

Abstract
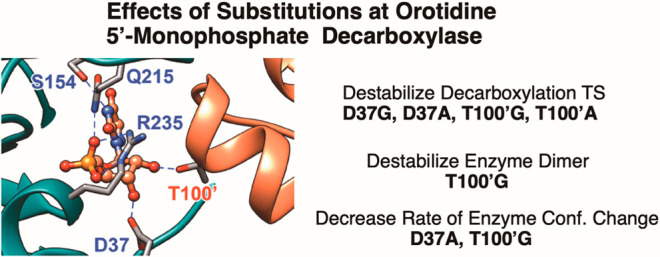
The D37 and T100′ side chains of orotidine 5′-monophosphate decarboxylase (OMPDC) interact with the C-3′ and C-2′ ribosyl hydroxyl groups, respectively, of the bound substrate. We compare the intra-subunit interactions of D37 with the inter-subunit interactions of T100′ by determining the effects of the D37G, D37A, T100′G, and T100′A substitutions on the following: (a) kcat and kcat/Km values for the OMPDC-catalyzed decarboxylations of OMP and 5-fluoroorotidine 5′-monophosphate (FOMP) and (b) the stability of dimeric OMPDC relative to the monomer. The D37G and T100′A substitutions resulted in 2 kcal mol–1 increases in ΔG† for kcat/Km for the decarboxylation of OMP, while the D37A and T100′G substitutions resulted in larger 4 and 5 kcal mol–1 increases, respectively, in ΔG†. The D37G and T100′A substitutions both resulted in smaller 2 kcal mol–1 decreases in ΔG† for the decarboxylation of FOMP compared to that of OMP. These results show that the D37G and T100′A substitutions affect the barrier to the chemical decarboxylation step while the D37A and T100′G substitutions also affect the barrier to a slow, ligand-driven enzyme conformational change. Substrate binding induces the movement of an α-helix (G′98–S′106) toward the substrate C-2′ ribosyl hydroxy bound at the main subunit. The T100′G substitution destabilizes the enzyme dimer by 3.5 kcal mol–1 compared to the monomer, which is consistent with the known destabilization of α-helices by the internal Gly side chains [Serrano, L., et al. (1992) Nature, 356, 453–455]. We propose that the T100′G substitution weakens the α-helical contacts at the dimer interface, which results in a decrease in the dimer stability and an increase in the barrier to the ligand-driven conformational change.
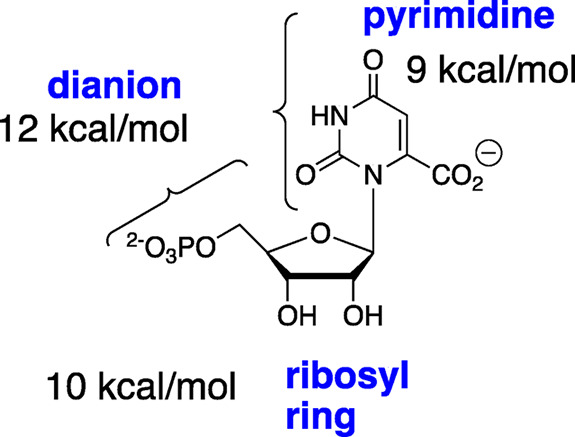
Orotidine 5′-monophosphate decarboxylase (OMPDC) is a dimeric enzyme composed of two identical monomers.1,2 The enzyme catalyzes the decarboxylation of OMP to form UMP through a vinyl carbanion reaction intermediate (Scheme 1).3−6 The enzyme provides an enormous 31 kcal mol–1 stabilization of the decarboxylation transition state.1,7,8 This stabilization has been partitioned into roughly equal contributions from protein interactions with the following three substrate fragments (Scheme 2):9 the nonreacting phosphodianion10 and ribosyl11 groups and the reacting pyrimidine ring.11 The binding interactions of the phosphodianion and ribosyl substrate pieces have been partitioned into interactions that are expressed at the Michaelis complex and interactions that only develop when approaching the reaction transition state.10−13
Scheme 1. OMPDC-Catalyzed Decarboxylation of OMP to Form a Vinyl Carbanion Reaction Intermediate.

Scheme 2. Partitioning of the Total 31 kcal mol–1 Stabilization of the Transition State for OMPDC Decarboxylation among the Three Contributing Substrate Fragments.
Scheme 3 shows the mechanism that provides the specificity for the binding interactions of the phosphodianion and the ribosyl hydroxyls at the decarboxylation transition state. The substrate binding interactions are utilized in order to drive an energetically unfavorable protein conformational change from the inactive open form of OMPDC (EO) to the active Michaelis complex (EC)12,13 where OMP is locked in a structured protein cage.14 The full intrinsic substrate binding energy, which is expressed by (Km)int for the binding of OMP to EC, is greater than the observed binding energy by the amount of binding energy that is utilized to drive the enzyme conformational change ((Km)int/Km = KC ≪ 1).15−17 The full intrinsic substrate binding energy is expressed at the rate determining transition state so that the value of kcat/Km for the enzyme turnover is the same as that for a second hypothetical reaction that proceeds through the closed enzyme complex EC·OMP without the requirement for an uphill enzyme conformational change.12,13
Scheme 3. Utilization of Intrinsic Substrate Binding Energy in Order to Drive an Unfavorable Enzyme Conformational Change.

We are working to define the roles of the active site amino acid side chains in OMPDC-catalyzed decarboxylation (Figure 1). We first examined the dianion gripper side chains Q215, Y217, and R235 as well as S154, which connects the phosphodianion gripper loop (P202–V220) to the pyrimidine umbrella loop (A151–T165).16−20 These side chain interactions provide a significant fraction of the driving force that activates the ligand-driven change in the enzyme conformation (Scheme 3), which results in the immobilization of two flexible protein loops by interactions with the substrate dianion and the pyrimidine ring.18,21
Figure 1.
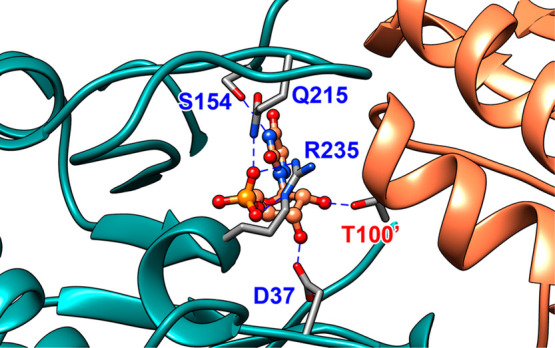
A representation (PDB 3GDL) of the interactions between the OMPDC active site side chains and the tight binding inhibitor 6-azauridine 5′-monophosphate (azaUMP) at the complex to the closed form of OMPDC (EC, Scheme 3). The enzyme active site is near the subunit interface, which is shown by the blue and orange shading of the two subunits.22 The inhibitor complex is stabilized by the following interactions: the Q215 and R235 side chains interact directly with the substrate dianion,17,20 the S154 side chain oxygen accepts a hydrogen bond from the pyrimidine −NH,20 the D37 side chain forms a hydrogen bond to the C-3′ ribosyl −OH, and the T100′ side chain from the second enzyme subunit forms a hydrogen bond to the C-2′ ribosyl −OH.18,23
In this paper, we focus on the interactions of the D37 and T100′ side chains with the C-3′ and C-2′ hydroxyls, respectively, of the substrate ribofuranosyl ring, where the D37 interaction is within a single enzyme subunit while the T100′ interaction spans the two subunits (Figure 1).2 Wolfenden and co-workers previously characterized the effects of D37A and T100′A substitutions on the kinetic parameters for the OMPDC-catalyzed decarboxylations of OMP and 2′-deoxyOMP.23 We revisit this problem and report the effects of a more extensive range of D37G, D37A, T100′A, and T100′G variants on the kinetic parameters for the OMPDC-catalyzed decarboxylations of OMP and 5-fluoroorotidine 5′-monophosphate (FOMP) and, for the first time, examine the effect of these substitutions on the association constant Kas for the formation of the active dimer of OMPDC from the inactive monomer.24 The D37G substitution is more conservative than that of D37A with respect to the effect on the activation barrier for the OMPDC-catalyzed decarboxylation. By contrast, the T100′A substitution is more conservative than that of T100′G by the same criteria, while the T100′G substitution results in an unusually large decrease in the stability of the active OMPDC dimer relative to the inactive monomer.
Methods
Materials
OMP25,26 and FOMP25,27 were prepared enzymatically by literature procedures. 3-(N-Morpholino)propanesulfonic acid (MOPS) and imidazole were purchased from Sigma (St. Louis, MO). Sodium hydroxide (1.0 N), hydrochloric acid (1.0 N), sodium chloride, and Amicon centrifugal filter units with a 10K molecular weight cutoff (MWCO) were purchased from Fisher (Hampton, NH). Nickel chloride hexahydrate was a generous gift from Prof. Andrew Murkin (University at Buffalo, Buffalo, NY). Chelating Sepharose Fast-Flow and Q-Sepharose were purchased from GE Healthcare (Marlborough, MA). Water was purified using a Milli-Q Academic purification system from EMD Millipore (Burlington, MA). All other commercial chemicals were reagent grade or better and were used without further purification.
Preparation of Wild Type and Variant Yeast Orotidine 5′-Monophosphate Decarboxylases
The plasmid pScODC-15b containing the gene encoding wild type orotidine 5′-monophosphate decarboxylase from Saccharomyces cerevisiae with an N-terminal His6 tag was available from earlier studies.20,28 The protein sequence differs from the published sequence for wild type OMPDC by the following substitutions: S2H, C155S, A160S, and N267D. The C155S substitution results in a more stable protein but does not affect the kinetic parameters or overall structure of the enzyme.29 Site-directed mutagenesis on pScODC-15b was carried out using the QuikChange II kit from Stratagene (San Diego, CA). The following primers, with the bold-face altered codons underlined, were used to prepare the new variant enzymes from wild type OMPDC: T100A:, GCT GAC ATT GGT AAT GCA GTC AAA TTG CAG TAC TCT GC; T100G, GCT GAC ATT GGT AAT GGA GTC AAA TTG CAG TAC TCT GCG GG; D37A, C TTG TGT GCT TCA TTG GCA GTT CGT ACC ACC AAG GAA TTA CTG G; and D37D, C TTG TGT GCT TCA TTG GGA GTT CGT ACC ACC AAG GAA TTA CTG G.
These variants of OMPDC were overexpressed after the transformation of Escherichia coli BL21 (DE3) with the appropriate plasmid. The proteins were purified as described previously.20 The N-terminal His6 tag was removed in the final step by treatment with thrombin, and the tag was separated from the protein by purification over a column of Q-Sepharose.20
Kinetic Parameters for the Decarboxylation of OMP and FOMP
The decarboxylation of OMP was monitored spectrophotometrically by following the decrease in the absorbance at 279 nm (0–0.12 mM OMP, Δε = 2400 M cm–1), 290 nm (0.12–0.48 mM OMP, Δε = 1620 M cm–1), and 295 nm (0.48–1.9 mM OMP, Δε = 840 M cm–1) as described in previous work.17 The decarboxylation of FOMP was monitored spectrophotometrically by following the decrease in absorbance at 282 nm (0.02–0.30 mM FOMP, Δε = 1380 M cm–1), 290 nm (0.3–0.5 mM FOMP, Δε = 1090 M cm–1), 295 nm (0.6 mM FOMP, Δε = 805 M cm–1), and 300 nm (0.9 mM FOMP, Δε = 490 M cm–1) as described in previous work.16
The initial velocity, v (M s–1), for the reaction of ≤10% of the substrate at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl) was determined as the slope of a linear plot of the absorbance vs time for reactions at several different values of [OMP] or [FOMP]. The decarboxylation of OMP that was catalyzed by the T100′G variant was monitored at 25 °C after mixing 80 μL of a 400 μM stock solution of enzyme variant with 0.920 μL of an assay solution that contained OMP in order to give final solutions of 32 μM OMPDC at pH 7.1 (30 mM MOPS) and I = 0.105 (NaCl). Similar procedures were followed at 25 °C in order to monitor the following: (a) the T100′A ([E] = 270 nM), D37A ([E] = 210 nM), and D37G ([E] = 110 nM) variant-catalyzed decarboxylations of FOMP at pH 7.1 (30 mM MOPS) and I = 0.105 (NaCl) at 279 nm and (b) the D37A ([E] = 60 nM) and D37G ([E] = 56 nM enzyme) variant-catalyzed decarboxylations of FOMP at pH 7.1 (30 mM MOPS) and I = 0.105 (NaCl) at 279 nm.16 The values of kcat and Km for the variant-catalyzed decarboxylation were determined from the nonlinear least-squares fits of plots of v/[E] (s–1) vs either [OMP] or [FOMP] to the Michaelis–Menten equation.
Apparent first-order rate constants kobs (s–1) for the T100′A ([E] = 130 nM, monitored at 282 nm) and T100′G ([E] = 35 μM, monitored at 292 nm) variants of the OMPDC-catalyzed decarboxylations of FOMP at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl) were determined from the fit to an exponential decay over at least 5 reaction half-life times at [FOMP] ≤ 0.1Km.18 The second-order rate constant, kcat/Km (M–1 s–1), for the variant OMPDC-catalyzed decarboxylation of FOMP was calculated from the relationship kcat/Km = kobs/[E].
Equilibrium Constants for Converting OMPDC Monomers to the Dimer
The initial velocity, v (M s–1), for the OMPDC-catalyzed decarboxylation of OMP was determined after ≥12-fold dilutions of a stock enzyme solution (0.5–6 μM, wild type OMPDC; 200–400 μM, T100′G variant; 2.6 μM, T100′A variant; or 5 μM, D37G variant) in order to give solutions at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl). The value of [v/[E]]obs determined over ≤10% of the reaction of total OMP increases with increasing values of [E] = [EM] + 2[ED] when dimeric OMPDC (ED) is in equilibrium with significant concentrations of the monomeric enzyme EM. The association constant Kas (eq 1 and Scheme 4) for the conversion of monomeric OMPDC (EM) to the dimer (ED) was determined from the nonlinear least-squares fit of the values of [v/[E]]obs against [E] to eq 2 (derived from Scheme 4), where [E] = [EM] + 2[ED]. The equation fD = 2[ED]/([EM] + 2[ED]) represents the fraction of OMPDC present as the dimer, and [v/[E]]max is the value of [v/[E]]obs for reactions at high values of [E] where OMPDC exists mainly in the dimeric form.30
| 1 |
| 2 |
Scheme 4. Conversion of the Inactive OMPDC Monomer (EM) to the Active Dimer (ED).

Results
The kinetic parameters for the variant OMPDC-catalyzed decarboxylations of OMP were determined at high [OMPDC], where the enzyme exists mainly as the dimer, so that v/[E] (s–1) was independent of [E]. The following are the Michaelis–Menten plots of v/[E] (s–1) against [OMP]: Figure 2 shows the decarboxylation of OMP catalyzed by the 32 μM T100′G variant and Figures S1A, S1B, and S1C show the decarboxylations of OMP catalyzed by the 110 nM D37G variant, the 210 nM D37A variant, and the 270 nM T100′A variant, respectively. The values of kcat and Km for the variant OMPDC-catalyzed decarboxylations of OMP, determined from the fits of these plots to the Michaelis–Menten equation, are reported in Table 1. The values of kcat and Km for the D37G (56 nM, Figure S2A) and D37A (60 nM, Figure S2B) variant-catalyzed decarboxylations of FOMP, determined from the fit of plots of v/[E] (s–1) against [FOMP] to the Michaelis–Menten equation, are reported in Table 1.
Figure 2.
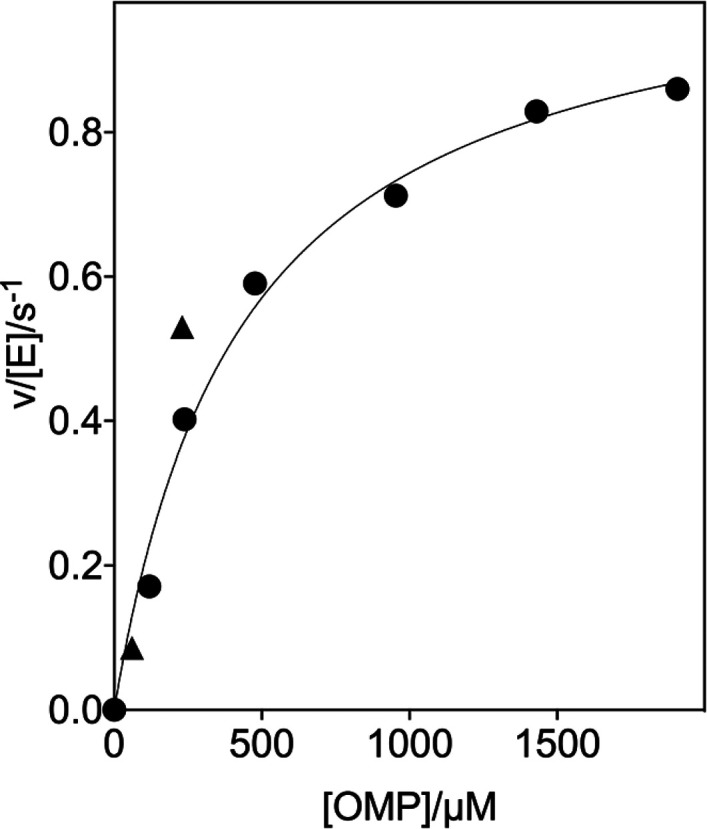
Dependence of v/[E] for the decarboxylation of OMP catalyzed by the T100′G variant of OMPDC for reactions at 25 °C, pH 7.1 (30 mM MOPS), I = 0.105 (NaCl), and 32 μM OMPDC (solid circles). The solid triangles show the limiting values of v/[E] determined for reactions catalyzed by high [OMPDC] at 60 μM (Figure 3A) and 230 μM OMP (Figure 3B).
Table 1. Kinetic Parameters for the Wild Type and Variant OMPDC-Catalyzed Decarboxylations of OMP and FOMP at pH 7.1 (10 mM MOPS), 25 °C, and I = 0.105 (NaCl).
| OMPa |
FOMPa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| OMPDC | kcat (s–1) | Km (M–1) | kcat/Km (M–1 s–1) | ΔΔG† (kcal mol–1)b | kcat (s–1) | Km (M–1) | kcat/Km (M–1 s–1) | ΔΔG† (kcal mol–1)b | ΔΔGOMP† – ΔΔGFOMP (kcal mol–1) |
| wild type | 15 ± 1 | (1.4 ± 0.2) × 10–6 | (1.1 ± 0.1) × 107 (6.3 × 107)c | 95 | 8 × 10–6 | 1.2 × 107 | 0.05 | ||
| D37A | 0.50 ± 0.03 | (4.2 ± 0.7) × 10–5 | (1.2 ± 0.2) × 104 (2.1 × 105)c | 4.0 ± 0.1 (3.4)c | 73 ± 15 | (1.1 ± 0.4) × 10–3 | (6.6 ± 2.5) × 104 | 3.0 ± 0.2 | 1.0 ± 0.3 |
| D37G | 4.2 ± 0.4 | (1.3 ± 0.3) × 10–5 | (3.2 ± 0.8) × 105 | 2.1 ± 0.2 | 134 ± 3 | (2.5 ± 0.2) × 10–5 | (5.4 ± 0.6) × 106 | 0.4 ± 0.1 | 1.7 ± 0.2 |
| T100′A | 3.3 ± 0.2 | (1.6 ± 0.4) × 10–5 | (2.1 ± 0.5) × 105 (1.1 × 106)c | 2.3 ± 0.2 (2.4)c | (4.3 ± 0.6) × 106 d | 0.6 ± 0.1 | 1.8 ± 0.2 | ||
| T100′G | 1.1 ± 0.1 | (4.4 ± 0.6) × 10–4 | (2.5 ± 0.4) × 103 | 5.0 ± 0.1 | (7.2 ± 0.2) × 103 d | 4.3 ± 0.1 | 0.6 ± 0.1 | ||
The kinetic parameters kcat and Km were determined from the nonlinear least-squares fits of plots of v/[E] against [OMP] or [FOMP] (Supporting Information) to the Michaelis–Menten equation, unless noted otherwise. The quoted uncertainty is the standard deviation obtained for the nonlinear least-squares fit of the experimental data.
The effect of the substitution on the activation barrier ΔG† for the wild type OMPDC-catalyzed decarboxylation reaction, calculated from the effect on kcat/Km.
Value was previously reported in ref (23).
kcat/Km = kobs/[E], where kobs is the observed first-order rate constant for the OMPDC-catalyzed decarboxylation determined at [FOMP] ≪ Km.
The decarboxylation of FOMP at [FOMP] ≪ Km that was catalyzed by the T100A′ (reaction of 0.16 mM FOMP monitored at 282 nm) and T100′G (reaction of 0.66 mM FOMP monitored at 297 nm) OMPDC variants was followed for >5 reaction half-life times. The apparent first-order rate constants kobs = 0.57 ± 0.01 s–1 (average of 3 runs) and kobs = 0.25 ± 0.01 s–1 (average of 2 runs) were determined from the fits of the exponential decay curves of absorbance vs time for the decarboxylations catalyzed by the T100′A ([E] = 130 nM) and T100′G ([E] = 35 μM) variants of OMPDC, respectively.16,18 The second-order rate constants kcat/Km (M–1 s–1) reported in Table 1 for the variant OMPDC-catalyzed decarboxylations of FOMP were calculated from the relationship kcat/Km = kobs/[E].
Association Constants for the Dimerization of OMPDC
Figure 3A shows the effect of increasing [E] on [v/[E]]obs for decarboxylation catalyzed by the T100′G variant of OMPDC at 25 °C, pH 7.1 (30 mM MOPS), I = 0.105 (NaCl), and 60 μM OMP, where [E] is the total concentration of the OMPDC monomer and [v/[E]]max is the limiting value observed when OMPDC exists essentially exclusively as the active dimer. The increase in [v/[E]]obs with increasing [E] is due to a shift in the position of the dimerization equilibrium (Kas, Scheme 4) toward the active dimer. Figure 3B shows the data for the decarboxylation of 230 μM OMP catalyzed by the T100′G variant. The solid lines for Figure 3A and B show the nonlinear least-squares fits of the experimental data to eq 2 (derived for Scheme 4)30 using values of Kas = 1.69 × 105 and 1.61 × 105 M–1, respectively, and [v/[E]]max = 0.086 ± 0.007 and 0.53 ± 0.05 s–1, respectively. The limiting values of [v/[E]]max determined for the decarboxylation of 60 and 230 μM OMP (Figure 3A and B) are shown by the solid triangles on the Michaelis–Menten plots (Figure 2) for the decarboxylation of OMP catalyzed by the T100′G variant.
Figure 3.

Effect of increasing concentrations of OMPDC on [v/[E]]obs for the decarboxylation of OMP catalyzed by the T100′G variant at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl). (A) The T100′G variant OMPDC-catalyzed decarboxylation of 60 μM OMP. (B) The T100′G variant-catalyzed decarboxylation of 230 μM OMP.
Figure 4A–C shows the effect of increasing [E] on [v/[E]]obs for the decarboxylation catalyzed by wild type OMPDC ([OMP] = 34 μM), the T100′A variant ([OMP] = 99 μM), and the D37G variant ([OMP] = 52 μM), respectively, at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl). The solid lines for Figure 4 show the nonlinear least-squares fit of the experimental data to eq 2 (derived from Scheme 4) using values of Kas reported in Table 2 and values of [v/[E]]max = 23 ± 2, 2.4 ± 0.1, and 3.1 ± 0.6 s–1 for the reactions catalyzed by the wild type and T100′A and D37G variants of OMPDC, respectively. The limiting values of [v/[E]]max determined for these variant enzyme-catalyzed decarboxylation reactions are shown as solid triangles on the following Michaelis–Menten plots: Figure S1A for the D37G variant ([OMP] = 52 μM) and Figure S1C for the T100′A variant ([OMP] = 99 μM).
Figure 4.

Effect of increasing concentrations of OMPDC on [v/[E]]obs for the decarboxylation of OMP catalyzed by the wild type and variant enzymes at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl). The solid line shows the fit of the experimental data to eq 2 where [E] is the concentration of the OMPDC monomers. (A) The wild type OMPDC-catalyzed decarboxylation reaction with 34 μM OMP. (B) The T100′A variant-catalyzed decarboxylation reaction of 99 μM OMP. (C) The D37G variant-catalyzed decarboxylation reaction of 52 μM OMP.
Table 2. Association Constants Kas (Scheme 4) for the Conversion of Monomeric Wild Type and Variant OMPDC to the Dimer Form at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl).
| enzyme | Kas (M–1) | ΔΔGo (kcal mol–1)a |
|---|---|---|
| wild type | (5.9 ± 0.6) × 107 b,c | |
| T100′G | (1.6 ± 0.6) × 105 d | 3.5 ± 0.2 |
| T100′A | (5.7 ± 0.6) × 107 c,e | <0.1 ± 0.1 |
| D37G | (1.4 ± 0.8) × 107 c,f | 0.9 ± 0.4 |
The effect of amino acid substitution on ΔGo for the association of OMPDC monomers in order to form the dimer.
Determined from the nonlinear least-squares fit of data from Figure 4A to eq 2 (derived from Scheme 4).
The quoted uncertainty is the standard deviation from the fitted correlation.
The average of the values of Kas determined for the T100′G variant-catalyzed decarboxylation of 60 μM (Figure 3A) and 230 μM (Figure 3B) OMP.
Discussion
Variant Enzyme-Catalyzed Decarboxylation of OMP and FOMP
The values of kcat/Km for the wild type and variant OMPDC-catalyzed decarboxylations of OMP at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl) reported in Table 1 are ca. 10-fold smaller than the values reported by Wolfenden and co-workers for the decarboxylation in solutions that contain no NaCl.23 Similar salt effects have been noted in earlier works.17,25 The 4.0 and 2.3 kcal mol–1 differences in the activation barriers ΔG† for the decarboxylations catalyzed by the D37A and T100′A variants, respectively, compared to that of wild type OMPDC (Table 1) are similar to the 3.4 and 2.4 kcal mol–1 (Table 1) differences from earlier work.23 Finally, the value for the association constant Kas = 6 × 107 M–1 reported in Table 2 is 15-fold larger than Kas = 4 × 106 M–1 for the dimerization of OMPDC at pH 7.2, 0.01 M MOPS buffer, and no NaCl, which was determined by a different method. A similar increase in the value of Kas with an increase in NaCl concentration was noted in ref (24).
X-ray crystal structures for the complexes formed between wild type yeast OMPDC and the tightly bound inhibitors show the D37 side chain from the main subunit hydrogen bonded to the C-3′ ribosyl-hydroxyl, and the T100′ side chain from the second subunit hydrogen bonded to the C-2′-hydoxyl.2,22 The contribution of these interactions to the stabilization of the decarboxylation transition state can be estimated by examining the enzyme variants that eliminate the interaction. Table 1 highlights the difficulties associated with this analysis. A comparison of the effects of the Gly and Ala substitutions on ΔG† for the wild type OMPDC-catalyzed decarboxylation of OMP (Table 1) shows that Gly is the more conservative substitution at D37 (ΔΔG† = 2.1 and 4.0 kcal mol–1 for the D37G and D37A variants, respectively) while Ala is the more conservative substitution at T100′ (ΔΔG† = 5.0 and 2.3 kcal mol–1 for the T100′G and T100′A variants, respectively). The results are consistent with the conclusion that the D37 and T100′ side chains each contribute ca. 2 kcal mol–1 to the total 31 kcal mol–1 transition state stabilization.
The decarboxylation of enzyme-bound OMP (kchem, Scheme 5) in order to form the vinyl carbanion reaction intermediate is partly rate determining for the decarboxylation of OMP.24 The 5-F of FOMP provides a ca. 4 kcal mol–1 stabilization of the vinyl carbanion-like transition state for kchem for the OMPDC-catalyzed decarboxylation16,31 but leads to only 1.1- and 6-fold increases in kcat/Km and kcat, respectively, for the decarboxylation of FOMP. This result shows that the large effect of the strongly electron-withdrawing 5-F group on kchem is only weakly expressed at the virtual transition state for kc (which is rate determining for kcat/Km) or k–c′ (which is rate determining for kcat) for the OMPDC-catalyzed decarboxylation of FOMP.16
Scheme 5. A Kinetic Scheme for OMPDC That Separates the Steps for Ligand Binding and the Enzyme Conformational Change from Open EO·S to Closed EC·S.

Amino acid substitutions have different effects on the values of ΔG† for the OMPDC-catalyzed decarboxylations of OMP and FOMP when the substitutions cause different changes to the barriers to kchem and (kc, k′–c), respectively, that control the activation barriers for the OMPDC-catalyzed decarboxylation of these substrates.16−18 For example, the conservative T100′A and D37G substitutions result in only small 0.5–0.6 kcal mol–1 increases in ΔG† to kcat/Km for the decarboxylation of FOMP and larger 2.1–2.3 kcal mol–1 increases in the barrier to the decarboxylation of OMP (Table 1). These results show that the T100′A and D37G substitutions cause only small changes in the barriers to kc, and that most of their effect (≈2 kcal mol–1) is on the barrier to kchem for the rate determining decarboxylation of the enzyme-bound substrate.
By comparison, the T100′G and D37A substitutions result in 5.0 and 4.0 kcal mol–1 increases, respectively, in the activation barrier ΔG† to kcat/Km for the enzyme-catalyzed decarboxylation of OMP, and in similar 4.4 and 3.1 kcal mol–1 increases in the barrier for the decarboxylation of FOMP (Table 1). This provides strong evidence that the effects of these substitutions are due largely to changes in the barrier to kc, which is partly rate determining for the OMPDC-catalyzed decarboxylation of OMP and strongly rate determining for the decarboxylation of FOMP.16
The small range of values of kcat determined for the decarboxylation of OMP catalyzed by the four variants (0.5–4.2 s–1) shows that these substitutions result in similar increases in ΔG† (kchem) for the decarboxylation of enzyme-bound OMP. We concluded that the D37A/G and T100′A/G substitutions each result in significant increases in ΔG† for the decarboxylation of OMP, and that the larger effects of the D37A and T100′G substitutions on the barrier to kcat/Km for the decarboxylation of OMP (Table 1) are due to their additional effects on the barrier to the enzyme conformational change (kc), which is partly rate determining for kcat/Km.24 The D37G and D37A substitutions result in small <2-fold changes in the value of kcat for the decarboxylation of FOMP. This shows that the substitutions result in only small changes in the barrier to k–c′, which is rate determining for kcat.
The T100′ side chain lies close to the N-terminal end of the α-helix (G′98–S′106, Figure 1).2 Substrate binding induces the movement of this α-helix toward the ribosyl ring at the main subunit and gives rise to an interaction between the T100′ side chain and the substrate C-2′ ribosyl hydroxyl. Internal Gly side chains are known to destabilize α-helices relative to Ala,32,33 which is consistent with the proposal that the larger barrier to kc for the T100′G variant compared to the T100′A variant is associated with the effect of the T100′G substitution on the helix stability.
The D37 side chain lies at the main subunit and interacts with an enzyme-bound water molecule (Figure 5) that bridges D37 and the substrate phosphodianion. A major driving force for the large substrate-induced enzyme conformational change is the formation of interactions between the phosphodianion of the substrate and the Q215, Y217, and R235 side chains. We suggest that the D37A and D37G substitutions cause different perturbations in the packing of the water that bridges D37 and the phosphodianion at wild type OMPDC, which are manifested as differences in the effects of these substitutions on kc for the enzyme conformational change (Scheme 5).
Figure 5.
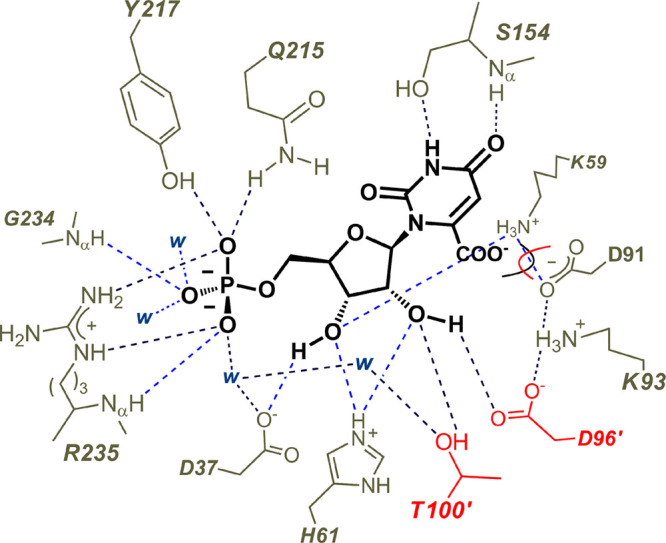
A pancake representation of the interactions between the amino acid side chains of OMPDC and the bound substrate OMP (PDB 1DQX, but with OMP inserted into the position of the 6-hydroxyuridine 5′-monophosphate inhibitor). The D96′ and T100′ side chains from the second enzyme subunit are shaded red. The D37 side chain is hydrogen bonded to the C-3′ ribosyl hydroxyl and interacts with the substrate phosphodianion through an intervening water molecule. The interactions of the substrate phosphodianion with the Q215, Y217, and R235 side chains develop during the conformational change from the open unliganded enzyme EO to the closed Michaelis complex EC·OMP (Scheme 3).
Effects of Substitutions on the Stability of Dimeric OMPDC
OMPDC is a homodimer with an active site that extends across the dimer interface. Most of the side chains that interact with the phosphodianion, ribosyl, and pyrimidine fragments of bound OMP (Figure 5) are located at a single subunit monomer; however, the T100′ and D96′ side chains are located at the second subunit (Figure 5). The dissociation of the dimer to the monomer gives rise to a protein that shows no activity for the decarboxylation of OMP24 so that the enzyme activity v/[E] is proportional to the fraction of OMPDC present as the dimer (fD, eq 2).
Table 2 reports the values of the association constant Kas for the dimerization of OMPDC monomers determined from the fits to eq 2 using data from Figures 3 and 4. The T100′A substitution results in little change in the value of Kas determined for wild type OMPDC, while the T100′G substitution results in a 3.5 kcal mol–1 destabilization of the active OMPDC dimer relative the inactive monomer. These results are consistent with a destabilization of the dimeric form of the T100′G variant due to the structural perturbation of the G′98–S′106 α-helix, which lies at the dimer interface. The 300-fold larger Km value for the dissociation of OMP from the T100′G variant compared to wild type OMPDC (Table 1) is also consistent with the large effect of this substitution on protein structure, which results in a decrease in the observed OMP binding energy. The smaller 15-fold effect of the T100′G substitution on the value of kcat for the decarboxylation of OMP (Table 1) suggests that OMP binds selectively to the small fraction of OMPDC with the wild type structure for the G′98–S′106 α-helix and that ca. 3 kcal mol–1 of the intrinsic OMP binding energy is utilized in order to restore this helix to the wild type conformation at the Michaelis complex to OMP.15 Additionally, the transition state for the decarboxylation of OMP bound to the T100′G variant is destabilized by 2 kcal mol–1 by the loss of the hydrogen bond to the C-2′ ribosyl hydroxyl.
The Partitioning of the OMPDC Active Site between Two Enzyme Subunits
Figure 6A shows the X-ray crystal structure for the open form of yeast OMPDC, but with a hypothetical 6-azauridine 5′-monophosphate (azaUMP) ligand at the position observed for the closed liganded enzyme.22Figure 6B shows the X-ray crystal structure of the closed form of OMPDC determined for the azaUMP complex, but with only one of the two bound ligands shown. Our previous work highlighted the ligand-driven movement of the phosphodianion gripper loop (P202–V220) toward the pyrimidine umbrella loop (A151–T165) and the clamping interaction between the −CH2OH and amide side chains of S154 and Q215,18,20 respectively, which forms a bridge between the two enzyme loops.2,22 We now note that the second subunit undergoes a large hinge motion upon ligand binding that is partly or entirely driven by interactions of the G′98–S′106 α-helix with the C-2′ ligand hydroxyl (the T100′ side chain) and the mobile pyrimidine umbrella loop (A151–T165) from the main subunit.2,22 In other words, the collective motion of two loops from the main subunit and the G′98–S′106 α-helix at the second subunit is driven by the formation of numerous stabilizing contacts with the bound substrate and intra-subunit interactions between the pyrimidine umbrella loop and the G′98–S′106 α-helix.
Figure 6.
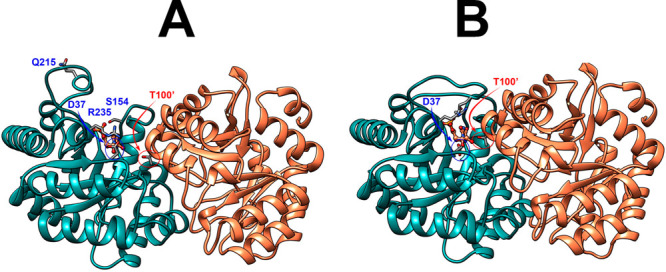
Representations of the open (A, PDB 3GDK) and the closed or liganded (B, PDB 3GDL) forms of yeast OMPDC where the azaUMP ligand is placed at structure A at the position determined for structure B. These representations show the movements of the phosphodianion gripper loop (P202–V220) toward the pyrimidine umbrella loop (A151–T165) and R235 toward the phosphodianion, as well as the movement of the G′98–S′106 α-helix from the second subunit toward the bound substrate and the pyrimidine umbrella of the main subunit. The bridging interaction between the −CH2OH side chain of S154 and the amide side chain of Q215 at the closed enzyme is not shown.
Substrate and allosteric binding sites are positioned at the subunit interfaces of phosphofructokinase; this has been linked to the allosteric activation and inhibition of the enzyme-catalyzed phosphorylation of fructose 6-phosphate by ATP in order to form 1,6-fructose diphosphate.34,35 We note that OMP binding to the “main” subunit of OMPDC induces the movement of the unoccupied subunit toward the closed enzyme conformation, and that this may result in an increase in the affinity for substrate binding to the second subunit. Such cooperativity in substrate binding has not been reported for OMPDC,17,18,23,24,36 but it is not clear that the data are of sufficient quality to rigorously demonstrate a similar affinity for the binding of the first and second substrate. The question of whether the large ligand-driven conformational change of OMPDC, which encompasses the two enzyme subunits, results in cooperativity in the substrate binding has important implications with respect to the mechanism of action of OMPDC and is deserving of further study.
Conclusions
The effects of the substitutions of D37 and T100′ on the activity of the OMPDC-catalyzed decarboxylations of OMP and FOMP reflect changes in the barriers to the formation of the decarboxylation transition state and to a rate determining enzyme conformational change. The D37A/G and T100′A/G mutations each resulted in a ca. 2 kcal mol–1 increase in the reaction barrier, which was proposed to be equal to the stabilization provided by the lost hydrogen bonds to the C-3′ and C-2′ ribosyl hydroxyls, respectively. The D37A and T100′A substitutions resulted in an additional 2–3 kcal mol–1 increase in the barrier to a slow enzyme conformational change.16 These results demonstrate the imperatives for OMPDC to minimize the barriers to both the formation of the decarboxylation transition state and the complex conformational change from the inactive open enzyme EO to the active closed Michaelis complex EO·OMP (Scheme 5).16 The T100′G substitution results in a 3.5 kcal mol–1 destabilization of dimeric OMPDC relative to the OMPDC monomer. We attribute this to the destabilization of the G′98–S′106 α-helix, which sits at the dimer interface. We propose that an ordered α-helix is also required for the efficient catalysis of the decarboxylation, and that the barrier to restoring the native α-helical structure contributes to the activation barrier for the rate determining conformational change for the T100′G variant.
Acknowledgments
We acknowledge Holly Ellis and Brian Miller for their helpful discussion.
Glossary
Abbreviations
- OMPDC
orotidine 5′-monophosphate decarboxylase
- OMP
orotidine 5′-monophosphate
- UMP
uridine 5′-monophosphate
- FOMP
5-fluoroorotidine 5′-monophosphate
- FUMP
5-fluorouridine 5′-monophosphate
- MOPS
3-(N-morpholino)propanesulfonic acid
- GlyGly
glycyl glycine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00241.
Michaelis–Menten plots of v/[E] vs [OMP] for the decarboxylation of OMP catalyzed by the D37G, D37A, and T100′A variants of OMPDC (Figures S1A–S1C, respectively) and Michaelis–Menten plots of v/[E] vs [FOMP] for the decarboxylation of FOMP catalyzed by the D37G and D37A variants of OMPDC (Figures S2A and S2B, respectively) (PDF)
Accession Codes
Yeast orotidine 5′-monophosphate decarboxylase (UnitProt P03962).
We acknowledge the National Institutes of Health Grants GM116921 and GM134881 for their generous support of this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Miller B. G.; Wolfenden R. (2002) Catalytic proficiency: the unusual case of OMP decarboxylase. Annu. Rev. Biochem. 71, 847–885. 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. (2000) Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Richard J. P. (2019) Role of the Carboxylate in Enzyme-Catalyzed Decarboxylation of Orotidine 5′-Monophosphate: Transition State Stabilization Dominates Over Ground State Destabilization. J. Am. Chem. Soc. 141, 13468–13478. 10.1021/jacs.9b04823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2011) OMP Decarboxylase: Phosphodianion Binding Energy Is Used To Stabilize a Vinyl Carbanion Intermediate. J. Am. Chem. Soc. 133, 6545–6548. 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. (2012) Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J. Am. Chem. Soc. 134, 14580–14594. 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. (2008) Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J. Am. Chem. Soc. 130, 1574–1575. 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R.; Snider M. J. (2001) The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. (1995) A proficient enzyme. Science 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Reyes A. C. (2018) Orotidine 5′-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis. Acc. Chem. Res. 51, 960–969. 10.1021/acs.accounts.8b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. (2005) Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. (2017) Enzyme Architecture: Erection of Active Orotidine 5′-Monophosphate Decarboxylase by Substrate-Induced Conformational Changes. J. Am. Chem. Soc. 139, 16048–16051. 10.1021/jacs.7b08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. (2019) Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc. 141, 3320–3331. 10.1021/jacs.8b10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2013) Specificity in transition state binding: The Pauling model revisited. Biochemistry 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. (2014) Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (2006) Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2015) Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. (2014) Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Plache D. C.; Koudelka A. P.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2018) Enzyme Architecture: Breaking Down the Catalytic Cage that Activates Orotidine 5′-Monophosphate Decarboxylase for Catalysis. J. Am. Chem. Soc. 140, 17580–17590. 10.1021/jacs.8b09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. (2012) Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. (2008) Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. (2010) A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. (2009) Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry 48, 5518–5531. 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Butterfoss G. L.; Short S. A.; Wolfenden R. (2001) Role of Enzyme-Ribofuranosyl Contacts in the Ground State and Transition State for Orotidine 5′-Phosphate Decarboxylase: A Role for Substrate Destabilization?. Biochemistry 40, 6227–6232. 10.1021/bi0028993. [DOI] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. (2000) Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2013) Role of a Guanidinium Cation–Phosphodianion Pair in Stabilizing the Vinyl Carbanion Intermediate of Orotidine 5′-Phosphate Decarboxylase-Catalyzed Reactions. Biochemistry 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vleet J. L.; Reinhardt L. A.; Miller B. G.; Sievers A.; Cleland W. W. (2008) Carbon isotope effect study on orotidine 5′-monophosphate decarboxylase: support for an anionic intermediate. Biochemistry 47, 798–803. 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- Gross A.; Abril O.; Lewis J. M.; Geresh S.; Whitesides G. M. (1983) Practical synthesis of 5-phospho-D-ribosyl α-1-pyrophosphate (PRPP): enzymatic routes from ribose 5-phosphate or ribose. J. Am. Chem. Soc. 105, 7428–7435. 10.1021/ja00363a037. [DOI] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. (2009) Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry 48, 5510–5517. 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers A.; Wolfenden R. (2005) The effective molarity of the substrate phosphoryl group in the transition state for yeast OMP decarboxylase. Bioorg. Chem. 33, 45–52. 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Carlier M. F.; Pantaloni D. (1978) Slow association-dissociation equilibrium of NADP-linked isocitrate dehydrogenase from beef liver in relation to catalytic activity. Eur. J. Biochem. 89, 511–516. 10.1111/j.1432-1033.1978.tb12555.x. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. (2013) Catalysis by Orotidine 5′-Monophosphate Decarboxylase: Effect of 5-Fluoro and 4′-Substituents on the Decarboxylation of Two-Part Substrates. Biochemistry 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. A.; Alonso D. O. V.; Sato S.; Fersht A. R.; Daggett V. (2007) Conformational entropy of alanine versus glycine in protein denatured states. Proc. Natl. Acad. Sci. U. S. A. 104, 2661–2666. 10.1073/pnas.0611182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano L.; Neira J. L.; Sancho J.; Fersht A. R. (1992) Effect of alanine versus glycine in alpha-helices on protein stability. Nature 356, 453–455. 10.1038/356453a0. [DOI] [PubMed] [Google Scholar]
- Fenton A. W.; Reinhart G. D. (2009) Disentangling the Web of Allosteric Communication in a Homotetramer: Heterotropic Inhibition in Phosphofructokinase from Escherichia coli. Biochemistry 48, 12323–12328. 10.1021/bi901456p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton A. W.; Paricharttanakul N. M.; Reinhart G. D. (2004) Disentangling the web of allosteric communication in a homotetramer: heterotropic activation in phosphofructokinase from Escherichia coli. Biochemistry 43, 14104–14110. 10.1021/bi048569q. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Snider M. J.; Short S. A.; Wolfenden R. (2000) Contribution of Enzyme-Phosphoribosyl Contacts to Catalysis by Orotidine 5′-Phosphate Decarboxylase. Biochemistry 39, 8113–8118. 10.1021/bi000818x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.