAECs, sputum) (219), and with tissue, blood and sputum eosinophils, sputum T2 gene expression, FeNO in exhaled breath and blood biomarkers, like periostin (141, 186, 219). Population-based identification of T2-Hi patients is limited by the availability, sensitivity, and specificity of these downstream biomarkers. Unfortunately, measuring the cytokines themselves in any compartment is limited by their very low and highly variable levels. Thus current biomarkers are rather nonspecific for T2 immune processes, with other factors, including treatments, contributing to their elevation and variability over time. Severe asthma patients with elevations in these T2 biomarkers also generally respond to T2-targeted biologics, confirming their overall relationships (41, 42, 52, 110, 199, 277). The best therapeutic responses to the T2/IL4Rα targeted drug dupilumab occur in patients with dual elevations in both FeNO and blood eosinophils (40). However, significant clinically meaningful responses to T2-targeted therapies also occur in patients with smaller elevations in FeNO or blood eosinophils, suggesting that widely used cut-points for T2-Hi and -Lo may be inaccurate. In fact, although most T2-targeted biologics used 300 eosinophils/μl as the initial threshold, responses are apparent at cut-points as low as 150 cells/μl (40). Thus, until better biomarkers are identified, assignment to a T2 molecular phenotype could also include a positive response to a specific T2 targeted therapy.
Consistent pathobiologic processes and clinical features are generally observed across this T2-Hi phenotype, such that, like asthma, it should also be thought of as an umbrella definition (FIGURE 5). As expected, T2 molecular phenotypes are consistently associated with eosinophilic inflammation in the lung/blood, elevations in FeNO, elevated IgE/atopy, and bronchodilator responsiveness (TABLE 4). Active T2 immune processes generate profound changes to the airway epithelium, driving goblet cell differentiation, ciliary dysfunction, and mucus abnormalities (163, 240, 303), linked to increased mucus plugging in T2-Hi asthma (66). Placebo arms from T2 targeted studies, as well as large-scale epidemiologic studies, show an increase in exacerbation rates in biomarker defined T2-Hi phenotypes compared with T2-Lo (22, 40, 61, 70, 224, 276). Clustering of inflammatory and physiological variables (together) report bronchodilator reversibility more strongly correlates with eosinophilic inflammation than FEV1 or forced vital capacity (FVC), measures of static lung function (186). As original definitions of asthma were based on clinical observations, followed by physiological definitions, it is conceivable the original perceptions/definitions of asthma were built around patients with T2-immune responses, and that some of the heterogeneity observed today relates to loosening of these original definitions, particularly in large-scale studies (see sect. I). Finally, most studies support stability of T2 inflammation over time (157, 176, 197, 271). The few long-term studies also suggest an association of persistent T2 inflammation with loss of lung function and persistent exacerbations (150, 197, 271).
Table 4.
Biologic and clinical characteristics consistently observed in T2-Hi molecular phenotypes
| 1. Elevated blood and/or sputum eosinophils |
| 2. Elevated FeNO |
| 3. Atopy/elevated IgE |
| 4. Bronchodilator reversibility (may be less in later-onset T2-Hi phenotype) |
| 5. Higher exacerbation rates |
| 6. Increased mucus on CT imaging (silent mucus) |
| 7. Greater response to corticosteroids |
CT, computed tomography; FeNO, fraction exhaled nitric oxide.
Despite many consistencies, patients and pathological processes under the broad T2-Hi molecular umbrella also vary dramatically one from another (FIGURE 5). The reasons for this are not clear, but likely include an oversimplification of T2 immune pathways (FIGURE 2). As is increasingly clear, the canonical T2 cytokines can be expressed by different cell types (Th2 cells, ILC2 cells, mast cells/basophils), in different proportions, in tissue compartments, and under varying regulatory control. In addition, accompanying innate [thymic stromal lymphopoietin (TSLP), IL-33] and adaptive [interferon-γ (IFN-γ), IL-17] immune pathway activity modifies these cytokines and their downstream pathways. Thus these variations may explain the spectrum of clinical subphenotypes associated with T2 immune activity.
2. Mild early-onset/allergic asthma (FIGURE 5)
Perhaps the earliest identified T2-Hi molecular phenotype is mild allergic asthma. Allergic responses have long been linked to Th2/T2 immunity in mouse models and human disease (153, 229), with the majority of transgenic mouse studies relying on Th2-skewed allergic models varyingly dependent on IgE. IgE, the allergic immunoglobulin, is produced by IL-4/13-induced isotype switching of B cells. It binds to high-affinity IgE receptors (FcεR1) on mast cells and basophils where crosslinking by allergen leads to activation (FIGURE 2). Human allergic/allergen challenge systems have consistently been inhibited by monoclonal antibody blockade of IgE (27, 68), T2 associated cytokines/receptors, including IL-13 (alone), the IL4Rα receptor (IL-4 and -13), and more recently by TSLP (90, 92, 279), confirming the importance of these pathways to both the immunologic and human physiological process. Additionally, inhalation of a DNAzyme against the canonical T2 transcription factor GATA-3, which controls expression of IL-4, -5, and -13, was also effective in a human allergen challenge setting (154). This allergic T2-Hi phenotype typically begins in childhood (before age 12), and the majority of patients are responsive to CS therapy (75, 107, 288). It is also associated with other T2-related diseases, including allergic rhinitis and dermatitis/eczema, as noted years ago in relation to extrinsic asthma (184, 226).
While eosinophilic inflammation is present in these patients, its relation to pathophysiology is less clear, as unlike the IL-4/-13 and IgE approaches, monoclonal antibodies towards IL-5 do not decrease allergen challenge responses, despite eliminating blood eosinophils (164). In contrast, the efficacy of anti-IgE in this subphenotype supports a role for mast cells and/or basophils (FIGURE 2). Mast cells are found in large numbers in proximal airway epithelium and submucosa where they can be activated by inhaled allergen, even in patients with atopic disease and no asthma (20, 91, 281). Mast cells are often considered the front line for innate immune defense/epithelial repair, such that movement of mast cells into the epithelium could be the initial response to the epithelial damage signal observed in many transcriptomic studies (186, 265, 275), even initiating mild allergic asthma. Exercise induced bronchospasm (EIB) is also associated with increases in and activation of mast cells in the epithelium, perhaps in response to epithelial damage induced by activation of TSLP and/or IL-33, or even by the overall vulnerability of the epithelium to hyperventilatory damage (98, 114, 160). It should be noted that the strongest and most consistent genetic associations with asthma are in the 17q12–21 asthma susceptibility locus (208). The region is specifically linked with childhood-onset, allergic asthma, increasing the rationale for identifying these early-onset patients as a separate T2-subphenotype, and questioning the overall importance of Th2 immunity.
3. Moderate to severe early-onset allergic asthma (FIGURE 5)
Patients with more severe allergic/early-onset asthma are found in most cluster analyses, often in association with higher levels of allergic sensitization (186, 203). These patients respond to both anti-IgE and anti-IL-4Rα therapies (40, 116, 276), although responses to anti-IgE appear less robust as disease severity increases, suggesting this allergic phenotype involves more than IgE (34, 115, 134). Interestingly, recent data suggest anti-IL-5/5R approaches may be less effective in early-onset allergic asthma, even when matched for baseline blood eosinophils, and consistent with the lack of efficacy of anti-IL-5 in human allergen challenges (23). In fact, treatment of moderate, likely predominantly allergic, asthmatic patients with anti-IL5R only modestly reduced exacerbations and led to only small improvements in lung function (69). Little is understood regarding the mechanisms which drive development of this more refractory T2-Hi allergic subphenotype, or its longitudinal trajectory, with studies suggesting this more severe disease may arise early in childhood, as opposed to progressing over time (237). The subphenotype may also transform rather acutely from milder to more severe disease in adulthood. Although ‘omic studies show consistent increases in epithelial T2 signatures, additional differences in epithelial differentiation, repair, mast cell signatures, and innate pathways may also determine more severe disease (140, 186, 187, 273).
4. Adult-onset T2-Hi asthma (FIGURE 5)
Another well-defined T2-Hi subphenotype is that of adult-onset, eosinophilic/T2-Hi asthma, typically associated with sinus disease, nasal polyposis, sometimes with AERD, and much less with allergies. Despite similar elevations in T2-biomarkers, numerous factors differentiate adult from childhood-onset asthma (TABLE 5). This phenotype has origins as intrinsic asthma and is identified in most clusters that include inflammatory variables (111, 186, 203). Unlike T2-Hi early-onset asthma, this subphenotype is less CS responsive, often requiring treatment with systemic CSs. Acute severe exacerbations requiring invasive or noninvasive ventilation (except in response to nonsteroidal anti-inflammatory drugs in susceptible patients) are less common. However, the T2 component of this phenotype is confirmed by robust responses to IL4Rα blockade, both clinically and biologically in nasal polyp tissue, where marked reductions in T2 chemokines were observed (15, 142). Importantly, IL-5 is a critical cytokine for this phenotype, as two reports now support greater efficacy of anti-IL-5 pathway inhibition in patients with this adult-onset T2-Hi eosinophilic phenotype as compared with early-onset disease (23, 29). Data with anti-IL-5R revealed the presence of nasal polyposis and systemic CS dependency as additional response predictors (22). Thus it is conceivable that an IL-5 dominant, adult-onset, nasal polyposis eosinophilic asthma endotype has emerged, with strong historical links to phenotypes identified by astute clinicians and more recently in cluster analyses. The high degree of responsiveness to anti-IL-5/5R (as relates to asthma), and blockade of IL-13 through anti IL-4Rα therapy, support the proposed mechanistic link to ILC2 cells, but identification of these cells in asthma remains controversial (31). While mast cells and/or basophils are also thought to play a role in these patients, particularly in relation to AERD, specific serum IgE levels are often low, weakening the link with traditional allergy (9, 186). Leukotrienes also play a role. They are abundantly generated in these patients, and 5-lipoxygenase inhibition improves outcomes, especially in those with aspirin sensitivity (136).
Table 5.
Differences in early childhood- versus adult-onset T2-Hi asthma
| Characteristic | Early Childhood Onset | Adult Onset |
|---|---|---|
| Age at onset | Typically <12 yr old | Begins to increase late 20s and beyond |
| Sex | In adults, women > men | Proportionally more men than early onset |
| Allergies (rhinitis, eczema) | Prominent | Often absent |
| Sinusitis | Often absent | Prominent |
| Eosinophilia | Modest | Prominent |
| Nasal polyps | Typically absent | Prominent |
| Genetic elements | ORMDL3, GSDMA/B, IL33/ST2, TSLP, IL18R/RAP | Less clear, possibly ALOX15 |
| Corticosteroid response | Moderate to good at lower doses | Often requires high/systemic doses |
| Biologic response | No known relation to anti-IgE response; lesser response to anti-IL-5/5R approaches | May be better response to anti-IL-5/5R approaches |
The epithelial and eosinophilic/T2-linked enzyme 15 lipoxygenase-1 (15LO1) also appears important to this phenotype. A recent single cell RNA-seq study supports a primary association with upper airway/nasal polyp epithelial cells. These mRNA data were confirmed at the protein level where 15LO1 was essential for the expression of the eosinophilic chemokine eotaxin-3/CCL26 (172, 209). In further support, a meta-analysis of genome-wide studies identified a coding single nucleotide polymorphism (SNP) in 15LO1, associated with a loss-of-function mutation, as highly protective against polyp development (152). Finally, local autoantibodies have been identified in some patients, but the relation to systemic immunity is less clear (193, 258).
5. Complex T2-Hi asthma (T2+) (FIGURE 5)
T2 immunity can be persistent despite increasingly severe CS-treated disease. In these cases, the T2 immunity may be impacted by additional immune pathways, different cell sources, and more distal lung inflammation. Distal lung studies of T2-Hi asthma are few. However, a persistent small airway signature of T2 immunity has been reported in distal airway epithelial cells, beyond the reach of most large particle inhaled CSs and only accessible to systemic CSs (243). Distal lung pathology also revealed increased small airway tissue inflammation, including the presence of mast cells in the inner and outer airway walls, in patients with more severe disease (19).
In patients with very severe systemic CS-dependent asthma, video-assisted thoracoscopic biopsy tissues have revealed noncaseating granulomas in the distal lung in a subset, without relation to eosinophilic granulomatosis with polyangitis. Thus, in addition to persistent T2 and mast cell presence, the granulomas suggest immunity beyond T2 alone (284). While asthma is traditionally associated with allergic as opposed to autoimmune inflammation, many of these very severe patients have a personal or family history of an autoimmune disease, supporting potential involvement of autoimmunity. BAL fluid/cell IFN-γ and downstream chemokines CXCL9 and -10, as well as the macrophage/dendritic cell transcription factor IRF5, in association with type 1 inflammation, have all been reported to be increased in severe asthma (89, 210, 227, 269). However, upstream processes that augment IRF5 or other factors in potentiating type 1 inflammation in severe asthma are unknown.
IL-6 pathways have also been reported to be activated in association with increased blood eosinophilia and airway T cell/macrophage infiltration in patients with more severe and exacerbation-prone disease, along with increases in innate immune pathways (IL-1β, Toll-like receptors) (140). Importantly, in contrast to studies linking obesity, asthma, and elevated serum IL-6 patients, this complex T2-Hi-IL-6 phenotype had no evidence for increases in systemic IL-6.
Some complex T2-Hi+ patients also have evidence for very high/‟ultra-high” levels of T2 gene expression in sputum, associated with older age, worse lung function, and more severe disease (217). This “ultra-Hi” T2-Hi phenotype was seen in association with increases in sputum CD11b+/CD103-/IRF4+ dendritic cells. If and how they are antigenically driving the “ultra-Hi” T2 phenotype and associated disease severity remains unclear.
While three of the current T2-directed biologics have shown efficacy in systemic CS-dependent (and refractory) asthma, supporting an important role for T2 immunity, some of these most severe patients remain refractory to these therapies, despite elevations in traditional T2 biomarkers (21, 200, 225). Inducible nitric oxide synthase (iNOS), the primary enzyme contributing to nitric oxide (NO) generation in human airways, is increased by IFN-γ, as well as IL-4/13 (48, 108), suggesting that elevated FeNO may not be due to T2 inflammation alone (269). Many additional factors cause blood eosinophilia, suggesting other non-T2 drivers may elevate these biomarkers, supporting the need for more specific T2 biomarkers, especially given the high cost of a trial of a T2 biologic.
6. Does the microbiome play a role in T2-Hi asthma phenotypes?
There is increasing evidence for a gut-lung or lung-microbiome axis in relation to disease and health. Current studies can generally be divided into those addressing disease development and those addressing persistence or severity. While the relationship of gut microbiota studies to T2-Hi (or Lo) asthma phenotypes is limited, many have addressed its role in development of atopy/allergy (FIGURE 6). It has been observed for years that birth by Caesarian section increases the child’s risk for atopy and asthma (reviewed in Sandall et al., Ref. 235), which led to enhanced Staphylococcus-associated microbiota in the infant. The presence of fecal Ruminococcus gnavus has also been associated with occurrence of allergy in children at 3 yr of age (51). Mouse models suggested this may occur through migration of gut ILC2 cells to the lung, but human studies are lacking. Having a dog in the home has also been associated with less risk of atopy/asthma. Mice exposed to dog-associated house dust developed increases in gut Lactobacillus johnsonii, which was protective against T2-lung responses (87). The presence of many other bacteria have been variably related to development of asthma and allergies (reviewed in Budden et al., Ref. 32), thus emerging data to support an association between the gut microbiome and human T2 (allergic) responses, and thereby asthma as well. However, cause and effect in humans remains unproven. In addition to gut microbiota, several studies have identified differences in the lung microbiota in relation to asthma phenotypes (FIGURE 6). Intriguingly, high diversity of organisms, in general, is observed to be protective in relation to disease. In patients with mild asthma, the presence of T2 inflammation (as measured by lung T2 biomarkers) was associated with nearly unmeasurable 16S rRNA for bacterial species in most cases, while modest differences existed between atopic asthmatic patients and those with atopy alone (67). Importantly, CS responsiveness appeared related to the presence of certain bacteria 16S rRNA, including Hemophilus, which was associated with low T2 (high neutrophilic) inflammation in a second study (261). Similar potentially pathogenic species (Streptococcus) have been identified in sputum from severe asthma, in association with sputum eosinophilia (T2-Hi) (301). Whether these studies truly represent the lung microbiome remains unclear, as sputum is highly contaminated with upper airway bacteria. While most studies have focused on bacterial rRNA, a recent bronchoscopic study also addressed fungal rRNA. Somewhat similar to bacterial rRNA, less fungal diversity was observed in asthma patients with evidence of T2-biomarker elevations, with Trichoderma species found in greater abundance in these patients (239). Yet, relations between fungal species and disease severity were more complex, including a positive relationship between Trichoderma and FEV1, but negative relationships with more traditional fungal allergens, including Cladosporium and Alternaria. How lung measurements of these fungal RNA relate to traditional (specific) allergen sensitization remains to be studied. While many associations have been observed with both gut and lung microbiota in relation to T2 (allergic and eosinophilic) asthma, further work (including ongoing studies of probiotics to alter gut microbiota) is required to determine whether these are causal or reactive relationships.
FIGURE 6.

Simplified schematic of potential microbiome-immune interactions in relation to molecular and inflammatory asthma phenotypes. Emerging data support the importance of gut microbiota/dysbiosis in the development of atopic-allergic disease, with certain bacteria including staphylococcus and ruminococcus associated with atopy, while Lactobacillus johnsonii has been considered protective. These alterations could promote the development of mild-moderate T2-Hi allergic asthma. In all cases, loss of diversity of the microbiome is believed to be a disadvantage and drives development of ILC2 and/or ILC3 cells which, under certain circumstances, can migrate to the airways. Abnormalities in the lung microbiome are more controversial and difficult to confirm. An abnormal airway epithelium could predispose to bacterial contamination, colonization, and infection with pathogenic bacteria being found more commonly in T2-Lo/neutrophilic asthma. Interestingly, in mild asthma, the presence of T2-Hi immune processes is associated with significantly less evidence for bacterial 16S RNA, suggesting the T2 environment/phenotype may inhibit bacterial presence. Similarly fungal RNA has also been observed in lower abundance.
7. Is T2 immunity the primary molecular abnormality?
Interestingly, a T2 transcriptomic signature has only been reported once in a fully unbiased manner (187). In contrast, unbiased clustering of genes reveals several other potentially relevant pathways. In fact, using the same WGCNA which identified the T2 gene signature by unbiased gene clustering, three other gene modules, including epithelial repair and mitochondrial and neuronal processes, were identified which more strongly linked to asthma diagnosis and severity than the T2 gene module. This study is complemented by genetic/genome-wide studies of asthma compared with healthy populations which have consistently identified asthma susceptibility loci in relation to epithelial genes (17q12–21), inflammasome related genes (2q), as well as other immune and tissue repair signals, but without relation to atopy-related genes (148, 188). No prominent and over-arching T2 or Th2 genetic signal has emerged from genetic studies. Importantly, a phase 2b trial of an anti-TSLP antibody in moderate to severe asthma, of mixed T2 background, showed reductions in T2 biomarkers, as well as improved clinical outcomes, even in patients without elevations in background T2 biomarkers (53). If confirmed in phase 3, a secondary, as opposed to primary, mechanism for T2-immunity in asthma would be further supported.
B. Molecular Phenotypes in the Absence of Prominent T2 Immune Pathways
1. Definition
Non or low T2 phenotypes are defined by the absence of T2 signatures as opposed to the presence/activation of other immune signatures. Unfortunately, this approach is highly dependent on definitions used for T2 signatures/immunity. Measuring sputum eosinophilia, blood eosinophils or FeNO are more likely to reveal elevated T2 levels as more measurements are collected or biomarkers analyzed. Furthermore, using cut points of ≥3 versus ≥1.5% for sputum eosinophils or FeNO >19 versus >25 or even >35 ppb provides very different percentages of T2-Hi vs Lo patients. Thus the percentage of asthma, which exists in the absence of any T2 biomarker elevation, or even granulocytic inflammation, over a period of time is unknown.
2. Associated characteristics (FIGURE 7)
FIGURE 7.

Schematic of the current understanding of T2-Lo molecular and clinical phenotypes. T2-Lo phenotypes are characterized by lack of clear biomarkers, obesity, and poor corticosteroid (CS) responsiveness, often despite high doses of CSs. Unlike the overlapping cellular and molecular characteristics which defined T2-Hi phenotypes, the characteristics of T2-Lo asthma are more variably overlapping. The subphenotypes include a broad range of severity from the most mild to severe patients, including elements of long disease duration (suppression of T2 immunity), a bronchitic, perhaps neutrophil associated phenotype, and a late-onset obese phenotype. Precision approaches to treating these patients remain poorly understood. IL, interleukin.
Non (or T2-Lo) asthma generally has been associated with later age at onset, long-term disease/high-dose CS use, milder and more stable disease, and, perhaps most consistently obesity (64, 118, 129, 264). Lung function is generally better, often with less obstruction as measured by FEV1/FVC, perhaps contributing to the lesser bronchodilator response typically seen in these patients (204). Patients may have a high degree of symptoms, but typically fewer life-threatening exacerbations (111, 118, 186). Most uniformly, they have a poor response to CSs, even when given systemically. Despite this, they are often treated with these drugs for long periods of time, only worsening obesity and metabolic dysfunction.
3. Obesity-associated T2-Lo asthma (FIGURE 7)
Obesity in association with asthma is a trait and not, by itself, a phenotype. While obesity almost certainly worsens symptoms and lung function in the majority of patients, the characteristics associated with this trait define the phenotypes. In many patients with severe asthma, obesity is associated with persistent T2 inflammation. This persistent T2 inflammation is treated with increasing doses of CSs, typically with modest improvements, but with increasing appetite, lower levels of physical activity, and more weight gain. In these patients, obesity is a confounder, not a cause of disease (64, 129) (FIGURE 8). Whether obesity worsens the T2 inflammation or drives a second metabolic or immune pathway, which additively or synergistically interacts with T2 inflammation, is poorly understood (63).
FIGURE 8.

Overview of the intersection of asthma and obesity. Obesity and asthma enjoy a complex relationship. Top left: persistence of severe, type 2 (T2) associated asthma, poorly responsive to standard treatment can lead to increasing obesity, worsening symptoms, and more exacerbations, which respond to improved asthma treatment (often with concurrent weight loss). Bottom right: however, an obesity-associated T2-Lo asthma arising in adulthood also exists in association with both metabolic and mechanical dysfunction. While balancing certain metabolic pathways may improve outcomes, weight loss appears to be the most effective therapy. FeNO, fraction exhaled nitric oxide; SOB, shortness of breath.
However, a second group, typically females with late-onset asthma and little evidence of T2 inflammation (111, 129), also is identifiable, who have profound symptoms, modest reductions in the FEV1/FVC (as well as FEV1), and low β2-agonist reversibility. Increasing data identify metabolic dysfunction in some patients which tracks with C-reactive protein and IL-6 levels in blood (218) (FIGURE 8). Alterations in the gut microbiome could additionally contribute to this metabolic dysfunction (reviewed in Shore et al., Ref. 241). Hypertension and diabetes are common, with lower lung function and exacerbations more likely in obese patients with elevated IL-6 levels. There is no association with T2 inflammation and no evidence for increased IL-6 expression in sputum cells. However, other studies have noted increases in IL-6 pathway-associated genes in epithelial brushings, such that details of this relationship are still unclear (140, 186). Finally, elevations in IL-6 also occur in nonobese patients and have been associated with similar worse outcomes. Thus, in addition to a pathological driver of disease, IL-6 could also be a biomarker for metabolic dysfunction. Trials are needed to determine whether specific blockade of this pathway in patients with elevated levels improves the clinical scenario.
Other studies have suggested T2-Lo obese asthma is associated with abnormalities in arginine metabolism, such that the traditional T2 biomarker FeNO may be spuriously low. In patients with adult-onset obese asthma, FeNO is inversely related to BMI (130). Low arginine levels associated with high asymmetric dimethyl arginine (ADMA) levels shift iNOS from generating NO to generating reactive oxygen species (12). In obese asthma, plasma ADMA levels are high relative to arginine, consistent with this hypothesis. When controlling for this ratio, the relationship between FeNO and obesity disappears. In vitro supplementation with l-citrulline, a precursor of l-arginine, restores the ability of iNOS to generate NO (286), but only in the presence of an active iNOS enzyme. Thus it is not clear whether this low arginine associated T2-Lo obese phenotype is, in reality, T2-Lo, or just traditional T2-biomarker low. In line with this, supplementing with l-citrulline should increase FeNO levels. This increase in FeNO could improve asthma outcomes concomitant with its known smooth muscle relaxation properties. However, increases in FeNO would also move these biomarker T2-Lo patients into a T2-Hi category, suggesting molecular studies of these patients are needed. In fact, it has been suggested that at least some obese asthma patients may have more tissue eosinophils and higher serum IL-5 than controls, consistent with a T2-Hi phenotype (63).
Other possible mechanisms for T2-Lo obese asthma include various mechanical abnormalities, such as premature small airway closure in relation to the increased fat-volume loading. Interestingly, waist circumference is associated with absolute FVC, but not methacholine-induced airway closure, while BMI is more strongly related (220). Thus some of this late-onset obesity-driven asthma may represent a relative fringe of the asthma umbrella. The associated drop in FVC during methacholine challenges, believed to be driven by airway closure, may not be reactive bronchoconstriction in the traditional sense, but rather compressive, metabolic, or even fatigue-related processes. When the 1980s ATS asthma definition was written, obesity represented a small section of society such that these patients were rarely encountered in asthma studies. Given the lack of T2 inflammation, it is unlikely these patients will respond to current asthma medications, with poor responses to both CSs and β2-agonists. Effective therapy appears to primarily be weight loss (64) (FIGURE 8). Thus some obese phenotypes could be considered obesity-induced thoracic disease with secondary airway effects, as opposed to a primary airway process more traditionally associated with asthma.
4. Other non-T2 molecular pathway phenotypes
One of the most consistent molecular pathways identified through various analyses are those related to the inflammasome, particularly in relation to IL-1β and IL-18 pathways (FIGURE 3). Genes in these pathways are consistently upregulated in both epithelial brushings and sputum, and related SNPs are consistently identified on genome-wide studies of asthma (208). However, some studies have shown IL-18 receptor upregulation in association with T2 gene expression (186, 283), while others have not (16, 156, 231), perhaps due to the complex relationship of IL-18 with type 1 or type 2 immunity, dependent on the associated IL-12 levels (296). Clustering genes related to sputum eosinophils and then using subsequent GSVA to identify pathways, the U-BIOPRED consortium identified a cluster associated with inflammasome expression, moderate T2 gene expression, and neutrophilic inflammation (231). This inflammasome cluster was associated with lower FEV1 and moderately high OCS use. They had the earliest age at onset and the highest C-reactive protein levels, suggesting T2 immune pathways may have been suppressed by CSs. A recent study from the SARP identified elevated sputum levels of extracellular nuclear DNA in 13% of its cohort (159). This increase in DNA associated with older, more severe disease, and bronchitic-like symptoms, and similar to the U-BIOPRED inflammasome cluster, more OCS use. Importantly, these higher DNA levels associated with identification of neutrophilic extracellular traps, sputum neutrophils, and apparent caspase-1 activation in the presence of higher sputum IL-1β levels, consistent with inflammasome activation. Unlike the U-BIOPRED study, there was no relation to type 2 biomarkers.
Neutrophilic asthma has also been proposed as a phenotype of asthma (97, 283). It remains one of the most frequently studied cellular phenotypes and is widely cited in review articles (137, 228). However, the specificity of this phenotype remains unclear. Cut-points for sputum neutrophilia depend on method, with high neutrophils ranging from >40 to >65% of total. Its consistency over time is also less than that of sputum eosinophilia (157). Importantly, two studies of a CXCR2 antagonist, the receptor for the neutrophilic chemokine IL-8, in neutrophilic asthma were negative on all asthma outcomes, despite requiring starting sputum neutrophilia, in one of the studies (198, 206). Target engagement occurred, however, as blood neutrophils decreased, with some subjects experiencing potentially dangerous reductions. Despite this lack of efficacy signal, the concept of neutrophilic asthma persists (17).
Increases in airway or sputum neutrophils also fostered interest in IL-17 in relation to asthma phenotypes. Two studies of an antibody to the IL-17RB, which neutralizes activation of the receptor by any IL-17 isoform, including IL17E/IL-25, considered a pro-type 2 innate cytokine have been completed. Despite an early suggestion that IL-17R may have therapeutic effects in those with a high degree of bronchodilator reversibility, a second study, enriching for that specific phenotype, was completely negative (35). However, sputum neutrophilia was not used as inclusion criteria in these studies. IL-17 pathway genes have occasionally been identified in transcriptomic studies, although overall their expression has been inconsistent (156, 227), with one study suggesting complete dissociation of T2 and Th17/type 17 inflammation (50). In that study, endobronchial IL-17 pathway gene expression was not associated with neutrophilia and was more closely linked to eosinophilia, particularly in the tissue, without difference from patients with T2-Hi asthma. The authors suggested that inhibition of one pathway could give rise to activation of the other. While the utility of dual blockade of IL-13 and IL-17 was suggested in a mouse model, validation of this approach is needed in humans, particularly in light of the negative trials with IL-17 specific therapies.
Smoking, or a history of smoking, has also been associated with neutrophilic (and IL-17-associated) asthma (47, 242). Whether the neutrophilic inflammation is any more clinically relevant in smokers as compared with non- or ex-smokers is unclear. Overall, the data to support a consistent neutrophilic driven (or IL-17-associated) phenotype of asthma are weak. Whether it could still be a biomarker for other pathogenic factors requires further study.
VI. PHENOTYPES AND MOLECULAR PHENOTYPES OF PEDIATRIC POPULATIONS: COMPARISONS WITH ADULTS AND SPECIFIC CHALLENGES
Important clinical differences are obvious in childhood as compared with adult asthma (TABLE 6). Whereas adults are likely to exhibit persistent symptoms, children with asthma tend to have more episodic disease. Many children have severe exacerbations triggered by viral infections and/or allergen exposure that result in healthcare utilization, but then remain asymptomatic between these episodes (88, 94). Depending on age cut-point, almost all childhood asthma is early onset, and the majority of children (>80%) have sensitization to aeroallergens (73, 81, 189, 262). Significant obstruction with air trapping is relatively uncommon (262), and lung function is often normal despite symptom burden and medication use (14, 139, 249). Instead, distal lung resistance may be increased in the absence of significant large airway involvement (128), and other measures such as FEV1/FVC, FEF25–75% predicted, or airway hyperresponsiveness to bronchodilators may relate better to asthma severity (14). While there are subsets of children with more hyperinflation, children tend to have less airway resistance than adults (250) and are generally reversible with bronchodilation (248).
Table 6.
Differences between adults and children with severe asthma
| Adults with Severe Asthma | Children with Severe Asthma | |
|---|---|---|
| Symptoms | Often persistent with significant burden | May be episodic (more common) or persistent (less common) |
| Exacerbations | Vary according to phenotypic cluster | More frequent and associated with greater healthcare utilization |
| Allergic sensitization | Varying degrees of atopy according to age of asthma onset and phenotypic cluster | Majority are sensitized |
| Airflow obstruction | Moderate to severe airflow obstruction often with incomplete reversal after bronchodilation | Typically mild to moderate airflow obstruction with near-complete reversal after bronchodilation; significant acceleration of airflow obstruction in some adolescents after puberty |
| Distal airways | Increased resistance that often accompanies large airway involvement, may not be reversible with bronchodilation | Less resistance than adults, may be increased in the absence of large airway involvement, largely reversible with bronchodilation |
One of the bigger differences in children from adults is change with age, particularly in adolescents who may remit or have worsening asthma (56, 100) (FIGURE 9). Indeed, some adolescents assume a more adult-like phenotype after puberty and develop significant airflow obstruction (80) that increases the risk of chronic obstructive pulmonary disease in later adulthood (256, 257) (FIGURE 9). Yet, the pattern or mechanisms for the transition to increasing airflow limitation in children remain poorly understood. Sex hormones may influence phenotypes and natural history, as asthma shifts from male predominance before puberty to female predominance after puberty. However, the mechanisms behind this shift are poorly understood. Girls maintain hyperresponsiveness and symptoms longer than males, where androgens may have beneficial effects on asthma symptoms and lung function (60, 86, 260). Longitudinal studies of the incidence and temporal stability of childhood phenotypes are needed, as are studies of sex as a biological variable in the transition from childhood to adulthood.
FIGURE 9.

General paradigm for development and progression of childhood-onset asthma. It is likely over 60% of all asthma phenotypes have their origins in childhood. Genetic predisposition to childhood asthma combined with environmental exposures/changes in gut microbiota lead to atopy, asthma persistence, and potential progression. While an IgE-dependent, likely T2-Hi asthma phenotype is recognized, the association with cellular and molecular profiles is less clear. T2-Lo asthma likely also exists in childhood, although its trajectory is even less clear. Finally, the immune or hormonal factors that drive the resolution, remission, or persistence of disease into adulthood remain largely unknown.
A. Clinical Clusters in Children
Cluster analyses of asthmatic children across severities identified multiple phenotypic groups differentiated by degree of allergic sensitization, asthma duration, controller medications, BMI, and pre-bronchodilator lung function (79, 131, 143, 144, 236, 305). These clusters only marginally conform to definitions of asthma severity proposed by current treatment guidelines, since children with severe asthma were present across the clusters. Not surprisingly, the majority of asthma clinical clusters identified before puberty are most aligned with the early-onset T2-Hi phenotype in adults. However, like adults, clusters associated with minimal atopy have also been described. For example, in children 6–11 yr with difficult-to-treat asthma, Schatz et al. (236) identified a moderately-prevalent (31%) group of children without allergic rhinitis or atopic dermatitis with significant airflow obstruction. However, mean IgE levels were elevated in this group, suggesting that at least some children had underlying T2 inflammation. Similarly, a cluster analysis of inner-city asthmatic children identified a low-prevalent (15%) cluster with low blood eosinophil, FeNO, and little evidence of specific IgE by allergen screening (28% of the cluster) (305). Unlike adults with modest evidence for T2 biomarkers or atopy, no other specific clinical features have been identified. Interestingly, unlike adults, obesity does not appear to distinguish or identify T2-Lo pediatric clusters. In fact, a meta-analysis of pediatric studies did not find an association between obesity and poor asthma control, although a small increase in exacerbation risk has been observed (6).
A consistent theme among these studies is that the greater the severity of childhood asthma, the more prominent the T2/allergic features. In all large-scale pediatric cluster studies, the clusters with the highest degree of aeroallergen sensitization and/or history of other allergic diseases had the highest medication requirements, greatest airflow obstruction and airway lability (often not fully reversible), and the highest prospective risk of exacerbations requiring treatment with systemic CSs (79, 131, 305). Interestingly, these clusters often tend to have the best treatment responses to CSs (45). However, as in adults, not all T2-Hi asthma is CS-responsive (221), with some more severe atopic early-onset clusters having the least response to guideline-based asthma treatment (45). These observations suggest that response to CS therapy may be an important phenotypic variable in children. Yet the mechanisms for these differences are unclear, emphasizing the need to include more inflammatory and molecular phenotyping.
B. Inflammatory Phenotypes in Children
Inflammatory and molecular phenotyping is only emerging in children. Studies to date have focused on peripheral blood cell counts, with three groups described in children (95, 96): 1) T2-Hi/eosinophilic, 2) pauci-granulocytic, and 3) neutrophilic (11, 104, 106). Like adult asthma, the eosinophilic phenotype likely falls under the umbrella of T2-Hi asthma. Importantly, it can be identified in the preschool or early school-age years (33, 233) and is associated with increased symptoms, less controlled disease with frequent exacerbations, more aeroallergen sensitization, and impaired lung function measures with increased airway hyperresponsiveness (96, 120). Like atopic clusters, the eosinophilic/T2-Hi phenotype is generally more CS-responsive (75, 298). One study of severe asthmatic children refractory to systemic CSs noted decreased expression of inflammatory mediators including IL-2, IL-5, IL-10, and tumor necrosis factor-α (TNFα) (77). The lower IL-5 expression observed in children with severe asthma who failed to achieve control with systemic CSs was present despite the absence of differences in baseline blood eosinophils, and consistent with other studies (24, 74). However, adult studies have also not observed increases in airway IL-5 in severe asthma, such that the importance of this finding in children is unclear. Other T2 biomarkers, such as FeNO, are equally variable, often discordant with symptoms and airway eosinophils, and may vary with age (25, 26). Interestingly, in contrast to adults, incorporation of sputum eosinophils into management of school-age severe asthmatic children failed to reduce exacerbations or improve asthma control (82, 101). Nonetheless, subsets of children with severe asthma have persistent symptoms, exacerbations, and airway eosinophilia after supervised systemic CS administration (25, 151, 214), consistent with refractory T2-Hi disease. These same children also had increased reticular basement membrane thickening and airway smooth muscle mass (109, 207, 215) despite relatively low airway T2 cytokines (24). Whether they will respond to T2 targeted biologics remains to be proven and will be essential to confirming (or not) whether pediatric T2-Hi asthma is similar to adult T2-Hi phenotypes. However, anti-IgE therapy improves asthma exacerbations in children even with modest elevations in IgE and atopy (162) and has greater impact in children with allergic sensitization to perennial allergens, including cockroach and dust mite, consistent with its mechanism of action and supporting a relationship with an atopic, likely T2-Hi phenotype (36). Until more detailed studies of T2-targeted biologics are performed in well-characterized children including those <12 yr of age, defining T2-Hi molecular phenotypes will remain controversial.
As T2-Hi asthma in children is poorly defined, T2-Lo asthma is even more poorly defined as it relies on the absence of T2 inflammation. However, like adults, children with a neutrophilic inflammatory phenotype have been identified, which may more accurately reflect activation of innate immune defects or environmental exposures such as smoking, which promote airway neutrophil accumulation and IL-8 release (65). Increased IL-8 expression has also been reported in children with exacerbation-prone asthma (119, 179, 180), but this observation may relate more to viral respiratory infections (125). An airway neutrophil phenotype was also described in a subset of children with severe asthma independent of clinically recognized fungal or bacterial infection (104, 168, 205), but this phenotype has not been replicated (11, 104, 106). As noted previously, a T2-Lo obese childhood asthma phenotype has not yet emerged.
Dysregulation of the airway antioxidant glutathione, in associations with increased lipid peroxidation and DNA nucleoside oxidation byproducts, has also been reported in severe asthmatic children with marked airflow obstruction (72, 78). Severe asthmatic children had decreased histone deacetylase activity in airway cells (78), as well as global disruption of thiol redox signaling (76, 212) associated with posttranslational modification of the transcription factor, nuclear factor (erythroid-derived 2)-like 2 (78). Importantly, these redox changes have not yet been associated with T2 biomarkers such that it is unclear whether the patterns of redox imbalance are primary or secondary factors to T2 skewing.
C. ‘Omic Studies in Children
Transcriptomic studies of childhood asthma have been reported utilizing peripheral blood mononuclear cell gene expression and nasal brushings, but not lower airway samples. One analysis identified a cluster of children with elevated eosinophil counts, both eosinophils and neutrophils, and one with elevated neutrophil counts, The most severe cluster of children was the neutrophilic cluster which associated with expression of genes for glucocorticoid signaling pathways and Th1/Th17 immune pathways (294). Nasal brushing samples may offer insight into molecular phenotypes in children, as nasal brushings bear reasonable relation to lower airway inflammation (223). In nasal brushings of children, 70 genes, including IL-13, IL-5, periostin (POSTN), calcium-activated chloride channel regulator 1 (CLCA1), and serpine peptidase inhibitor, clade B (SERPINB2), were differentially expressed between asthmatic and healthy children (223). Children with T2-Hi nasal brushing samples were more likely to have had a history of exacerbations, allergy, and blood eosinophilia, consistent with adult phenotypes. Combining nasal and peripheral blood cell samples in children with asthma exacerbations (viral or not) identified a network of 46 genes associated with the transcription factor SMAD3 and transforming growth factor-β signaling as well as gene networks associated with eosinophil activation, extracellular matrix production, and epidermal growth factor receptor signaling, but with little evidence for lymphocyte involvement. These pathways were dynamically expressed in relation to exacerbations and more predominantly focused on epithelial growth and repair pattern as opposed to T2-associated, questioning the primary role of T2 immunity in pediatric asthma (8). Further molecular investigations, in coordination with studies of T2 targeted biologics, are warranted, particularly in children with more severe disease, incorporating trajectories over time as pediatric phenotypes transition into adulthood.
VII. ANIMAL MODELS OF MOLECULAR PHENOTYPES OF ASTHMA
Several animal species have been used to model immunological and pathophysiological features of human asthma, although mouse models remain the most common (13). Advantages of mouse models include availability of genetically altered mice (both transgenic and gene knockout) and mouse-specific reagents to study mechanisms of inflammation, airway hyperresponsiveness (AHR), and remodeling. Many reviews of this topic have focused on both usefulness and limitations of these models (59, 99, 155, 278). Limitations include the absence of spontaneous development of disease or disease exacerbations in mice, striking differences in airway structure and cell types including lack of eosinophil degranulation in tissue, as well as poor replication of airway remodeling features of human disease. However, mouse models remain potentially useful for modeling the full spectrum of asthma phenotypes, including their variable response to different treatment modalities.
A. Modeling Allergic Asthma
Mouse models have been used to study Th2-high asthma for years, beginning with ovalbumin (OVA) as a model allergen (30, 93). Since then, variations on the OVA-based model have been developed, all of which are essentially acute models of the mild allergic asthma molecular phenotype. These models variably induce an allergic Th2-dominated response with associated eosinophilic airway inflammation and AHR that responds to CS therapy, consistent with the human phenotype. BALB/c and C57BL/6 are the most common inbred mouse strains used, although the A/J strain, which is more prone to AHR, has also been used. These allergen-based mouse studies have collectively associated IL-4 as a driver of the Th2 phenotype, IL-5 as the key cytokine associated with eosinophilic response, and IL-13 as the one that promotes goblet cell hyperplasia, mucus production, and AHR. IL-4 and IL-13 both bind to the type 2 IL-4 receptor comprising IL-4Rα and IL-13Rα1, with IL-4Rα as the common signaling subunit mediating the response to both cytokines (123). The development of the IL-4Rα antibody dupilumab was influenced by studies using STAT6−/− mice, which showed that lack of this key transcription factor downstream of IL-4Rα attenuated Th2 responses, airway inflammation, and AHR in an allergen-driven model (7, 158). Similarly, association of IL-5 with eosinophilic responses was demonstrated using IL-5−/− (84) and IL-5 transgenic mice (165) in mouse models of allergic asthma prompting the development of monoclonal antibodies against IL-5 and its receptor. Although some studies reported a decrease in AHR using IL-5−/− mice (84) or upon antibody-mediated neutralization of IL-5 (112), another study targeting IL-5 did not reduce AHR, consistent with human studies targeting IL-5 (55, 110). IL-5 also associated with airway remodeling using a mouse model (49). While many of these early studies investigating the various arms of Th2-induced allergic airway disease relied on OVA as a model allergen, Th2-driven immune response can be also induced in mice by allergens relevant to human asthma. These allergens include house dust mite (HDM) (102, 124, 210, 252), cockroach (38, 202, 211), ragweed (46, 255), and fungal allergens (127, 182a, 194, 247, 267) (TABLE 7). However, as described below, most of these allergens elicit a more complex immune response than just Th2 cells.
Table 7.
Mouse models of asthma
| Agent | Immune Response (Cell Type) | Airway Inflammation | AHR | Airway Remodeling | CS Response | Reference Nos. |
|---|---|---|---|---|---|---|
| OVA+alum (sensitization)/OVA (challenge) | Th2 | Eosinophilic | + | + | + | 30, 55, 84, 103, 285, 300 |
| HDM | Mixed Th cell response (Th2, Th1, Th17) and ILC2 | Mixed eosinophilic/neutrophilic | + | + | + | 102, 124, 210, 252 |
| Cockroach | Th2 and Th17 | Mixed eosinophilic/neutrophilic | + | + | + | 38, 202, 211 |
| Aspergillus fumigatus | Th1, Th2, Th17 | Mixed eosinophilic/neutrophilic | + | + | + | 127, 182a, 194 |
| Alternaria alternata | Th2, ILC2 | Eosinophilic | + | + | + | 247, 267 |
| HDM+c-di-GMP | Th1, Th2, Th17 | Mixed eosinophilic/neutrophilic | + | + (unpublished) | Poor | 210, 227 |
| OVA+bacteria | Th2, Th17 | Mixed eosinophilic/neutrophilic | + | Not studied | Poor | 149 |
AHR, airway hyperresponsiveness; HDM, house dust mite; CS, corticosteroid; OVA, ovalbumin.
B. Non-Th2 Models of Eosinophilic Asthma
Cells other than T cells also secrete IL-4, IL-5, and IL-13. One such cell is the ILC2 cell, which upon activation secretes IL-5, IL-9, and IL-13 but likely not IL-4. Studies have associated ILC2s with human asthma (126). However, caution needs to be exercised in ILC2 identification by flow cytometry particularly because of their low abundance (126). If ILC2s have a role in eosinophilic inflammation, then developing mouse models to promote release of cytokines such as IL-33, TSLP, or IL-25 from lung epithelial cells, in turn causing release of T2 cytokines from ILC2s, may be useful to better understand the adult-onset eosinophilic phenotype. Many complex allergens present in HDM, cockroach, and fungi such as Aspergillus and Alternaria have protease activity, and instillation of these allergens into mouse airways triggers the release of IL-33 and TSLP from airway epithelial cells with downstream activation of ILC2s (18, 43, 145, 146, 222). Outcome measures were attenuated in ST2−/− mice, although not by CS treatment, supporting a role for this pathway in persistent CS-resistant eosinophilic asthma phenotypes (39). Human trials are underway to assess ST2 as a target to attenuate disease. ILC2 cells have also been reported in nasal polyps associated with human late-onset eosinophilic asthma (29, 245), although no mouse models are available that recapitulate this association.
The epithelial-derived cytokines IL-33 and TSLP also activate basophils and mast cells, both of which have been implicated in asthma (98). While the growth factors stem cell factor and IL-3 promote mast cell and basophil development, respectively, the concept of in situ hematopoiesis (ISH) in driving mast cell and basophil expansion in tissues from hematopoietic CD34+ progenitor cells in response to IL-33 and TSLP is increasingly appreciated (FIGURE 10) (133). An increase in numbers of eosinophil-basophil progenitors was initially described during seasonal allergies with expansion of CD34+IL-5Rα+ cells in the nasal mucosa during the exposure season, and with allergen inhalation (173, 238). Mouse models suggest that IL-33, TSLP, and IL-25 may promote ISH (232, 244). Interestingly, allergen challenge induces recruitment of progenitor cells expressing TSLPR, CD127, and ST2 in the sputum of asthmatics, which was facilitated by preexposure to IL-33 and TSLP (245). Humans treated with anti-IL-5 showed reduced eosinophil maturation in the bone marrow as well as in the bronchial mucosa, suggesting that local tissue eosinophil development may also be partly IL-5 dependent (183). However, because of redundancy among the hematopoietic growth factors, IL-3, IL-5, and granulocyte-macrophage colony stimulating factor, targeting IL-5 alone may only achieve partial benefit where ISH drives local hematopoiesis. Promising results of anti-TSLP in reducing baseline eosinophil counts (92) begs for development of new animal models where the sole inciting stimulus is epithelial injury inflicted by pathogens or pathogen products such as (but not limited to) proteases that can induce release of TSLP and IL-33 from the damaged epithelium.
FIGURE 10.
In situ hematopoiesis (ISH) as a potential mechanism for expansion of T2 cytokine-producing cells in the airways of nonallergic asthmatics. The airway epithelium at the interface of the environment and the respiratory system is vulnerable to breaching by pathogens and allergens. Factors such as thymic stromal lymphopoietin (TSLP), interleukin (IL)-33, and TSLP released by disrupted and dysfunctional epithelial cells can be expected to induce IL-5 and IL-13 production by tissue-resident ILC2 cells. TSLP and IL-33 may also promote expansion of hematopoietic stem and progenitor cells (HSPCs) that are resident in the submucosa or are recruited from the periphery. Upon activation stimulation by these cytokines and potentially other mediators, the HSPCs would undergo differentiation into various myeloid lineages that have the ability to produce T2 cytokines. Eos, eosinophil; Baso, basophil; MC, mast cell.
C. Modeling Complex T2-Hi+ Asthma
Infections with bacteria, viruses, and fungi have been associated with asthma (246). Infections impact all arms of immunity and can rapidly activate one or more cells involved in innate immunity including macrophages, mast cells, basophils, and ILCs. To mimic infections, combinations of allergens and infectious agents or their components have been used in mouse models. These models have attempted to mimic more complex immune responses in asthma that include expression of T1, T2, and T17 cytokines, alone or in combination, as well as participation of innate immune responses. Identification of higher numbers of IFN-γ+ T cells in the BAL cells of severe asthmatic patients combined with the association of multiple bacterial species with more severe disease, prompted development of a mouse model involving combinations of the complex allergen HDM and the cyclic dinucleotide cyclic di-GMP (c-di-GMP), an intracellular messenger produced by bacteria. Furthermore, c-di-GMP activates the STING pathway, a target of type I IFNs induced subsequent to viral infections and implicated in asthma exacerbations. A complex immune response is elicited involving production of IFN-γ, T2 cytokines, and IL-17 from T cells as well as increases in airway mast cells. With the use of this model, a role for IFN-γ, but not IL-17, in increased AHR was demonstrated (227). Furthermore, airway inflammation and AHR as well as the level of inflammatory cytokines were only partially suppressed by CSs, similar to CS-refractory severe asthma. A different study also demonstrated the ability of IFN-γ to promote AHR when mice were exposed to OVA in the context of bacterial lipopolysaccharide (LPS) or respiratory syncytial virus (170). Additionally, an IFN-γ-mast cell axis was implicated in AHR in a model of chronic asthma in which mice were repeatedly exposed to intraperitoneal OVA (297). How mast cells and type 1 immune responses connect in the context of infections or otherwise will be interesting to examine further given a central role of mast cells in infections and the fact that once differentiated they are long-lived tissue-resident cells that are difficult to target. In similar models, a combination of OVA and bacterial infection resulted in inflammasome activation and neutrophil-dominated airway inflammation, which was poorly responsive to CS treatment (149). Thus many opportunities exist to iteratively model complex T2-immunity in mice, based on evaluation of reciprocating information from humans and mice.
D. Modeling T2-Lo Asthma
Limited attempts have been made to establish models that exclusively reflect T2-Lo driven mechanisms such as those associated with obesity and metabolic dysfunction. In one model, obesity was developed in mice by feeding a high-fat diet, which induced inflammasome activation, airway inflammation, and AHR, dependent on IL-1β and IL-17 production by ILC3s (147). The study did not report whether the model caused neutrophil accumulation in the airways. Models such as these will be helpful to understand mechanisms underlying T2-Lo, non-eosinophilic asthma.
1. Conclusions about mouse models
Use of overexpression and gene knockout approaches revealed specific roles for mediators controlling features of asthma. Most models have focused on CS responsive allergic asthma, with models of poor CS responsiveness only recently described. The mechanisms and treatment responses behind these newer models await further exploration, fueled by iterative information from human ‘omic studies.
VIII. CONTROVERSIES AND POTENTIAL SOLUTIONS
What percentage of the asthma population should be termed T2-Hi asthma, and how should it be defined? Broad integrated evaluation of elevations in known T2-biomarkers over time will be required in large cohorts or replicated in multiple cohorts. However, as these biomarkers are not sensitive or specific, additional biomarkers will need to be identified. Better predictive biomarkers should be identified in relation to responses to T2 biologics.
Will molecular phenotypes/endotypes be identified which respond differently to IL-4/13 targeted versus IL-5 pathway blockade? This question becomes increasingly important as more and more targeted expensive therapies become available. However, it is unlikely head to head comparative studies will be performed. Molecular understanding of responders and nonresponders, which utilize ‘omics data to refine the impact of the different interventions on known and unknown pathways and then relate them to clinical outcomes, could give important clues to better biomarkers for each therapy. With these improved biomarkers, it is conceivable that “smaller” biomarker-driven adaptive trial designs could be undertaken to confirm those results.
Will biomarkers be identified for T2-Lo asthma? Currently T2-Lo asthma is defined by the absence of T2 biomarkers. However, current treatments and cut-points are all likely to impact current T2 biomarkers, suggesting that specific biomarkers which track with pathobiology and clinical characteristics are needed to better understand T2-Lo asthma.
Will precision medicine in asthma be cost-effective? The goals of precision medicine are to optimize care for specific groups of patients with specific disease phenotypes, accomplished through improvement in disease outcomes such as hospitalizations, patient-reported outcomes (enabling return to work), and minimization of risks (side effects of drugs). In chronic, potentially life-long diseases, including patients with some severe asthma phenotypes, accomplishing these goals through selective use of targeted biologics is already cost-neutral, if not cost beneficial. However, markets cannot sustain broad use of medications with yearly costs of 30–50,000 USD. It is incumbent upon academia, industry, and regulators to rapidly develop predictive biomarkers for “at risk” patients with the most need. Additionally, as pathway importance is confirmed by expensive biologic therapies, newer technologies and platforms are needed to reduce costs of targeted therapies. Until precision medicine is delivered in the way it was envisioned, it will not be cost effective.
Will precise identification of molecular phenotypes/endotypes enable “curesˮ of asthma endotypes to occur? Development of targeted molecular therapies has reintroduced the concept of disease modification and even cure. As of now, no therapies have sustained impact on the underlying disease once the therapy is discontinued. However, as outlined in FIGURE 1, uncovering the complex networks underlying molecular phenotypes, “learning” the portions of the networks which targeted biologic approaches impact, “relearning” from improved/customized animal models, continuing trials of targeted human interventions, and finally “perpetually relearning” the data as it evolves, has the potential to eventually identify the core drivers of the disease and enable cures.
IX. CONCLUSIONS AND FUTURE NEEDS
Due to the findings of the last 10 yr, it is likely the term asthma will never be viewed in the same manner as it was even 20 yr ago. These findings have increasing application to the clinicians treating asthma patients (see call-out box). In fact, we would propose that “asthma” officially becomes relegated to a lowercase “a” noun, such that its use always requires a phenotypic modifier, as in T2-Hi asthma, similar in concept to terms such as rheumatoid arthritis and iron-deficiency anemia. At this point, the modifiers are limited and cumbersome (T2-Hi, early-onset mild asthma). Despite that, even our coarse ability to identity T2-Hi asthma phenotypes has led to marked improvements in treatments for some severely impacted patients. Unfortunately, most current subphenotypes only modestly meet criteria for molecular phenotypes, and all are associated (or not) with a broad definition of T2 inflammation. With the application of the molecular phenotyping approaches outlined in this review and summarized in FIGURE 1, more granularity (and precision) will be identified, linking the precise molecular pathways to clinical phenotypes and targeting them for intervention. Thus the impressive results achieved in only 10 yr of phenotyping these lowercase “asthmas” requires continued learning to fully achieve the goals of precision medicine.
GRANTS
This work was supported by National Institutes of Health Grants P01AI106684, HL69174, HL103453, and HL113956.
DISCLOSURES
S. Wenzel has consulted for and served as an investigator on multicenter clinical trials from the following companies: AstraZeneca, Boehringer Ingelheim, GSK, Novartis, and Sanofi Aventis. She has never personally received more than $10,000 from any particular company in a given year.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: S. E. Wenzel, 130 Desoto St. Public Health, Pittsburgh, PA 15261 (e-mail: swenzel@pitt.edu).
REFERENCES
- 1.Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am Rev Respir Dis 136: 225–244, 1987. [DOI] [PubMed] [Google Scholar]
- 2.Aaron SD, Boulet LP, Reddel HK, Gershon AS. Underdiagnosis and Overdiagnosis of Asthma. Am J Respir Crit Care Med 198: 1012–1020, 2018. doi: 10.1164/rccm.201804-0682CI. [DOI] [PubMed] [Google Scholar]
- 3.Aaron SD, Vandemheen KL, FitzGerald JM, Ainslie M, Gupta S, Lemière C, Field SK, McIvor RA, Hernandez P, Mayers I, Mulpuru S, Alvarez GG, Pakhale S, Mallick R, Boulet LP; Canadian Respiratory Research Network . Reevaluation of Diagnosis in Adults With Physician-Diagnosed Asthma. JAMA 317: 269–279, 2017. doi: 10.1001/jama.2016.19627. [DOI] [PubMed] [Google Scholar]
- 4.Agbetile J, Bourne M, Fairs A, Hargadon B, Desai D, Broad C, Morley J, Bradding P, Brightling CE, Green RH, Haldar P, Pashley CH, Pavord ID, Wardlaw AJ. Effectiveness of voriconazole in the treatment of Aspergillus fumigatus-associated asthma (EVITA3 study). J Allergy Clin Immunol 134: 33–39, 2014. doi: 10.1016/j.jaci.2013.09.050. [DOI] [PubMed] [Google Scholar]
- 5.Agusti A, Bel E, Thomas M, Vogelmeier C, Brusselle G, Holgate S, Humbert M, Jones P, Gibson PG, Vestbo J, Beasley R, Pavord ID. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J 47: 410–419, 2016. doi: 10.1183/13993003.01359-2015. [DOI] [PubMed] [Google Scholar]
- 6.Ahmadizar F, Vijverberg SJ, Arets HG, de Boer A, Lang JE, Kattan M, Palmer CN, Mukhopadhyay S, Turner S, Maitland-van der Zee AH. Childhood obesity in relation to poor asthma control and exacerbation: a meta-analysis. Eur Respir J 48: 1063–1073, 2016. doi: 10.1183/13993003.00766-2016. [DOI] [PubMed] [Google Scholar]
- 7.Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, Takeda K, Akira S. Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med 187: 1537–1542, 1998. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altman MC, Gill MA, Whalen E, Babineau DC, Shao B, Liu AH, Jepson B, Gruchalla RS, O’Connor GT, Pongracic JA, Kercsmar CM, Khurana Hershey GK, Zoratti EM, Johnson CC, Teach SJ, Kattan M, Bacharier LB, Beigelman A, Sigelman SM, Presnell S, Gern JE, Gergen PJ, Wheatley LM, Togias A, Busse WW, Jackson DJ. Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat Immunol 20: 637–651, 2019. doi: 10.1038/s41590-019-0347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amelink M, de Groot JC, de Nijs SB, Lutter R, Zwinderman AH, Sterk PJ, ten Brinke A, Bel EH. Severe adult-onset asthma: a distinct phenotype. J Allergy Clin Immunol 132: 336–341, 2013. doi: 10.1016/j.jaci.2013.04.052. [DOI] [PubMed] [Google Scholar]
- 10.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 372: 1107–1119, 2008. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- 11.Andersson CK, Adams A, Nagakumar P, Bossley C, Gupta A, De Vries D, Adnan A, Bush A, Saglani S, Lloyd CM. Intraepithelial neutrophils in pediatric severe asthma are associated with better lung function. J Allergy Clin Immunol 139: 1819–1829.e11, 2017. doi: 10.1016/j.jaci.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, Cunnington C, Tousoulis D, Pillai R, Ratnatunga C, Stefanadis C, Channon KM. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 30: 1142–1150, 2009. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 13.Aun MV, Bonamichi-Santos R, Arantes-Costa FM, Kalil J, Giavina-Bianchi P. Animal models of asthma: utility and limitations. J Asthma Allergy 10: 293–301, 2017. doi: 10.2147/JAA.S121092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bacharier LB, Strunk RC, Mauger D, White D, Lemanske RF Jr, Sorkness CA. Classifying asthma severity in children: mismatch between symptoms, medication use, and lung function. Am J Respir Crit Care Med 170: 426–432, 2004. doi: 10.1164/rccm.200308-1178OC. [DOI] [PubMed] [Google Scholar]
- 15.Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, Gevaert P, Hellings P, Jiao L, Wang L, Evans RR, Pirozzi G, Graham NM, Swanson B, Hamilton JD, Radin A, Gandhi NA, Stahl N, Yancopoulos GD, Sutherland ER. Effect of Subcutaneous Dupilumab on Nasal Polyp Burden in Patients With Chronic Sinusitis and Nasal Polyposis: A Randomized Clinical Trial. JAMA 315: 469–479, 2016. doi: 10.1001/jama.2015.19330. [DOI] [PubMed] [Google Scholar]
- 16.Baines KJ, Simpson JL, Wood LG, Scott RJ, Fibbens NL, Powell H, Cowan DC, Taylor DR, Cowan JO, Gibson PG. Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol 133: 997–1007, 2014. doi: 10.1016/j.jaci.2013.12.1091. [DOI] [PubMed] [Google Scholar]
- 17.Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol 127: 153–160.e9, 2011. doi: 10.1016/j.jaci.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 18.Balenga NA, Klichinsky M, Xie Z, Chan EC, Zhao M, Jude J, Laviolette M, Panettieri RA Jr, Druey KM. A fungal protease allergen provokes airway hyper-responsiveness in asthma. Nat Commun 6: 6763, 2015. doi: 10.1038/ncomms7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med 171: 431–439, 2005. doi: 10.1164/rccm.200407-949OC. [DOI] [PubMed] [Google Scholar]
- 20.Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, Castro M, Gaston B, Israel E, Schwartz LB, Curran-Everett D, Moore CG, Wenzel SE. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med 183: 299–309, 2011. doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bel EH, Wenzel SE, Thompson PJ, Prazma CM, Keene ON, Yancey SW, Ortega HG, Pavord ID; SIRIUS Investigators . Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med 371: 1189–1197, 2014. doi: 10.1056/NEJMoa1403291. [DOI] [PubMed] [Google Scholar]
- 22.Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, Sproule S, Gilmartin G, Aurivillius M, Werkström V, Goldman M; SIROCCO Study Investigators . Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet 388: 2115–2127, 2016. doi: 10.1016/S0140-6736(16)31324-1. [DOI] [PubMed] [Google Scholar]
- 23.Bleecker ER, Wechsler ME, FitzGerald JM, Menzies-Gow A, Wu Y, Hirsch I, Goldman M, Newbold P, Zangrilli JG. Baseline patient factors impact on the clinical efficacy of benralizumab for severe asthma. Eur Respir J 52: 1800936, 2018. doi: 10.1183/13993003.00936-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bossley CJ, Fleming L, Gupta A, Regamey N, Frith J, Oates T, Tsartsali L, Lloyd CM, Bush A, Saglani S. Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J Allergy Clin Immunol 129: 974–982.e13, 2012. doi: 10.1016/j.jaci.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bossley CJ, Fleming L, Ullmann N, Gupta A, Adams A, Nagakumar P, Bush A, Saglani S. Assessment of corticosteroid response in pediatric patients with severe asthma by using a multidomain approach. J Allergy Clin Immunol 138: 413–420.e6, 2016. doi: 10.1016/j.jaci.2015.12.1347. [DOI] [PubMed] [Google Scholar]
- 26.Bossley CJ, Saglani S, Kavanagh C, Payne DN, Wilson N, Tsartsali L, Rosenthal M, Balfour-Lynn IM, Nicholson AG, Bush A. Corticosteroid responsiveness and clinical characteristics in childhood difficult asthma. Eur Respir J 34: 1052–1059, 2009. doi: 10.1183/09031936.00186508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulet LP, Chapman KR, Côté J, Kalra S, Bhagat R, Swystun VA, Laviolette M, Cleland LD, Deschesnes F, Su JQ, DeVault A, Fick RB Jr, Cockcroft DW. Inhibitory effects of an anti-IgE antibody E25 on allergen-induced early asthmatic response. Am J Respir Crit Care Med 155: 1835–1840, 1997. doi: 10.1164/ajrccm.155.6.9196083. [DOI] [PubMed] [Google Scholar]
- 28.Brightling CE, Woltmann G, Wardlaw AJ, Pavord ID. Development of irreversible airflow obstruction in a patient with eosinophilic bronchitis without asthma. Eur Respir J 14: 1228–1230, 1999. doi: 10.1183/09031936.99.14512289. [DOI] [PubMed] [Google Scholar]
- 29.Brusselle G, Germinaro M, Weiss S, Zangrilli J. Reslizumab in patients with inadequately controlled late-onset asthma and elevated blood eosinophils. Pulm Pharmacol Ther 43: 39–45, 2017. doi: 10.1016/j.pupt.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Brusselle GG, Kips JC, Tavernier JH, van der Heyden JG, Cuvelier CA, Pauwels RA, Bluethmann H. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy 24: 73–80, 1994. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- 31.Brusselle GG, Maes T, Bracke KR. Eosinophils in the spotlight: eosinophilic airway inflammation in nonallergic asthma. Nat Med 19: 977–979, 2013. doi: 10.1038/nm.3300. [DOI] [PubMed] [Google Scholar]
- 32.Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, Hansbro PM. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol 15: 55–63, 2017. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 33.Bush A. How early do airway inflammation and remodeling occur? Allergol Int 57: 11–19, 2008. doi: 10.2332/allergolint.R-07-155. [DOI] [PubMed] [Google Scholar]
- 34.Busse WW. Anti-immunoglobulin E (omalizumab) therapy in allergic asthma. Am J Respir Crit Care Med 164, Suppl 1: S12–S17, 2001. doi: 10.1164/ajrccm.164.supplement_1.2103026. [DOI] [PubMed] [Google Scholar]
- 35.Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, Lin SL. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med 188: 1294–1302, 2013. doi: 10.1164/rccm.201212-2318OC. [DOI] [PubMed] [Google Scholar]
- 36.Busse WW, Morgan WJ, Gergen PJ, Mitchell HE, Gern JE, Liu AH, Gruchalla RS, Kattan M, Teach SJ, Pongracic JA, Chmiel JF, Steinbach SF, Calatroni A, Togias A, Thompson KM, Szefler SJ, Sorkness CA. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med 364: 1005–1015, 2011. doi: 10.1056/NEJMoa1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camiz S, Pillar VD. Comparison of single and complete linkage clustering with the hierarchical factor classification of variables. Community Ecol 8: 25–30, 2007. doi: 10.1556/ComEc.8.2007.1.4. [DOI] [Google Scholar]
- 38.Campbell EM, Kunkel SL, Strieter RM, Lukacs NW. Temporal role of chemokines in a murine model of cockroach allergen-induced airway hyperreactivity and eosinophilia. J Immunol 161: 7047–7053, 1998. [PubMed] [Google Scholar]
- 39.Castanhinha S, Sherburn R, Walker S, Gupta A, Bossley CJ, Buckley J, Ullmann N, Grychtol R, Campbell G, Maglione M, Koo S, Fleming L, Gregory L, Snelgrove RJ, Bush A, Lloyd CM, Saglani S. Pediatric severe asthma with fungal sensitization is mediated by steroid-resistant IL-33. J Allergy Clin Immunol 136: 312–22.e7, 2015. doi: 10.1016/j.jaci.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, Busse WW, Ford L, Sher L, FitzGerald JM, Katelaris C, Tohda Y, Zhang B, Staudinger H, Pirozzi G, Amin N, Ruddy M, Akinlade B, Khan A, Chao J, Martincova R, Graham NMH, Hamilton JD, Swanson BN, Stahl N, Yancopoulos GD, Teper A. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N Engl J Med 378: 2486–2496, 2018. doi: 10.1056/NEJMoa1804092. [DOI] [PubMed] [Google Scholar]
- 41.Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, Young J, Wilkins HJ, Henkel T, Nair P; Res-5-0010 Study Group . Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med 184: 1125–1132, 2011. doi: 10.1164/rccm.201103-0396OC. [DOI] [PubMed] [Google Scholar]
- 42.Castro M, Wenzel SE, Bleecker ER, Pizzichini E, Kuna P, Busse WW, Gossage DL, Ward CK, Wu Y, Wang B, Khatry DB, van der Merwe R, Kolbeck R, Molfino NA, Raible DG. Benralizumab, an anti-interleukin 5 receptor α monoclonal antibody, versus placebo for uncontrolled eosinophilic asthma: a phase 2b randomised dose-ranging study. Lancet Respir Med 2: 879–890, 2014. doi: 10.1016/S2213-2600(14)70201-2. [DOI] [PubMed] [Google Scholar]
- 43.Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, Burlet-Schiltz O, Gonzalez-de-Peredo A, Girard JP. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol 19: 375–385, 2018. doi: 10.1038/s41590-018-0067-5. [DOI] [PubMed] [Google Scholar]
- 44.Chang P, Gohain M, Yen MR, Chen PY. Computational Methods for Assessing Chromatin Hierarchy. Comput Struct Biotechnol J 16: 43–53, 2018. doi: 10.1016/j.csbj.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang TS, Lemanske RF Jr, Mauger DT, Fitzpatrick AM, Sorkness CA, Szefler SJ, Gangnon RE, Page CD, Jackson DJ; Childhood Asthma Research and Education (CARE) Network Investigators . Childhood asthma clusters and response to therapy in clinical trials. J Allergy Clin Immunol 133: 363–369.e3, 2014. doi: 10.1016/j.jaci.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapoval SP, Nabozny GH, Marietta EV, Raymond EL, Krco CJ, Andrews AG, David CS. Short ragweed allergen induces eosinophilic lung disease in HLA-DQ transgenic mice. J Clin Invest 103: 1707–1717, 1999. doi: 10.1172/JCI6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaudhuri R, Livingston E, McMahon AD, Lafferty J, Fraser I, Spears M, McSharry CP, Thomson NC. Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. Am J Respir Crit Care Med 174: 127–133, 2006. doi: 10.1164/rccm.200510-1589OC. [DOI] [PubMed] [Google Scholar]
- 48.Chibana K, Trudeau JB, Mustovich AT, Hu H, Zhao J, Balzar S, Chu HW, Wenzel SE. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin Exp Allergy 38: 936–946, 2008. doi: 10.1111/j.1365-2222.2008.02969.x. [DOI] [PubMed] [Google Scholar]
- 49.Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, McElwain K, McElwain S, Friedman S, Broide DH. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest 113: 551–560, 2004. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, Jia G, Ohri CM, Doran E, Vannella KM, Butler CA, Hargadon B, Sciurba JC, Gieseck RL, Thompson RW, White S, Abbas AR, Jackman J, Wu LC, Egen JG, Heaney LG, Ramalingam TR, Arron JR, Wynn TA, Bradding P. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 7: 301ra129, 2015. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- 51.Chua HH, Chou HC, Tung YL, Chiang BL, Liao CC, Liu HH, Ni YH. Intestinal Dysbiosis Featuring Abundance of Ruminococcus gnavus Associates With Allergic Diseases in Infants. Gastroenterology 154: 154–167, 2018. doi: 10.1053/j.gastro.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Corren J, Lemanske RF Jr, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab treatment in adults with asthma. N Engl J Med 365: 1088–1098, 2011. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 53.Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, van der Merwe R. Tezepelumab in Adults with Uncontrolled Asthma. N Engl J Med 377: 936–946, 2017. doi: 10.1056/NEJMoa1704064. [DOI] [PubMed] [Google Scholar]
- 54.Corrigan CJ, Hamid Q, North J, Barkans J, Moqbel R, Durham S, Gemou-Engesaeth V, Kay AB. Peripheral blood CD4 but not CD8 t-lymphocytes in patients with exacerbation of asthma transcribe and translate messenger RNA encoding cytokines which prolong eosinophil survival in the context of a Th2-type pattern: effect of glucocorticoid therapy. Am J Respir Cell Mol Biol 12: 567–578, 1995. doi: 10.1165/ajrcmb.12.5.7742019. [DOI] [PubMed] [Google Scholar]
- 55.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. [Correction in J Exp Med 185: 1715, 1997.] J Exp Med 183: 109–117, 1996. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Covar RA, Strunk R, Zeiger RS, Wilson LA, Liu AH, Weiss S, Tonascia J, Spahn JD, Szefler SJ; Childhood Asthma Management Program Research Group . Predictors of remitting, periodic, and persistent childhood asthma. J Allergy Clin Immunol 125: 359–366.e3, 2010. doi: 10.1016/j.jaci.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies JO, Oudelaar AM, Higgs DR, Hughes JR. How best to identify chromosomal interactions: a comparison of approaches. Nat Methods 14: 125–134, 2017. doi: 10.1038/nmeth.4146. [DOI] [PubMed] [Google Scholar]
- 58.Day WHEE, Edelsbrunner H. H. Efficient algorithms for agglomerative hierarchical clustering methods. J Classif 1: 7–24, 1984. doi: 10.1007/BF01890115. [DOI] [Google Scholar]
- 59.Debeuf N, Haspeslagh E, van Helden M, Hammad H, Lambrecht BN. Mouse Models of Asthma. Curr Protoc Mouse Biol 6: 169–184, 2016. doi: 10.1002/cpmo.4. [DOI] [PubMed] [Google Scholar]
- 60.DeBoer MD, Phillips BR, Mauger DT, Zein J, Erzurum SC, Fitzpatrick AM, Gaston BM, Myers R, Ross KR, Chmiel J, Lee MJ, Fahy JV, Peters M, Ly NP, Wenzel SE, Fajt ML, Holguin F, Moore WC, Peters SP, Meyers D, Bleecker ER, Castro M, Coverstone AM, Bacharier LB, Jarjour NN, Sorkness RL, Ramratnam S, Irani AM, Israel E, Levy B, Phipatanakul W, Gaffin JM, Teague WG. Effects of endogenous sex hormones on lung function and symptom control in adolescents with asthma. BMC Pulm Med 18: 58, 2018. doi: 10.1186/s12890-018-0612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denlinger LC, Phillips BR, Ramratnam S, Ross K, Bhakta NR, Cardet JC, Castro M, Peters SP, Phipatanakul W, Aujla S, Bacharier LB, Bleecker ER, Comhair SA, Coverstone A, DeBoer M, Erzurum SC, Fain SB, Fajt M, Fitzpatrick AM, Gaffin J, Gaston B, Hastie AT, Hawkins GA, Holguin F, Irani AM, Israel E, Levy BD, Ly N, Meyers DA, Moore WC, Myers R, Opina MT, Peters MC, Schiebler ML, Sorkness RL, Teague WG, Wenzel SE, Woodruff PG, Mauger DT, Fahy JV, Jarjour NN; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program-3 Investigators . Inflammatory and Comorbid Features of Patients with Severe Asthma and Frequent Exacerbations. Am J Respir Crit Care Med 195: 302–313, 2017. doi: 10.1164/rccm.201602-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Denning DW, O’Driscoll BR, Powell G, Chew F, Atherton GT, Vyas A, Miles J, Morris J, Niven RM. Randomized controlled trial of oral antifungal treatment for severe asthma with fungal sensitization: The Fungal Asthma Sensitization Trial (FAST) study. Am J Respir Crit Care Med 179: 11–18, 2009. doi: 10.1164/rccm.200805-737OC. [DOI] [PubMed] [Google Scholar]
- 63.Desai D, Newby C, Symon FA, Haldar P, Shah S, Gupta S, Bafadhel M, Singapuri A, Siddiqui S, Woods J, Herath A, Anderson IK, Bradding P, Green R, Kulkarni N, Pavord I, Marshall RP, Sousa AR, May RD, Wardlaw AJ, Brightling CE. Elevated sputum interleukin-5 and submucosal eosinophilia in obese individuals with severe asthma. Am J Respir Crit Care Med 188: 657–663, 2013. doi: 10.1164/rccm.201208-1470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, Garudathri J, Raymond D, Poynter ME, Bunn JY, Irvin CG. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol 128: 508–515.e2, 2011. doi: 10.1016/j.jaci.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax 57: 643–648, 2002. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, Raymond WW, Lachowicz-Scroggins ME, Di Maio S, Hoffman EA, Castro M, Fain SB, Jarjour NN, Israel E, Levy BD, Erzurum SC, Wenzel SE, Meyers DA, Bleecker ER, Phillips BR, Mauger DT, Gordon ED, Woodruff PG, Peters MC, Fahy JV; National Heart Lung and Blood Institute (NHLBI) Severe Asthma Research Program (SARP) . Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest 128: 997–1009, 2018. doi: 10.1172/JCI95693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durack J, Lynch SV, Nariya S, Bhakta NR, Beigelman A, Castro M, Dyer AM, Israel E, Kraft M, Martin RJ, Mauger DT, Rosenberg SR, Sharp-King T, White SR, Woodruff PG, Avila PC, Denlinger LC, Holguin F, Lazarus SC, Lugogo N, Moore WC, Peters SP, Que L, Smith LJ, Sorkness CA, Wechsler ME, Wenzel SE, Boushey HA, Huang YJ; National Heart, Lung and Blood Institute’s “AsthmaNet” . Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J Allergy Clin Immunol 140: 63–75, 2017. doi: 10.1016/j.jaci.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fahy JV, Fleming HE, Wong HH, Liu JT, Su JQ, Reimann J, Fick RB Jr, Boushey HA. The effect of an anti-IgE monoclonal antibody on the early- and late-phase responses to allergen inhalation in asthmatic subjects. Am J Respir Crit Care Med 155: 1828–1834, 1997. doi: 10.1164/ajrccm.155.6.9196082. [DOI] [PubMed] [Google Scholar]
- 69.Ferguson GT, FitzGerald JM, Bleecker ER, Laviolette M, Bernstein D, LaForce C, Mansfield L, Barker P, Wu Y, Jison M, Goldman M; BISE Study Investigators . Benralizumab for patients with mild to moderate, persistent asthma (BISE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 5: 568–576, 2017. doi: 10.1016/S2213-2600(17)30190-X. [DOI] [PubMed] [Google Scholar]
- 70.FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, Ferguson GT, Busse WW, Barker P, Sproule S, Gilmartin G, Werkström V, Aurivillius M, Goldman M; CALIMA Study Investigators . Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 388: 2128–2141, 2016. doi: 10.1016/S0140-6736(16)31322-8. [DOI] [PubMed] [Google Scholar]
- 71.Fitzhugh DJ, Lockey RF. Allergen immunotherapy: a history of the first 100 years. Curr Opin Allergy Clin Immunol 11: 554–559, 2011. doi: 10.1097/ACI.0b013e32834c3134. [DOI] [PubMed] [Google Scholar]
- 72.Fitzpatrick AM, Brown LA, Holguin F, Teague WG; National Institutes of Health/National Heart, Lung, and Blood Institute Severe Asthma Research Program . Levels of nitric oxide oxidation products are increased in the epithelial lining fluid of children with persistent asthma. J Allergy Clin Immunol 124: 990–996.e91, 2009. doi: 10.1016/j.jaci.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fitzpatrick AM, Gaston BM, Erzurum SC, Teague WG; National Institutes of Health/National Heart, Lung, and Blood Institute Severe Asthma Research Program . Features of severe asthma in school-age children: Atopy and increased exhaled nitric oxide. J Allergy Clin Immunol 118: 1218–1225, 2006. doi: 10.1016/j.jaci.2006.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fitzpatrick AM, Higgins M, Holguin F, Brown LA, Teague WG; National Institutes of Health/National Heart, Lung, and Blood Institute’s Severe Asthma Research Program . The molecular phenotype of severe asthma in children. J Allergy Clin Immunol 125: 851–857.e18, 2010. doi: 10.1016/j.jaci.2010.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fitzpatrick AM, Jackson DJ, Mauger DT, Boehmer SJ, Phipatanakul W, Sheehan WJ, Moy JN, Paul IM, Bacharier LB, Cabana MD, Covar R, Holguin F, Lemanske RF Jr, Martinez FD, Pongracic JA, Beigelman A, Baxi SN, Benson M, Blake K, Chmiel JF, Daines CL, Daines MO, Gaffin JM, Gentile DA, Gower WA, Israel E, Kumar HV, Lang JE, Lazarus SC, Lima JJ, Ly N, Marbin J, Morgan W, Myers RE, Olin JT, Peters SP, Raissy HH, Robison RG, Ross K, Sorkness CA, Thyne SM, Szefler SJ; NIH/NHLBI AsthmaNet . Individualized therapy for persistent asthma in young children. J Allergy Clin Immunol 138: 1608–1618.e12, 2016. doi: 10.1016/j.jaci.2016.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fitzpatrick AM, Park Y, Brown LA, Jones DP. Children with severe asthma have unique oxidative stress-associated metabolomic profiles. J Allergy Clin Immunol 133: 258–261.e8, 2014. doi: 10.1016/j.jaci.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fitzpatrick AM, Stephenson ST, Brown MR, Nguyen K, Douglas S, Brown LAS. Systemic Corticosteroid Responses in Children with Severe Asthma: Phenotypic and Endotypic Features. J Allergy Clin Immunol Pract 5: 410–419.e4, 2017. doi: 10.1016/j.jaip.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fitzpatrick AM, Stephenson ST, Hadley GR, Burwell L, Penugonda M, Simon DM, Hansen J, Jones DP, Brown LA. Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J Allergy Clin Immunol 127: 1604–1611, 2011. doi: 10.1016/j.jaci.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fitzpatrick AM, Teague WG, Meyers DA, Peters SP, Li X, Li H, Wenzel SE, Aujla S, Castro M, Bacharier LB, Gaston BM, Bleecker ER, Moore WC; National Institutes of Health/National Heart, Lung, and Blood Institute Severe Asthma Research Program . Heterogeneity of severe asthma in childhood: confirmation by cluster analysis of children in the National Institutes of Health/National Heart, Lung, and Blood Institute Severe Asthma Research Program. J Allergy Clin Immunol 127: 382–389.e13, 2011. doi: 10.1016/j.jaci.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fitzpatrick AM, Teague WG; National Institutes of Health/National Heart, Lung, and Blood Institute’s Severe Asthma Research Program . Progressive airflow limitation is a feature of children with severe asthma. J Allergy Clin Immunol 127: 282–284, 2011. doi: 10.1016/j.jaci.2010.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fleming L, Murray C, Bansal AT, Hashimoto S, Bisgaard H, Bush A, Frey U, Hedlin G, Singer F, van Aalderen WM, Vissing NH, Zolkipli Z, Selby A, Fowler S, Shaw D, Chung KF, Sousa AR, Wagers S, Corfield J, Pandis I, Rowe A, Formaggio E, Sterk PJ, Roberts G; U-BIOPRED Study Group . The burden of severe asthma in childhood and adolescence: results from the paediatric U-BIOPRED cohorts. [Correction in Eur Respir J 49: 1550780, 2017.] Eur Respir J 46: 1322–1333, 2015. doi: 10.1183/13993003.00780-2015. [DOI] [PubMed] [Google Scholar]
- 82.Fleming L, Wilson N, Regamey N, Bush A. Use of sputum eosinophil counts to guide management in children with severe asthma. Thorax 67: 193–198, 2012. doi: 10.1136/thx.2010.156836. [DOI] [PubMed] [Google Scholar]
- 83.Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, Robinson D, Wenzel S, Busse W, Hansel TT, Barnes NC; International Mepolizumab Study Group . A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med 176: 1062–1071, 2007. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- 84.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med 183: 195–201, 1996. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu L, Freishtat RJ, Gordish-Dressman H, Teach SJ, Resca L, Hoffman EP, Wang Z. Natural progression of childhood asthma symptoms and strong influence of sex and puberty. Ann Am Thorac Soc 11: 939–944, 2014. doi: 10.1513/AnnalsATS.201402-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujimura KE, Demoor T, Rauch M, Faruqi AA, Jang S, Johnson CC, Boushey HA, Zoratti E, Ownby D, Lukacs NW, Lynch SV. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc Natl Acad Sci USA 111: 805–810, 2014. doi: 10.1073/pnas.1310750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galant SP, Morphew T, Amaro S, Liao O. Current asthma guidelines may not identify young children who have experienced significant morbidity. Pediatrics 117: 1038–1045, 2006. doi: 10.1542/peds.2005-1076. [DOI] [PubMed] [Google Scholar]
- 89.Gauthier M, Chakraborty K, Oriss TB, Raundhal M, Das S, Chen J, Huff R, Sinha A, Fajt M, Ray P, Wenzel SE, Ray A. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight 2: e94580, 2017. doi: 10.1172/jci.insight.94580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gauvreau GM, Boulet LP, Cockcroft DW, FitzGerald JM, Carlsten C, Davis BE, Deschesnes F, Duong M, Durn BL, Howie KJ, Hui L, Kasaian MT, Killian KJ, Strinich TX, Watson RM, Y N, Zhou S, Raible D, O’Byrne PM. Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med 183: 1007–1014, 2011. doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]
- 91.Gauvreau GM, Lee JM, Watson RM, Irani AM, Schwartz LB, O’Byrne PM. Increased numbers of both airway basophils and mast cells in sputum after allergen inhalation challenge of atopic asthmatics. Am J Respir Crit Care Med 161: 1473–1478, 2000. doi: 10.1164/ajrccm.161.5.9908090. [DOI] [PubMed] [Google Scholar]
- 92.Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, FitzGerald JM, Boedigheimer M, Davis BE, Dias C, Gorski KS, Smith L, Bautista E, Comeau MR, Leigh R, Parnes JR. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med 370: 2102–2110, 2014. doi: 10.1056/NEJMoa1402895. [DOI] [PubMed] [Google Scholar]
- 93.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol 10: 587–593, 1994. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 94.Gelfand EW, Kraft M. The importance and features of the distal airways in children and adults. J Allergy Clin Immunol 124, Suppl: S84–S87, 2009. doi: 10.1016/j.jaci.2009.07.062. [DOI] [PubMed] [Google Scholar]
- 95.Gibson PG, Henry RL, Thomas P. Noninvasive assessment of airway inflammation in children: induced sputum, exhaled nitric oxide, and breath condensate. Eur Respir J 16: 1008–1015, 2000. [PubMed] [Google Scholar]
- 96.Gibson PG, Simpson JL, Hankin R, Powell H, Henry RL. Relationship between induced sputum eosinophils and the clinical pattern of childhood asthma. Thorax 58: 116–121, 2003. doi: 10.1136/thorax.58.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest 119: 1329–1336, 2001. doi: 10.1378/chest.119.5.1329. [DOI] [PubMed] [Google Scholar]
- 98.Gordon ED, Simpson LJ, Rios CL, Ringel L, Lachowicz-Scroggins ME, Peters MC, Wesolowska-Andersen A, Gonzalez JR, MacLeod HJ, Christian LS, Yuan S, Barry L, Woodruff PG, Ansel KM, Nocka K, Seibold MA, Fahy JV. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad Sci USA 113: 8765–8770, 2016. doi: 10.1073/pnas.1601914113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gorska MM. Mouse Models of Asthma. Methods Mol Biol 1809: 351–362, 2018. doi: 10.1007/978-1-4939-8570-8_23. [DOI] [PubMed] [Google Scholar]
- 100.Granell R, Henderson AJ, Sterne JA. Associations of wheezing phenotypes with late asthma outcomes in the Avon Longitudinal Study of Parents and Children: a population-based birth cohort. J Allergy Clin Immunol 138: 1060–1070.e11, 2016. doi: 10.1016/j.jaci.2016.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, Wardlaw AJ, Pavord ID. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet 360: 1715–1721, 2002. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 102.Gregory LG, Lloyd CM. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol 32: 402–411, 2011. doi: 10.1016/j.it.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grünig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282: 2261–2263, 1998. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grunwell JR, Stephenson ST, Tirouvanziam R, Brown LAS, Brown MR, Fitzpatrick AM. Children with Neutrophil-Predominant Severe Asthma Have Proinflammatory Neutrophils With Enhanced Survival and Impaired Clearance. J Allergy Clin Immunol Pract 7: 516–525.e6, 2019. doi: 10.1016/j.jaip.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guénoche A, Hansen P, Jaumard B. Efficient algorithms for divisive hierarchical clustering with the diameter criterion. J Classif 8: 5–30, 1991. doi: 10.1007/BF02616245. [DOI] [Google Scholar]
- 106.Guiddir T, Saint-Pierre P, Purenne-Denis E, Lambert N, Laoudi Y, Couderc R, Gouvis-Echraghi R, Amat F, Just J. Neutrophilic Steroid-Refractory Recurrent Wheeze and Eosinophilic Steroid-Refractory Asthma in Children. J Allergy Clin Immunol Pract 5: 1351–1361.e2, 2017. doi: 10.1016/j.jaip.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 107.Guilbert TW, Morgan WJ, Zeiger RS, Mauger DT, Boehmer SJ, Szefler SJ, Bacharier LB, Lemanske RF Jr, Strunk RC, Allen DB, Bloomberg GR, Heldt G, Krawiec M, Larsen G, Liu AH, Chinchilli VM, Sorkness CA, Taussig LM, Martinez FD. Long-term inhaled corticosteroids in preschool children at high risk for asthma. N Engl J Med 354: 1985–1997, 2006. doi: 10.1056/NEJMoa051378. [DOI] [PubMed] [Google Scholar]
- 108.Guo FH, Uetani K, Haque SJ, Williams BR, Dweik RA, Thunnissen FB, Calhoun W, Erzurum SC. Interferon gamma and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. J Clin Invest 100: 829–838, 1997. doi: 10.1172/JCI119598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gupta A, Sjoukes A, Richards D, Banya W, Hawrylowicz C, Bush A, Saglani S. Relationship between serum vitamin D, disease severity, and airway remodeling in children with asthma. Am J Respir Crit Care Med 184: 1342–1349, 2011. doi: 10.1164/rccm.201107-1239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, Pavord ID. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med 360: 973–984, 2009. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, Wardlaw AJ, Green RH. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 178: 218–224, 2008. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hamelmann E, Cieslewicz G, Schwarze J, Ishizuka T, Joetham A, Heusser C, Gelfand EW. Anti-interleukin 5 but not anti-IgE prevents airway inflammation and airway hyperresponsiveness. Am J Respir Crit Care Med 160: 934–941, 1999. doi: 10.1164/ajrccm.160.3.9806029. [DOI] [PubMed] [Google Scholar]
- 113.Han G, Spitzer MH, Bendall SC, Fantl WJ, Nolan GP. Metal-isotope-tagged monoclonal antibodies for high-dimensional mass cytometry. Nat Protoc 13: 2121–2148, 2018. doi: 10.1038/s41596-018-0016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Han NR, Oh HA, Nam SY, Moon PD, Kim DW, Kim HM, Jeong HJ. TSLP induces mast cell development and aggravates allergic reactions through the activation of MDM2 and STAT6. J Invest Dermatol 134: 2521–2530, 2014. doi: 10.1038/jid.2014.198. [DOI] [PubMed] [Google Scholar]
- 115.Hanania NA, Alpan O, Hamilos DL, Condemi JJ, Reyes-Rivera I, Zhu J, Rosen KE, Eisner MD, Wong DA, Busse W. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med 154: 573–582, 2011. doi: 10.7326/0003-4819-154-9-201105030-00002. [DOI] [PubMed] [Google Scholar]
- 116.Hanania NA, Wenzel S, Rosén K, Hsieh HJ, Mosesova S, Choy DF, Lal P, Arron JR, Harris JM, Busse W. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med 187: 804–811, 2013. doi: 10.1164/rccm.201208-1414OC. [DOI] [PubMed] [Google Scholar]
- 117.Hartigan JA, Wong MA. Algorithm AS 136: A K-Means Clustering Algorithm. J R Stat Soc Ser C Appl Stat 28: 100–108, 1979. doi: 10.2307/2346830. [DOI] [Google Scholar]
- 118.Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, Bleecker ER; National Heart, Lung, and Blood Institute Severe Asthma Research Program . Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol 125: 1028–1036.e13, 2010. doi: 10.1016/j.jaci.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hauk PJ, Krawiec M, Murphy J, Boguniewicz J, Schiltz A, Goleva E, Liu AH, Leung DY. Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatr Pulmonol 43: 916–923, 2008. doi: 10.1002/ppul.20880. [DOI] [PubMed] [Google Scholar]
- 120.He XY, Simpson JL, Wang F. Inflammatory phenotypes in stable and acute childhood asthma. Paediatr Respir Rev 12: 165–169, 2011. doi: 10.1016/j.prrv.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 121.Hekking PP, Loza MJ, Pavlidis S, De Meulder B, Lefaudeux D, Baribaud F, Auffray C, Wagener AH, Brinkman P, Lutter R, Bansal AT, Sousa AR, Bates SA, Pandis I, Fleming LJ, Shaw DE, Fowler SJ, Guo Y, Meiser A, Sun K, Corfield J, Howarth P, Bel EH, Adcock IM, Chung KF, Djukanovic R, Sterk PJ; U-BIOPRED Study Group . Transcriptomic gene signatures associated with persistent airflow limitation in patients with severe asthma. Eur Respir J 50: 1602298, 2017. doi: 10.1183/13993003.02298-2016. [DOI] [PubMed] [Google Scholar]
- 122.Hekking PP, Loza MJ, Pavlidis S, de Meulder B, Lefaudeux D, Baribaud F, Auffray C, Wagener AH, Brinkman P, Lutter R, Bansal AT, Sousa AR, Bates SA, Pandis Y, Fleming LJ, Shaw DE, Fowler SJ, Guo Y, Meiser A, Sun K, Corfield J, Howarth PH, Bel EH, Adcock IM, Chung KF, Djukanovic R, Sterk PJ; U-BIOPRED Study Group . Pathway discovery using transcriptomic profiles in adult-onset severe asthma. J Allergy Clin Immunol 141: 1280–1290, 2018. doi: 10.1016/j.jaci.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 123.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol 111: 677–690, 2003. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 124.Herz U, Lumpp U, Da Palma JC, Enssle K, Takatsu K, Schnoy N, Daser A, Köttgen E, Wahn U, Renz H. The relevance of murine animal models to study the development of allergic bronchial asthma. Immunol Cell Biol 74: 209–217, 1996. doi: 10.1038/icb.1996.30. [DOI] [PubMed] [Google Scholar]
- 125.Heymann PW, Platts-Mills TA, Johnston SL. Role of viral infections, atopy and antiviral immunity in the etiology of wheezing exacerbations among children and young adults. Pediatr Infect Dis J 24, Suppl: S217–S222, 2005. doi: 10.1097/01.inf.0000188164.33856.f9. [DOI] [PubMed] [Google Scholar]
- 126.Hinks TSC, Batty P, Klenerman P, Pavord ID, Xue L. Cytometric Gating Stringency Impacts Studies of Type 2 Innate Lymphoid Cells in Asthma. Am J Respir Cell Mol Biol 57: 745–747, 2017. doi: 10.1165/rcmb.2017-0201LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hogaboam CM, Blease K, Mehrad B, Steinhauser ML, Standiford TJ, Kunkel SL, Lukacs NW. Chronic airway hyperreactivity, goblet cell hyperplasia, and peribronchial fibrosis during allergic airway disease induced by Aspergillus fumigatus. Am J Pathol 156: 723–732, 2000. doi: 10.1016/S0002-9440(10)64775-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 278: 1355–1360, 1968. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 129.Holguin F, Bleecker ER, Busse WW, Calhoun WJ, Castro M, Erzurum SC, Fitzpatrick AM, Gaston B, Israel E, Jarjour NN, Moore WC, Peters SP, Yonas M, Teague WG, Wenzel SE. Obesity and asthma: an association modified by age of asthma onset. J Allergy Clin Immunol 127: 1486–1493.e2, 2011. doi: 10.1016/j.jaci.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Holguin F, Comhair SA, Hazen SL, Powers RW, Khatri SS, Bleecker ER, Busse WW, Calhoun WJ, Castro M, Fitzpatrick AM, Gaston B, Israel E, Jarjour NN, Moore WC, Peters SP, Teague WG, Chung KF, Erzurum SC, Wenzel SE. An association between l-arginine/asymmetric dimethyl arginine balance, obesity, and the age of asthma onset phenotype. Am J Respir Crit Care Med 187: 153–159, 2013. doi: 10.1164/rccm.201207-1270OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Howrylak JA, Fuhlbrigge AL, Strunk RC, Zeiger RS, Weiss ST, Raby BA; Childhood Asthma Management Program Research Group . Classification of childhood asthma phenotypes and long-term clinical responses to inhaled anti-inflammatory medications. J Allergy Clin Immunol 133: 1289–1300.e12, 2014. doi: 10.1016/j.jaci.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 133.Hui CC, McNagny KM, Denburg JA, Siracusa MC. In situ hematopoiesis: a regulator of TH2 cytokine-mediated immunity and inflammation at mucosal surfaces. Mucosal Immunol 8: 701–711, 2015. doi: 10.1038/mi.2015.17. [DOI] [PubMed] [Google Scholar]
- 134.Humbert M, Beasley R, Ayres J, Slavin R, Hébert J, Bousquet J, Beeh KM, Ramos S, Canonica GW, Hedgecock S, Fox H, Blogg M, Surrey K. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 60: 309–316, 2005. doi: 10.1111/j.1398-9995.2004.00772.x. [DOI] [PubMed] [Google Scholar]
- 135.Humbert M, Durham SR, Ying S, Kimmitt P, Barkans J, Assoufi B, Pfister R, Menz G, Robinson DS, Kay AB, Corrigan CJ. IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients with atopic and nonatopic asthma: evidence against “intrinsic” asthma being a distinct immunopathologic entity. Am J Respir Crit Care Med 154: 1497–1504, 1996. doi: 10.1164/ajrccm.154.5.8912771. [DOI] [PubMed] [Google Scholar]
- 136.Israel E, Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Shapiro J, Cohn J, Rubin P, Drazen JM. The pivotal role of 5-lipoxygenase products in the reaction of aspirin-sensitive asthmatics to aspirin. Am Rev Respir Dis 148: 1447–1451, 1993. doi: 10.1164/ajrccm/148.6_Pt_1.1447. [DOI] [PubMed] [Google Scholar]
- 137.Israel E, Reddel HK. Severe and Difficult-to-Treat Asthma in Adults. N Engl J Med 377: 965–976, 2017. doi: 10.1056/NEJMra1608969. [DOI] [PubMed] [Google Scholar]
- 138.Jain AK. Data clustering: 50 years beyond K-means. Pattern Recognit Lett 31: 651–666, 2010. doi: 10.1016/j.patrec.2009.09.011. [DOI] [Google Scholar]
- 139.Jenkins HA, Cherniack R, Szefler SJ, Covar R, Gelfand EW, Spahn JD. A comparison of the clinical characteristics of children and adults with severe asthma. Chest 124: 1318–1324, 2003. doi: 10.1378/chest.124.4.1318. [DOI] [PubMed] [Google Scholar]
- 140.Jevnikar Z, Östling J, Ax E, Calvén J, Thörn K, Israelsson E, Öberg L, Singhania A, Lau LCK, Wilson SJ, Ward JA, Chauhan A, Sousa AR, De Meulder B, Loza MJ, Baribaud F, Sterk PJ, Chung KF, Sun K, Guo Y, Adcock IM, Payne D, Dahlen B, Chanez P, Shaw DE, Krug N, Hohlfeld JM, Sandström T, Djukanovic R, James A, Hinks TSC, Howarth PH, Vaarala O, van Geest M, Olsson H, Adcock IM, Ahmed H, Auffray C, Bakke P, Bansal AT, Baribaud F, Bates S, Bel EH, Bigler J, Bisgaard H, Boedigheimer MJ, Bønnelykke K, Brandsma J, Brinkman P, Bucchioni E, Burg D, Bush A, Caruso M, Chaiboonchoe A, Chanez P, Chung FK, Compton CH, Corfield J, D’Amico A, Dahlen SE, De Meulder B, Djukanovic R, Erpenbeck VJ, Erzen D, Fichtner K, Fitch N, Fleming LJ, Formaggio E, Fowler SJ, Frey U, Gahlemann M, Geiser T, Goss V, Guo Y, Hashimoto S, Haughney J, Hedlin G, Hekking PW, Higenbottam T, Hohlfeld JM, Holweg C, Horváth I, James AJ, Knowles R, Knox AJ, Krug N, Lefaudeux D, Loza MJ, Manta A, Matthews JG, Mazein A, Meiser A, Middelveld RJM, Miralpeix M, Montuschi P, Mores N, Murray CS, Musial J, Myles D, Pahus L, Pandis I, Pavlidis S, Postle A, Powel P, Praticò G, Rao N, Riley J, Roberts A, Roberts G, Rowe A, Sandström T, Schofield JPR, Seibold W, Selby A, Shaw DE, Sigmund R, Singer F, Skipp PJ, Sousa AR, Sterk PJ, Sun K, Thornton B, van Aalderen WM, van Geest M, Vestbo J, Vissing NH, Wagener AH, Wagers SS, Weiszhart Z, Wheelock CE, Wilson SJ; Unbiased Biomarkers in Prediction of Respiratory Disease Outcomes Study Group . Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol 143: 577–590, 2019. doi: 10.1016/j.jaci.2018.05.026. [DOI] [PubMed] [Google Scholar]
- 141.Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, Shikotra A, Carter R, Audusseau S, Hamid Q, Bradding P, Fahy JV, Woodruff PG, Harris JM, Arron JR; Bronchoscopic Exploratory Research Study of Biomarkers in Corticosteroid-refractory Asthma (BOBCAT) Study Group . Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol 130: 647–654.e10, 2012. doi: 10.1016/j.jaci.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jonstam K, Swanson BN, Mannent L, Cardell LO, Tian N, Wang Y, Zhang D, Fan C, Holtappels G, Hamilton JD, Grabher A, Graham NMH, Pirozzi G, Bachert C. Dupilumab reduces local type 2 pro-inflammatory biomarkers in chronic rhinosinusitis with nasal polyposis. Allergy 74: 743–752, 2019. doi: 10.1111/all.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Just J, Gouvis-Echraghi R, Rouve S, Wanin S, Moreau D, Annesi-Maesano I. Two novel, severe asthma phenotypes identified during childhood using a clustering approach. Eur Respir J 40: 55–60, 2012. doi: 10.1183/09031936.00123411. [DOI] [PubMed] [Google Scholar]
- 144.Just J, Saint-Pierre P, Gouvis-Echraghi R, Laoudi Y, Roufai L, Momas I, Annesi Maesano I. Childhood allergic asthma is not a single phenotype. J Pediatr 164: 815–820, 2014. doi: 10.1016/j.jpeds.2013.11.037. [DOI] [PubMed] [Google Scholar]
- 145.Kale SL, Agrawal K, Gaur SN, Arora N. Cockroach protease allergen induces allergic airway inflammation via epithelial cell activation. Sci Rep 7: 42341, 2017. doi: 10.1038/srep42341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145a.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38, Suppl_1: D355–D360, 2010. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol 169: 5904–5911, 2002. doi: 10.4049/jimmunol.169.10.5904. [DOI] [PubMed] [Google Scholar]
- 147.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20: 54–61, 2014. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim KW, Ober C. Lessons Learned From GWAS of Asthma. Allergy Asthma Immunol Res 11: 170–187, 2019. doi: 10.4168/aair.2019.11.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, Mayall JR, Ali MK, Starkey MR, Hansbro NG, Hirota JA, Wood LG, Simpson JL, Knight DA, Wark PA, Gibson PG, O’Neill LAJ, Cooper MA, Horvat JC, Hansbro PM. Role for NLRP3 Inflammasome-mediated, IL-1β-Dependent Responses in Severe, Steroid-Resistant Asthma. Am J Respir Crit Care Med 196: 283–297, 2017. doi: 10.1164/rccm.201609-1830OC. [DOI] [PubMed] [Google Scholar]
- 150.Kimura H, Konno S, Makita H, Taniguchi N, Shimizu K, Suzuki M, Kimura H, Goudarzi H, Nakamaru Y, Ono J, Ohta S, Izuhara K, Ito YM, Wenzel SE, Nishimura M; Hi-CARAT investigators . Prospective predictors of exacerbation status in severe asthma over a 3-year follow-up. Clin Exp Allergy 48: 1137–1146, 2018. doi: 10.1111/cea.13170. [DOI] [PubMed] [Google Scholar]
- 151.Koo S, Gupta A, Fainardi V, Bossley C, Bush A, Saglani S, Fleming L. Ethnic Variation in Response to IM Triamcinolone in Children With Severe Therapy-Resistant Asthma. Chest 149: 98–105, 2016. doi: 10.1378/chest.14-3241. [DOI] [PubMed] [Google Scholar]
- 152.Kristjansson RP, Benonisdottir S, Davidsson OB, Oddsson A, Tragante V, Sigurdsson JK, Stefansdottir L, Jonsson S, Jensson BO, Arthur JG, Arnadottir GA, Sulem G, Halldorsson BV, Gunnarsson B, Halldorsson GH, Stefansson OA, Oskarsson GR, Deaton AM, Olafsson I, Eyjolfsson GI, Sigurdardottir O, Onundarson PT, Gislason D, Gislason T, Ludviksson BR, Ludviksdottir D, Olafsdottir TA, Rafnar T, Masson G, Zink F, Bjornsdottir G, Magnusson OT, Bjornsdottir US, Thorleifsson G, Norddahl GL, Gudbjartsson DF, Thorsteinsdottir U, Jonsdottir I, Sulem P, Stefansson K. A loss-of-function variant in ALOX15 protects against nasal polyps and chronic rhinosinusitis. Nat Genet 51: 267–276, 2019. doi: 10.1038/s41588-018-0314-6. [DOI] [PubMed] [Google Scholar]
- 153.Kroegel C, Häfner D, Walker C, Luttmann W, Matthys H, Virchow JC Jr. [Immunopathogenesis of allergic bronchial asthma. Detection of activated CD25-CD4 lymphocytes and release of cytokines in the bronchoalveolar space following segmental allergen challenge]. Dtsch Med Wochenschr 120: 10–17, 1995. doi: 10.1055/s-2008-1043192. [DOI] [PubMed] [Google Scholar]
- 154.Krug N, Hohlfeld JM, Kirsten AM, Kornmann O, Beeh KM, Kappeler D, Korn S, Ignatenko S, Timmer W, Rogon C, Zeitvogel J, Zhang N, Bille J, Homburg U, Turowska A, Bachert C, Werfel T, Buhl R, Renz J, Garn H, Renz H. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N Engl J Med 372: 1987–1995, 2015. doi: 10.1056/NEJMoa1411776. [DOI] [PubMed] [Google Scholar]
- 155.Kumar RK, Herbert C, Foster PS. Mouse models of acute exacerbations of allergic asthma. Respirology 21: 842–849, 2016. doi: 10.1111/resp.12760. [DOI] [PubMed] [Google Scholar]
- 156.Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, Sousa A, Corfield J, Djukanovic R, Lutter R, Sterk PJ, Auffray C, Guo Y, Adcock IM, Chung KF; U-BIOPRED Study Group . T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J 49: 1602135, 2017. doi: 10.1183/13993003.02135-2016. [DOI] [PubMed] [Google Scholar]
- 157.Kupczyk M, Dahlén B, Sterk PJ, Nizankowska-Mogilnicka E, Papi A, Bel EH, Chanez P, Howarth PH, Holgate ST, Brusselle G, Siafakas NM, Gjomarkaj M, Dahlén SE; BIOAIR investigators . Stability of phenotypes defined by physiological variables and biomarkers in adults with asthma. Allergy 69: 1198–1204, 2014. doi: 10.1111/all.12445. [DOI] [PubMed] [Google Scholar]
- 158.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med 187: 939–948, 1998. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lachowicz-Scroggins ME, Dunican EM, Charbit AR, Raymond W, Looney MR, Peters MC, Gordon ED, Woodruff PG, Lefrancais E, Phillips BR, Mauger DT, Comhair SA, Erzurum SC, Johansson MW, Jarjour NN, Coverstone AM, Castro M, Hastie AT, Bleecker ER, Fajt ML, Wenzel SE, Israel E, Levy BD, Fahy JV; National Heart, Lung, and Blood Institute Severe Ashtma Research Program-3 Investigators . Extracellular DNA, Neutrophil Extracellular Traps, and Inflammasome Activation in Severe Asthma. Am J Respir Crit Care Med 199: 1076–1085, 2019. doi: 10.1164/rccm.201810-1869OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lai Y, Altemeier WA, Vandree J, Piliponsky AM, Johnson B, Appel CL, Frevert CW, Hyde DM, Ziegler SF, Smith DE, Henderson WR Jr, Gelb MH, Hallstrand TS. Increased density of intraepithelial mast cells in patients with exercise-induced bronchoconstriction regulated through epithelially derived thymic stromal lymphopoietin and IL-33. J Allergy Clin Immunol 133: 1448–1455, 2014. doi: 10.1016/j.jaci.2013.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559, 2008. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF. Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol 124: 1210–1216, 2009. doi: 10.1016/j.jaci.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 163.Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, Coste A, Jorissen M, Marano F, Caput D, Tournier F. IL-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest 108: 1817–1824, 2001. doi: 10.1172/JCI200113557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, Hansel TT, Holgate ST, Sterk PJ, Barnes PJ. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356: 2144–2148, 2000. doi: 10.1016/S0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 165.Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, Brenneise IE, Horton MA, Haczku A, Gelfand EW, Leikauf GD, Lee NA. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med 185: 2143–2156, 1997. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, Bansal AT, Lutter R, Sousa AR, Corfield J, Pandis I, Bakke PS, Caruso M, Chanez P, Dahlén SE, Fleming LJ, Fowler SJ, Horvath I, Krug N, Montuschi P, Sanak M, Sandstrom T, Shaw DE, Singer F, Sterk PJ, Roberts G, Adcock IM, Djukanovic R, Auffray C, Chung KF; U-BIOPRED Study Group . U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol 139: 1797–1807, 2017. doi: 10.1016/j.jaci.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 167.Leimgruber A. [Widal triad (Asthma-Nasal polyposis-aspirin intolerance): an inflammatory metabolism abnormality]. Rev Med Suisse 1: 15–18, 2005. [PubMed] [Google Scholar]
- 168.Lex C, Payne DN, Zacharasiewicz A, Li AM, Wilson NM, Hansel TT, Bush A. Sputum induction in children with difficult asthma: safety, feasibility, and inflammatory cell pattern. Pediatr Pulmonol 39: 318–324, 2005. doi: 10.1002/ppul.20159. [DOI] [PubMed] [Google Scholar]
- 169.Li E, Landers CT, Tung HY, Knight JM, Marshall Z, Luong AU, Rodriguez A, Kheradmand F, Corry DB. Fungi in Mucoobstructive Airway Diseases. Ann Am Thorac Soc 15, Suppl 3: S198–S204, 2018. doi: 10.1513/AnnalsATS.201803-154AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li JJ, Wang W, Baines KJ, Bowden NA, Hansbro PM, Gibson PG, Kumar RK, Foster PS, Yang M. IL-27/IFN-γ induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J Immunol 185: 4401–4409, 2010. doi: 10.4049/jimmunol.1001039. [DOI] [PubMed] [Google Scholar]
- 171.Li Y, Wu FX, Ngom A. A review on machine learning principles for multi-view biological data integration. Brief Bioinform 19: 325–340, 2018. doi: 10.1093/bib/bbw113. [DOI] [PubMed] [Google Scholar]
- 172.Li Z, Zeng M, Deng Y, Zhao J, Zhou X, Trudeau JB, Goldschmidt E, Moore JA, Chu H, Zhang W, Yin S, Liu Z, Di YP, Lee SE, Wenzel SE. 15-Lipoxygenase 1 in nasal polyps promotes CCL26/eotaxin 3 expression through extracellular signal-regulated kinase activation. J Allergy Clin Immunol 144: 1228–1241.e9, 2019. doi: 10.1016/j.jaci.2019.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Linden M, Svensson C, Andersson M, Greiff L, Andersson E, Denburg JA, Persson CG. Circulating eosinophil/basophil progenitors and nasal mucosal cytokines in seasonal allergic rhinitis. Allergy 54: 212–219, 1999. doi: 10.1034/j.1398-9995.1999.00756.x. [DOI] [PubMed] [Google Scholar]
- 174.Lötvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, Lemanske RF Jr, Wardlaw AJ, Wenzel SE, Greenberger PA. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 127: 355–360, 2011. doi: 10.1016/j.jaci.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 175.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Loza MJ, Djukanovic R, Chung KF, Horowitz D, Ma K, Branigan P, Barnathan ES, Susulic VS, Silkoff PE, Sterk PJ, Baribaud F; ADEPT (Airways Disease Endotyping for Personalized Therapeutics) and U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome Consortium) Investigators . Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res 17: 165, 2016. doi: 10.1186/s12931-016-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mair F, Prlic M. OMIP-044: 28-color immunophenotyping of the human dendritic cell compartment. [Correction in Cytometry A 95: 925–926, 2019.] Cytometry A 93: 402–405, 2018. doi: 10.1002/cyto.a.23331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Marguet C, Bocquel N, Benichou J, Basuyau JP, Hellot MF, Couderc L, Mallet E, Macé B. Neutrophil but not eosinophil inflammation is related to the severity of a first acute epidemic bronchiolitis in young infants. Pediatr Allergy Immunol 19: 157–165, 2008. doi: 10.1111/j.1399-3038.2007.00600.x. [DOI] [PubMed] [Google Scholar]
- 180.Marguet C, Jouen-Boedes F, Dean TP, Warner JO. Bronchoalveolar cell profiles in children with asthma, infantile wheeze, chronic cough, or cystic fibrosis. Am J Respir Crit Care Med 159: 1533–1540, 1999. doi: 10.1164/ajrccm.159.5.9805028. [DOI] [PubMed] [Google Scholar]
- 181.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110, 2006. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181a.McGrath KE, Bushnell TP, Palis J. Multispectral imaging of hematopoietic cells: where flow meets morphology. J Immunol Methods 336: 91–97, 2008. doi: 10.1016/j.jim.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McLachlan GJ, Bean RW, Peel D. A mixture model-based approach to the clustering of microarray expression data. Bioinformatics 18: 413–422, 2002. doi: 10.1093/bioinformatics/18.3.413. [DOI] [PubMed] [Google Scholar]
- 182a.Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, Oettgen HC. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci USA 94: 1344–1349, 1997. doi: 10.1073/pnas.94.4.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Menzies-Gow A, Flood-Page P, Sehmi R, Burman J, Hamid Q, Robinson DS, Kay AB, Denburg J. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J Allergy Clin Immunol 111: 714–719, 2003. doi: 10.1067/mai.2003.1382. [DOI] [PubMed] [Google Scholar]
- 184.Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol 113: 101–108, 2004. doi: 10.1016/j.jaci.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 185.Modena BD, Bleecker ER, Busse WW, Erzurum SC, Gaston BM, Jarjour NN, Meyers DA, Milosevic J, Tedrow JR, Wu W, Kaminski N, Wenzel SE. Gene Expression Correlated with Severe Asthma Characteristics Reveals Heterogeneous Mechanisms of Severe Disease. Am J Respir Crit Care Med 195: 1449–1463, 2017. doi: 10.1164/rccm.201607-1407OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Modena BD, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Wu W, Bar-Joseph Z, Erzurum SC, Gaston BM, Busse WW, Jarjour NN, Kaminski N, Wenzel SE. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am J Respir Crit Care Med 190: 1363–1372, 2014. doi: 10.1164/rccm.201406-1099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Modena BD, Wenzel SE. Consistency of T2 Gene Signatures in Severe Asthma. Key to Effective Treatments or Merely the Tip of the Iceberg? Am J Respir Crit Care Med 195: 411–412, 2017. doi: 10.1164/rccm.201609-1854ED. [DOI] [PubMed] [Google Scholar]
- 188.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448: 470–473, 2007. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 189.Moore WC. The natural history of asthma phenotypes identified by cluster analysis. Looking for chutes and ladders. Am J Respir Crit Care Med 188: 521–522, 2013. doi: 10.1164/rccm.201307-1203ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, Calhoun WJ, Castro M, Chung KF, Clark MP, Dweik RA, Fitzpatrick AM, Gaston B, Hew M, Hussain I, Jarjour NN, Israel E, Levy BD, Murphy JR, Peters SP, Teague WG, Meyers DA, Busse WW, Wenzel SE; National Heart, Lung, Blood Institute’s Severe Asthma Research Program . Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol 119: 405–413, 2007. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, Wenzel SE, Peters SP, Meyers DA, Bleecker ER; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program . Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol 133: 1557–1563.e5, 2014. doi: 10.1016/j.jaci.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136: 2348–2357, 1986. [PubMed] [Google Scholar]
- 193.Mukherjee M, Bulir DC, Radford K, Kjarsgaard M, Huang CM, Jacobsen EA, Ochkur SI, Catuneanu A, Lamothe-Kipnes H, Mahony J, Lee JJ, Lacy P, Nair PK. Sputum autoantibodies in patients with severe eosinophilic asthma. J Allergy Clin Immunol 141: 1269–1279, 2018. doi: 10.1016/j.jaci.2017.06.033. [DOI] [PubMed] [Google Scholar]
- 194.Murdock BJ, Shreiner AB, McDonald RA, Osterholzer JJ, White ES, Toews GB, Huffnagle GB. Coevolution of TH1, TH2, and TH17 responses during repeated pulmonary exposure to Aspergillus fumigatus conidia. Infect Immun 79: 125–135, 2011. doi: 10.1128/IAI.00508-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Murtagh F, Contreras P. Algorithms for hierarchical clustering: an overview. Wiley Interdiscip Rev Data Min Knowl Discov 2: 86–97, 2012. doi: 10.1002/widm.53. [DOI] [Google Scholar]
- 196.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D1: D336–D342, 2016. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nagasaki T, Matsumoto H, Kanemitsu Y, Izuhara K, Tohda Y, Kita H, Horiguchi T, Kuwabara K, Tomii K, Otsuka K, Fujimura M, Ohkura N, Tomita K, Yokoyama A, Ohnishi H, Nakano Y, Oguma T, Hozawa S, Ito I, Oguma T, Inoue H, Tajiri T, Iwata T, Izuhara Y, Ono J, Ohta S, Yokoyama T, Niimi A, Mishima M. Integrating longitudinal information on pulmonary function and inflammation using asthma phenotypes. J Allergy Clin Immunol 133: 1474–1477.e2, 2014. doi: 10.1016/j.jaci.2013.12.1084. [DOI] [PubMed] [Google Scholar]
- 198.Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, Stryszak P, Gann L, Sadeh J, Chanez P; Study Investigators . Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy 42: 1097–1103, 2012. doi: 10.1111/j.1365-2222.2012.04014.x. [DOI] [PubMed] [Google Scholar]
- 199.Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O’Byrne PM. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med 360: 985–993, 2009. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 200.Nair P, Wenzel S, Rabe KF, Bourdin A, Lugogo NL, Kuna P, Barker P, Sproule S, Ponnarambil S, Goldman M; ZONDA Trial Investigators . Oral Glucocorticoid-Sparing Effect of Benralizumab in Severe Asthma. N Engl J Med 376: 2448–2458, 2017. doi: 10.1056/NEJMoa1703501. [DOI] [PubMed] [Google Scholar]
- 201.Nakamura Y, Ghaffar O, Olivenstein R, Taha RA, Soussi-Gounni A, Zhang DH, Ray A, Hamid Q. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol 103: 215–222, 1999. doi: 10.1016/S0091-6749(99)70493-8. [DOI] [PubMed] [Google Scholar]
- 202.Narala VR, Ranga R, Smith MR, Berlin AA, Standiford TJ, Lukacs NW, Reddy RC. Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma. Respir Res 8: 90, 2007. doi: 10.1186/1465-9921-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Newby C, Heaney LG, Menzies-Gow A, Niven RM, Mansur A, Bucknall C, Chaudhuri R, Thompson J, Burton P, Brightling C; British Thoracic Society Severe Refractory Asthma Network . Statistical cluster analysis of the British Thoracic Society Severe Refractory Asthma Registry: clinical outcomes and phenotype stability. PLoS One 9: e102987, 2014. doi: 10.1371/journal.pone.0102987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ntontsi P, Loukides S, Bakakos P, Kostikas K, Papatheodorou G, Papathanassiou E, Hillas G, Koulouris N, Papiris S, Papaioannou AI. Clinical, functional and inflammatory characteristics in patients with paucigranulocytic stable asthma: comparison with different sputum phenotypes. Allergy 72: 1761–1767, 2017. doi: 10.1111/all.13184. [DOI] [PubMed] [Google Scholar]
- 205.O’Brien CE, Tsirilakis K, Santiago MT, Goldman DL, Vicencio AG. Heterogeneity of lower airway inflammation in children with severe-persistent asthma. Pediatr Pulmonol 50: 1200–1204, 2015. doi: 10.1002/ppul.23165. [DOI] [PubMed] [Google Scholar]
- 206.O’Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, Larsson B, Cullberg M, Nair P. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 4: 797–806, 2016. doi: 10.1016/S2213-2600(16)30227-2. [DOI] [PubMed] [Google Scholar]
- 207.O’Reilly R, Ullmann N, Irving S, Bossley CJ, Sonnappa S, Zhu J, Oates T, Banya W, Jeffery PK, Bush A, Saglani S. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J Allergy Clin Immunol 131: 1024–1032.e16, 2013. doi: 10.1016/j.jaci.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 208.Ober C. Asthma Genetics in the Post-GWAS Era. Ann Am Thorac Soc 13, Suppl 1: S85–S90, 2016. doi: 10.1513/AnnalsATS.201507-459MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, Wadsworth MH II, Hughes TK, Kazer SW, Yoshimoto E, Cahill KN, Bhattacharyya N, Katz HR, Berger B, Laidlaw TM, Boyce JA, Barrett NA, Shalek AK. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560: 649–654, 2018. doi: 10.1038/s41586-018-0449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Oriss TB, Raundhal M, Morse C, Huff RE, Das S, Hannum R, Gauthier MC, Scholl KL, Chakraborty K, Nouraie SM, Wenzel SE, Ray P, Ray A. IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI Insight 2: e91019, 2017. doi: 10.1172/jci.insight.91019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Page K, Lierl KM, Hughes VS, Zhou P, Ledford JR, Wills-Karp M. TLR2-mediated activation of neutrophils in response to German cockroach frass. J Immunol 180: 6317–6324, 2008. doi: 10.4049/jimmunol.180.9.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Park YH, Fitzpatrick AM, Medriano CA, Jones DP. High-resolution metabolomics to identify urine biomarkers in corticosteroid-resistant asthmatic children. J Allergy Clin Immunol 139: 1518–1524.e4, 2017. doi: 10.1016/j.jaci.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 213.Pavord ID, Beasley R, Agusti A, Anderson GP, Bel E, Brusselle G, Cullinan P, Custovic A, Ducharme FM, Fahy JV, Frey U, Gibson P, Heaney LG, Holt PG, Humbert M, Lloyd CM, Marks G, Martinez FD, Sly PD, von Mutius E, Wenzel S, Zar HJ, Bush A. After asthma: redefining airways diseases. Lancet 391: 350–400, 2018. doi: 10.1016/S0140-6736(17)30879-6. [DOI] [PubMed] [Google Scholar]
- 214.Payne DN, Adcock IM, Wilson NM, Oates T, Scallan M, Bush A. Relationship between exhaled nitric oxide and mucosal eosinophilic inflammation in children with difficult asthma, after treatment with oral prednisolone. Am J Respir Crit Care Med 164: 1376–1381, 2001. doi: 10.1164/ajrccm.164.8.2101145. [DOI] [PubMed] [Google Scholar]
- 215.Payne DN, Qiu Y, Zhu J, Peachey L, Scallan M, Bush A, Jeffery PK. Airway inflammation in children with difficult asthma: relationships with airflow limitation and persistent symptoms. Thorax 59: 862–869, 2004. doi: 10.1136/thx.2003.017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Peters MC, Kerr S, Dunican EM, Woodruff PG, Fajt ML, Levy BD, Israel E, Phillips BR, Mauger DT, Comhair SA, Erzurum SC, Johansson MW, Jarjour NN, Coverstone AM, Castro M, Hastie AT, Bleecker ER, Wenzel SE, Fahy JV; National Heart, Lung, and Blood Institute Severe Asthma Research Program 3 . Refractory airway type 2 inflammation in a large subgroup of asthmatic patients treated with inhaled corticosteroids. J Allergy Clin Immunol 143: 104–113.e14, 2019. doi: 10.1016/j.jaci.2017.12.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, Phillips BR, Mauger DT, Comhair SA, Erzurum SC, Johansson MW, Jarjour NN, Coverstone AM, Castro M, Holguin F, Wenzel SE, Woodruff PG, Bleecker ER, Fahy JV; National Heart, Lung, and Blood Institute Severe Asthma Research Program . Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med 4: 574–584, 2016. doi: 10.1016/S2213-2600(16)30048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol 133: 388–394.e5, 2014. doi: 10.1016/j.jaci.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Peters U, Subramanian M, Chapman DG, Kaminsky DA, Irvin CG, Wise RA, Skloot GS, Bates JHT, Dixon AE. BMI but not central obesity predisposes to airway closure during bronchoconstriction. Respirology 24: 543–550, 2019. doi: 10.1111/resp.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Phipatanakul W, Mauger DT, Sorkness RL, Gaffin JM, Holguin F, Woodruff PG, Ly NP, Bacharier LB, Bhakta NR, Moore WC, Bleecker ER, Hastie AT, Meyers DA, Castro M, Fahy JV, Fitzpatrick AM, Gaston BM, Jarjour NN, Levy BD, Peters SP, Teague WG, Fajt M, Wenzel SE, Erzurum SC, Israel E; Severe Asthma Research Program . Effects of Age and Disease Severity on Systemic Corticosteroid Responses in Asthma. Am J Respir Crit Care Med 195: 1439–1448, 2017. doi: 10.1164/rccm.201607-1453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Pichavant M, Charbonnier AS, Taront S, Brichet A, Wallaert B, Pestel J, Tonnel AB, Gosset P. Asthmatic bronchial epithelium activated by the proteolytic allergen Der p 1 increases selective dendritic cell recruitment. J Allergy Clin Immunol 115: 771–778, 2005. doi: 10.1016/j.jaci.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 223.Poole A, Urbanek C, Eng C, Schageman J, Jacobson S, O’Connor BP, Galanter JM, Gignoux CR, Roth LA, Kumar R, Lutz S, Liu AH, Fingerlin TE, Setterquist RA, Burchard EG, Rodriguez-Santana J, Seibold MA. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol 133: 670–8.e12, 2014. doi: 10.1016/j.jaci.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Price DB, Rigazio A, Campbell JD, Bleecker ER, Corrigan CJ, Thomas M, Wenzel SE, Wilson AM, Small MB, Gopalan G, Ashton VL, Burden A, Hillyer EV, Kerkhof M, Pavord ID. Blood eosinophil count and prospective annual asthma disease burden: a UK cohort study. Lancet Respir Med 3: 849–858, 2015. doi: 10.1016/S2213-2600(15)00367-7. [DOI] [PubMed] [Google Scholar]
- 225.Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, Zhu H, Hamilton JD, Swanson BN, Khan A, Chao J, Staudinger H, Pirozzi G, Antoni C, Amin N, Ruddy M, Akinlade B, Graham NMH, Stahl N, Yancopoulos GD, Teper A. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N Engl J Med 378: 2475–2485, 2018. doi: 10.1056/NEJMoa1804093. [DOI] [PubMed] [Google Scholar]
- 226.Rackemann FM. A working classification of asthma. Am J Med 3: 601–606, 1947. doi: 10.1016/0002-9343(47)90204-0. [DOI] [PubMed] [Google Scholar]
- 227.Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, Huff R, Pilewski J, Holguin F, Kolls J, Wenzel S, Ray P, Ray A. High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest 125: 3037–3050, 2015. doi: 10.1172/JCI80911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ray A, Kolls JK. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol 38: 942–954, 2017. doi: 10.1016/j.it.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Robinson D, Hamid Q, Bentley A, Ying S, Kay AB, Durham SR. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J Allergy Clin Immunol 92: 313–324, 1993. doi: 10.1016/0091-6749(93)90175-F. [DOI] [PubMed] [Google Scholar]
- 230.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med 326: 298–304, 1992. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 231.Rossios C, Pavlidis S, Hoda U, Kuo CH, Wiegman C, Russell K, Sun K, Loza MJ, Baribaud F, Durham AL, Ojo O, Lutter R, Rowe A, Bansal A, Auffray C, Sousa A, Corfield J, Djukanovic R, Guo Y, Sterk PJ, Chung KF, Adcock IM; Unbiased Biomarkers for the Prediction of Respiratory Diseases Outcomes (U-BIOPRED) Consortia Project Team . Sputum transcriptomics reveal upregulation of IL-1 receptor family members in patients with severe asthma. J Allergy Clin Immunol 141: 560–570, 2018. doi: 10.1016/j.jaci.2017.02.045. [DOI] [PubMed] [Google Scholar]
- 232.Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, Peterson LW, Wherry EJ, Goldrath AW, Bhandoola A, Artis D. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med 210: 1823–1837, 2013. doi: 10.1084/jem.20122332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, Jeffery PK. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med 176: 858–864, 2007. doi: 10.1164/rccm.200702-212OC. [DOI] [PubMed] [Google Scholar]
- 234.Samter M, Beers RF Jr. Concerning the nature of intolerance to aspirin. J Allergy 40: 281–293, 1967. doi: 10.1016/0021-8707(67)90076-7. [DOI] [PubMed] [Google Scholar]
- 235.Sandall J, Tribe RM, Avery L, Mola G, Visser GH, Homer CS, Gibbons D, Kelly NM, Kennedy HP, Kidanto H, Taylor P, Temmerman M. Short-term and long-term effects of caesarean section on the health of women and children. Lancet 392: 1349–1357, 2018. doi: 10.1016/S0140-6736(18)31930-5. [DOI] [PubMed] [Google Scholar]
- 236.Schatz M, Hsu JW, Zeiger RS, Chen W, Dorenbaum A, Chipps BE, Haselkorn T. Phenotypes determined by cluster analysis in severe or difficult-to-treat asthma. J Allergy Clin Immunol 133: 1549–1556, 2014. doi: 10.1016/j.jaci.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 237.Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, Cowan JO, Herbison GP, Silva PA, Poulton R. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med 349: 1414–1422, 2003. doi: 10.1056/NEJMoa022363. [DOI] [PubMed] [Google Scholar]
- 238.Sergejeva S, Malmhäll C, Lötvall J, Pullerits T. Increased number of CD34+ cells in nasal mucosa of allergic rhinitis patients: inhibition by a local corticosteroid. Clin Exp Allergy 35: 34–38, 2005. doi: 10.1111/j.1365-2222.2004.02038.x. [DOI] [PubMed] [Google Scholar]
- 239.Sharma A, Laxman B, Naureckas ET, Hogarth DK, Sperling AI, Solway J, Ober C, Gilbert JA, White SR. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol 144: 1214–1227.e7, 2019. doi: 10.1016/j.jaci.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol 280: L134–L140, 2001. doi: 10.1152/ajplung.2001.280.1.L134. [DOI] [PubMed] [Google Scholar]
- 241.Shore SA, Cho Y. Obesity and Asthma: Microbiome-Metabolome Interactions. Am J Respir Cell Mol Biol 54: 609–617, 2016. doi: 10.1165/rcmb.2016-0052PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Siew LQC, Wu SY, Ying S, Corrigan CJ. Cigarette smoking increases bronchial mucosal IL-17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clin Exp Allergy 47: 740–750, 2017. doi: 10.1111/cea.12907. [DOI] [PubMed] [Google Scholar]
- 243.Singhania A, Rupani H, Jayasekera N, Lumb S, Hales P, Gozzard N, Davies DE, Woelk CH, Howarth PH. Altered Epithelial Gene Expression in Peripheral Airways of Severe Asthma. PLoS One 12: e0168680, 2017. doi: 10.1371/journal.pone.0168680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Siracusa MC, Saenz SA, Wojno ED, Kim BS, Osborne LC, Ziegler CG, Benitez AJ, Ruymann KR, Farber DL, Sleiman PM, Hakonarson H, Cianferoni A, Wang ML, Spergel JM, Comeau MR, Artis D. Thymic stromal lymphopoietin-mediated extramedullary hematopoiesis promotes allergic inflammation. Immunity 39: 1158–1170, 2013. doi: 10.1016/j.immuni.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Smith SG, Gugilla A, Mukherjee M, Merim K, Irshad A, Tang W, Kinoshita T, Watson B, Oliveria JP, Comeau M, O’Byrne PM, Gauvreau GM, Sehmi R. Thymic stromal lymphopoietin and IL-33 modulate migration of hematopoietic progenitor cells in patients with allergic asthma. J Allergy Clin Immunol 135: 1594–1602, 2015. doi: 10.1016/j.jaci.2014.12.1918. [DOI] [PubMed] [Google Scholar]
- 246.Smits HH, Hiemstra PS, Prazeres da Costa C, Ege M, Edwards M, Garn H, Howarth PH, Jartti T, de Jong EC, Maizels RM, Marsland BJ, McSorley HJ, Müller A, Pfefferle PI, Savelkoul H, Schwarze J, Unger WW, von Mutius E, Yazdanbakhsh M, Taube C. Microbes and asthma: Opportunities for intervention. J Allergy Clin Immunol 137: 690–697, 2016. doi: 10.1016/j.jaci.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 247.Snelgrove RJ, Gregory LG, Peiró T, Akthar S, Campbell GA, Walker SA, Lloyd CM. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol 134: 583–592.e6, 2014. doi: 10.1016/j.jaci.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sorkness RL, Teague WG, Penugonda M, Fitzpatrick AM; National Institutes of Health, National Heart, Lung, and Blood Institute’s Severe Asthma Research Program . Sex dependence of airflow limitation and air trapping in children with severe asthma. J Allergy Clin Immunol 127: 1073–1074, 2011. doi: 10.1016/j.jaci.2010.12.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Spahn JD, Cherniack R, Paull K, Gelfand EW. Is forced expiratory volume in one second the best measure of severity in childhood asthma? Am J Respir Crit Care Med 169: 784–786, 2004. doi: 10.1164/rccm.200309-1234OE. [DOI] [PubMed] [Google Scholar]
- 250.Spahn JD, Covar RA, Jain N, Gleason M, Shimamoto R, Szefler SJ, Gelfand EW. Effect of montelukast on peripheral airflow obstruction in children with asthma. Ann Allergy Asthma Immunol 96: 541–549, 2006. doi: 10.1016/S1081-1206(10)63548-X. [DOI] [PubMed] [Google Scholar]
- 251.Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 16: 133–145, 2015. doi: 10.1038/nrg3833. [DOI] [PubMed] [Google Scholar]
- 252.Stewart GA, Holt PG. Immunogenicity and tolerogenicity of a major house dust mite allergen, Der p I from Dermatophagoides pteronyssinus, in mice and rats. Int Arch Allergy Appl Immunol 83: 44–51, 1987. doi: 10.1159/000234329. [DOI] [PubMed] [Google Scholar]
- 253.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14: 865–868, 2017. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sur S, Wild JS, Choudhury BK, Sur N, Alam R, Klinman DM. Long term prevention of allergic lung inflammation in a mouse model of asthma by CpG oligodeoxynucleotides. J Immunol 162: 6284–6293, 1999. [PubMed] [Google Scholar]
- 256.Tai A, Tran H, Roberts M, Clarke N, Wilson J, Robertson CF. The association between childhood asthma and adult chronic obstructive pulmonary disease. Thorax 69: 805–810, 2014. doi: 10.1136/thoraxjnl-2013-204815. [DOI] [PubMed] [Google Scholar]
- 257.Tai A, Tran H, Roberts M, Clarke N, Wilson J, Robertson CF. Trends in eczema, rhinitis, and rye grass sensitization in a longitudinal asthma cohort. Ann Allergy Asthma Immunol 112: 437–440, 2014. doi: 10.1016/j.anai.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 258.Tan BK, Li QZ, Suh L, Kato A, Conley DB, Chandra RK, Zhou J, Norton J, Carter R, Hinchcliff M, Harris K, Peters A, Grammer LC, Kern RC, Mohan C, Schleimer RP. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 128: 1198–1206.e1, 2011. doi: 10.1016/j.jaci.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tantisira KG, Colvin R, Tonascia J, Strunk RC, Weiss ST, Fuhlbrigge AL; Childhood Asthma Management Program Research Group . Airway responsiveness in mild to moderate childhood asthma: sex influences on the natural history. Am J Respir Crit Care Med 178: 325–331, 2008. doi: 10.1164/rccm.200708-1174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Taylor SL, Leong LEX, Choo JM, Wesselingh S, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, Peters MJ, Baraket M, Marks GB, Gibson PG, Simpson JL, Rogers GB. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol 141: 94–103.e15, 2018. doi: 10.1016/j.jaci.2017.03.044. [DOI] [PubMed] [Google Scholar]
- 262.Teague WG, Phillips BR, Fahy JV, Wenzel SE, Fitzpatrick AM, Moore WC, Hastie AT, Bleecker ER, Meyers DA, Peters SP, Castro M, Coverstone AM, Bacharier LB, Ly NP, Peters MC, Denlinger LC, Ramratnam S, Sorkness RL, Gaston BM, Erzurum SC, Comhair SAA, Myers RE, Zein J, DeBoer MD, Irani AM, Israel E, Levy B, Cardet JC, Phipatanakul W, Gaffin JM, Holguin F, Fajt ML, Aujla SJ, Mauger DT, Jarjour NN. Baseline Features of the Severe Asthma Research Program (SARP III) Cohort: Differences with Age. J Allergy Clin Immunol Pract 6: 545–554.e4, 2018. doi: 10.1016/j.jaip.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc Series B Stat Methodol 63: 411–423, 2001. doi: 10.1111/1467-9868.00293. [DOI] [Google Scholar]
- 264.Tliba O, Panettieri RA Jr. Paucigranulocytic asthma: Uncoupling of airway obstruction from inflammation. J Allergy Clin Immunol 143: 1287–1294, 2019. doi: 10.1016/j.jaci.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Trautmann A, Toksoy A, Engelhardt E, Bröcker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol 190: 100–106, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 266.Urich MA, Nery JR, Lister R, Schmitz RJ, Ecker JR. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc 10: 475–483, 2015. doi: 10.1038/nprot.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Valladao AC, Frevert CW, Koch LK, Campbell DJ, Ziegler SF. STAT6 Regulates the Development of Eosinophilic versus Neutrophilic Asthma in Response to Alternaria alternata. J Immunol 197: 4541–4551, 2016. doi: 10.4049/jimmunol.1600007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Van der Laan M, Pollard K, Bryan J. A new partitioning around medoids algorithm. J Stat Comput Simul 73: 575–584, 2003. doi: 10.1080/0094965031000136012. [DOI] [Google Scholar]
- 268a.Van der Maaten L, Hinton GE. Visualizing Data using t-SNE. J Mach Learn Res 9: 2579–2605, 2008. [Google Scholar]
- 269.Voraphani N, Gladwin MT, Contreras AU, Kaminski N, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Ray A, Ray P, Erzurum SC, Busse WW, Zhao J, Trudeau JB, Wenzel SE. An airway epithelial iNOS-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. Mucosal Immunol 7: 1175–1185, 2014. doi: 10.1038/mi.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Walker C, Kaegi MK, Braun P, Blaser K. Activated T cells and eosinophilia in bronchoalveolar lavages from subjects with asthma correlated with disease severity. J Allergy Clin Immunol 88: 935–942, 1991. doi: 10.1016/0091-6749(91)90251-I. [DOI] [PubMed] [Google Scholar]
- 271.Walsh CJ, Zaihra T, Benedetti A, Fugère C, Olivenstein R, Lemière C, Hamid Q, Martin JG. Exacerbation risk in severe asthma is stratified by inflammatory phenotype using longitudinal measures of sputum eosinophils. Clin Exp Allergy 46: 1291–1302, 2016. doi: 10.1111/cea.12762. [DOI] [PubMed] [Google Scholar]
- 272.Ward JH., Jr Hierarchical Grouping to Optimize an Objective Function. J Am Stat Assoc 58: 236–244, 1963. doi: 10.1080/01621459.1963.10500845. [DOI] [Google Scholar]
- 273.Weathington N, O’Brien ME, Radder J, Whisenant TC, Bleecker ER, Busse WW, Erzurum SC, Gaston B, Hastie AT, Jarjour NN, Meyers DA, Milosevic J, Moore WC, Tedrow JR, Trudeau JB, Wong HP, Wu W, Kaminski N, Wenzel SE, Modena BD. BAL Cell Gene Expression in Severe Asthma Reveals Mechanisms of Severe Disease and Influences of Medications. Am J Respir Crit Care Med 200: 837–856, 2019. doi: 10.1164/rccm.201811-2221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Weller K, Foitzik K, Paus R, Syska W, Maurer M. Mast cells are required for normal healing of skin wounds in mice. FASEB J 20: 2366–2368, 2006. doi: 10.1096/fj.06-5837fje. [DOI] [PubMed] [Google Scholar]
- 276.Wenzel S, Castro M, Corren J, Maspero J, Wang L, Zhang B, Pirozzi G, Sutherland ER, Evans RR, Joish VN, Eckert L, Graham NM, Stahl N, Yancopoulos GD, Louis-Tisserand M, Teper A. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 388: 31–44, 2016. doi: 10.1016/S0140-6736(16)30307-5. [DOI] [PubMed] [Google Scholar]
- 277.Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, Wang L, Kirkesseli S, Rocklin R, Bock B, Hamilton J, Ming JE, Radin A, Stahl N, Yancopoulos GD, Graham N, Pirozzi G. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med 368: 2455–2466, 2013. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 278.Wenzel S, Holgate ST. The mouse trap: it still yields few answers in asthma. Am J Respir Crit Care Med 174: 1173–1176, 2006. doi: 10.1164/rccm.2609002. [DOI] [PubMed] [Google Scholar]
- 279.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet 370: 1422–1431, 2007. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 280.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716–725, 2012. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 281.Wenzel SE, Fowler AA III, Schwartz LB. Activation of pulmonary mast cells by bronchoalveolar allergen challenge. In vivo release of histamine and tryptase in atopic subjects with and without asthma. Am Rev Respir Dis 137: 1002–1008, 1988. doi: 10.1164/ajrccm/137.5.1002. [DOI] [PubMed] [Google Scholar]
- 282.Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, Chu HW. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 160: 1001–1008, 1999. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 283.Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med 156: 737–743, 1997. doi: 10.1164/ajrccm.156.3.9610046. [DOI] [PubMed] [Google Scholar]
- 284.Wenzel SE, Vitari CA, Shende M, Strollo DC, Larkin A, Yousem SA. Asthmatic granulomatosis: a novel disease with asthmatic and granulomatous features. Am J Respir Crit Care Med 186: 501–507, 2012. doi: 10.1164/rccm.201203-0476OC. [DOI] [PubMed] [Google Scholar]
- 285.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261, 1998. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 286.Winnica D, Que LG, Baffi C, Grasemann H, Fiedler K, Yang Z, Etling E, Wasil K, Wenzel SE, Freeman B, Holguin F. l-Citrulline prevents asymmetric dimethylarginine-mediated reductions in nitric oxide and nitrosative stress in primary human airway epithelial cells. Clin Exp Allergy 47: 190–199, 2017. doi: 10.1111/cea.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA 104: 15858–15863, 2007. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Arron JR, Koth LL, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180: 388–395, 2009. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wu W, Bang S, Bleecker ER, Castro M, Denlinger L, Erzurum SC, Fahy JV, Fitzpatrick AM, Gaston BM, Hastie AT, Israel E, Jarjour NN, Levy BD, Mauger DT, Meyers DA, Moore WC, Peters M, Phillips BR, Phipatanakul W, Sorkness RL, Wenzel SE. Multiview Cluster Analysis Identifies Variable Corticosteroid Response Phenotypes in Severe Asthma. Am J Respir Crit Care Med 199: 1358–1367, 2019. doi: 10.1164/rccm.201808-1543OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wu W, Bleecker E, Moore W, Busse WW, Castro M, Chung KF, Calhoun WJ, Erzurum S, Gaston B, Israel E, Curran-Everett D, Wenzel SE. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J Allergy Clin Immunol 133: 1280–1288, 2014. doi: 10.1016/j.jaci.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wysocki K, Park SY, Bleecker E, Busse W, Castro M, Chung KF, Gaston B, Erzurum S, Israel E, Teague WG, Moore CG, Wenzel S. Characterization of factors associated with systemic corticosteroid use in severe asthma: data from the Severe Asthma Research Program. J Allergy Clin Immunol 133: 915–918, 2014. doi: 10.1016/j.jaci.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Xie X, Rigor P, Baldi P. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics 25: 167–174, 2009. doi: 10.1093/bioinformatics/btn605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yan X, Chu JH, Gomez J, Koenigs M, Holm C, He X, Perez MF, Zhao H, Mane S, Martinez FD, Ober C, Nicolae DL, Barnes KC, London SJ, Gilliland F, Weiss ST, Raby BA, Cohn L, Chupp GL. Noninvasive analysis of the sputum transcriptome discriminates clinical phenotypes of asthma. Am J Respir Crit Care Med 191: 1116–1125, 2015. doi: 10.1164/rccm.201408-1440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Yeh YL, Su MW, Chiang BL, Yang YH, Tsai CH, Lee YL. Genetic profiles of transcriptomic clusters of childhood asthma determine specific severe subtype. Clin Exp Allergy 48: 1164–1172, 2018. doi: 10.1111/cea.13175. [DOI] [PubMed] [Google Scholar]
- 295.Ying S, Durham SR, Corrigan CJ, Hamid Q, Kay AB. Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am J Respir Cell Mol Biol 12: 477–487, 1995. doi: 10.1165/ajrcmb.12.5.7742012. [DOI] [PubMed] [Google Scholar]
- 296.Yoshimoto T, Okamura H, Tagawa YI, Iwakura Y, Nakanishi K. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-gamma production from activated B cells. Proc Natl Acad Sci USA 94: 3948–3953, 1997. doi: 10.1073/pnas.94.8.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest 116: 1633–1641, 2006. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Zacharasiewicz A, Wilson N, Lex C, Erin EM, Li AM, Hansel T, Khan M, Bush A. Clinical use of noninvasive measurements of airway inflammation in steroid reduction in children. Am J Respir Crit Care Med 171: 1077–1082, 2005. doi: 10.1164/rccm.200409-1242OC. [DOI] [PubMed] [Google Scholar]
- 298a.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P. Ensembl 2018. Nucleic Acids Res 46, D1: D754–D761, 2018. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem 272: 21597–21603, 1997. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 300.Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray P, Ray A. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity 11: 473–482, 1999. doi: 10.1016/S1074-7613(00)80122-3. [DOI] [PubMed] [Google Scholar]
- 301.Zhang Q, Cox M, Liang Z, Brinkmann F, Cardenas PA, Duff R, Bhavsar P, Cookson W, Moffatt M, Chung KF. Airway Microbiota in Severe Asthma and Relationship to Asthma Severity and Phenotypes. PLoS One 11: e0152724, 2016. doi: 10.1371/journal.pone.0152724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89: 587–596, 1997. doi: 10.1016/S0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 303.Zhou X, Kinlough CL, Hughey RP, Jin M, Inoue H, Etling E, Modena BD, Kaminski N, Bleecker ER, Meyers DA, Jarjour NN, Trudeau JB, Holguin F, Ray A, Wenzel SE. Sialylation of MUC4β N-glycans by ST6GAL1 orchestrates human airway epithelial cell differentiation associated with type-2 inflammation. JCI Insight 4: e122475, 2019. doi: 10.1172/jci.insight.122475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhu J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine 75: 14–24, 2015. doi: 10.1016/j.cyto.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Zoratti EM, Krouse RZ, Babineau DC, Pongracic JA, O’Connor GT, Wood RA, Khurana Hershey GK, Kercsmar CM, Gruchalla RS, Kattan M, Teach SJ, Sigelman SM, Gergen PJ, Togias A, Visness CM, Busse WW, Liu AH. Asthma phenotypes in inner-city children. J Allergy Clin Immunol 138: 1016–1029, 2016. doi: 10.1016/j.jaci.2016.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]