

ABSTRACT
DNA repair is essential for cell survival. In all domains of life, error-prone and error-free repair pathways ensure maintenance of genome integrity under stress. Mutagenic, low-fidelity repair mechanisms help avoid potential lethality associated with unrepaired damage, thus making them important for genome maintenance and, in some cases, the preferred mode of repair. However, cells carefully regulate pathway choice to restrict activity of these pathways to only certain conditions. One such repair mechanism is translesion synthesis (TLS), where a low-fidelity DNA polymerase is employed to synthesize across a lesion. In bacteria, TLS is a potent source of stress-induced mutagenesis, with potential implications in cellular adaptation as well as antibiotic resistance. Extensive genetic and biochemical studies, predominantly in Escherichia coli, have established a central role for TLS in bypassing bulky DNA lesions associated with ongoing replication, either at or behind the replication fork. More recently, imaging-based approaches have been applied to understand the molecular mechanisms of TLS and how its function is regulated. Together, these studies have highlighted replication-independent roles for TLS as well. In this review, we discuss the current status of research on bacterial TLS, with emphasis on recent insights gained mostly through microscopy at the single-cell and single-molecule level.
Keywords: bacteria, translesion synthesis, error-prone polymerases, mutagenesis, DNA repair, live-cell imaging, single-molecule microscopy
In this review we highlight the application of microscopy-based approaches to unravel the in vivo mechanism and regulation of bacterial translesion synthesis.
ABBREVIATIONS
- Translesion synthesis
(TLS)
- Nucleotide excision repair
(NER)
- Single-particle tracking
(spt)
- Photoactivable localization microscopy
(PALM)
- Total internal reflection fluorescence microscopy
(TIRF)
INTRODUCTION
Faithful duplication and segregation of genetic material is fundamental to life. In bacteria, DNA replication initiates at the ‘origin’ of replication and the replication machinery synthesizes DNA bi-directionally until the ‘terminus’ where the process is completed (Reyes-Lamothe and Sherratt 2019). Both initiation and termination are highly regulated events. For example, initiation is coupled to nutrient status in some bacteria (for example, via sensing ATP levels), while cell division can be co-regulated with replication termination and chromosome dimer resolution (Badrinarayanan, Le and Laub 2015; Leslie et al. 2015; Katayama, Kasho and Kawakami 2017; Kleckner et al. 2018). Replication is carried out by a multi-subunit machinery called the ‘replisome’. A fundamental component of the replisome is the replicative polymerase (Pol III in most bacteria) that faithfully copies DNA with the help of the processivity factor, the β-clamp (McHenry 2011a; Beattie and Reyes-Lamothe 2015; Xu and Dixon 2018). Pol III has a high synthesis rate of ∼550–750 nucleotides per second at 37°C in Escherichia coli (Pham et al. 2013). Additionally, proofreading activity of the polymerase minimizes base misincorporations. Errors that arise despite this are fixed by mismatch repair, thus maintaining a robust check on mutation rates (Kunkel and Erie 2005, 2015; Jiricny 2013), which are estimated to be in the range of 10−9–10−10 in optimal growth conditions (Foster et al. 2015; Schroeder et al. 2018).
DNA damage from endogenous and exogenous sources, including reaction intermediates of biochemical pathways or UV rays (Fu, Calvo and Samson 2012; Ippoliti et al. 2012; Fuchs and Fujii 2013), can hamper basic cellular processes including transcription and replication (Cox et al. 2000; Cohen, Godoy and Walker 2009; Lang and Merrikh 2018). Under such conditions, replisome progression can be perturbed. For example, studies have observed that mitomycin C-mediated lesions lead to dissociation of the replicative polymerase, PolC, in Bacillus subtilis (Li et al. 2019), while UV exposure causes delay in replication (Rudolph, Upton and Lloyd 2007) and replisome slowdown in E. coli (Soubry, Wang and Reyes-Lamothe 2019). This slowdown is attributed to the inability of Pol III to accommodate bulky DNA modifications and synthesize across them (Yang and Gao 2018). Several outcomes have been proposed for replisome stalling at a lesion: (i) the replicative polymerase is replaced by a low-fidelity polymerase that synthesizes across the lesion, (ii) the replisome skips the lesion, re-primes downstream and continues DNA synthesis or (iii) the replisome disassembles, which can result in a double-strand break (Cox et al. 2000; Yeeles and Marians 2011, 2013; Gabbai, Yeeles and Marians 2014). Situations that perturb replication progression lead to accumulation of single-stranded DNA, which could subsequently elicit the SOS response in bacteria (Cohen et al. 2008; Baharoglu and Mazel 2014; Marians 2018).
Activation of the SOS response leads to expression of multiple repair-associated genes including various mutagenic DNA polymerases that synthesize across DNA lesions in a process called translesion synthesis (TLS). Unlike the replicative polymerase, TLS polymerases often lack proofreading activity and have structural conformations suitable for accommodating bulky lesions, thus enabling DNA synthesis across them (Napolitano et al. 2000; Fuchs and Fujii 2013; Baharoglu and Mazel 2014; Fitzgerald, Hastings and Rosenberg 2017; Yang and Gao 2018). Stress-induced mutagenesis is a potent source for increase in bacterial mutation rates (Fitzgerald, Hastings and Rosenberg 2017; Fitzgerald and Rosenberg 2019) and hence, processes such as TLS are tightly regulated through temporal control of expression of the TLS polymerases and, in some cases, via post-translational modifications of its components specifically in response to damage. In addition, biochemical as well as structural constraints of TLS polymerases limit DNA access and synthesis (Yeiser et al. 2002; Vaisman, McDonald and Woodgate 2012; Corzett, Goodman and Finkel 2013; Robinson et al. 2015; Jaszczur et al. 2016; Vaisman and Woodgate 2017; Yang and Gao 2018; Chang et al. 2019). The types and frequency of mutations vary based on the class of TLS polymerase as well as the type of DNA lesion the polymerase bypasses (summarized in Table 1).
Table 1.
Bacterial translesion synthesis repair polymerases.
| Polymerase | Lesions | Mutations | Other bacteria studied | References |
|---|---|---|---|---|
| Pol II B-family (E. coli) | G-AAF εC | -2 frameshifts C:G → T:A | Salmonella typhimurium | (Paz-Elizur et al. 1996; Napolitano et al. 2000; Becherel and Fuchs 2001; Al Mamun and Humayun 2006; Koskiniemi and Andersson 2009; Fuchs and Fujii 2013; Goodman and Woodgate 2013) |
| Pol IV (DinB) Y-family (E. coli) | G-AAF ROS NFZ 4-NQ0 BaP MMS | -1 frameshifts A:T → G:C | S. typhimurium B. subtilis (UvrX, YqjH)C. crescentusMycobacterium smegmatis Mycobacterium tuberculosis Pseudomonas aeruginosa Pseudomonas putida | (Kim et al. 2001; Boshoff et al. 2003; Galhardo et al. 2005; Jarosz et al. 2006; Sanders et al. 2006; Bjedov et al. 2007; Koskiniemi and Andersson 2009; Hori et al. 2010; Kana et al. 2010; Williams, Hetrick and Foster 2010; Ippoliti et al. 2012; Sharma and Nair 2012; Fuchs and Fujii 2013; Kath et al. 2014; Ordonez and Shuman 2014; Million-Weaver et al. 2015; Jatsenko et al. 2017; Thrall et al. 2017; Lang and Merrikh 2018; Johnson, Kottur and Nair 2019) |
| Pol V (UmuDC) Y-family (E. coli) | UV G-AAF ROS | T:A → C:G | S. typhimurium B. subtilis (YqjW) | (Tang et al. 2000; Duigou et al. 2004; Fujii, Gasser and Fuchs 2004; Neeley et al. 2007; Koskiniemi and Andersson 2009; Ippoliti et al. 2012; Fuchs and Fujii 2013) |
| DnaE2 (ImuC) C-family C. crescentus | UV MMC | G:C → C:G G:C → A:T | M. tuberculosis M. smegmatis B. subtilis P. aeruginosa P. putida Myxococcus xanthus | (Boshoff et al. 2003; Le Chatelier et al. 2004; Galhardo et al. 2005; Warner et al. 2010; Ippoliti et al. 2012; Fuchs and Fujii 2013; Lopes-Kulishev et al. 2015; Jatsenko et al. 2017; Martins-Pinheiro et al. 2017; Peng et al. 2017) |
Abbreviations used in the table: N 2-deoxyguanosine-acetylaminofluorene (G-AAF), N4-ethenocytosine (εC), reactive oxygen species (ROS), nitrofurazone (NFZ), 4-nitroquinoline N-oxide (4-NQO), benzo(a)pyrene (BaP), methyl methanesulfonate (MMS), mitomycin C (MMC).
The best characterized bacterial TLS polymerases are the B-family polymerase Pol II, and the Y-family polymerases Pol IV (DinB) and Pol V (UmuD'2C) from E. coli (Napolitano et al. 2000; Goodman 2002; Cohen et al. 2008; Sutton 2010; Ippoliti et al. 2012; Vaisman, McDonald and Woodgate 2012; Fuchs and Fujii 2013; Goodman and Woodgate 2013; Thrall et al. 2017; Henrikus, van Oijen and Robinson 2018). In contrast to E. coli, B. subtilis utilizes a distinct set of error-prone polymerases: (i) DnaE, the lagging-strand polymerase for primer synthesis, is SOS-inducible and participates in mutagenic repair (Dervyn et al. 2001; Le Chatelier et al. 2004). (ii) Two Y-family polymerases, YqjH (PolY1) and YqjW (PolY2), contribute specifically to UV-induced mutagenesis (Sung et al. 2003). (iii) Additionally, YqjH is also responsible for stationary phase mutagenesis and lagging-strand mutagenesis associated with transcription-replication collisions (Sung et al. 2003; Million-Weaver et al. 2015; Lang and Merrikh 2018). Several bacteria with GC-rich genomes, such as Mycobacterium, Pseudomonas and Caulobacter, possess two C-family polymerases: constitutively expressed Pol III (DnaE), which performs high-fidelity replication, and damage-induced TLS polymerase, DnaE2, which is responsible for bypass of UV- and MMC-induced lesions (Boshoff et al. 2003; Galhardo et al. 2005; Warner et al. 2010; McHenry 2011b; Lopes-Kulishev et al. 2015) (See Table 1).
We refer readers to excellent comprehensive reviews over the last decade for additional information on types, occurrence, substrate preferences and mutational impact of TLS polymerases (Ippoliti et al. 2012; Vaisman, McDonald and Woodgate 2012; Fuchs and Fujii 2013; Goodman and Woodgate 2013; Vaisman and Woodgate 2017; Henrikus, van Oijen and Robinson 2018; Yang and Gao 2018). Here, we will discuss the mechanism(s) of TLS and its interplay with replication, with emphasis on insights recently gained from imaging-based approaches (examples of tools used for visualizing these dynamics are provided in Fig. 1 and referred in specific contexts in later sections of this review). Most of these studies have focused their efforts on TLS polymerases in E. coli. TLS polymerases in other bacterial systems are less characterized and their in vivo regulation and mechanism(s) of action are exciting avenues for future research. We highlight some open questions in this regard in later sections.
Figure 1.

Visualizing repair across scales. (A)In vitro single-molecule imaging of TLS polymerase activity. (i) Schematic showing the setup used by Kath et al. (2014), for visualizing polymerase switching. (ii) Representative trajectories showing Pol IV or Pol III synthesis over time on individual DNA molecules (Kath et al. 2014). Images reprinted with permission from PNAS.(B)In vivo single-molecule imaging of TLS activity. (i) Cartoon representation of the method of single-particle tracking with photoactivable localization microscopy (spt-PALM) (adapted from Stracy et al. 2014). Photoconvertible fluorophores are used and only one molecule is activated per cell at any point in time. These molecules are then imaged over time to capture single-molecule trajectories. Populations of freely diffusing, slow diffusing and bound molecules are identified and their characteristics studied. (ii) and (iii) Application of spt-PALM to study Pol IV activity in E. coli reveals association of Pol IV with DNA under damage-induced conditions. Images reprinted with permission from Thrall et al. (2017) and shared under Creative Commons public license (creativecommons.org/policies). (C) Tracking replication in single cells (from Aakre et al. 2013). (i) In this example, the β-clamp (DnaN) in Caulobacter crescentus is fluorescently tagged with YFP. Replication initiates at one cell pole and is tracked over time until completion at the opposite cell pole, where DnaN dissociates from DNA and its localization is lost. (ii) Schematic representation of the replication fork. (iii) DnaN position over time for cell in (i) is shown. τfocus represents time from focus formation to loss (Aakre et al. 2013). Image reproduced with permission from Elsevier. (D) Visualizing mutations in single cells. (i) and (ii) Microfluidics devices such as the ‘mother-machine’ allow the visualization of >105 cells in a single experiment. This can be combined with fluorescence imaging to follow activity and regulation of repair pathways, such as TLS, in single cells. (iii) This approach has been used to track mutations in real-time by following MutL-YFP foci that denote the position of DNA mismatches (Uphoff 2018). Image reproduced with permission from PNAS.
MECHANISMS OF TLS REPAIR
Replication-associated TLS
Although replication is stable and processive, replisome components themselves undergo dynamic exchange through the replication process. For example, in vitro total internal reflection fluorescence microscopy was used to visualize real-time exchange of fluorescently tagged single polymerase molecules within actively synthesizing individual replisomes of T7 bacteriophage and E. coli. These experiments indicated that components of the replication machinery, including the replicative polymerase, are dynamically exchanged with free molecules in solution (Loparo et al. 2011; Lewis et al. 2017). Real-time tracking of leading-strand DNA synthesis from single replisomes in vitro further showed that polymerases exchanged even on the leading-strand during synthesis, contrary to the long-held assumption of continuous synthesis (Graham, Marians and Kowalczykowski 2017). Consistent with these observations, in vivo single-particle tracking with photoactivable localization microscopy (spt-PALM) and fluorescence recovery after photobleaching experiments in E. coli and B. subtilis revealed few seconds dwell times for most replisome components including Pol III (Liao et al. 2016; Beattie et al. 2017).
Given these results, it is plausible that upon encountering a DNA lesion, the replisome stalls and the replicative polymerase is replaced by a TLS polymerase to synthesize across a lesion (‘TLS at the fork’ model via the ‘Toolbelt’ or ‘Mass-action’ mechanism) (Fujii and Fuchs 2004; Indiani et al. 2009; Kath et al. 2014; Zhao, Gleave and Lamers 2017). It is equally possible that the replisome is capable of skipping over the lesion, leaving behind a β-clamp and a single-stranded DNA gap, across which the TLS polymerase synthesizes (‘TLS behind the fork’ model) (Yeeles and Marians 2011; Marians 2018). In both cases, the β-clamp plays a pivotal role in recruitment of low-fidelity polymerases to the site of a lesion (described in detail below and schematically represented in Fig. 2).
Figure 2.

Proposed models for replication-associated TLS. (A) TLS at the fork. (i) Replication stalls since the replicative polymerase fails to synthesize past the DNA lesion. (ii) Replicative polymerase switches with TLS polymerase on the β-clamp, while the helicase continues to unwind DNA. (iii) TLS polymerase synthesizes across the lesion albeit with increased probability of incorporating mutations and subsequently the replicative polymerase switches back to continue DNA synthesis. (B) TLS behind the fork. (i) Same as in A(i). (ii) Replicative polymerase skips past the lesion, re-primes downstream and continues synthesis, leaving a single-stranded gap behind. (iii) Gap-filling behind the fork is mediated by the error-prone TLS polymerase.
The β-clamp is a central component of the replisome that holds the replicative polymerase in place for DNA synthesis. High processivity of replicative polymerases is mediated by specific interactions with the ring-shaped β-clamp, which encircles and slides on the DNA (Kong et al. 1992; Lamers et al. 2006; Fernandez-Leiro et al. 2015). All TLS polymerases studied so far in bacteria also interact with β-clamp (either directly or indirectly) and this interaction is important for modulating the activity of the polymerase (Wagner et al. 2000, 2009; Becherel 2002; Warner et al. 2010; Patoli, Winter and Bunting 2013). Indeed, it was recently shown that mutating the β-clamp binding pocket on Pol IV resulted in reduction in both binding events and co-localization with single-stranded binding protein SSB (Thrall et al. 2017). β-clamp has two pockets that bind polymerases; one is bound by the replicative polymerase synthesis subunit, α, and the other is bound by the proofreading exonuclease subunit, ε, thus preventing TLS polymerases from accessing the replisome during ongoing replication in steady state conditions (Jergic et al. 2013; Toste Rêgo et al. 2013). This restriction appears to be bypassed under DNA damage (discussed in detail below).
TLS at the fork
Based on in vitro biochemical assays and genetic studies it was proposed that when the replisome stalls at a lesion, a TLS polymerase can replace the replicative polymerase and efficiently synthesize across the lesion in a process referred to as ‘TLS at the fork’ (Indiani et al. 2005, 2009; Heltzel et al. 2012; Kath et al. 2014) (Fig. 2A). Given that TLS polymerases including Pol IV and Pol V are induced under DNA damage, initial models suggested that they gain access to the stalled replisome via ‘mass-action-based’ displacement of the replicative polymerase from the β-clamp. For example, under steady state, there are ∼250 copies of Pol IV, which increases ∼10-fold during SOS response (Kuban et al. 2004); Pol IV overexpression can also result in replication inhibition when induced to high enough levels (Uchida et al. 2008). In line with this, multicolor co-localization single-molecule spectroscopy (CoSMoS) experiments showed that Pol III and the TLS polymerase did not bind the β-clamp simultaneously and instead, competed for binding after stochastic dissociation of one of them (Zhao, Gleave and Lamers 2017).
However, as Pol IV is expressed to moderate levels even during normal growth, its access to DNA cannot simply be dictated by laws of ‘mass-action’; another layer of regulation is imperative. Consistently, it was observed that mere increase in Pol IV was not sufficient for mutagenesis and that DNA damage was also essential (Moore et al. 2017). Furthermore, in vivo single-molecule imaging showed that in the absence of damage Pol IV molecules diffused across the entire length of the cell and did not show enrichment along specific cellular positions (Thrall et al. 2017). It was also observed that overexpression of Pol IV without DNA damage could not result in persistent foci as seen during DNA damage (Henrikus et al. 2018). Taken together, another mechanism for ‘TLS at the fork’ can be considered. A ‘Toolbelt’ mechanism suggests that replicative and TLS polymerases could be simultaneously bound to the two different polymerase binding pockets on the β-clamp. This can then facilitate fast switching of polymerases to an active DNA synthesis conformation (Indiani et al. 2005, 2009).
In addition to ensemble biochemical assays, this model was supported by in vitro reconstitution and single-molecule imaging of TLS, where synthesis rates of Pol III and Pol IV on individual DNA molecules containing a replication blocking lesion were measured. The switch in synthesis rates when either of the polymerases accessed the DNA provided insights about kinetics of polymerase exchange, which supported a ‘toolbelt’ model; exchange was faster than recruitment of free molecules from solution and exchange slowed down when the binding pocket for the polymerase on the β-clamp was reduced to one (Kath et al. 2014). A detailed study of multiple Pol IV mutants also revealed that interaction of the TLS polymerase with replicative polymerase is required for its function, suggesting that they could be simultaneously bound to the β-clamp (Scotland et al. 2015), albeit in a transient manner (Zhao, Gleave and Lamers 2017).
It is important to note though, that constant engagement of an error-prone polymerase with β-clamp can be detrimental if it accesses DNA in an unregulated manner. In support of this idea, a recent study proposed a modified version of the toolbelt mechanism. Consistent with previous studies it was shown that exonuclease subunit (ε) associates with the secondary polymerase binding site on the β-clamp. This binding is critical for the gate-keeping function of ε to regulate promiscuous access of β-clamp by Pol IV (Chang et al. 2019). Levels of Pol IV would then dictate whether the TLS polymerase gains access to the replisome at the site of the lesion itself or if the replisome would skip the lesion and TLS would then occur behind the replication fork.
TLS behind the fork
The dynamic nature of the replisome is consistent with the possibility that obstacles can be bypassed by dissociation of the replisome followed by its re-association downstream of the block (Beattie et al. 2017) (Fig. 2B). Using an in vitro plasmid-based assay, it was previously shown that a DNA lesion on the leading strand only resulted in a temporary stall of replication (Yeeles and Marians 2011). Replication was reinitiated downstream of the lesion, which was dependent on the presence of the primase (DnaG), but independent of replication restart proteins, suggesting that restart is an inherent property of the replisome (Yeeles and Marians 2011, 2013; Yeeles et al. 2013). It was further proposed that Pol IV-mediated lesion bypass at the replisome vs lesion skipping by downstream re-initiation could be competing mechanisms (Gabbai, Yeeles and Marians 2014; Marians 2018). This is supported by a recent study that suggests that the gate-keeping function of ε determines the pathway choice between TLS at and behind the fork (Chang et al. 2019).
Indeed, several lines of evidence are in favor of the idea that a majority of TLS repair occurs post-replication. It was recently observed that, upon UV exposure, there is an increase in the number of fluorescently tagged Pol III foci that localize away from active replisomes (denoted by the localization of helicase) (Soubry, Wang and Reyes-Lamothe 2019). Some of these foci could be repair centers performing post-replicative TLS at the lesions that were skipped by active replisomes. This is consistent with observations in methyl methanesulfonate-treated cells, where 61% cells showed an increase in β-clamp localizations (Thrall et al. 2017). In another study, time-lapse imaging was performed to follow the dynamics of fluorescently tagged Pol IV during DNA damage by ciprofloxacin, methyl methanesulfonate and UV. It was observed that, under DNA damage, only 5–10% of Pol IV molecules are associated with active centers of replication (tracked using fluorescently labeled τ or ε subunits of the Pol III holoenzyme). The minority of foci that did associate with the replisome showed a broad range of co-localization distances indicating that a significant proportion of these foci could be involved in post-replicative TLS (Henrikus et al. 2018; Henrikus, van Oijen and Robinson 2018). Interestingly, Pol IV localization close to active sites of replication dropped dramatically after 90 min of DNA damage, which corresponds to the time when Pol V levels peak, raising the possibility that Pol IV is involved in TLS during early phases, while Pol V takes over this function in later phases of SOS response (Henrikus et al. 2018). This is in conjunction with the emerging idea of DNA damage response being chronologically modulated by different sets of proteins and repair pathways that act at discrete time zones of the response (Naiman et al. 2014; Fuchs 2016; Fujii, Isogawa and Fuchs 2018; Uphoff 2018).
TLS beyond the replication fork
Taken together, the widely cited model for TLS suggests that it occurs along with replication, either at or behind the fork (Indiani et al. 2009; Yeeles and Marians 2011; Heltzel et al. 2012; Kath et al. 2014; Marians 2018). It is probable that these are competing mechanisms; a combination of both occurs, possibly dictated by the levels of the TLS polymerase at a given instance (Gabbai, Yeeles and Marians 2014; Chang et al. 2019). Apart from this, efficiency with which the polymerase can bypass different types of lesions would also have a significant impact on the mechanism of repair (Thrall et al. 2017). It becomes important to ask whether these models hold true for other bacteria as well, that encode for different families of TLS polymerases and have distinct rates and mechanisms of DNA replication when compared with E. coli.
In contrast to this ‘replication-centric’ view of TLS, several lines of work (predominantly genetic) highlight a role for this form of repair outside the replisome as well (Fig. 3). A classic, replication-independent function of TLS proposed almost a decade ago is in transcription-coupled TLS (Fig. 3A). If RNA polymerase (RNAP) stalls at a gap on the transcribed strand (most likely generated due to replisome stalling at a lesion on the non-transcribed strand), gap filling mediated by TLS is required to relieve the halted transcription machinery (Cohen, Godoy and Walker 2009; Cohen et al. 2010; Cohen and Walker 2011). Additionally, studies have shown that Pol IV and Pol V interact with the transcriptional regulator, NusA (Cohen, Godoy and Walker 2009).
Figure 3.
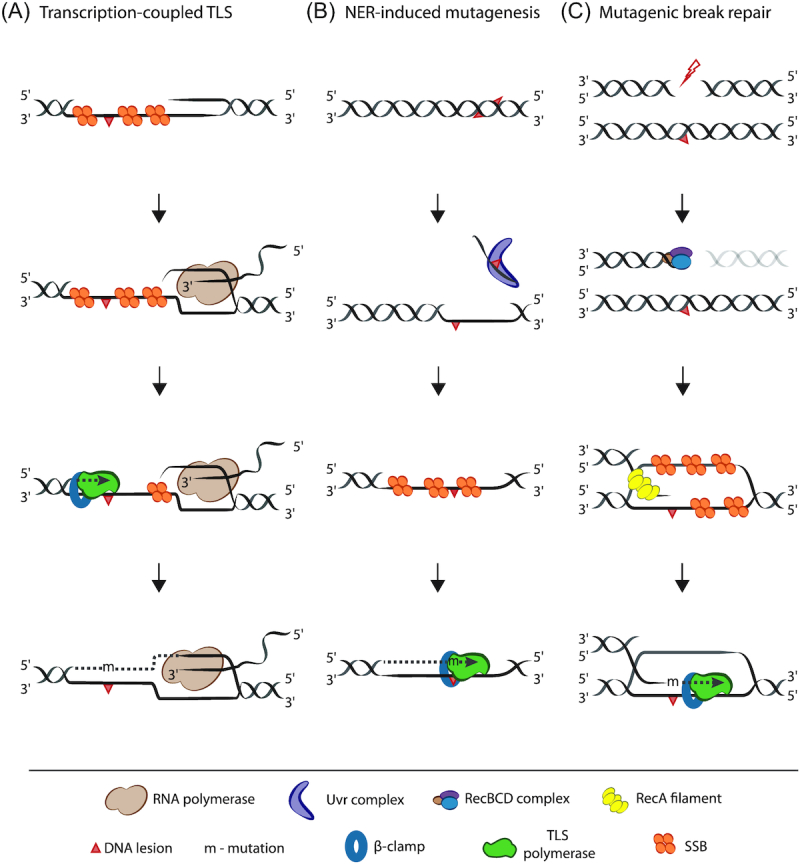
Proposed roles for TLS beyond replication. (A) Transcription-coupled TLS. RNA polymerase stalls at a single-stranded gap (likely generated due to perturbation in replication progression) on the transcribed strand. TLS polymerase is then required for gap filling, in the event that a DNA lesion is present on the opposite strand. (B) Nucleotide excision repair (NER)-induced mutagenesis. Closely spaced opposing lesions on DNA can result in incision of one of the lesions by NER. Due to persistence of lesion on the opposite strand DNA Pol I fails to fill the gap, thus requiring TLS repair instead. (C) Mutagenic break repair. DNA double-strand breaks are processed by the RecBCD (or AddAB) complex to reveal 3′ overhang on which RecA filaments and leads to strand invasion and subsequent repair. In this case, TLS polymerase is required if a lesion is present on the template strand.
Another interesting role for TLS is resolving collisions between transcription and replication machineries (Million-Weaver et al. 2015; Sankar et al. 2016; Lang and Merrikh 2018). Transcription and replication machineries traversing from opposite or same directions can crash into each other leading to head-on or co-directional collisions, respectively. Pol IV homolog in B. subtilis PolY1 (YqjH) is responsible for increased mutagenesis on the lagging strand in the presence of head-on collisions (Paul et al. 2013; Million-Weaver et al. 2015). Though head-on collisions are more mutagenic than co-directional ones, the mutation spectra associated with both of them are considerably different suggesting that they occur via distinct mechanistic modes (Srivatsan et al. 2010; Sankar et al. 2016).
Apart from transcription-associated TLS, homologous recombination is another repair mechanism that has been linked to TLS, and multiple studies show cooperativity between these two pathways (Lovett 2006, 2017; Williams, Hetrick and Foster 2010) leading to mutagenic break repair (Fitzgerald and Rosenberg 2019) (Fig. 3C). It was found that Pol IV can extend D-loops reconstituted in vitro and result in -1 frameshifts even in the absence of DNA lesions. Though the exonuclease activity of Pol II suppresses this activity under physiological conditions, Pol IV supersedes Pol II under stress-induced concentrations (Pomerantz et al.2013). In a parallel study, the same group found that Pol I is error prone at RecA-bound recombination intermediates (Pomerantz, Goodman and O'Donnell 2013). Pol IV can also contribute to mutagenic repair of I-Sce1 endonuclease mediated site-specific double-stranded breaks on the E. coli chromosome (Shee et al. 2011). Consistently, it was observed that fluorescently tagged Pol IV co-localized with RecA at a site-specific double-stranded break in both exponential and stationary phase E. coli cells (Mallik et al. 2015).
More recently, studies have uncovered cross-talk between TLS and NER (Fig. 3B). Using elegant genetic experiments in combination with mathematical modeling, it was shown that NER and Pol V can act together in processing closely spaced opposing lesions in E. coli. Importantly, this type of repair, termed NER-induced mutagenesis, can occur independent of active replication and results in mutations even in stationary phase E. coli cells (Janel-Bintz et al. 2017). This may be a conserved mechanism of repair across domains of life as coordinated function of NER and TLS has been observed in eukaryotes as well (Kozmin and Jinks-Robertson 2013; Sertic et al. 2018).
Finally, in addition to SOS, starvation-induced general stress response also leads to overexpression of TLS polymerases (Shee et al. 2011; Fitzgerald, Hastings and Rosenberg 2017). However, overexpression of Pol IV via SOS or general stress responses alone cannot result in mutagenesis. It also requires persistent DNA damage, most likely induced by ROS-mediated oxidative damage (Moore et al. 2017). This raises an intriguing possibility of heterogeneity in TLS activity and subsequent mutagenesis, based on the amount of ROS accumulation in individual cells. A recent study has substantiated this by showing increased mutagenesis in a subpopulation of cells with high ROS induction during ciprofloxacin treatment (Pribis et al. 2019). Recently developed microscopy assays applied to other repair pathways such as the adaptive response (Uphoff et al. 2016) could be used in these contexts to study TLS activity and its regulation in single cells and outside the context of replication.
IMPLICATIONS OF TLS AND OPEN QUESTIONS
In contrast with replicative polymerases that perform high-fidelity synthesis of long stretches of DNA, TLS polymerases perform error-prone, small gap-filling repair synthesis (Fuchs and Fujii 2013; Goodman and Woodgate 2013; Vaisman and Woodgate 2017; Yang and Gao 2018). Low fidelity of polymerases offers the ability to generate genetic variability during DNA synthesis. For TLS polymerases that are induced under stress responses, this feature directly feeds into efficient bypass of a large spectrum of lesions and stress-induced mutagenesis, generating genetic variants that can be resistant to antibiotics or better adapted as pathogens (Galhardo et al. 2005; Jarosz et al. 2006; Warner et al. 2010; Foti et al. 2012; Robinson et al. 2015; Janel-Bintz et al. 2017; Moore et al. 2017; Cole et al. 2018; Henrikus et al. 2018; Isogawa et al. 2018; Pribis et al. 2019).
TLS polymerases are also responsible for certain types of spontaneous mutagenesis in cells such as during replication–transcription conflicts (Million-Weaver et al. 2015; Lang and Merrikh 2018). There is ample data to suggest the role of TLS polymerases in stationary phase mutagenesis (Yeiser et al. 2002; Sung et al. 2003; Corzett, Goodman and Finkel 2013; Janel-Bintz et al. 2017; Moore et al. 2017). In the past, these assessments have mostly been carried out using fluctuation tests based on forward or reverse genetic approaches, which rely on loss- or gain-of-function mutations of particular selectable markers (Lee et al. 2012; Sankar et al. 2016; Schroeder et al. 2018, Pribis et al. 2019) (Table 1). However, these assays are again at the level of population and calculate ensemble averages, ignoring variations at the level of single cells.
In a pioneering approach to track mutagenesis in vivo, Elez et al. (2010) fluorescently tagged a component of the mismatch repair system (MutL) and visualized its localization in live E. coli cells. Persistent MutL foci should bind to mismatches that get converted to mutations in the subsequent generation (Elez et al. 2010). Combining this with microfluidics-based approaches, it was possible to simultaneously study mutations by tracking MutL foci in single cells across a significantly large population (>105 cells) (Uphoff et al. 2016; Robert et al. 2018; Uphoff 2018). This strategy can be used to understand when and where TLS is active in a population of cells, its propensity for mutagenic or non-mutagenic repair and subsequent impact on cellular survival.
The need to study the error-prone nature of TLS in vivo is highlighted by a recent study that revealed the extent of the mutagenic potential of these polymerases. Using deep sequencing techniques, it was shown that Pol V-mediated lesion bypass of a site-specific lesion results in mutations spread across hundreds of nucleotides upstream and downstream of the actual lesion. Interestingly, Pol V creates multiple TLS (mutagenic) patches interspersed with small error-free patches after the lesion, with decreasing mutation frequencies as the patch distance from the lesion increases (Isogawa et al. 2018). It is suggested that this mutation patchwork is caused by unique interactions and regulation of Pol V by the 3′ end of RecA nucleoprotein filament, which results in repeated re-association of an otherwise distributive polymerase (Goodman et al. 2016; Isogawa et al. 2018). It would now be very informative to employ imaging-based tools in this experimental regime to understand the mechanisms by which TLS components are recruited to sites of repair and how their activity may be regulated.
In sum, mechanistic and regulatory investigations in bacteria using imaging-based approaches have been pivotal in understanding the process of TLS. Given the conserved nature of this pathway, these studies have also provided insights into TLS-based repair across domains of life (Fitzgerald and Rosenberg 2019; Xia et al. 2019). Most studies have focused their efforts on specific model systems like E. coli; in future, it would be important to consider the diversity of TLS polymerases across bacterial species as well (Table 1). For example, M. smegmatis has a unique Y-family polymerase Dpo4, which is capable of incorporating rNTPs during DNA synthesis (Ordonez and Shuman 2014; Johnson, Kottur and Nair 2019). Further, presence of distinct families of polymerases (C-family vs Y-family, Table 1) across bacteria highlights the probable role of these polymerases in targeted repair or bypass of lesions that may be faced by cells in their environmental niches or based on their genome GC content. A comprehensive understanding of the specificity, regulation and activity of these polymerases, such as DnaE2 (Zhao et al. 2007; Timinskas et al. 2014), still remains unexplored, especially at the single-cell and single-molecule level.
In addition, while mechanisms of TLS in the context of replication are well studied, in vivo understanding of replication-independent TLS is limited. Studies have only recently begun to focus imaging-based efforts on TLS outside the context of the replisome and/or in response to different DNA lesions. Thrall et al. (2017) used spt-PALM to uncover that the type of lesion determined the cellular localization patterns of TLS polymerases. Under methyl methanesulfonate damage, Pol IV molecules were enriched at mid cell, strongly overlapping with localization of single-stranded DNA binding protein, SSB. On the other hand, under nitrofurazone damage, Pol IV molecules were enriched near cell poles (away from SSB). Furthermore, Pol IV localization under nitrofurazone was independent of its β-clamp binding motif and catalytic activity (Thrall et al. 2017). Similar two-color imaging of Pol IV with Pol III subunits under various DNA damaging conditions showed that 90% of Pol IV molecules localized away from active sites of replication (Henrikus et al. 2018; Henrikus, van Oijen and Robinson 2018). The impact of replication-independent TLS could be significant with respect to mutagenesis in cells devoid of active replication, such as slow-growing bacteria or dormant spores (Janel-Bintz et al. 2017). In combination with genetic and biochemical findings, these microscopy-based studies highlight the need to consider the mechanisms and modes of regulation of TLS polymerases that function outside the realms of active replication through cross-talks with other cellular processes and repair pathways.
FUNDING
Work in the AB lab is funded by Human Frontier of Sciences Programme (HFSP CDA (00051/ 2017-C) (AB), intramural funding from NCBS-TIFR (Department of Atomic Energy, Government of India, under project no. 12-R&D-TFR-5.04-0800) (AB), Department of Biotechnology (BT/12/IYBA/2019/10) (AB) and Department of Science and Technology, DST-SERB (PDF/2018/001164) (AMJ).
ACKNOWLEDGEMENTS
We apologize to authors whose work we have not cited. We are grateful to Stephan Uphoff, Rodrigo Reyes-Lamothe, Joseph J. Loparo and members of the AB lab for critical feedback and comments on the review. We acknowledge Stephan Uphoff, Joseph J. Loparo and journals for giving us permissions to use published figure panels to highlight examples of technologies or experimental details.
Contributor Information
Asha Mary Joseph, National Centre for Biological Sciences (Tata Institute of Fundamental Research), Bangalore, Karnataka 560065, India.
Anjana Badrinarayanan, National Centre for Biological Sciences (Tata Institute of Fundamental Research), Bangalore, Karnataka 560065, India.
Conflicts of Interest
None declared.
REFERENCES
- Aakre CD, Phung TN, Huang D, et al. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol Cell. 2013;52:617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Mamun AAM, Humayun MZ. Escherichia coli DNA polymerase II can efficiently bypass 3,N(4)-ethenocytosine lesionsin vitro and in vivo. Mutat Res. 2006;593:164–76. [DOI] [PubMed] [Google Scholar]
- Badrinarayanan A, Le TBK, Laub MT. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 2015;31:171–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Mazel D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev. 2014;38:1126–45. [DOI] [PubMed] [Google Scholar]
- Beattie TR, Kapadia N, Nicolas E, et al. Frequent exchange of the DNA polymerase during bacterial chromosome replication. eLife. 2017;6:e21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie TR, Reyes-Lamothe R. A replisome's journey through the bacterial chromosome. Front Microbiol. 2015;6:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Fuchs RP, Wagner J. Pivotal role of the beta-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair. 2002;1:703–8. [DOI] [PubMed] [Google Scholar]
- Becherel OJ, Fuchs RP. Mechanism of DNA polymerase II-mediated frameshift mutagenesis. Proc Natl Acad Sci USA. 2001;98:8566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjedov I, Dasgupta CN, Slade D, et al. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics. 2007;176:1431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshoff HIM, Reed MB, Barry CE, et al. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell. 2003;113:183–93. [DOI] [PubMed] [Google Scholar]
- Chang S, Naiman K, Thrall ES, et al. A gatekeeping function of the replicative polymerase controls pathway choice in the resolution of lesion-stalled replisomes. Proc Natl Acad Sci USA. 2019;116:25591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SE, Foti JJ, Simmons LA, et al. The SOS regulatory network. EcoSal Plus. 2008, DOI: 10.1128/ecosalplus.5.4.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SE, Godoy VG, Walker GC. Transcriptional modulator NusA interacts with translesion DNA polymerases in Escherichia coli. J Bacteriol. 2009;191:665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SE, Lewis CA, Mooney RA, et al. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc Natl Acad Sci USA. 2010;107:15517–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SE, Walker GC. New discoveries linking transcription to DNA repair and damage tolerance pathways. Transcription. 2011;2:37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JM, Acott JD, Courcelle CT, et al. Limited capacity or involvement of excision repair, double-strand breaks, or translesion synthesis for psoralen cross-link repair in Escherichia coli. Genetics. 2018;210:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corzett CH, Goodman MF, Finkel SE. Competitive fitness during feast and famine: how SOS DNA polymerases influence physiology and evolution in Escherichia coli. Genetics. 2013;194:409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, et al. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. [DOI] [PubMed] [Google Scholar]
- Dervyn E, Suski C, Daniel R, et al. Two essential DNA polymerases at the bacterial replication fork. Science. 2001;294:1716–9. [DOI] [PubMed] [Google Scholar]
- Duigou S, Ehrlich SD, Noirot P, et al. Distinctive genetic features exhibited by the Y-family DNA polymerases in Bacillus subtilis. Mol Microbiol. 2004;54:439–51. [DOI] [PubMed] [Google Scholar]
- Elez M, Murray AW, Bi L-J, et al. Seeing Mutations in Living Cells. Curr Biol. 2010;20:1432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R, Conrad J, Scheres SH, et al. cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife. 2015;4:e11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DM, Hastings PJ, Rosenberg SM. Stress-induced mutagenesis: implications in cancer and drug resistance. Annu Rev Cancer Biol. 2017;1:119–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DM, Rosenberg SM. What is mutation? A chapter in the series: how microbes “jeopardize” the modern synthesis. PLOS Genet. 2019;15:e1007995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Lee H, Popodi E, et al. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc Natl Acad Sci USA. 2015;112:E5990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Devadoss B, Winkler JA, et al. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RP, Fujii S. Translesion DNA synthesis and mutagenesis in prokaryotes. Cold Spring Harb Perspect Biol. 2013;5:a012682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RP. Tolerance of lesions in E. coli: chronological competition between translesion synthesis and damage avoidance. DNA Repair. 2016;44:51–8. [DOI] [PubMed] [Google Scholar]
- Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Gasser V, Fuchs RP. The biochemical requirements of DNA polymerase V-mediated translesion synthesis revisited. J Mol Biol. 2004;341:405–17. [DOI] [PubMed] [Google Scholar]
- Fujii S, Isogawa A, Fuchs RP. Chronological switch from translesion synthesis to homology-dependent gap repair in vivo. Toxicol Res. 2018;34:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbai CB, Yeeles JTP, Marians KJ. Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms. J Biol Chem. 2014;289:32811–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galhardo RS, Rocha RP, Marques MV, et al. An SOS-regulated operon involved in damage-inducible mutagenesis in Caulobacter crescentus. Nucleic Acids Res. 2005;33:2603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF, McDonald JP, Jaszczur MM, et al. Insights into the complex levels of regulation imposed on Escherichia coli DNA polymerase V. DNA Repair. 2016;44:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5:a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF. Error-Prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. [DOI] [PubMed] [Google Scholar]
- Graham JE, Marians KJ, Kowalczykowski SC. Independent and stochastic action of DNA polymerases in the replisome. Cell. 2017;169:1201–13.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltzel JMH, Maul RW, Wolff DW, et al. Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III replicase. J Bacteriol. 2012;194:3589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrikus SS, van Oijen AM, Robinson A. Specialised DNA polymerases in Escherichia coli: roles within multiple pathways. Curr Genet. 2018;64:1189–96. [DOI] [PubMed] [Google Scholar]
- Henrikus SS, Wood EA, McDonald JP, et al. DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli. PLoS Genet. 2018;14:e1007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori M, Yonekura S-I, Nohmi T, et al. Error-prone translesion DNA synthesis by Escherichia coli DNA polymerase IV (DinB) on templates containing 1,2-dihydro-2-oxoadenine. J Nucleic Acids. 2010;2010:807579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiani C, Langston LD, Yurieva O, et al. Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc Natl Acad Sci USA. 2009;106:6031–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiani C, McInerney P, Georgescu R, et al. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol Cell. 2005;19:805–15. [DOI] [PubMed] [Google Scholar]
- Ippoliti PJ, DeLateur NA, Jones KM, et al. Multiple strategies for translesion synthesis in bacteria. Cells. 2012;1:799–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogawa A, Ong JL, Potapov V, et al. Pol V-mediated translesion synthesis elicits localized untargeted mutagenesis during post-replicative gap repair. Cell Rep. 2018;24:1290–300. [DOI] [PubMed] [Google Scholar]
- Janel-Bintz R, Napolitano RL, Isogawa A, et al. Processing closely spaced lesions during nucleotide excision repair triggers mutagenesis in E. coli. PLOS Genet. 2017;13:e1006881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Godoy VG, Delaney JC, et al. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–8. [DOI] [PubMed] [Google Scholar]
- Jaszczur M, Bertram JG, Robinson A, et al. Mutations for worse or better: low-fidelity DNA synthesis by SOS DNA polymerase V is a tightly regulated double-edged sword. Biochemistry. 2016;55:2309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jatsenko T, Sidorenko J, Saumaa S, et al. DNA polymerases ImuC and DinB are involved in DNA alkylation damage tolerance in Pseudomonas aeruginosaandPseudomonas putida. PLOS ONE. 2017;12:e0170719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jergic S, Horan NP, Elshenawy MM, et al. A direct proofreader–clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 2013;32:1322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. 2013;5:a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MK, Kottur J, Nair DT. A polar filter in DNA polymerases prevents ribonucleotide incorporation. Nucleic Acids Res. 2019;47:10693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kana BD, Abrahams GL, Sung N, et al. Role of the DinB homologs Rv1537 and Rv3056 in Mycobacterium tuberculosis. J Bacteriol. 2010;192:2220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Kasho K, Kawakami H. The DnaA cycle in Escherichia coli: activation, function and inactivation of the initiator protein. Front Microbiol. 2017;8:2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kath JE, Jergic S, Heltzel JMH, et al. Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc Natl Acad Sci USA. 2014;111:7647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, et al. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics. 2001;266:207–15. [DOI] [PubMed] [Google Scholar]
- Kleckner NE, Chatzi K, White MA, et al. Coordination of growth, chromosome replication/segregation, and cell division in E. coli. Front Microbiol. 2018;9:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XP, Onrust R, O'Donnell M, et al. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–37. [DOI] [PubMed] [Google Scholar]
- Koskiniemi S, Andersson DI. Translesion DNA polymerases are required for spontaneous deletion formation in Salmonella typhimurium. Proc Natl Acad Sci. 2009;106:10248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmin SG, Jinks-Robertson S. The mechanism of nucleotide excision repair-mediated UV-induced mutagenesis in nonproliferating cells. Genetics. 2013;193:803–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuban W, Jonczyk P, Gawel D, et al. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J Bacteriol. 2004;186:4802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 2015;49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers MH, Georgescu RE, Lee S-G, et al. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell. 2006;126:881–92. [DOI] [PubMed] [Google Scholar]
- Lang KS, Merrikh H. The clash of macromolecular Titans: replication–transcription conflicts in bacteria. Annu Rev Microbiol. 2018;72:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chatelier E, Bécherel OJ, d'Alençon E, et al. Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J Biol Chem. 2004;279:1757–67. [DOI] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, et al. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci. 2012;109:E2774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie DJ, Heinen C, Schramm FD, et al. Nutritional control of DNA replication initiation through the proteolysis and regulated translation of DnaA. PLoS Genet. 2015;11:e1005342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JS, Spenkelink LM, Jergic S, et al. Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife. 2017;6:e23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Li Y, Schroeder JW, et al. Single-molecule DNA polymerase dynamics at a bacterial replisome in live cells. Biophys J. 2016;111:2562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen Z, Matthews LA, et al. Dynamic exchange of two essential DNA polymerases during replication and after fork arrest. Biophys J. 2019;116:684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loparo JJ, Kulczyk AW, Richardson CC, et al. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc Natl Acad Sci USA. 2011;108:3584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Kulishev CO, Alves IR, Valencia EY, et al. Functional characterization of two SOS-regulated genes involved in mitomycin C resistance in Caulobacter crescentus. DNA Repair. 2015;33:78–89. [DOI] [PubMed] [Google Scholar]
- Lovett ST. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair. 2006;5:1421–7. [DOI] [PubMed] [Google Scholar]
- Lovett ST. Template-switching during replication fork repair in bacteria. DNA Repair. 2017;56:118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik S, Popodi EM, Hanson AJ, et al. Interactions and localization of Escherichia coli error-prone DNA polymerase IV after DNA damage. J Bacteriol. 2015;197:2792–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ. Lesion bypass and the reactivation of stalled replication forks. Annu Rev Biochem. 2018;87:217–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Pinheiro M, Oliveira AR, Valencia AO, et al. Molecular characterization of Caulobacter crescentusmutator strains. Gene. 2017;626:251–7. [DOI] [PubMed] [Google Scholar]
- McHenry CS. Bacterial replicases and related polymerases. Curr Opin Chem Biol. 2011a;15:587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry CS. Breaking the rules: bacteria that use several DNA polymerase IIIs. EMBO Rep. 2011b;12:408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Million-Weaver S, Samadpour AN, Moreno-Habel DA, et al. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. Proc Natl Acad Sci. 2015;112:E1096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JM, Correa R, Rosenberg SM, et al. Persistent damaged bases in DNA allow mutagenic break repair in Escherichia coli. PLoS Genet. 2017;13:e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiman K, Philippin G, Fuchs RP, et al. Chronology in lesion tolerance gives priority to genetic variability. Proc Natl Acad Sci USA. 2014;111:5526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano R, Janel-Bintz R, Wagner J, et al. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeley WL, Delaney S, Alekseyev YO, et al. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J Biol Chem. 2007;282:12741–8. [DOI] [PubMed] [Google Scholar]
- Ordonez H, Shuman S. Mycobacterium smegmatis DinB2 misincorporates deoxyribonucleotides and ribonucleotides during templated synthesis and lesion bypass. Nucleic Acids Res. 2014;42:12722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patoli AA, Winter JA, Bunting KA. The UmuC subunit of the E. coli DNA polymerase V shows a unique interaction with the β-clamp processivity factor. BMC Struct Biol. 2013;13:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Million-Weaver S, Chattopadhyay S, et al. Accelerated gene evolution through replication-transcription conflicts. Nature. 2013;495:512–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Elizur T, Takeshita M, Goodman M, et al. Mechanism of translesion DNA synthesis by DNA polymerase II. Comparison to DNA polymerases I and III core. J Biol Chem. 1996;271:24662–9. [DOI] [PubMed] [Google Scholar]
- Peng R, Chen J-H, Feng W-W, et al. Error-prone DnaE2 balances the genome mutation rates in Myxococcus xanthus DK1622. Front Microbiol. 2017;8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TM, Tan KW, Sakumura Y, et al. A single-molecule approach to DNA replication in Escherichia coli cells demonstrated that DNA polymerase III is a major determinant of fork speed. Mol Microbiol. 2013;90:584–96. [DOI] [PubMed] [Google Scholar]
- Pomerantz RT, Goodman MF, O'Donnell ME. DNA polymerases are error-prone at RecA-mediated recombination intermediates. Cell Cycle. 2013;12:2558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, Kurth I, Goodman MF, et al. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat Struct Mol Biol. 2013;20:748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribis JP, García-Villada L, Zhai Y, et al. Gamblers: an antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol Cell. 2019;74:785–800.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ. The bacterial cell cycle, chromosome inheritance and cell growth. Nat Rev Microbiol. 2019;17:467–78. [DOI] [PubMed] [Google Scholar]
- Robert L, Ollion J, Robert J, et al. Mutation dynamics and fitness effects followed in single cells. Science. 2018;359:1283–6. [DOI] [PubMed] [Google Scholar]
- Robinson A, McDonald JP, Caldas VEA, et al. Regulation of mutagenic DNA polymerase V activation in space and time. PLOS Genet. 2015;11:e1005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Lloyd RG. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007;21:668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders LH, Rockel A, Lu H, et al. Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J Bacteriol. 2006;188:8573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar TS, Wastuwidyaningtyas BD, Dong Y, et al. The nature of mutations induced by replication–transcription collisions. Nature. 2016;535:178–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JW, Yeesin P, Simmons LA, et al. Sources of spontaneous mutagenesis in bacteria. Crit Rev Biochem Mol Biol. 2018;53:29–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland MK, Heltzel JMH, Kath JE, et al. A genetic selection for dinB mutants reveals an interaction between DNA polymerase IV and the replicative polymerase that is required for translesion synthesis. PLoS Genet. 2015;11:e1005507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sertic S, Mollica A, Campus I, et al. Coordinated activity of Y family TLS polymerases and EXO1 protects non-S phase cells from UV-induced cytotoxic lesions. Mol Cell. 2018;70:34–47..e4. [DOI] [PubMed] [Google Scholar]
- Sharma A, Nair DT. MsDpo4-a DinB homolog from Mycobacterium smegmatis is an error-prone DNA polymerase that can promote G:T and T:G mismatches. J Nucleic Acids. 2012;2012:285481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee C, Gibson JL, Darrow MC, et al. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci USA. 2011;108:13659–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubry N, Wang A, Reyes-Lamothe R. Replisome activity slowdown after exposure to ultraviolet light in Escherichia coli. Proc Natl Acad Sci. 2019;116:11747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan A, Tehranchi A, MacAlpine DM, et al. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010;6:e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracy M, Uphoff S, Garza de Leon F, et al. In vivo single-molecule imaging of bacterial DNA replication, transcription, and repair. FEBS Lett. 2014;588:3585–94. [DOI] [PubMed] [Google Scholar]
- Sung H-M, Yeamans G, Ross CA, et al. Roles of YqjH and YqjW, homologs of the Escherichia coli UmuC/DinB or Y superfamily of DNA polymerases, in stationary-phase mutagenesis and UV-induced mutagenesis ofBacillus subtilis. J Bacteriol. 2003;185:2153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MD. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim Biophys Acta. 2010;1804:1167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Pham P, Shen X, et al. Roles of E. coliDNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–8. [DOI] [PubMed] [Google Scholar]
- Thrall ES, Kath JE, Chang S, et al. Single-molecule imaging reveals multiple pathways for the recruitment of translesion polymerases after DNA damage. Nat Commun. 2017;8:2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timinskas K, Balvočiūtė M, Timinskas A, et al. Comprehensive analysis of DNA polymerase III α subunits and their homologs in bacterial genomes. Nucleic Acids Res. 2014;42:1393–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toste Rêgo A, Holding AN, Kent H, et al. Architecture of the Pol III–clamp–exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J. 2013;32:1334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Furukohri A, Shinozaki Y, et al. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol Microbiol. 2008;70:608–22. [DOI] [PubMed] [Google Scholar]
- Uphoff S, Lord ND, Okumus B, et al. Stochastic activation of a DNA damage response causes cell-to-cell mutation rate variation. Science. 2016;351:1094–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff S. Real-time dynamics of mutagenesis reveal the chronology of DNA repair and damage tolerance responses in single cells. Proc Natl Acad Sci USA. 2018;115:E6516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A, McDonald JP, Woodgate R. Translesion DNA synthesis. EcoSal Plus. 2012;5, DOI: 10.1128/ecosalplus.7.2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A, Woodgate R. Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol. 2017;52:274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Etienne H, Fuchs RPet al. . Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol Microbiol. 2009;74:1143–51. [DOI] [PubMed] [Google Scholar]
- Wagner J, Fujii S, Gruz Pet al. . The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000;1:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DF, Ndwandwe DE, Abrahams GL, et al. Essential roles for imuA’- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2010;107:13093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AB, Hetrick KM, Foster PL. Interplay of DNA repair, homologous recombination, and DNA polymerases in resistance to the DNA damaging agent 4-nitroquinoline-1-oxide in Escherichia coli. DNA Repair. 2010;9:1090–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Chiu L-Y, Nehring RB, et al. Bacteria-to-human protein networks reveal origins of endogenous DNA damage. Cell. 2019;176:127–43..e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z-Q, Dixon NE. Bacterial replisomes. Curr Opin Struct Biol. 2018;53:159–68. [DOI] [PubMed] [Google Scholar]
- Yang W, Gao Y. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu Rev Biochem. 2018;87:239–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Marians KJ. Dynamics of leading-strand lesion skipping by the replisome. Mol Cell. 2013;52:855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Marians KJ. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011;334:235–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Poli J, Marians KJ, et al. Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol. 2013;5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeiser B, Pepper ED, Goodman MF, et al. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci USA. 2002;99:8737–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Gleave ES, Lamers MH. Single-molecule studies contrast ordered DNA replication with stochastic translesion synthesis. eLife. 2017;6:e32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zhang Z, Yan J, et al. GC content variability of eubacteria is governed by the pol III α subunit. Biochem Biophys Res Commun. 2007;356:20–5. [DOI] [PubMed] [Google Scholar]