

Abstract
Bacterial biofilms contain subpopulations of cells that are dormant and highly tolerant to antibiotics. While dormant, the bacteria must maintain the integrity of macromolecules required for resuscitation. Previously, we showed that hibernation promoting factor (HPF) is essential for protecting Pseudomonas aeruginosa from ribosomal loss during dormancy. In this study, we mapped the genetic components required for hpf expression. Using 5ʹ-RACE and fluorescent protein reporter fusions, we show that hpf is expressed as part of the rpoN operon, but that hpf also has a second promoter (Phpf) within the rpoN gene. Phpf is active when the cells enter stationary phase, and expression from Phpf is modulated, but not eliminated, in mutant strains impaired in stationary phase transition (ΔdksA2, ΔrpoS and ΔrelA/ΔspoT mutants). The results of reporter gene studies and mRNA folding predictions indicated that the 5ʹ end of the hpf mRNA may also influence hpf expression. Mutations that opened or that stabilized the mRNA hairpin loop structures strongly influenced the amount of HPF produced. The results demonstrate that hpf is expressed independently of rpoN, and that hpf regulation includes both transcriptional and post-transcriptional processes, allowing the cells to produce sufficient HPF during stationary phase to maintain viability while dormant.
1. Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that causes chronic biofilm-associated infections (Costerton et al., 1999; Lyczak et al., 2002). P. aeruginosa causes pulmonary infections in patients with cystic fibrosis, and reduces wound healing in diabetic ulcers (Stewart et al., 2015; Malone et al., 2017). Biofilm-associated infections are often highly tolerant to antibiotic treatments (Stewart and Costerton, 2001). One mechanism for the enhanced resistance of biofilms to antibiotics is that biofilms harbor physiologically heterogeneous subpopulations of cells (Haagensen et al., 2007; Stewart and Franklin, 2008; Williamson et al., 2012). While some cells may be sensitive to antibiotics, subpopulations of the bacteria may be highly tolerant to the treatments. In particular, when bacteria enter a dormant state due to oxygen or nutrient limitation, the dormant subpopulation of cells is able to resist antibiotics that target active metabolism or growth (Stewart et al., 2015; Williamson et al., 2012). This subpopulation of cells is able to survive treatment, then repopulate the biofilm when conditions become favorable, ultimately resulting in persistent infections (Lewis, 2007).
In most environments, heterotrophic bacteria experience conditions that vary from nutrient abundance to nutrient starvation. Both of these conditions may occur at different regions within biofilms, since nutrient availability is limited by diffusion into the biofilm, and utilization of the nutrients by the bacteria (Stewart and Franklin, 2008). Cells may become nutrient-starved in certain regions of the biofilms, with the starved cells entering a low-metabolic or dormant state (Stewart et al., 2015). In order for the bacteria to resuscitate from starvation-induced dormancy, the cells must maintain the integrity of certain macromolecules, including ribosomes, so that they may perform anabolic processes, including de novo protein synthesis, required for resuscitation (Akiyama et al., 2017). Our prior transcriptomics analyses of P. aeruginosa biofilm subpopulations showed high abundance of mRNA encoding the HPF, in the dormant antibiotic-tolerant subpopulation of cells (Williamson et al., 2012). A deletion of the hpf gene resulted in impaired recovery of starvation-induced dormant cells, as well as loss of ribosomal RNAs in the starved cells (Akiyama et al., 2017). Those results demonstrated that HPF is required for ribosome protection in the dormant subpopulation of cells.
HPF structure and ribosome hibernation have been well characterized in E. coli and several other species of bacteria (Maki et al., 2000; Ueta et al., 2005; Sato et al., 2009; Kato et al., 2010; Kline et al., 2015; Beckert et al., 2017; Khusainov et al., 2017). In E. coli, ribosome hibernation requires the small ribosome binding proteins, hibernation factor (HPF), or its paralog, YfiA, and ribosome modulation factor (RMF) (Maki et al., 2000; Ueta et al., 2005;Yoshida et al., 2009). The model for ribosome hibernation involves binding of HPF and RMF to the sites where mRNA and tRNA bind at the mRNA exit channel (Polikanov et al., 2012). Binding of these factors causes formation of inactive 70S ribosomes, or inactive100S dimers (Vila-Sanjurjo et al., 2004; Kato et al., 2010), with YfiA competing with HPF, and forming an inactive 70S ribosome (Ueta et al., 2005). In P. aeruginosa PAO1, RMF does not appear to be required for recovery from starvation or for ribosome integrity during starvation (Akiyama et al., 2017). P. aeruginosa PAO1 does not encode a YfiA homolog. Therefore, HPF is the crucial ribosome preservation factor in P. aeruginosa (Akiyama et al., 2017). Unlike RMF, HPF homologs are widely distributed in bacteria (Ueta et al., 2008), suggesting that HPF may be important for most bacteria that live in environments where nutrient availability fluctuates. Sequence homologs for HPF and RMF have yet to be discovered in Archaea.
While HPF structure and activity have been characterized for several species of bacteria (Maki et al., 2000; Ueta et al., 2005; Sato et al., 2009; Kato et al., 2010; Kline et al., 2015; Beckert et al., 2017; Khusainov et al., 2017), little is known about the regulatory mechanisms that control hpf expression. Based on its function, hpf is likely expressed as the bacteria begin to enter a starvation state. The hpf gene is located downstream of rpoN and was originally recognized as part of the rpoN operon (Jones et al., 1994). However, recent transcriptomic analysis of P. aeruginosa growing planktonically and in biofilms suggested that hpf may be expressed independently of rpoN (Dotsch et al., 2012; Gill et al., 2018). Our previous microarray data show that under biofilm conditions, hpf is expressed at much higher levels than rpoN or than the downstream genes of the rpoN operon (Williamson et al., 2012). Recent RNA-Seq-based transcriptomics studies of P. aeruginosa indicate that hpf may have three transcriptional start sites (TSSs) (Dotsch et al., 2012; Gill et al., 2018). Gill et al. (2018), using RNA-Seq which could differentiate naïve mRNA from processed RNA, based on the presence of 5ʹ triphosphate group of the mRNA, identified the presence of two naïve transcriptional start sites (TSSs) upstream of the hpf gene. Collectively, the transcriptomics results suggest that hpf may be transcribed as part of the rpoN operon, but that hpf may also contain its own promoter and possibly be expressed as a monocistronic mRNA. This promoter arrangement may allow independent expression of hpf even under conditions where rpoN expression is not induced.
In this study, we identified and characterized transcriptional factors and post-transcriptional regulatory sequences that are involved in the regulation of HPF production. To study transcriptional regulation, we used a Tn7 transposon-based single-copy fluorescent reporter system where the hpf gene was fused in-frame to the gene for the yellow fluorescent protein (yfp) (Choi et al., 2005). From the results, we demonstrate that hpf is expressed from its own promoter, in addition to being expressed at low levels by transcriptional read-through from PrpoN. We tested the roles of stress and stringent response factors on the regulation of hpf expression, and found that stationary phase factors, RpoS and DksA2, play minor roles in expression from Phpf. However, deletions of rpoS or dksA2 do not eliminate hpf expression. The stringent response factor, ppGpp, also plays a small role in regulation of hpf expression. We identified hairpin loop structures (HPL) that overlapped the 5ʹ untranslated region (UTR) and the 5ʹ end of the coding region of the hpf mRNA. The 5ʹ UTR plays a major role in regulation of hpf expression. Collectively, the results indicate that hpf is regulated at both the transcriptional and post-transcriptional levels, and that this complex regulatory control plays a role in ribosome hibernation and maintenance of cell viability during dormancy.
2. Experimental Methods
2.1. Bacterial strains and growth conditions
Pseudomonas aeruginosa strain PAO1 and its Δhpf mutant strain (Williamson et al., 2012) were used in this study. Transposon insertion mutant strains of rpoS and dksA2 were obtained from the Pseudomonas aeruginosa PAO1 transposon mutant library (Held et al., 2012; Jacobs et al., 2003). The ΔrelA/ΔspoT double knockout mutant strain was kindly provided by Dr. Pradeep K. Singh (University of Washington). The rpoN deletion mutant was kindly provided by Dr. Michael Schurr (University of Colorado). Cells were cultured in Luria Bertani (LB) broth (Lennox), with antibiotic concentrations as follows: for E. coli, Ampicillin 100 μg/mL; for P. aeruginosa, Carbenicillin 150 μg/mL, Gentamicin 30 μg/mL, Trimethoprim 500 μg/mL.
2.2. Generation of reporter constructs
All reporter constructs were generated using a Tn7-based single copy reporter system described by Choi et al. (2005). A schematic description of transcriptional and translational reporters to the yfp is shown in Figs 1 and S2. A complete list of strains, plasmid constructs and transposon fusion constructs is shown in Table S1. Sequences of DNA primers used for all constructs, 5ʹ-RACE and RT-qPCR are shown in Table S2.
Fig. 1.
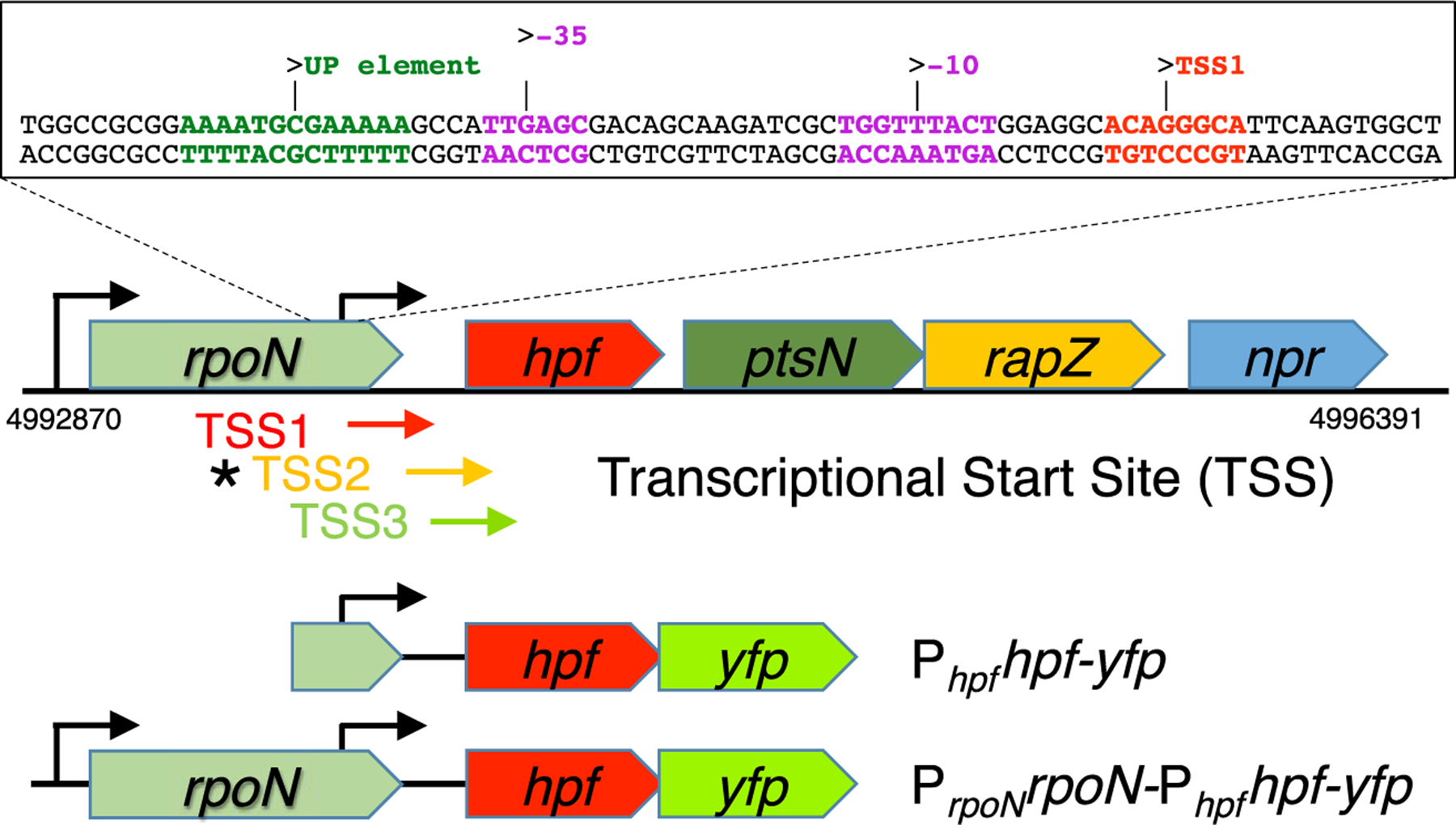
Schematic representation of the rpoN operon of P. aeruginosa PAO1, showing the position of the predicted Phpf promoter sequence (located within the rpoN gene). Shown are the positions of the three transcriptional start sites (TSS1, TSS2 and TSS3) detected by RNA-Seq analysis of P. aeruginosa PA14 (Dotsch et al., 2012; Gill et al., 2018). The asterisk indicates that the TSS2 sequence was detected here using 5ʹ-RACE. Schematic representation of two of the single-copy hpf-yfp reporter constructs used to assay the relative promoter strengths of the PrpoN and Phpf promoters (all reporter constructs are shown in Fig S2).
DNA fragments were amplified from genomic DNA isolated from P. aeruginosa PAO1 using the Phusion PCR kit (NEB) with a gene specific primer set. Amplicons were either directly ligated into the Tn7 vector, or ligated into the TA cloning vector pCR2.1, using the TA cloning kit (Thermo Fisher), prior to ligating into Tn7 vector. The Tn7-YFP vector (pUC18T-mini-Tn7T-Gm-eyfp) (Choi et al., 2005) and inserts were digested with the appropriate restriction enzymes and ligated using the Quick Ligation kit (NEB). Ligation reactions were transformed into chemically competent cells and plated onto LB agar plates with appropriate antibiotics. Plasmids were screened by restriction digestion and insert sequences were confirmed by sequencing (GenScript). Arabinose-inducible reporter constructs (PBADTSS1–3-hpf-yfp) were generated by extraction of TSS-hpf-yfp fragments from promoterless reporters by restriction digestion and cloned into the arabinose-inducible vector pTJ1 (Damron et al., 2013). Overlap extension PCR was used to generate hpf-yfp reporters with modified HPLs. Insert direction was confirmed by restriction digestion and sequencing. The Tn7 reporter constructs were introduced into the P. aeruginosa PAO1 genome by transposition as described (Choi et al., 2005) using electrocompetent cells and the transposase helper plasmid (pTNS1). After recovery in 1 mL SOC medium for 1 h at 37°C, transformants were plated onto LB agar plates with gentamicin or trimethoprim.
2.3. Assay of gene expression by YFP fluorescence
YFP fluorescence and cell growth were assayed over time using a 96 well plate reader, Synergy 2 Multi-Mode Microplate Reader (BioTek). Overnight cultures of the reporter strains were diluted in LB broth and adjusted to an optical density of 0.1 at 600 nm (OD600). 100 μL of diluted samples and 100 μL of LB broth were mixed in a 96 well clear bottom black plate (Corning). The edges of the plates were sealed with parafilm to help prevent evaporation, and the plates were incubated in the Synergy 2 Multi-Mode Microplate Reader with medium speed at 37°C. Fluorescence and OD600 were measured from the bottom of the wells every 15 min for 48 h. The fluorescence filter set used for YFP detection was: Excitation at 485 nm/20 and emission at 528 nm/20 with sensitivity set to 50. All experiments were performed with at least three independent biological replicates and three technical replicates per plate. The average and standard deviations were calculated from the biological replicates after first averaging the values of the technical replicates. The growth rates for every 30 min were calculated as follows: growth rate (h−1) = [In(OD(t + 30 min)) – In(OD(t))]/0.5 h. The gene expression rate for every 30 min was calculated based on the equation developed by Leveau and Lindow (2001) with promoter activity P = Fss × μ × (1 + u/m), where Fss = ∂FL/∂OD, μ = growth rate (h−1), and m = maturation time for the fluorescent protein (h−1). Maturation time of eYFP was set for 0.66 h–1 (Gordon et al., 2007). The slopes of change in fluorescence of Phpfhpf-yfp and PrpoNrpoN-Phpfhpf-yfp between 10–20 and 10–30 h were determined using a trend line function of Microsoft Excel. Statistical significance of fluorescence level was determined at 28 h of incubation (unless otherwise indicated) using a two-tailed two-sample t-test or by analysis of variance (ANOVA) with Fisher least significant difference. The 28 h time point was chosen for most experiments, because that is the time when expression rate reached a steady state. Since the T score of the pair-wise comparison of the fluorescence level between PrpoNrpoN-Phpfhpf-yfp and PrpoNrpoN147-Phpfhpf-yfp was the highest at 37 h of incubation, the p-value at for these strains was determined at that time point.
2.4. Epifluorescence microscopy
An overnight culture of PAO1Δhpf::Phpfhpf-yfp was diluted 1:100 in 3 mL of LB broth (Lennox) in a test tube and grown on a roller at 37°C. Cells were sampled every hour and placed between a microscope slide and a coverslip. Cells were visualized and recorded using a Zeiss Axio Imager.A1 with AioCam MRc5 (Zeiss).
2.5. 5ʹ Rapid Amplification of cDNA Ends (5ʹ-RACE)
An overnight culture of PAO1Δhpf::Phpfhpf-yfp was diluted 1:100 in 25 mL of LB broth (Lennox). Following 6 h of growth at 37°C with shaking, cells were collected by centrifugation, and RNA was extracted, cleaned and turbo DNase treated as described previously (Guragain et al., 2016). Quality was assessed using the Bioanalyzer 2100 RNA 6000 Nanoassay (Agilent). Rapid-amplification of 5′-complementary DNA ends (5′-RACE) analysis was performed similar to the method described previously (Nygaard et al., 2010) using the primers listed in Table S2. The product was visualized with the Bioanalyzer 2100 DNA 12000 assay (Agilent) and sequenced (GenScript).
2.6. Analysis of mRNA levels by RT-qPCR
RNA was extracted from the wild-type cells and mutant strains that were cultured in microtiter plates for 3, 10 and 28 h as described previously (Guragain et al., 2016). The RNA was purified and DNase treated as described (Guragain et al., 2016). One-step reverse transcription-quantitative PCR (RT-qPCR) was carried out as described (Williamson et al., 2012). Three biological replicates for each strain at each time point were assayed with three technical replicates per RNA sample, using 2 ng of RNA and 250 nM primers. Primer sets were designed to assay hpf-yfp expression and expression of the housekeeping gene acpP, which was used to normalize RNA abundance. Primer sequences used for RT-qPCR are shown in Table S2. Primer set validation revealed that the primers had similar efficiencies (EacpP = 1.03, EeYFP = 1.04) over four orders of magnitude, with r2 > 0.995. Controls lacking reverse transcriptase revealed that all 36 biological samples were negative for DNA contamination. Abundance of hpf-yfp mRNA in each strain was calculated relative to that produced from the native promoter Phpf at each time point using the formula Fold Change = 2–ΔΔCt. A one-tailed t-test was performed on the normalized Δct values to determine if hpf-yfp mRNA was produced in greater amounts from the PBAD promoter compared to the native promoter at each time point. A one-way ANOVA was performed to determine if the fold change in expression in PBAD-TSS2 strain was greater than that in PBAD-TSS1 or PBAD-TSS3 at each time point. An alpha level of 0.10 was used for all statistical tests.
To determine if hpf mRNA is produced by transcriptional read-through from PrpoN, RT-qPCR was performed on mRNA isolated from wild type cells, using a primer upstream of the predicted Phpf transcriptional start site, TSS1 (Primer UP-TSS1-F) or downstream of the predicted Phpf TSS1 (Primer Down-TSS1-F) and an hpf reverse primer (hpf-R) (Table S2). PAO1 wild type cells were cultured in LB for 9 h at 37°C, from a 1:100 dilution of an overnight culture. RNA was extracted, treated and analyzed as described above. Absolute quantitation was performed using DNA standards generated from PCR of genomic DNA from strain PAO1. Standard curves, constructed from 10-fold serial dilutions of the PCR products, were linear over five orders of magnitude (R2 > 0.998). Copy number was calculated using RotorGene Software version 1.7, and mean and standard deviation were plotted using GraphPad Prism version 7.00. Controls lacking reverse transcriptase or lacking template did not generate PCR products, as confirmed by melt curve analysis and the Bioanalyzer 2100 DNA12000 assay (Agilent).
2.7. Immunoblot analysis
Strains for immunoblot analysis were cultured in microtiter plates for 3, 10 and 28 h. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 12% resolving gels (Laemmli, 1970). Proteins were electroblotted onto nitrocellulose membranes and then probed with anti-GFP antibodies at 1:10,000 dilution. Horseradish peroxidase-conjugated anti-rabbit immunoglobulin G was used as the secondary antibody. Immunoblots were developed using chemiluminescence detection (Ausubel et al., 1993). Representative gels from three biological replicates of the immunoblot analyses are shown.
2.8. Bioinformatic analyses
The hpf gene and its neighboring genes for different bacterial species were retrieved using the STRING database (Szklarczyk et al., 2017) and BioCyc (Caspi et al., 2016). Promoter sequence predictions were made using BPROM (Solovyev and Salamov, 2011). Hairpin loop predictions were generated using the RNAfold server (Lorenz et al., 2011).
3. Results
3.1. HPF is widely distributed in bacteria and is often genetically linked to the rpoN operon in proteobacteria
In most bacterial taxa, hpf is immediately downstream of rpoN (Figs 1 and S1), which encodes the alternative sigma factor required for expression of nitrogen scavenging genes (Jin et al., 2003; Potvin et al., 2008). In most Proteobacteria, the rpoN operon also includes two other genes involved in carbon and nitrogen metabolism: the ptsN gene encoding Phosphoenolpyruvate-dependent phosphotransferase, and the rapZ gene encoding a P-loop containing nucleoside triphosphate hydrolase- RNase adaptor protein. In some species of gamma Proteobacteria, the operon also contains npr, encoding a phosphocarrier HPr protein-like protein. Expression of hpf and ptsN may be translationally coupled, since there is no obvious ribosomal binding site (RBS) upstream of ptsN in P. aeruginosa. At this time, it is not clear if there is a physiological association of HPF with the other proteins encoded on the rpoN operon, or why hpf is usually linked to the rpoN operon (Fig. S1).
3.2. The hpf promoter is independent of the rpoN promoter and within the rpoN coding sequence
Our previous expression studies indicate that in biofilms, hpf may be highly expressed even when rpoN is not expressed at high levels (Williamson et al., 2012). Therefore, hpf may contain its own regulatory components that are independent of rpoN, and hpf may be monocistronic. Analysis of RNAseq data from Dötsch et al. (2012), indicated that there are three putative transcriptional start sites (TSS) upstream of hpf. Here, we refer to those TSSs as TSS1, TSS2 and TSS3, with TSS3 closest to the hpf RBS and the hpf start codon (Fig. 1). An RNA-Seq study by Gill et al. (2018) that differentiated 5ʹ-triphosphate groups indicated that TSS1 and TSS3 produce naïve unprocessed mRNAs, containing 5ʹ-tri-phosphate groups, suggested that hpf transcriptional starts at TSS1 and/or TSS3. Using a promoter prediction program, BPROM (Solovyev and Salamov, 2011), we analyzed the sequence upstream of hpf, and predicted the presence of an RpoD/RpoS-like promoter sequence approximately 12 base pairs upstream of TSS1 (Fig. 1). The putative Phpf promoter sequence is within the protein coding sequence of rpoN. RpoD-like promoters are often associated with AT rich-UP elements, where the C-terminus of alpha subunits of RNA holoenzyme interact with the DNA (Ross et al., 1993). The putative Phpf identified by BPROM also contains a predicted UP-element (Fig. 1).
To determine if hpf is expressed from the promoter predicted by BPROM, we constructed a series of reporter fusions that generated translational fusions of HPF to the yellow fluorescent protein (HPF-YFP fusions) using a single copy Tn7-based reporter system (Choi et al., 2005) (Figs 1 and S2). We then assayed YFP-fluorescence over time, while monitoring cell growth as optical density, using a 96 well plate reader. The HPF-YFP reporter fusions either contained the rpoN promoter (PrpoNrpoN-Phpfhpf-yfp), or lacked PrpoN, but contained the predicted Phpf (Phpfhpf-yfp) (Fig. 1). The results showed that fluorescence from the Phpfhpf-yfp reporter was much higher than background fluorescence (t-test, p < 0.00001) (Fig. 2A), supporting the RNA-Seq results (Gill et al., 2018) that hpf contains its own promoter that is independent of the PrpoN promoter. The fluorescence from the strain containing the PrpoNrpoN-Phpfhpf-yfp reporter occurred at a faster rate (p < 0.00001 for slope at 10–20 h) and peaked at a higher level than the strain lacking PrpoN (p < 0.0001, 28 h)(Fig. 2A), indicating that in addition to the Phpf promoter, there is likely transcriptional read through of hpf from PrpoN.
Fig. 2.
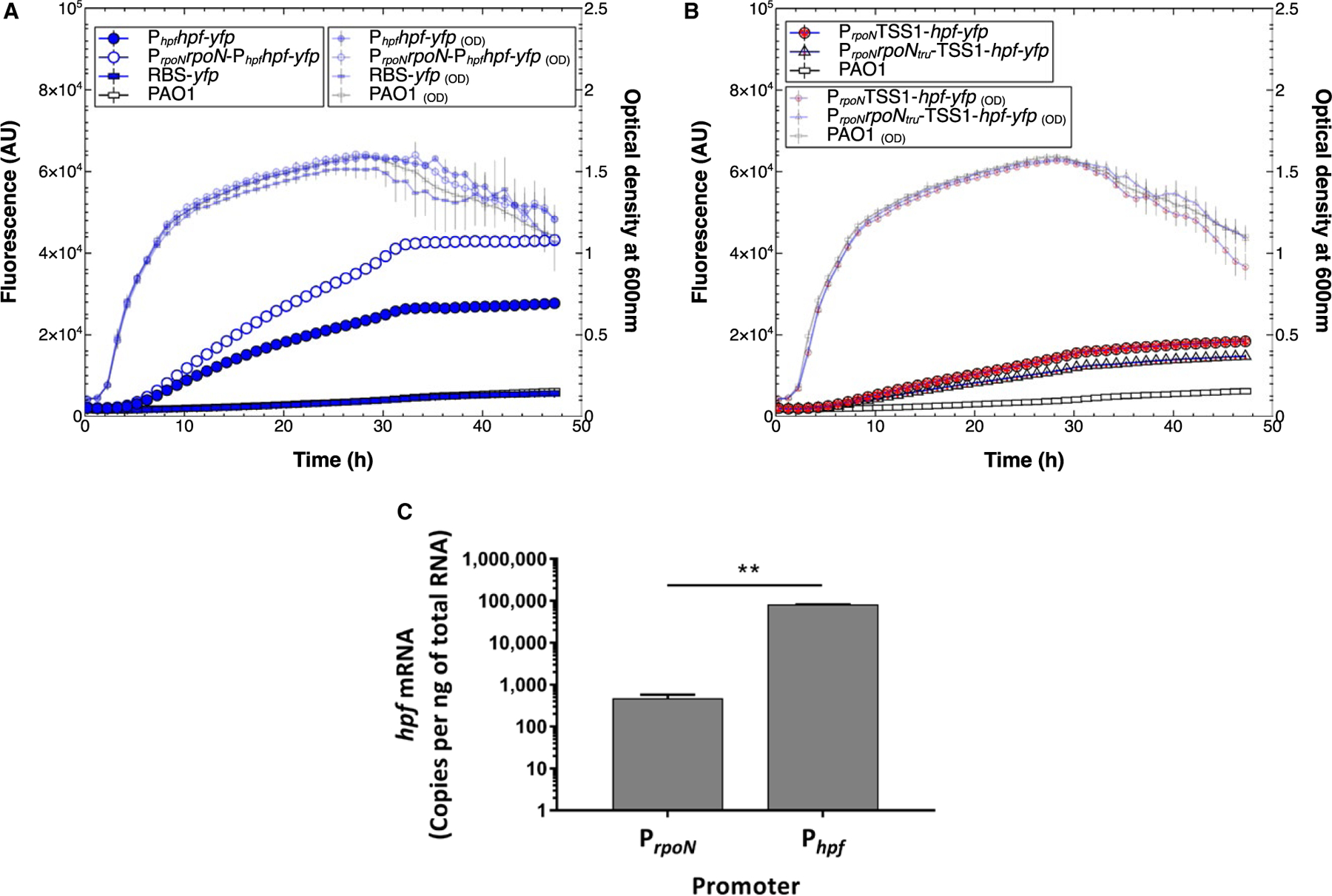
A. hpf expression from PrpoN and Phpf promoters. Fluorescence (large symbols) and optical density (small symbols) of P. aeruginosa PAO1 containing single-copy Tn7 translational hfp-yfp fusions, during 48 h of growth. The control strains are wild-type P. aeruginosa PAO1 lacking a reporter construct (open rectangles), and PAO1 with an hpf-yfp translational fusion, but lacking a promoter sequence (closed rectangles). The Phpfhpf-yfp reporter (closed blue circles) contains the putative Phpf promoter sequence and UP element. The reporter containing both the PrpoN and putative Phpf promoters (open blue circles) had greater expression than the reporter containing Phpf alone.
B. hpf expression from PrpoN promoter, but lacking the Phpf promoter. The reporter constructs contained the PrpoN promoter and either a deleted rpoN gene (closed red circles) or a truncated rpoN gene (open red triangles).
C. RT-qPCR analysis of hpf expression due to transcriptional read through from PrpoN versus expression from Phpf. Results show the average of three biological replicates. Error bars show standard deviation.
To determine whether higher expression of PrpoNrpoNPhpfhpf-yfp compared to Phpfhpf-yfp is due to polycistronic expression from the PrpoN promoter, reporter constructs were generated that contained PrpoN but lacked Phpf. The PrpoN was fused to the hpf TSS1, and Phpf was deleted. The reporter constructs had either a deleted rpoN gene (PrpoNTSS1-hpf-yfp) (Fig. S2, construct C) or a truncated rpoN gene (PrpoNrpoNtru-TSS1-hpf-yfp) (Fig. S2, construct D). Expression of hpf from PrpoN was low, but higher than the control lacking both the PrpoN and the Phpf promoters (p < 0.0003 for both C and D constructs) (Fig. 2B), indicating that hpf is expressed primarily from Phpf but also from PrpoN at low levels. To confirm hpf expression by transcriptional read-through from PrpoN, we performed reverse transcription quantitative PCR (RT-qPCR) on hpf mRNA produced from PrpoN using a primer upstream of Phpf and a reverse primer within the hpf coding sequence (Table S2). The amount of mRNA was compared to the amount when the RT-qPCR primer was downstream of Phpf (Table S2). At 9 h of growth, there was significantly more hpf mRNA produced from Phpf than from PrpoN (p < 0.001) (Fig. 2C). However, low levels of mRNA were produced from PrpoN, as indicated by the presence of an RT-qPCR product (Fig. S3A), and by confirmation of the PCR product by melt curve analysis (Fig. S3B).
Expression of hpf was higher in the presence of both PrpoN and Phpf than for Phpf alone (Fig. 2A). The increased fluorescence in the PrpoNrpoN-Phpfhpf-yfp strain may have been due to a gene dosage effect, since the PrpoNrpoN-Phpfhpf-yfp construct contains an extra copy of the rpoN gene. To determine if the additional copy of rpoN affects Phpf expression, and to determine if Phpf is under RpoN regulatory control, we truncated the rpoN coding sequence, producing a reporter strain with an inactive rpoN (PrpoNrpoN147-Phpfhpf-yfp) (Fig. S2 construct E). Little difference in expression from Phpf was observed between strains containing full-length rpoN and truncated rpoN (Fig. S4A). Next, we introduced the Phpfhpf-yfp fusion into P. aeruginosa PAO1 ΔrpoN. The YFP-fluorescence intensity from the Phpf promoter was similar in the ΔrpoN mutant as for the wild-type strain (p = 0.27)(Fig. S4B). However, the growth rate and growth yield were reduced in the ΔrpoN mutant compared to the wild-type strain (Fig. S3B). Therefore, RpoN may have an inhibitory effect (either directly or indirectly) on expression from Phpf. Taken together, the results above demonstrate that: (i) hpf contains its own promoter that is downstream of the rpoN promoter, (ii) hpf is also expressed at low levels as part of a polycistronic RNA from PrpoN, and (iii) Phpf is not under positive control of the RpoN sigma factor.
3.3. Mapping the sequence of the Phpf promoter region
Based on the predictions from BPROM, we mapped the position of the Phpf promoter sequence, by generating HPF-YFP reporter fusions that lacked the Phpf or its associated UP-element. The hpf-yfp fusions were constructed so that transcription would begin at each of the three predicted TSSs, but each lacking the predicted Phpf (Fig. S2 constructs F,G,H). Each of the strains lacking Phpf had low levels of fluorescence that was less than the strain containing the predicted Phpf (p < 0.0001 for all three TSS) (Fig. 3). To test the role of the predicted UP-element, we deleted the UP-element by starting the construct four bases upstream of the −35 consensus sequence, leaving the −35 and −10 consensus sequences intact (Fig. S2 construct I). The reporter strain lacking the UP-element had approximately twofold reduced Phpf expression compared to the strain containing the intact Phpf and UP-element (p < 0.00002) (Fig. 3). The results show that both the predicted promoter sequence and the UP-element are necessary for maximum hpf expression from Phpf. The Phpf -promoterless constructs had fluorescence levels that were slightly higher than the background control strains, indicating that there may be a weak secondary promoter downstream of Phpf.
Fig. 3.
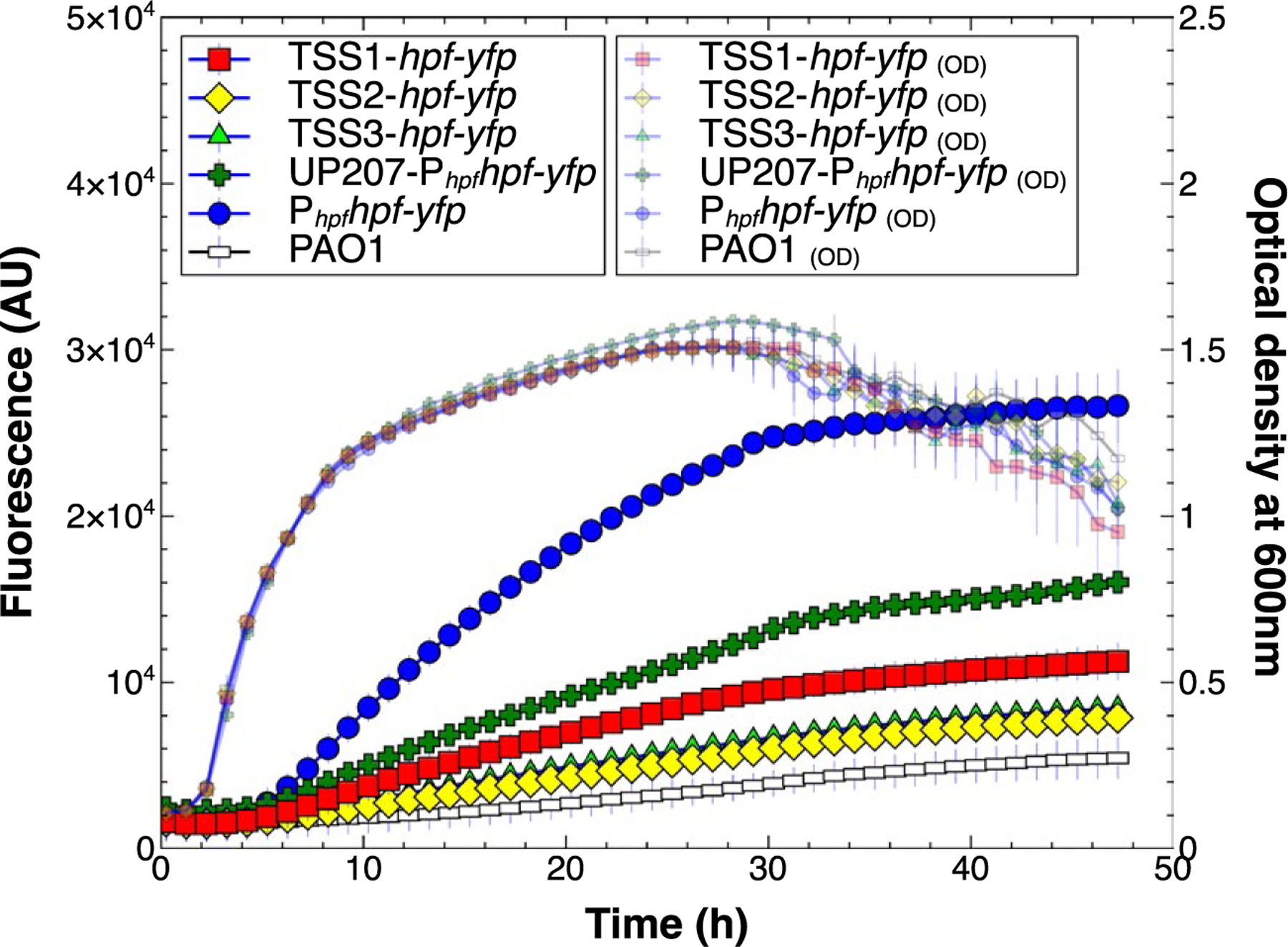
Expression and growth of P. aeruginosa PAO1 containing hpf-yfp reporter fusions that lacked the Phpf promoter or its UP-element (constructs F, G, H of Fig S2). The promoterless reporter constructs were generated so that transcription would start at each of the three putative transcriptional start sites; TSS1 (red squares), TSS2 (yellow diamonds) and TSS3 (green triangles). The results show comparison to the strain containing the Phpf (closed blue circles). The control strain shows background fluorescence (open rectangles). The strain containing Phpf, but lacking the UP-element (green crosses) had reduced expression compared to Phpf. Data represent the mean and standard deviation of three independent biological replicates.
3.4. Growth phase dependence of hpf expression
The growth and fluorescence results above indicated that there was very little hpf expression during exponential phase, as was predicted based on its function as a dormancy hibernation factor. We used the equation developed by Leveau and Lindow (2001) to estimate the expression rate per cell, based on the fluorescence of the reporter, the optical density of cultures, and the bacterial growth rate. The results confirmed that the expression rate of hpf increased as cells entered stationary phase (Fig. S5A). We also analyzed expression of hpf using the Phpfhpf-yfp constructs at the cellular level using epifluorescence microscopy. In the stationary phase inoculum, cells were fluorescent, (Fig. S5B). Cell fluorescence dimmed after 1 and 2 h of growth then increased at 5 h of growth, with most cells containing the Phpfhpf-yfp reporter being highly fluorescent at 5 h. These results indicated that hpf is low during exponential phase, and increases as cells transition to stationary phase.
3.5. Effect of stationary phase factors on hpf expression
Since Phpf appears to be active during the transition to stationary phase, we next tested the effect of mutants with defects in genes required for bacterial transition to stationary phase on hpf expression. We introduced the Phpfhpf-yfp translational reporter into P. aeruginosa strains that were mutants of the stationary phase sigma factor RpoS, the RNA polymerase-interacting protein DksA2, and proteins that produce guanidine tetra- and penta-phosphate ((p)ppGpp), RelA/SpoT. The rpoS and dksA2 mutants gave similar expression profiles for Phpfhpf-yfp as the wild-type strain for the first five hours of growth. However, the expression plateaued at a lower level for the rpoS and dksA2 mutants as they entered stationary phase (p < 0.00004) (Fig. 4). The fluorescence rates per cell, characterized by the Leveau equation (Leveau and Lindow, 2001), confirmed similar expression rates for the rpoS and dksA2 mutants compared to wild-type for the first five hours of growth, but reduced expression per cell in stationary phase (Fig. S6). The expression patterns and growth curves of the rpoS and dksA2 mutant strains were almost identical to each other, indicating that the regulatory pathways of RpoS and DksA2 may overlap.
Fig. 4.
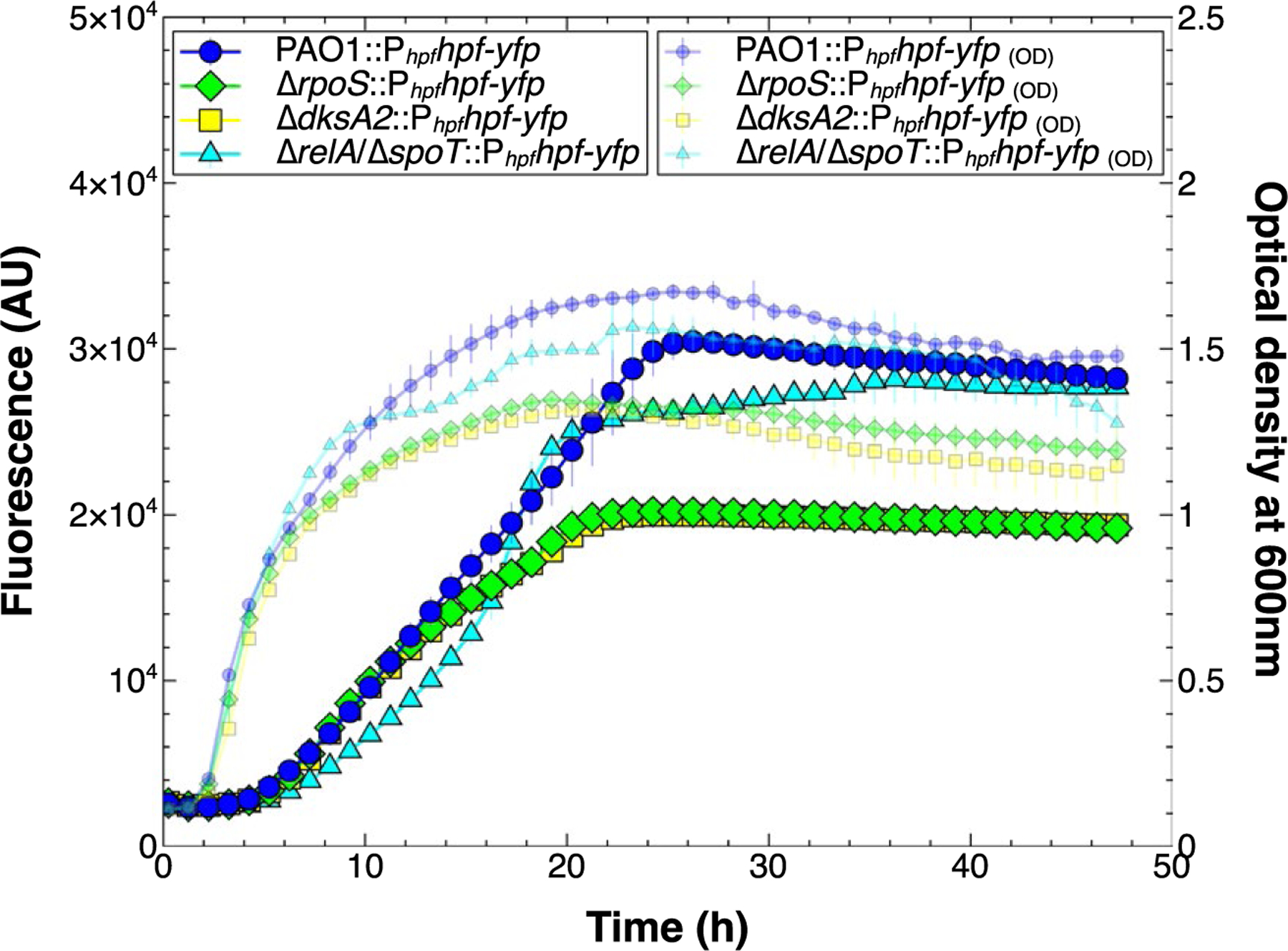
Effect of stationary phase factors on growth (small symbols) and expression (large symbols) from Phpf. Expression of Phpf hpf-yfp in an rpoS mutant (green diamonds) and dksA2 mutant (yellow squares) reached a lower level than the wild-type control (blue circles). The mutant defective in ppGpp production (cyan triangles) had reduced initial expression compared to the control, then equivalent expression when in stationary phase.
The ΔrelA/ΔspoT mutant, which is impaired for (p) ppGpp production, had a Phpfhpf-yfp expression pattern that differed from that of the wild type (Fig. 4). Initially, the expression rate was lower than in the wild type strain (p = 0.01 at 12 h), but reached the wild type level as the cells entered late stationary phase (p = 0.84, 48 h). The expression rate per cell was initially slower than wild-type in stationary phase, but peaked as the cells were in late stationary phase (Fig. S6). The results indicate that ppGpp may modulate expression from Phpf, but expression is not eliminated from the Phpf promoter in the ΔrelA/ΔspoT mutant. We also tested the effect of RpoS, DksA2 and RelA/SpoT on expression of hpf from both the PrpoN and Phpf using the construct PrpoNrpoN-Phpfhpf-yfp (Fig. S2, construct B). The results showed a similar pattern of expression as the construct with Phpf alone, with the rpoS and dksA2 mutants showing reduced expression when cells are in stationary phase (p < 0.0003, 28 h), and the ΔrelA/ΔspoT mutant having a modulatory effect on hpf expression primarily during stationary phase (Fig. S7A, S7B).
3.6. Mapping the transcriptional start site for the hpf mRNA
The RNAseq results of Gill et al. (2018) indicated that TSS1 and TSS3 are unprocessed (naïve) transcriptional start sites containing 5ʹ triphosphate groups on the mRNA, whereas TSS2 is a processed site. We performed 5ʹ rapid amplification of cDNA ends (5ʹ-RACE) to identify the major mRNA species produced from the wild type strain with the Phpfhpf-yfp reporter in the hpf deletion strain. The results showed a single mRNA species that corresponded to starting at TSS2 using a primer that targets the hpf gene of the reporter construct (Table S2). Since TSS1 is likely the naïve form of the mRNA transcript, and since TSS1 is in an optimal position with respect to the Phpf promoter, mRNA starting at TSS2 as determined by 5ʹ-RACE indicates that TSS2 may be a processed form of the hpf transcript.
To determine if the position of the transcriptional start site (TSS1, TSS2 or TSS3) affects the degree of hpf expression, we replaced the native Phpf promoter with the arabinose inducible promoter (PBAD), so that transcription would begin near each of the three putative TSSs (Fig. S2, constructs J,K,L). In the absence of arabinose addition, expression from each of the three TSSs was lower than transcription from the native Phpf promoter (p < 0.00001) (Fig. S8). However, when arabinose was added to the medium, expression was up to ten fold greater than expression from the native Phpf promoter (p < 0.00001, for all three TSS) (Fig. 5A). Surprisingly, the expression level varied greatly depending on the position of the PBAD promoter, even though the positions of the TSS sites varied by only 110 nucleotides from TSS1 and TSS2, and 31 nucleotides from TSS2 and TSS3. Expression from TSS2 showed the greatest expression level (ANOVA, p < 0.0001) and was approximately two fold greater than expression from TSS1 (t-test, p < 0.0001) and three fold greater than expression from TSS3 (t-test, p < 0.00001).
Fig. 5.
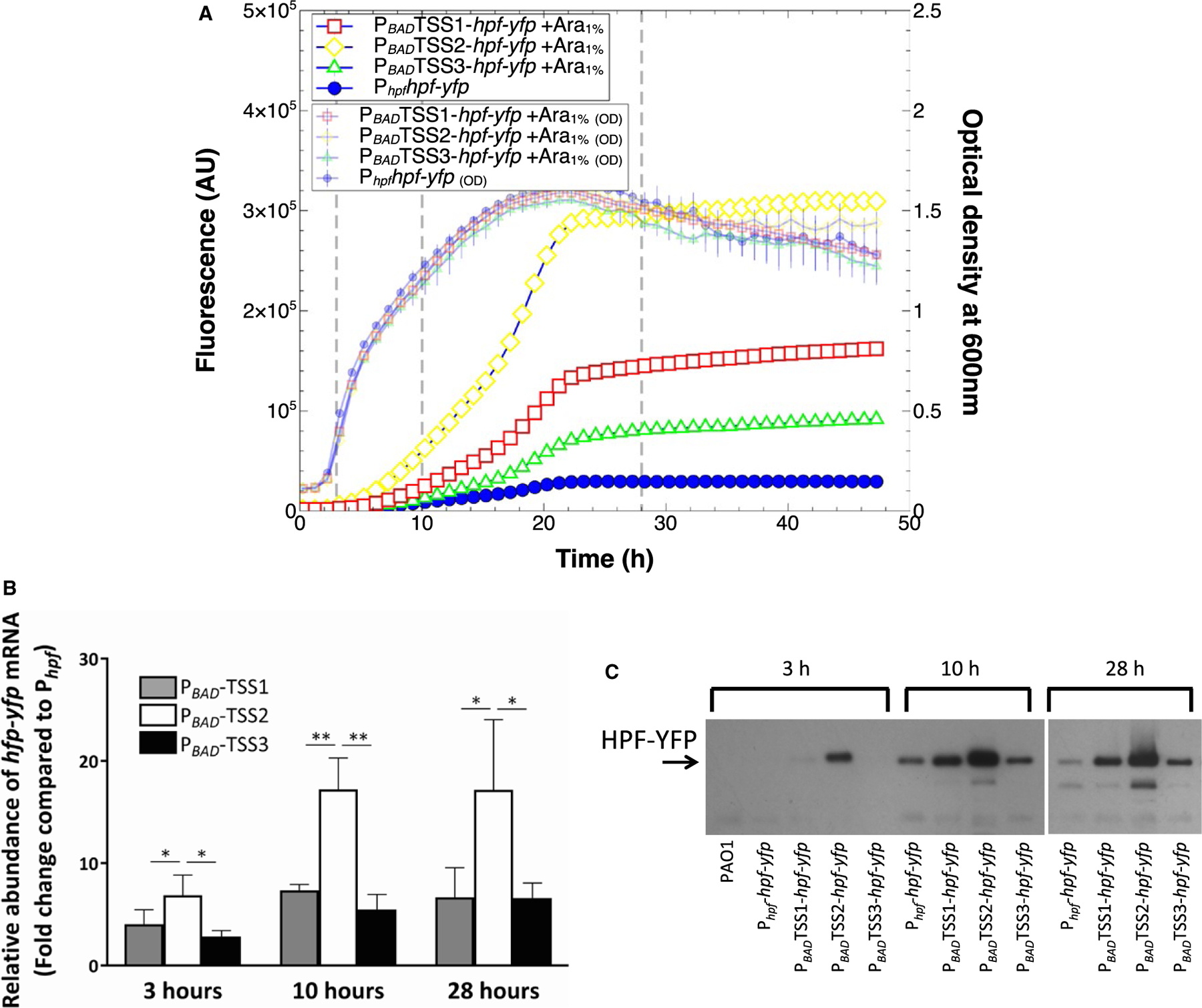
Expression of hpf-yfp reporter with the Phpf promoter replaced by the PBAD promoter, in the presence of arabinose induction.
A. Growth (small symbols) and expression of hpf-yfp reporter when the PBAD promoter was placed in orientation for expression from TSS1 (red squares), TSS2 (yellow diamonds) and TSS3 (green triangles). Expression from Phpf (blue circles) is shown as a positive control. (note that the Y-axis scale is 10-fold greater than in Fig. 1).
B. RT-qPCR of the hpf-yfp mRNA expressed from the PBAD promoter upstream of TSS1, TSS2 and TSS3, showing fold change compared to mRNA expressed from the Phpf promoter (t-test, *p < 0.1, **p < 0.01).
C. Immunoblots of the HPF-YFP fusion protein, when expressed from the Phpf promoter, compared to the protein amounts produced from the PBAD promoter, positioned upstream of TSS1, TSS2, and TSS3. Data for A and B show the mean and standard deviation from three biological replicates. Data from C shows a representative immunoblot from three replicates.
To determine if the differences in fluorescence between the arabinose inducible promoters, positioned at the three different TSSs, was due to changes in mRNA abundance or protein amounts, we assayed hpf-yfp mRNA levels using reverse transcription-quantitative PCR (RT-qPCR) and HPF-YFP protein levels using immunoblots. The RT-qPCR and immunoblot analyses were performed at three time points, 3, 10 and 28 h. The abundance of hpf-yfp mRNA in the PBAD constructs was calculated relative to that produced from the native promoter (Phpf) (Fig. 5B). The RT-qPCR results show that hpf mRNA is produced at greater amounts from the PBAD promoter, compared to the native promoter, at all three timepoints (Fig. 5B) (p < 0.10).
In addition, the hpf-yfp mRNA abundances varied depending on the position of the PBAD promoter with respect to the putative TSSs. The greatest mRNA amounts occurred when PBAD was positioned for expression from TSS2 (for all three growth phases) (ANOVA, p < 0.10).
The immunoblot analysis showed that the amount of HPF-YFP protein produced from the Phpf native promoter was negligible at 3 h, and increased during stationary phase at 10 h (Fig. 5C). Similar to the RT-qPCR results, the immunoblot analyses also showed that HFPYFP was produced maximally when the native promoter was replaced by the PBAD promoter, and when PBAD was positioned optimally with expression from TSS2. Overall, the results show that (i) Phpf is a relatively weak promoter compared to the PBAD promoter, (ii) transcription of hpf likely begins at TSS1, however, (iii) the greatest hpf expression is derived when transcription begins at TSS2 rather than TSS1. The results indicate that there may be a regulatory sequence(s) between TSS1 and TSS2 that affects hpf mRNA production or stability. The 5ʹ-RACE results suggest that this regulatory sequence between TSS1 and TSS2 may be cleaved to give the mature hpf mRNA product.
3.7. Post-transcriptional regulation of hpf expression
We constructed transcriptional reporters that lacked the hpf coding sequence (Fig. S2, constructs M,N). Surprisingly, YFP fluorescence from these transcriptional reporters lacking the hpf coding sequence was much less than for the translational reporter that contained the hpf coding sequence (Fig. S2, construct A) (p < 0.00002) (Fig. 6A). Expression of the transcriptional reporters was less than the translational reporter, both when the constructs contained the hpf 5ʹUTR (Phpf5ʹUTR-yfp) (Fig. S2, construct M) or lacked the hpf 5ʹUTR (Phpfyfp) (Fig. S2, construct N). The results suggest that the 5ʹUTR alone does not influence expression of hpf, and that the 5ʹ end of the hpf coding sequence may be necessary for optimal hpf expression.
Fig. 6.
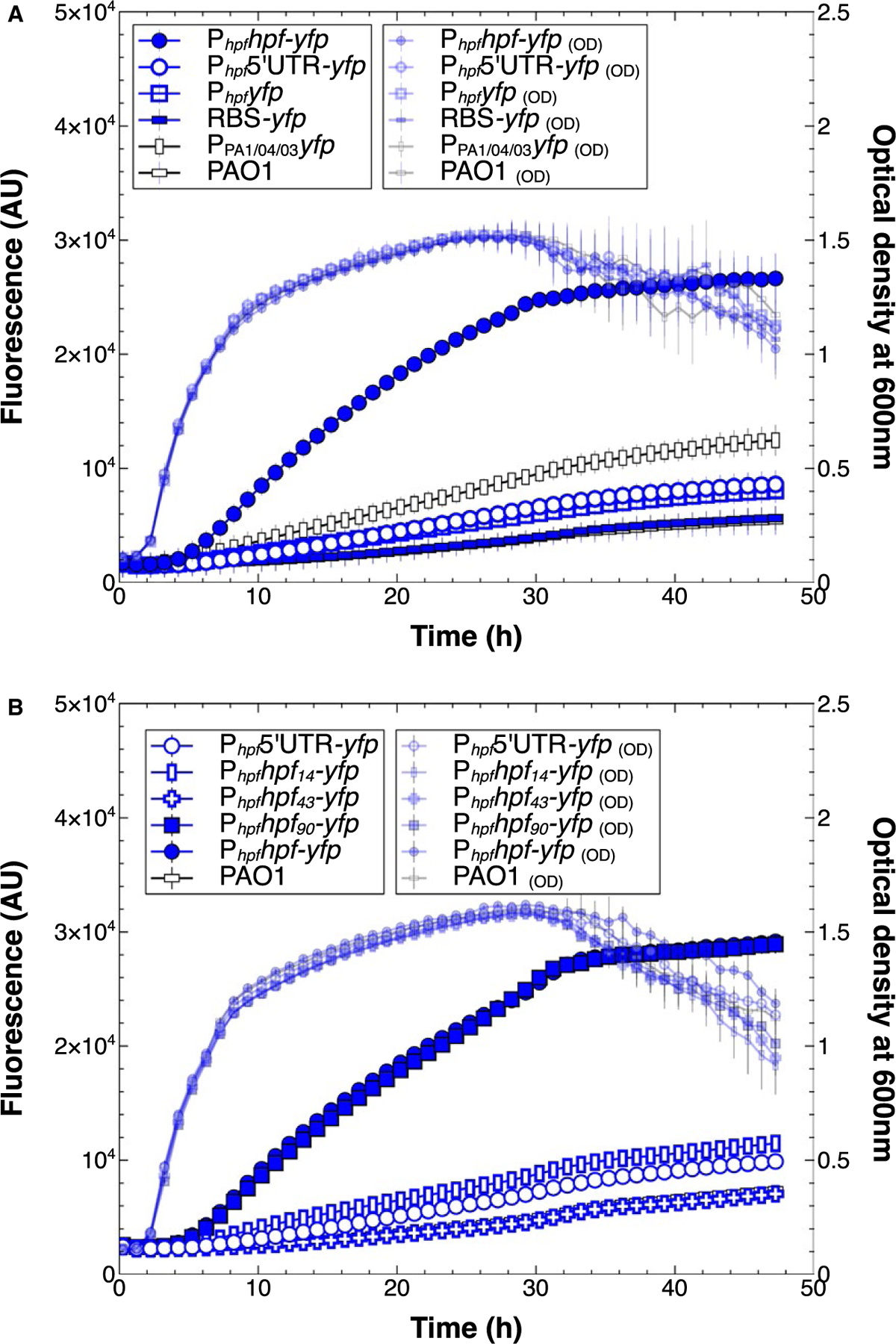
Growth (small symbols) and expression from Phpf reporters that lack the hpf coding sequence.
A. Expression patterns of transcriptional reporters of Phpf -yfp containing (open blue circles) or lacking the hpf 5′UTR (open blue squares), compared to the control strain containing the translational fusion (closed blue circles). Expression levels are compared to a control that lacks the Phpf (open rectangles).
B. Expression patterns of translational fusions that contain part of the N-terminus of HPF; 14 amino acids (blue rectangles), 43 amino acids (blue crosses), 90 amino acids (closed blue squares) or full length HPF (closed blue circles). All data show the mean and standard deviation of three biological replicates.
To identify the portion of the hpf coding sequence required for optimal expression, we generated a series of translational fusion reporters, containing different lengths of the HPF N-terminus. The in-frame translational fusions were constructed to contain 14, 43 or 90 amino acids of the HPF N-terminus (Fig. S2, constructs P,Q,R) or the full-length HPF. Full expression from Phpf only occurred when at least 90 amino acids were included in the construct (p = 0.89)(Fig. 6B). These results, combined with the different expression levels from each of the TSSs, indicated some aspect the 5ʹUTR and the hpf coding region are required for optimal hpf expression, possibly involving mRNA folding.
We used the RNAfold server (Lorenz et al., 2011) to identify potential HPL in the hpf 5ʹUTR. When we included the first 100 nucleotides of the hpf coding sequence in the RNAfold analysis, the analysis showed at least two stable HPLs: one located in the 5ʹUTR between TSS2 and TSS3 and a second HPL overlapping the first 50 nucleotides of the hpf coding region, approximately seven nucleotides downstream of the ATG start codon (Fig. 7A). To determine if these putative HPLs play a role in hpf expression, we substituted nucleotides that would disrupt the HPLs, but not affect the amino acid sequence of HPF. Mod1 was designed to create an open less-stable HPL in the coding region (Fig. S2, construct S, Fig. S9B), and mod3 was designed to create a closed stable HPL (Fig. S2, construct T, Fig. S9C). Mod1 (open) resulted in a twofold increase in expression from Phpfhpf-yfp (p < 0.00001), whereas mod3 resulted in reduced expression of hpf by approximately twofold (p < 0.00001) (Fig. 7B). Modifications 4 and 5 (mod 4 and 5) were designed to disrupt the HPL in the 5ʹUTR (Fig. S2, constructs U, V). Both of these modifications caused a reduction in hpf expression (p < 0.0001) (Fig. 7B).
Fig. 7.
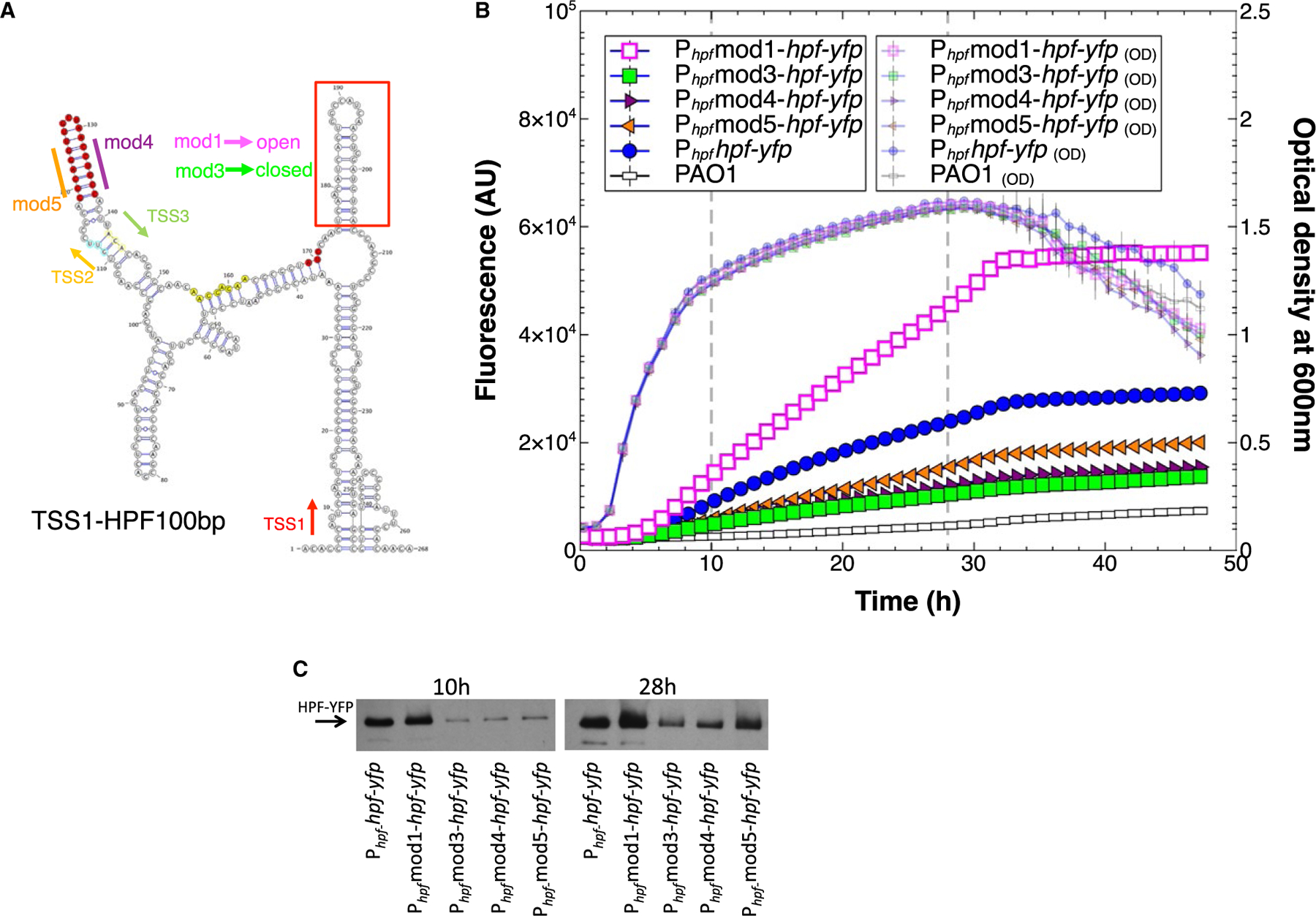
A. Secondary structure prediction of the hpf mRNA starting from TSS1 through the first 100 nucleotides of the hpf structural gene, predicted by RNAfold server and visualized using VARNA (Lorenz et al., 2011). Shown are the sites of the predicted transcriptional start sites (TSS1,2,3), and the position of the hpf start codon (red). The hairpin loop (HPL) at 5′ end of the hpf coding sequence (red box) was mutated by nucleotide substitutions without modifying the amino acid sequence of HPF, creating open (mod1) or closed (mod3) HPLs. The 5ʹ and 3′ ends of the HPL stem located between TSS2 and TSS3 (highlighted in red) were altered to disrupt the HPL structure (mod 4 and mod5).
B. Growth and expression from Phpf containing modifications; mod1 (pink squares) mod3 (green squares) mod4 (maroon triangles) mod5 (orange triangles), compared to the wild-type sequence (blue circles).
C. Immunoblot analysis of the HPF-YFP fusion protein produced at 10 and 24 h of growth, when the HPLs were modified as in Fig. 7B.
The effect of the four HPL modifications on protein production was tested by immunoblot analysis on cells cultured for 10 and 28 h. As with the fluorescence assay, the HPF-YFP protein amount was greater with the mod1 modification, which formed the open complex, than for the wild-type sequence at both time points (Fig. 7C).The least protein production occurred with mod3, which formed the closed complex in the coding region HPL. Mod4 and mod5, which disrupted the HPL in the 5ʹUTR also resulted in reduced protein amounts, compared to the wild-type sequence, similar to the reporter construct fluorescence data.
4. Discussion
In bacteria, the number of ribosomes per cell varies depending on the growth phase of the bacteria (Nomura et al., 1984). Ribosomes are abundant in exponentially growing cells, but decrease on a per cell basis when cells enter stationary phase (Deutscher, 2003). However, for bacteria to recover from long-term starvation, they must maintain a sufficient number of ribosomes to allow the de novo protein synthesis required for resuscitation. Previously, we showed that P. aeruginosa cultured in biofilms maintain a constant ribosome supply (approximately 15,000 per cell) even for the cells deep in the biofilms that have very low metabolic activity (Pérez-Osorio et al., 2010). HFP plays a role in maintenance of this ribosome supply in the low metabolic activity cells. When the hpf gene of P. aeruginosa is deleted, the ribosomal supply of cells cultured under starvation conditions falls below a level required for optimal resuscitation (Akiyama et al., 2017). Therefore, HPF likely evolved as a factor to help prevent complete ribosome loss during starvation.
The hpf gene is widely distributed in bacteria (Fig. S1). Some species of bacteria encode hpf paralogs (such as hpf and yfiA), while others, particularly the Gram-positive bacteria, encode a long HPF, that has an extended C-terminal tail (Ueta et al., 2010). P. aeruginosa and many other species of Proteobacteria encode a short hpf, that is genetically linked to the rpoN operon. Here, in order to define the timing of expression of hpf, we describe the regulatory factors involved in production of HPF. In P. aeruginosa, this regulation includes both transcriptional and post-transcriptional processes.
Early work characterizing the rpoN operon in E. coli and P. aeruginosa indicated that hpf and ptsN are likely transcribed as part of the rpoN operon (Jin et al., 2003; Jones et al., 1994). Kohler et al. (1994) using Northern blotting showed that in Pseudomonas putida, hpf is separately transcribed when the rpoN operon was expressed on a plasmid. Using RT-PCR Ancona et al. (2014) supported that hpf is part of the rpoN operon in Erwinia amylovora. Here, we show that in P. aeruginosa, hpf is expressed from both its own promoter and as part of the rpoN operon. Under the conditions used here, hpf is primarily expressed from its own promoter with some read-through from the PrpoN. The −35 and −10 promoter consensus sequence of Phpf was mapped to be within the rpoN coding sequence. In addition to the consensus sequence, the Phpf promoter also contains an UP element, that when deleted, reduced hpf expression. This dual promoter arrangement (PrpoN followed by Phpf) may allow differential expression of the rpoN and hpf genes depending on the environmental conditions. For example, hpf mRNA may be produced under nitrogen stress conditions from the PrpoN promoter, but hpf mRNA amounts can increase independently of rpoN under certain conditions, such as for cells deep in biofilms that are undergoing hypoxia, as we observed previously. This dual promoter arrangement is consistent with RNA-seq results that show abundant hpf mRNA under conditions where the amount of rpoN mRNA is low (Gill et al., 2018).
Based on the physiological role of HPF as a starvation-responsive factor, and on the promoter consensus sequence, which is similar to RpoD/RpoS promoters, we predicted that hpf expression would be expressed maximally when cells enter stationary phase. The results indicate that hpf is expressed primarily when the bacteria transition to stationary phase. Surprisingly, hpf expression is only moderately affected by the stationary phase and/or stringent response factors, RpoS, DksA2 and (p) ppGpp (Figs 4, and S7). The dksA2 mutant and the rpoS mutant showed similar patterns of growth and hpf expression in P. aeruginosa, with slightly less expression of hpf in stationary phase cells, but similar expression profiles in exponential phase cells. In E. coli, DksA influences RpoS abundance at the post-translation level, probably through expression of anti-RpoS adapter proteins (Brown et al., 2002; Bougdour and Gottesman, 2007). Therefore, the effect of DksA2 disruption on hpf expression is probably RpoS-dependent. P. aeruginosa encodes at least two DksA paralogs (Blaby-Haas et al., 2011; Furman et al., 2013). The DksA paralogs have unique features, with dksA listed as essential for growth of P. aeruginosa PAO1 in MOPS-succinate medium, while DksA2 is not essential (Turner et al., 2015). DksA2 is a zinc-independent DksA which is expressed during metal depletion and can complement the ΔdksA mutant of E. coli (Blaby-Haas et al., 2011).
The stringent response signaling molecule (p)ppGpp influences expression of many genes during nutrient depletion and the stringent response through interaction with DksA and the RNA polymerase (Ross et al., 2016). In E. coli, these genes include rRNA/tRNA synthesis genes, metabolic genes and genes for amino acid biosynthesis (Durfee et al., 2008). The effect of a ΔrelA/ΔspoT mutant on expression from Phpf in P. aeruginosa was minor (Fig. 4). However, the expression from Phpf in the ΔrelA/ΔspoT mutant was distinct from the wild-type strain. In particular, the expression rate from Phpf was reduced in the ΔrelA/ΔspoT mutant during exponential phase, but then increased as the cells entered stationary phase. This expression pattern differed from our prediction. If expression from Phpf was controlled by the stringent response, we expected that expression would be reduced in the ΔrelA/ΔspoT mutant. The results indicate that regulation of hpf production by (p)ppGpp differs from other ribosomal proteins. The results presented here do not preclude the possibility that other, yet to be identified, regulatory proteins or DNA-binding proteins influence hpf expression at the transcriptional level.
Riboswitches and the secondary and tertiary structures of RNA can influence translation primarily by blocking access of the RBS on mRNAs (Henkin, 2008).The results of the Phpf expression studies presented here indicate that the 5ʹ UTR of the hpf mRNA has a strong effect on translation of HPF. The hpf riboswitch appears to extend into the 5ʹ coding region of the hpf gene. Several lines of evidence indicate that mRNA folding influences HPF production at the post-transcriptional level. First, expression from Phpf differs, depending on whether or not the hpf coding region is included in the reporter constructs (Fig. 6). Second, HPF mRNA and protein production differ when the Phpf promoter is replaced with the PBAD promoter, and the PBAD promoter is positioned to delete part of the 5ʹUTR region (Fig. 5A). Third, stable hairpin loop structures are predicted in the 5ʹUTR and the adjacent coding region of hpf (Fig. 7). When the HPL structure predicted to be within the 5ʹUTR is modified, HPF translation is impaired. When the hairpin loop that occludes the RBS s is altered to create an open structure, HPF production increases, while a modification that forms a stable hairpin structure in this region causes decreased HPF production. Therefore, the sequence associated with the native HPL overlapping the 5ʹend of the hpf gene plays an inhibitory role on HPF production, while the HPL in the 5ʹUTR of hpf has a positive regulatory role on HPF production. Post-transcriptional regulation through modification of mRNA stability and translation efficiency is a major regulatory process that allows HPF production under certain environmental conditions.
In this study, we show that regulation of HPF is complex, and involves both transcriptional and post-transcriptional processes. This complex regulatory network may allow cells to respond to the conditions that cells experience as they enter a dormant state, allowing them to protect ribosomes from complete functional loss during conditions of low metabolic activity.
Supplementary Material
Acknowledgements
We thank Phil Stewart for his helpful discussions of this work. We thank Dr. Pradeep K. Singh for graciously providing the ΔrelA/ΔspoT mutant, and Dr. Michael Schurr for graciously providing the ΔrpoN mutant.
Footnotes
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- Akiyama T, Williamson KS, Schaefer R, Pratt S, Chang CB, and Franklin MJ (2017) Resuscitation of Pseudomonas aeruginosa from dormancy requires hibernation promoting factor (PA4463) for ribosome preservation. Proc Nat Acad Sci 114, 3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancona V, Li W, and Zhao Y (2014) Alternative sigma factor RpoN and its modulation protein YhbH are indispensable for Erwinia amylovora virulence. Mol Plant Pathol 15, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, and Smith J (1993) Current Protocols in Molecular Biology (Vol. 2).New York, NY: Greene Publishing Associates, Inc. and John Wiley & Sons, Inc. [Google Scholar]
- Beckert B, Abdelshahid M, Schafer H, Steinchen W, Arenz S, Berninghausen O, et al. (2017) Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J 36, 2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaby-Haas CE, Furman R, Rodionov DA, Artsimovitch I, and de Crecy-Lagard V (2011) Role of a Zn-independent DksA in Zn homeostasis and stringent response. Mol Microbiol 79, 700–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougdour A, and Gottesman S (2007) ppGpp regulation of RpoS degradation via anti-adaptor protein IraP. Proc Natl Acad Sci U S A 104, 12896–12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Gentry D, Elliott T, and Cashel M (2002) DksA affects ppGpp induction of RpoS at a translational level. J Bacteriol 184, 4455–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, et al. (2016) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 44(D1): D471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, et al. (2005) A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2, 443–448. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, and Greenberg EP (1999) Bacterial biofilms: A common cause of persistent infections. Science 284: 1318–1322. [DOI] [PubMed] [Google Scholar]
- Damron FH, McKenney ES, Schweizer HP, and Goldberg JB (2013) Construction of a broad-host-range Tn7-based vector for single-copy P(BAD)-controlled gene expression in gram-negative bacteria. Appl Environ Microbiol 79, 718–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher MP (2003) Degradation of stable RNA in bacteria. J Biol Chem 278, 45041–45044. [DOI] [PubMed] [Google Scholar]
- Dotsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, et al. (2012) The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One 7, e31092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T, Hansen AM, Zhi H, Blattner FR, and Jin DJ (2008) Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman R, Biswas T, Danhart EM, Foster MP, Tsodikov OV, and Artsimovitch I (2013) DksA2, a zinc-independent structural analog of the transcription factor DksA. FEBS Lett 587, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill EE, Chan LS, Winsor GL, Dobson N, Lo R, Ho Sui SJ, et al. (2018) High-throughput detection of RNA processing in bacteria. BMC Genomics 19, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Colman-Lerner A, Chin TE, Benjamin K., Yu RC, and Brent R (2007) Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat Methods 4, 175–181. [DOI] [PubMed] [Google Scholar]
- Guragain M, King MM, Williamson KS, Perez-Osorio AC, Akiyama T, Khanam S, et al. (2016) The Pseudomonas aeruginosa PAO1 two-component regulator CarSR regulates calcium homeostasis and calcium-induced virulence factor production through its regulatory targets CarO and CarP. J Bacteriol 198, 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagensen JA, Klausen M, Ernst RK, Miller SI, Folkesson A, Tolker-Nielsen T, et al. (2007) Differentiation and distribution of colistin- and sodium dodecyl sulfate-tolerant cells in Pseudomonas aeruginosa biofilms. J Bacteriol 189, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held K, Ramage E, Jacobs M, Gallagher L, and Manoil C (2012) Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J Bacteriol 194, 6387–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkin TM (2008) Riboswitch RNAs: Using RNA to sense cellular metabolism. Genes Dev 22, 3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. (2003) Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100, 14339–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Ishimoto K, and Lory S (1994) Nucleotide sequence of the rpoN gene and characterization of two downstream open reading frames in Pseudomonas aeruginosa. J Bacteriol 176, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DH, Franklin FC, and Thomas CM (1994) Molecular analysis of the operon which encodes the RNA polymerase sigma factor sigma 54 of Escherichia coli. Microbiology 140(Pt 5): 1035–1043. [DOI] [PubMed] [Google Scholar]
- Kato T, Yoshida H, Miyata T, Maki Y, Wada A, and Namba K (2010) Structure of the 100S ribosome in the hibernation stage revealed by electron cryomicroscopy. Structure 18, 719–724. [DOI] [PubMed] [Google Scholar]
- Khusainov I, Vicens Q, Ayupov R, Usachev K, Myasnikov A, Simonetti A, et al. (2017) Structures and dynamics of hibernating ribosomes from Staphylococcus aureus mediated by intermolecular interactions of HPF. EMBO J 36, 2073–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline BC, McKay SL, Tang WW, and Portnoy DA (2015) The Listeria monocytogenes hibernation-promoting factor is required for the formation of 100S ribosomes, optimal fitness, and pathogenesis. J Bacteriol 197, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler T, Alvarez JF, and Harayama S (1994) Regulation of the rpoN, ORF102 and ORF154 genes in Pseudomonas putida. FEMS Microbiol Lett 115 177–184. [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Leveau JH, and Lindow SE (2001) Predictive and interpretive simulation of green fluorescent protein expression in reporter bacteria. J Bacteriol 183, 6752–6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K (2007) Persister cells. Annu Rev Microbiol 64, 357–372. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, et al. (2011) ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, and Pier GB (2002) Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15, 194–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki Y, Yoshida H, and Wada A (2000) Two proteins, YfiA and YhbH, associated with resting ribosomes in stationary phase Escherichia coli. Genes Cells 5, 965–974. [DOI] [PubMed] [Google Scholar]
- Malone M, Bjarnsholt T, McBain AJ, James GA, Stoodley P, Leaper D, et al. (2017) The prevalence of biofilms in chronic wounds: A systematic review and meta-analysis of published data. J Wound Care 26, 20–25. [DOI] [PubMed] [Google Scholar]
- Nomura M, Gourse R, and Baughman G (1984) Regulation of the synthesis of ribosomes and ribosomal components. Ann Rev Biochem 53: 75–117. [DOI] [PubMed] [Google Scholar]
- Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, and Voyich JM (2010) SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J Infect Dis 201, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Osorio AC, Williamson KS, and Franklin MJ (2010) Heterogeneous rpoS and rhlR mRNA levels and 16S rRNA/rDNA (rRNA gene) ratios within Pseudomonas aeruginosa biofilms, sampled by laser capture microdis-section. J Bacteriol 192, 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Blaha GM, and Steitz TA (2012) How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336, 915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potvin E, Sanschagrin F, and Levesque RC (2008) Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev 32, 38–55. [DOI] [PubMed] [Google Scholar]
- Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, et al. (1993) A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, and Gourse RL (2016) ppGpp binding to a Site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol Cell 62, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Watanabe T, Maki Y, Ueta M, Yoshida H, Ito Y et al. (2009) Solution structure of the E. coli ribosome hibernation promoting factor HPF: Implications for the relationship between structure and function. Biochem Biophys Res Commun 389, 580–585. [DOI] [PubMed] [Google Scholar]
- Solovyev V, and Salamov A (2011) Automatic annotation of microbial genomes and metagenomic sequences In Metagenomics and its applications in agriculture, biomedicine and environmental studies. Li RW (ed). Nova Science Publishers, pp. 61–78. [Google Scholar]
- Stewart PS, and Costerton JW (2001) Antibiotic resistance of bacteria in biofilms. Lancet 358, 135–138. [DOI] [PubMed] [Google Scholar]
- Stewart PS, and Franklin MJ (2008) Physiological heterogeneity in biofilms. Nat Rev Microbiol 6, 199–210. [DOI] [PubMed] [Google Scholar]
- Stewart PS, Franklin MJ, Williamson KS, Folsom JP, Boegli L, and James GA (2015) Contribution of Stress Responses to Antibiotic Tolerance in Pseudomonas aeruginosa Biofilms. Antimicrob Agents Chemother 59, 3838–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. (2017) The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45(D1): D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KH, Wessel AK, Palmer GC, Murray JL, and Whiteley M (2015) Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A 112, 4110–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta M, Ohniwa RL, Yoshida H, Maki Y, Wada C, and Wada A (2008) Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli. J Biochem 143, 425–433. [DOI] [PubMed] [Google Scholar]
- Ueta M, Wada C, and Wada A (2010) Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells 15, 43–58. [DOI] [PubMed] [Google Scholar]
- Ueta M, Yoshida H, Wada C, Baba T, Mori H, and Wada A (2005) Ribosome binding proteins YhbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli. Genes Cells 10, 1103–1112. [DOI] [PubMed] [Google Scholar]
- Vila-Sanjurjo A, Schuwirth BS, Hau CW, and Cate JH (2004) Structural basis for the control of translation initiation during stress. Nat Struct Mol Biol 11, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Williamson KS, Richards LA, Perez-Osorio AC, Pitts B, McInnerney K, Stewart PS, and Franklin MJ (2012) Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of ribosome hibernation factors in the antibiotic-tolerant subpopulation and hypoxia-induced stress response in the metabolically active population. J Bacteriol 194, 2062–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Ueta M, Maki Y, Sakai A, and Wada A (2009) Activities of Escherichia coli ribosomes in IF3 and RMF change to prepare 100S ribosome formation on entering the stationary growth phase. Genes Cells 14, 271–280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.