

 We describe the synthesis and evaluation of Strathclyde Minor Groove Binders (S-MGBs) as selective anti-cancer agents which act through a non-alkylating mechanism.
We describe the synthesis and evaluation of Strathclyde Minor Groove Binders (S-MGBs) as selective anti-cancer agents which act through a non-alkylating mechanism.
Abstract
Traditional cytotoxic agents which act through a DNA-alkylating mechanism are relatively non-specific, resulting in a small therapeutic window and thus limiting their effectiveness. In this study, we evaluate a panel of 24 non-alkylating Strathclyde Minor Groove Binders (S-MGBs), including 14 novel compounds, for in vitro anti-cancer activity against a human colon carcinoma cell line, a cisplatin-sensitive ovarian cancer cell line and a cisplatin-resistant ovarian cancer cell line. A human non-cancerous retinal epithelial cell line was used to measure selectivity of any response. We have identified several S-MGBs with activities comparable to cis-platin and carboplatin, but with better in vitro selectivity indices, particularly S-MGB-4, S-MGB-74 and S-MGB-317. Moreover, a comparison of the cis-platin resistant and cis-platin sensitive ovarian cancer cell lines reveals that our S-MGBs do not show cross resistance with cisplatin or carboplatin and that they likely have a different mechanism of action. Finally, we present an initial investigation into the mechanism of action of one compound from this class, S-MGB-4, demonstrating that neither DNA double strand breaks nor the DNA damage stress sensor protein p53 are induced. This indicates that our S-MGBs are unlikely to act through an alkylating or DNA damage response mechanism.
1. Introduction
Cancer is the second leading cause of death globally and was responsible for 8.8 million deaths in 2015.1 Whilst cancer research is one of the largest and most well-funded fields of scientific research in the world, proportionately little progress has been made in improving patient outcomes for certain cancers and in the development of more effective chemotherapeutic treatments. Particular challenges include i) the heterogeneity and plasticity of cancer cells resulting in different types of chemotherapeutic drug resistance and ii) the difficulty in achieving selective toxicity to cancer cells and not adversely affecting normal, healthy cells.2 Traditional chemotherapeutic agents such as the platinates cisplatin and carboplatin mostly act through exploiting the rapid proliferation of cancer cells and induction of extensive DNA damage that is insufficiently repaired. Such agents are still heavily used in the clinic today and are a vital part of chemotherapy for many cancers. Whilst potent, however, these traditional cytotoxic agents are relatively non-specific which limits their effectiveness and the therapeutic window is small.3 More recently, advances in our understanding of the molecular and cellular biology of cancers has led to development of targeted therapies that exploit molecular differences between cancer and non-cancer cells. However, whilst these new approaches offer improved selectivity, limitations include targeting only cancer cells with particular lesions, reduced potency and being too targeted in nature enabling acquisition of resistance.4 It is clear that more effective agents that offer both selectivity and potency are necessary.
Minor Groove Binders (MGBs) are a class of compound which bind to the minor groove of DNA and exert their biological effects through interference of DNA-related processes. Whilst the modes of action of MGBs are likely to vary depending on their specific structure, reported effects include induction of sequence-specific DNA alkylation damage, inhibition of DNA replication and gene transcription, perturbation of the cell cycle and interference in DNA repair.5 The MGBs reported here, developed at the Universities of Strathclyde, Strathclyde-MGBs (S-MGBs), are based on the polyamide natural product distamycin (Fig. 1).6 We have extensively investigated the anti-infective properties of distamycin-based S-MGBs through systematic structural modifications of the distamycin framework. Our team has developed several potent anti-infective compounds, selectively targeting various infectious organisms such as bacteria,7 parasites,8 and fungi.9 This includes the discovery of the drug MGB-BP-3 which, in collaboration with MGB Biopharma, is about to progress into phase II clinical trials for the treatment of Clostridium difficile infections. In this study, a panel of our distamycin-based S-MGBs are investigated for potential cytotoxic activity towards different cancer cells and whether they show selectivity towards cancer cells compared to non-cancer cells.
Fig. 1. Molecular structure of distamycin with the key structural components highlighted.

The platinate cisplatin (Fig. 2) is one of a number of platinum-based compounds currently in clinical use for treatment of a number of cancers including bladder, testicular and ovarian cancers and its mechanism of action involves irreversible covalent binding to nucleotides, causing direct damage to DNA via inter- and intra-strand crosslinking. This is achieved through the initial exchange of chloride ligands with water inside the cell, which is then displaced by the nucleophilic attack of N-heterocyclic DNA bases, causing direct linkage of cisplatin to DNA.10 Tallimustine (Fig. 2) is an MGB derived from distamycin that acts through a DNA alkylating mechanism similar to cisplatin and which has undergone extensive evaluation as a potential anti-cancer agent. Tallimustine induces DNA alkylation through its benzoic acid mustard (BAM) group. Many anti-cancer compounds work in this fashion, directly damaging DNA, however, compounds that work through this mechanism of action commonly suffer from significant toxicity issues associated with damage to DNA of healthy cells. Whilst alkylating MGBs show more specific DNA alkylation than conventional alkylators, tallimustine failed in the clinic due to severe bone marrow toxicity.11
Fig. 2. Left – Cisplatin, a common chemotherapy agent that is alkylating-like and induces DNA crosslinking. Right – Tallimustine, a minor groove binder with anticancer activity derived from distamycin which has an alkylating mode of action.

Importantly, our S-MGBs do not alkylate DNA but instead bind non-covalently through interaction with DNA bases and the sugar-phosphate backbone. This is due to the absence of any functional groups within the molecule that could perceivably alkylate or react with DNA. This non-covalent binding is facilitated by the concave curvature of MGBs, which is complementary to the shape and curvature of the DNA minor groove. Other non-covalent binding MGBs of a different class to distamycin have been explored as potential tools for recognition of specific genes via sequence-specific MGB binding raising the possibility that non-covalent binding MGBs could be more selective as therapeutics.12
We have previously reported that some of our S-MGBs inhibit the proliferation of mouse melanoma B16-F10-luc cells. This is a cell line derived from cells that metastasized to the lung and were isolated from lung metastatic nodules.13 In that study, we identified that subtle structural variations significantly influenced the activity profile of the S-MGBs. Significantly, we were able to identify an S-MGB with around a 70-fold greater activity than gemcitabine, a current treatment option used in the clinic for non-small cell lung cancer.
In this study, we evaluate a panel of S-MGBs, including 14 novel S-MGBs, for potential activity against several different human cancer cell lines including cisplatin-resistant cells. Activity against healthy, human retinal epithelial cells was also assessed providing an in vitro indication of any preferential selectivity towards cancer cells compared to non-cancer cells. The main structural modifications of the S-MGBs reported here compared to distamycin are: substitution of the N-(1-methyl-1H-pyrrol-3-yl)formamide head group with a more lipophilic aryl system; substitution of the amidine tail group with either dimethylaminopropyl or morpholine; substitution of the amide linker of the head group with either an amidine or alkene; and, the modification of one of the N-methylpyrrole heterocyclic subunits to a more lipophilic isopentylthiazole (Fig. 3).
Fig. 3. Different structural modifications made to the S-MGB framework. The tail group (green) varies in basicity and lipophilicity with the exchange of dimethylaminopropyl with ethyl-morpholine. The first heterocycle (pink) can be either a substituted thiazole or N-methylpyrrole, which has a drastic effect on the overall lipophilicity of the molecule. Various head groups (red) and head group linkers (blue) have been experimented with which, again, has a significant effect on the overall physicochemical properties of the molecule as well as having an impact on DNA binding.

2. Results and discussion
2.1. Synthesis
A library of 24 S-MGBs was synthesised for investigation in this study (ESI† Table S1), 14 of which were new (Fig. 4 and 5). S-MGBs were synthesised systematically through sequential coupling of the various subunits and were purified by HPLC and isolated as the mono or di-TFA salts. The protocols used were similar to those in our previous work, with the synthesis for the novel compounds described below.9
Fig. 4. Synthesis of novel amide and amidine-linked S-MGBs with yields obtained as indicated. The structure of the head and tail groups for the different MGBs is shown.

Fig. 5. Synthesis of novel alkene-linked MGBs with yields obtained as indicated. The structure of the head and tail groups for the different MGBs is shown.

To prepare compounds with a pyrrole-amidine or pyrrole-amide linked head group, the appropriate nitro dimer was reduced to the corresponding amine by hydrogenation with palladium on carbon as a catalyst in methanol at room temperature. The amine was then reacted with 1-methyl-4-nitro-1H-pyrrole-2-carbonyl chloride (prepared by refluxing the corresponding carboxylic acid in thionyl chloride) in the presence of triethylamine at room temperature. This nitro trimer was then reduced to the amine as described previously and then reacted with the appropriate commercially available methyl carbimidothioate hydroiodide or carboxylic acid functionalised head group at room temperature yielding the full S-MGB with yields ranging from 26–50% (Fig. 4).
To prepare the remaining compounds containing a phenyl-amide, phenyl-alkene or pyridyl-alkene linked head groups, the appropriate nitro dimer was reduced to the amine as described above and reacted with the desired carboxylic acid-functionalised head group in the presence of a coupling agent at room temperature, with yields ranging from 24–42% (Fig. 5). Synthesis details for the phenyl-amide, phenyl-alkene or pyridyl-alkene linked head groups are previously published (see Experimental section).
The structures of all new MGBs, and relevant analogues synthesised in previous studies, are presented in Table 1.
Table 1. Collection of S-MGBs under examination in this study.
| S-MGB ID | Structure | S-MGB ID | Structure |
| 351 |
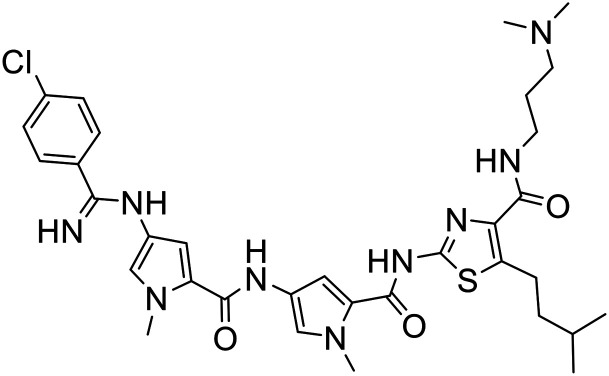
|
380 |
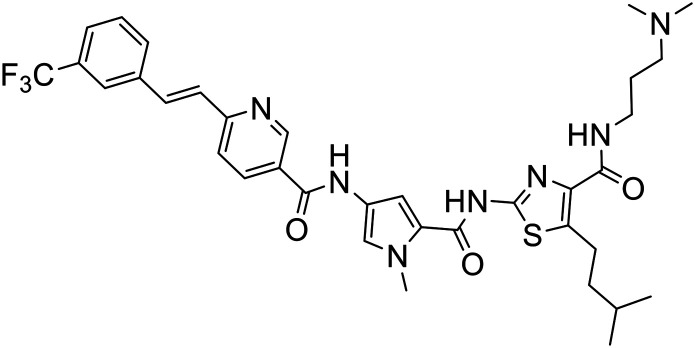
|
| 352 |
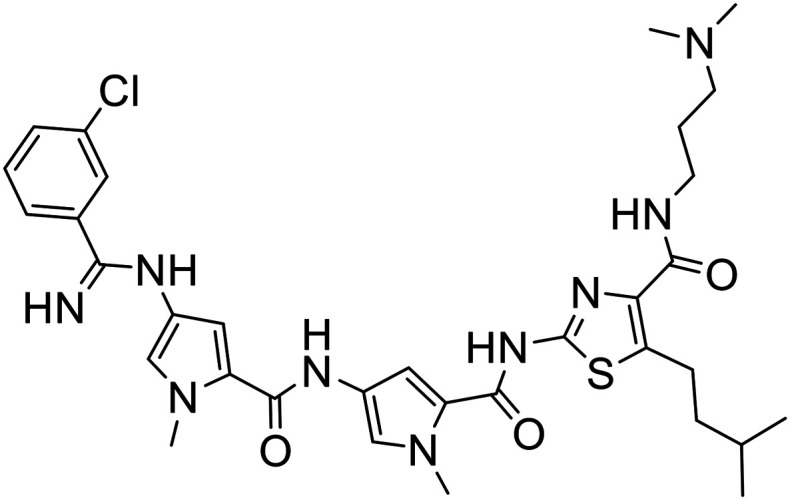
|
381 |
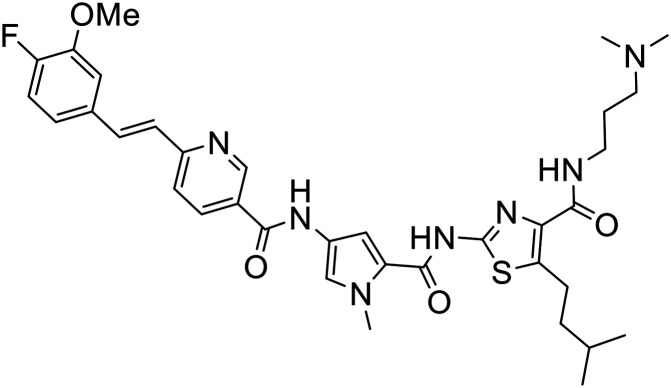
|
| 353 |
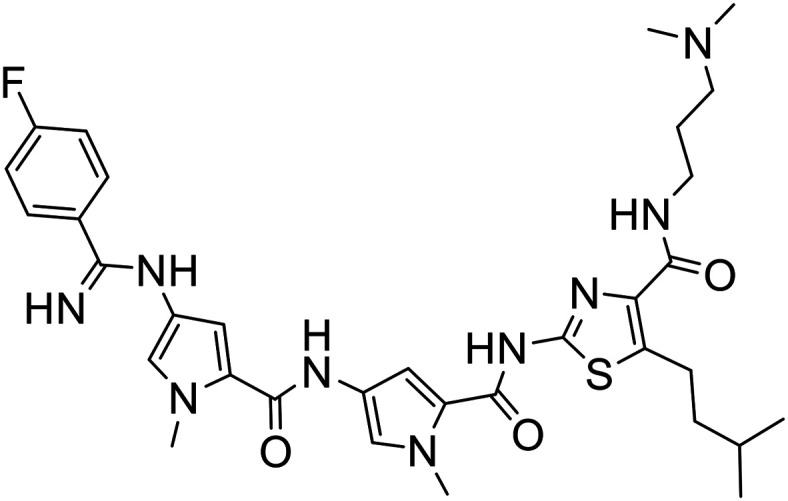
|
382 |
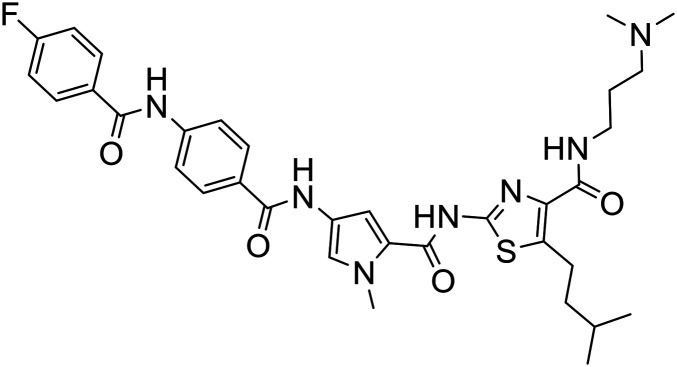
|
| 354 |
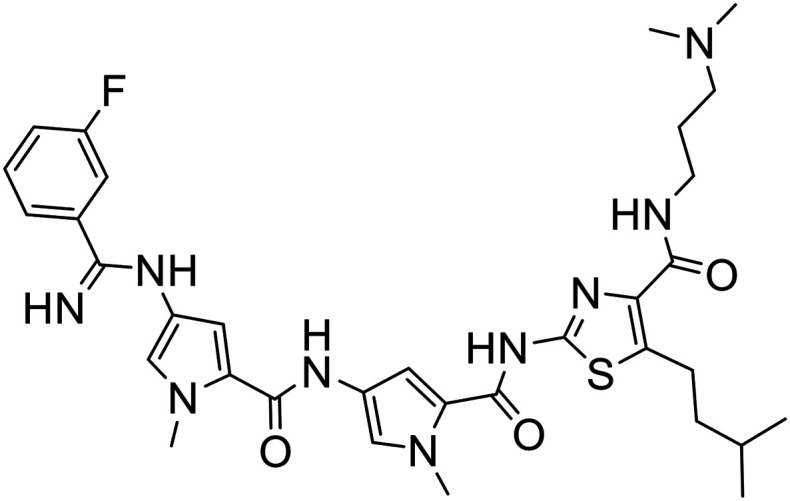
|
385 |
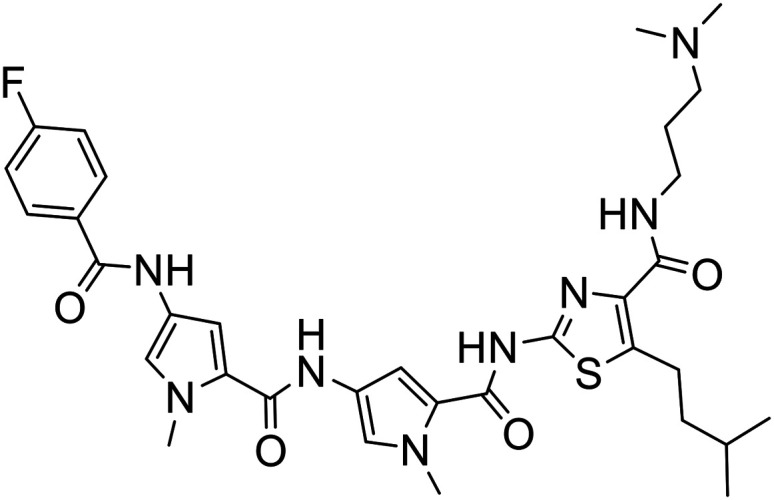
|
| 371 |
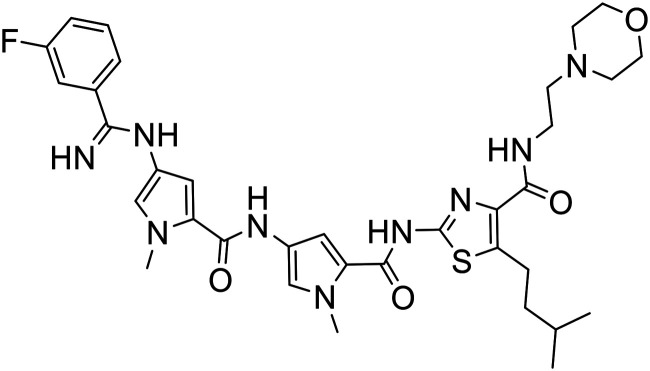
|
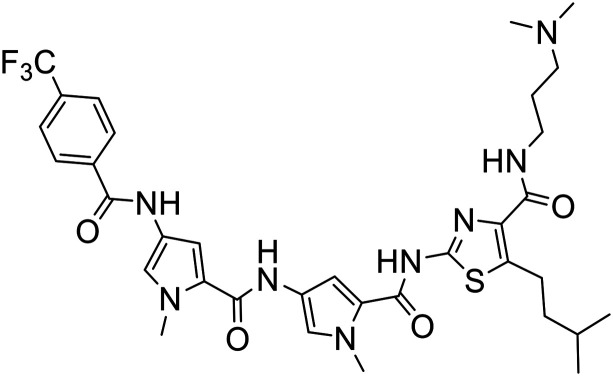
386 |
|
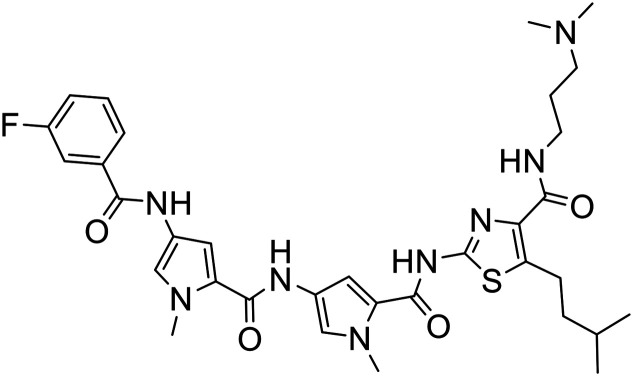
| 372 |
|
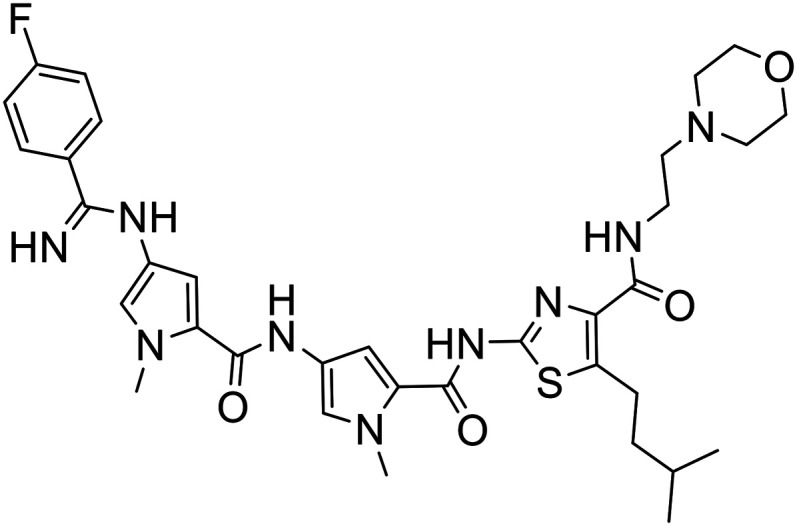
387 |
|
| 373 |
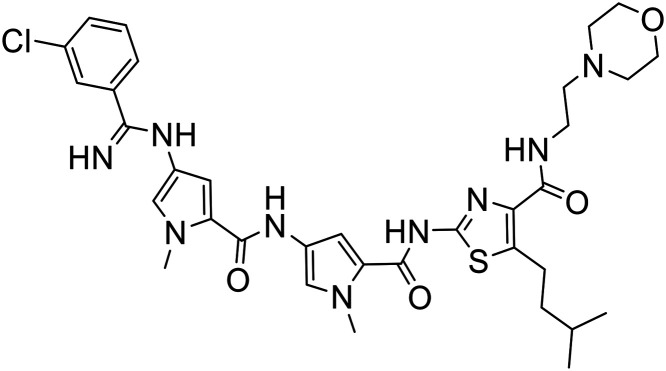
|
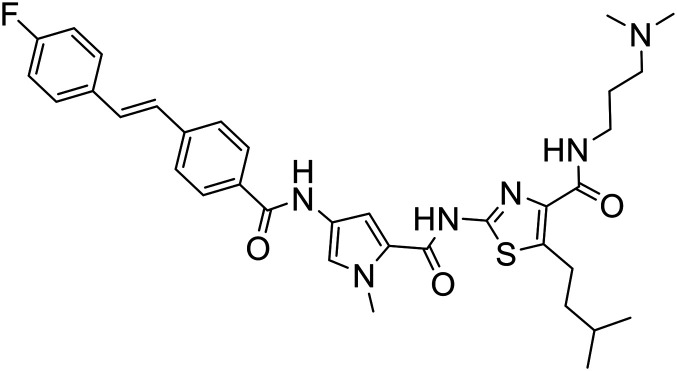
4 |
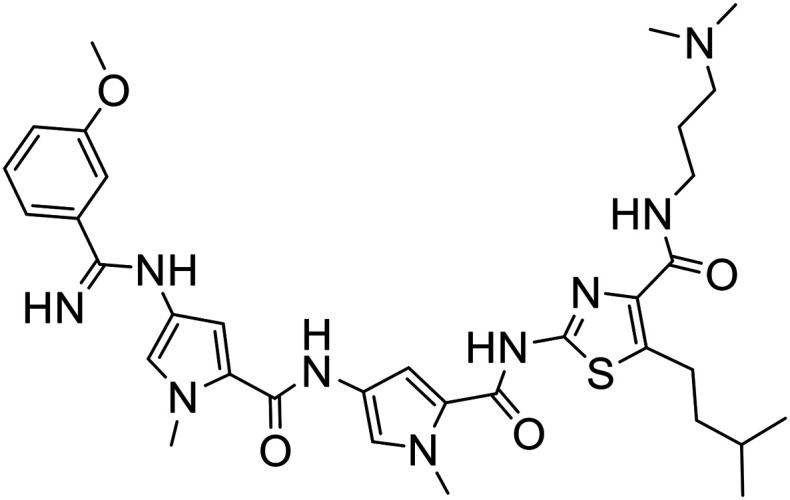
|
| 374 |
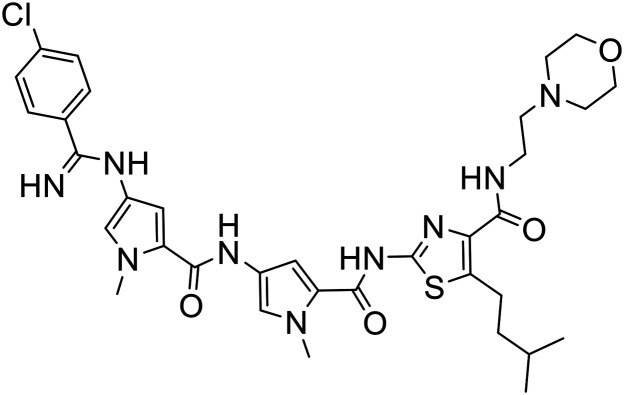
|
317 |
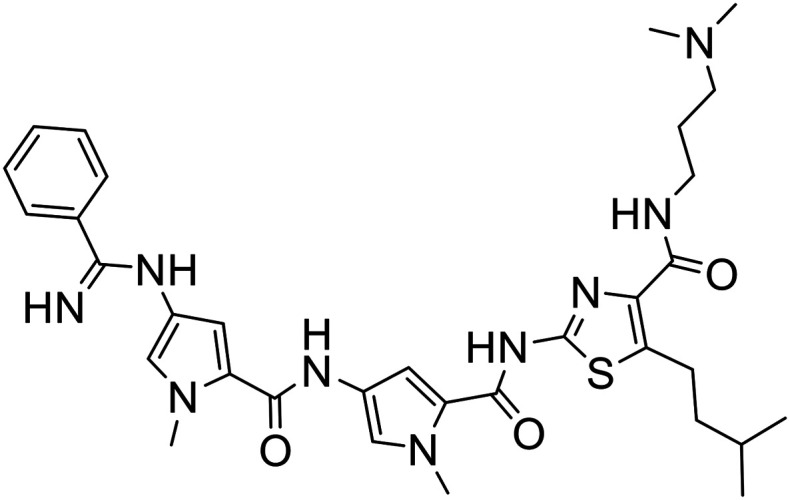
|
| 376 |
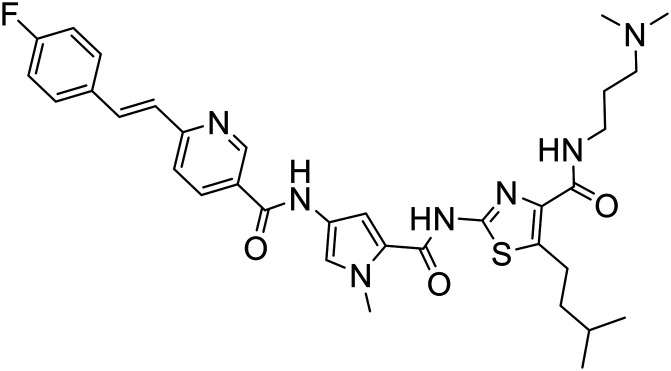
|
74 |
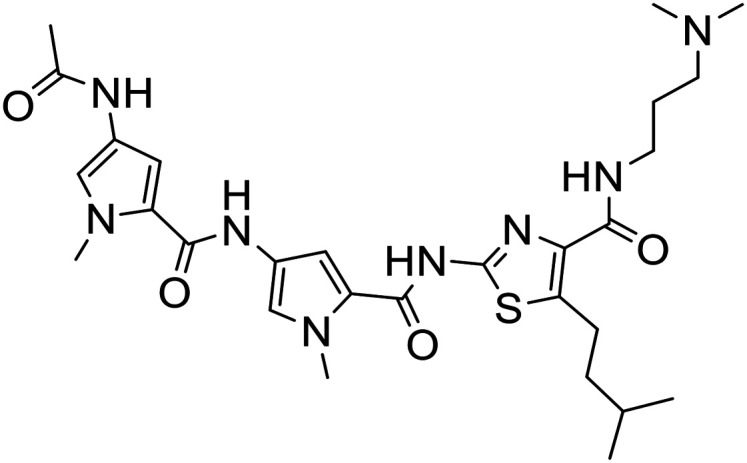
|
| 377 |
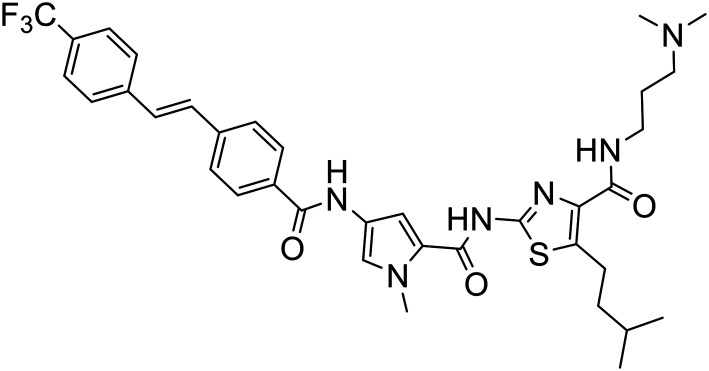
|
67 |
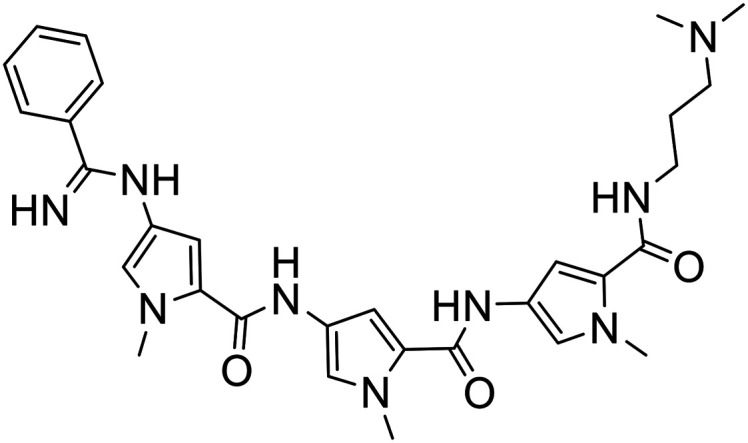
|
| 378 |
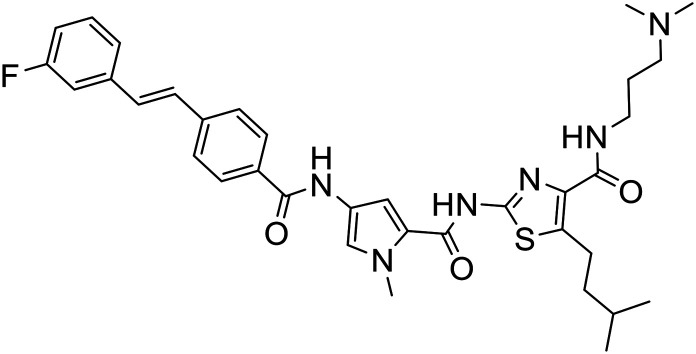
|
68 |
|
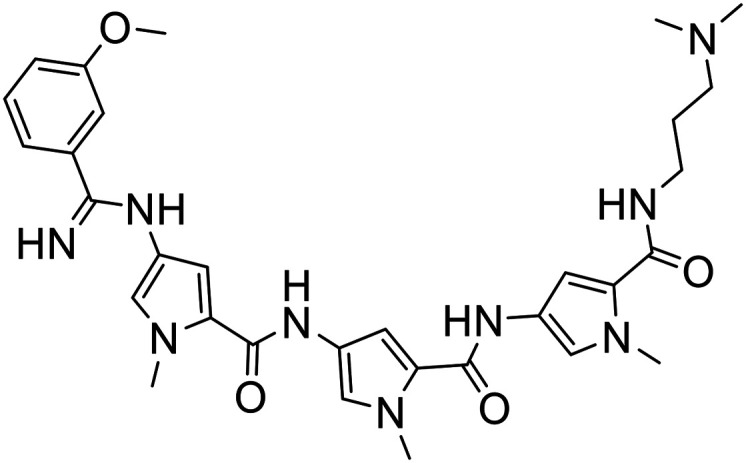
| 379 |
|
338 |
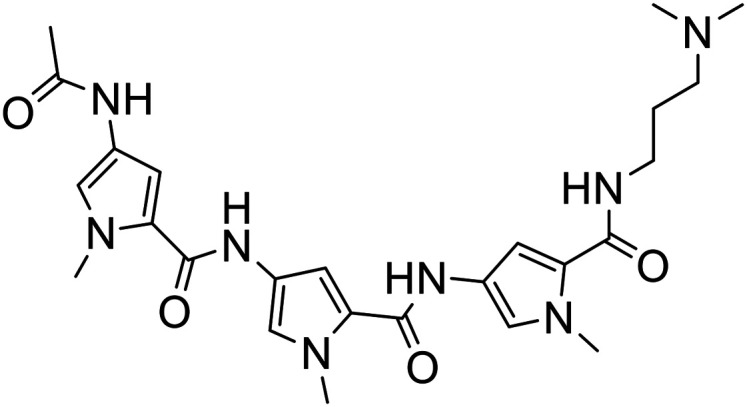
|
2.2. Evaluation of S-MGBs for in vitro anti-cancer activity and cancer cell selectivity
The 24 S-MGBs were tested for cytotoxic activity towards cancer cells in vitro through chemosensitivity screening and determination of IC50 values as previously described.14,15 Cancer cell lines tested included the human colorectal carcinoma cell line HCT116 and cisplatin-sensitive and cisplatin-resistant A2780 human ovarian cancer cells.16 The activity of the individual S-MGBs was also tested against ARPE19 human retinal epithelial non-cancer cells which have been extensively utilised as a non-cancer cell model.15–18 As a comparative benchmark, the clinically used platinates cisplatin and carboplatin were also evaluated with IC50 values summarised in Table 2 as well as calculated log D values at pH 7.4 providing an estimation of lipophilicity for the different S-MGBs. log D values at pH 6.5 and 5.0 are presented in the supplementary information. Selectivity indices (SI: IC50 against non-cancer cells divided by IC50 against the cancer cell line) were also calculated providing a preliminary indication of cancer cell selectivity of the different S-MGBs; a SI > 1 indicating selectivity for cancer cells in vitro (Table 2).
Table 2. IC50 values (μM) of the library of S-MGBs, associated log D7.4 values and cancer cell selectivity indices. Each IC50 value represents the mean ± SD of three independent experiments.
| S-MGB ID | A2780 (ovarian cancer) | A2780cis (cisplatin resistant) | HCT116 (colon cancer) | ARPE19 (non-cancer) | log D7.4 | Selectivity index (A2780, ovarian cancer) | Selectivity index (A2780cis, cisplatin resistant ovarian cancer) | Selectivity index (HCT116, colon cancer) |
| 351 | 2.90 + 1.4 | 3.14 + 2.20 | 1.17 + 0.20 | 2.85 + 0.09 | 3.13 | 0.98 | 0.91 | 2.44 |
| 352 | 2.76 + 0.27 | 1.23 + 0.38 | 1.27 + 0.09 | 3.53 + 1.60 | 3.28 | 1.28 | 2.87 | 2.78 |
| 353 | 1.89 + 0.29 | 1.26 + 0.09 | 1.27 + 0.48 | 1.80 + 0.10 | 2.56 | 0.95 | 1.43 | 1.42 |
| 354 | 3.83 + 0.30 | 0.99 + 0.07 | 0.90 + 0.20 | 3.50 + 0.70 | 2.91 | 0.91 | 3.54 | 3.89 |
| 371 | 2.82 + 0.90 | 1.11 + 0.13 | 1.30 + 0.05 | 2.35 + 0.04 | 4.51 | 0.81 | 2.12 | 1.81 |
| 372 | 2.15 + 0.50 | 0.62 + 0.08 | 0.68 + 0.06 | 2.40 + 0.20 | 4.17 | 1.12 | 3.87 | 3.53 |
| 373 | 0.84 + 0.18 | 1.02 + 0.29 | 0.88 + 0.21 | 1.11 + 0.18 | 4.88 | 1.32 | 1.09 | 1.26 |
| 374 | 1.04 + 0.20 | 1.10 + 0.23 | 1.25 + 0.13 | 1.37 + 0.40 | 4.74 | 1.32 | 1.25 | 1.10 |
| 376 | 1.09 + 0.07 | 0.96 + 0.14 | 2.07 + 0.63 | 1.47 + 0.54 | 4.13 | 1.35 | 1.53 | 0.71 |
| 377 | 1.04 + 0.01 | 1.16 + 0.04 | 2.14 + 0.63 | 1.41 + 0.44 | 5.85 | 1.36 | 1.22 | 0.66 |
| 378 | 0.94 + 0.18 | 0.92 + 0.11 | 1.78 + 0.14 | 1.65 + 0.89 | 5.12 | 1.76 | 1.79 | 0.93 |
| 379 | 1.04 + 0.19 | 1.05 + 0.22 | 1.85 + 0.64 | 2.14 + 1.25 | 5.12 | 2.06 | 2.04 | 1.16 |
| 380 | 2.55 + 1.35 | 1.28 + 0.42 | 1.32 + 0.07 | 2.06 + 1.50 | 4.87 | 0.81 | 1.61 | 1.56 |
| 381 | 5.00 + 1.60 | 1.86 + 0.32 | 1.25 + 0.02 | 4.26 + 1.90 | 3.98 | 0.85 | 2.29 | 3.41 |
| 382 | 4.59 + 1.80 | 2.28 + 0.41 | 1.40 + 0.06 | 4.55 + 1.70 | 3.87 | 0.99 | 2.00 | 3.25 |
| 385 | 4.30 + 1.57 | 2.43 + 0.16 | 1.27 + 0.13 | 1.75 + 0.15 | 3.09 | 0.41 | 0.72 | 1.38 |
| 386 | 3.08 + 0.02 | 2.79 + 0.60 | 1.11 + 0.04 | 1.70 + 0.70 | 3.09 | 0.55 | 0.61 | 1.53 |
| 387 | 5.00 + 1.86 | 2.00 + 0.50 | 1.04 + 0.30 | 1.69 + 0.33 | 3.83 | 0.34 | 0.85 | 1.63 |
| 4 | 0.89 + 0.20 | 1.12 + 0.27 | 1.33 + 0.12 | >10 | 2.48 | >11.24 | >8.93 | >7.52 |
| 317 | 1.34 + 0.07 | 1.08 + 0.05 | 2.12 + 0.75 | >10 | 2.42 | >7.46 | >9.26 | >4.72 |
| 74 | 1.46 + 0.01 | 1.51 + 0.17 | >10 | >10 | 1.05 | >6.85 | >6.62 | >1 |
| 67 | >10 | >10 | >10 | >10 | –0.35 | >1 | >1 | >1 |
| 68 | >10 | >10 | 5.64 + 3.70 | >10 | –0.29 | >1 | >1 | >1.77 |
| 338 | >10 | >10 | >10 | >10 | –1.67 | >1 | >1 | >1 |
| Cis-platin | 1.47 ± 0.04 a | 10.27 ± 1.77 a | 3.26 ± 0.38 a | 6.41 ± 0.95 a | N/A | 4.36 | 0.62 | 1.97 |
| Carboplatin | 2.50 + 0.30 | 44.90 + 3.20 | 35.37 ± 11.14 a | 77.73 ± 10.52 a | N/A | 31.09 | 1.73 | 2.20 |
aIC50 as reported in ref. 14.
As shown in Table 2, the majority of the S-MGBs had notable single digit micromolar IC50 values against each of the cancer cell lines tested indicating good anti-cancer activity in vitro with the exception of S-MGB-67, S-MGB-68 and S-MGB-338 that were much less active (IC50 > 5 or > 10 μM). Many of the MGBs showed comparable or better activity to the clinically used agents cisplatin and carboplatin. Furthermore, over half of the S-MGBs were found to be more active against the cisplatin- and carboplatin-resistant A2780cis ovarian cancer cells than the parental cis-sensitive A2780 cells as indicated by a resistance factor of <1 (IC50 against A2780cis cells divided by the IC50 against A2780 parental cells). Whereas cisplatin was ∼7 fold less active against the A2780cis cells than against the parental A2780 cells and carboplatin was ∼18 fold less active, S-MGB-354 and S-MGB-372 were >3.4 fold more active towards the cisplatin resistant cells (Fig. 6). This indicates that these S-MGBs, as well as the other S-MGBs with a resistance factor <1, do not show cross resistance with cisplatin or carboplatin and that they likely have a different mechanism of action.
Fig. 6. Bar chart showing the activity of the S-MGBs against the A2780cis ovarian cancer cells compared to the A2780 parental cells and in comparison with the platinates cisplatin and carboplatin. Values are expressed as the resistance factor which is the IC50 against A2780cis cells divided by the IC50 against A2780 parental cells. A resistance factor >1 indicates the compound is less active towards the A2780cis cells than A2780 parental cells, a resistance factor equal to 1 indicates equitoxicity whereas a factor <1 indicates the compound is more active towards the A2780cis cells.

In addition to potency and lack of cross-drug resistance, another critical consideration in the identifying of potential lead compounds was whether any of these S-MGBs showed preferential cytotoxic activity towards cancer cells or whether they were generally toxic. As the selectivity indices in Fig. 7 indicate, many of the S-MGBs appeared to have good in vitro cancer cell selectivity, however, S-MGB-4 and S-MGB-317 emerged as the two lead compounds that were selectively potent against all three cancer cell lines with little activity against the non-cancer cells. S-MGB-74 showed good cancer cell selectivity and potency towards the A2780 and A2780cis ovarian cancer cells but was largely inactive (IC50 > 10 μM) towards the HCT116 cells.
Fig. 7. Selectivity indices for the panel of S-MGBs. A selectivity index >1 indicates that the MGB is more active against the indicated cancer cells than the ARPE19 non-cancer cells. Where applicable, values plotted are the lower bounds.

Further analysis of the data indicates some structure–activity relationships (SAR). Firstly, discrete structural modifications to the head group and head group-linker were extensively investigated with analogues S-MGB-351–387. These modifications had no clear structure–activity effect in significantly enhancing or reducing the activity or SI of the S-MGBs with most of the activities for these analogues being similar across the cell lines. This would suggest that the specific structure of the head group, for example whether it is an amide or an alkene, is not a major determinant of anti-cancer activity or selectivity although some subtle differences against particular cell lines were observed. This is distinct from previous observations of some of our S-MGBs tested in different therapeutic contexts where the head group profoundly affected activity against different organisms. The presence of an alkene link in the head group resulted in compounds active against bacteria, fungi and parasites, however, when replaced with an amide link, only significantly active compounds against the parasite Trypanosoma brucei brucei have been observed.6 Furthermore, replacement of the alkene link with an amidine resulted in significantly active compounds against the fungus Cryptococcus neoformans only.9
The data shows that there is a significant effect on activity due to changing one of the heterocyclic subunits from a pyrrole to a much more lipophilic isopentylthiazole. Specifically, S-MGBs 4, 74 and 317, our three identified lead compounds (Table 2; Fig. 7), are analogous to S-MGBs 67, 68 and 338 respectively, with the only major structural difference being the heterocyclic subunit. S-MGBs 4, 74 and 317 contain the isopentylthiazole subunit and show promising selective activities against all three cancer cell lines tested whilst being inactive against the non-cancer cell model. However, S-MGBs 67, 68 and 338, where the isopentylthiazole has been replaced with a pyrrole, were inactive (IC50 > 5 or 10 μM) against all of the cell lines. Fig. 8 shows the log D values at different pHs for these six compounds. It can be seen that the more active isopentylthiazole containing S-MGBs, 4, 74 and 317, are much more lipophilic than the inactive pyrrole containing MGBs, 67, 68 and 338.
Fig. 8. Influence of the heterocyclic subunit on log DpH.

This may suggest that the isopentylthiazole is necessary for anti-cancer activity for these types of compounds. However, rather than being an effect specific to the isopentyl thiazole per se, it may simply be due to the increased lipophilicity of the resulting compounds. To explore this further, the log D values for each S-MGB was compared against the IC50 values and also the selectivity indices (SI), as shown in Fig. 9 and 10, respectively.
Fig. 9. Relationship between log D7.4 and IC50 (μM).

Fig. 10. Relationship between log D7.4 and selectivity index.

A minimum level of lipophilicity appears to be required for S-MGB activity (a log D7.4 of ∼1 to 2; Fig. 9), however further increases in lipophilicity do not seem to be directly linked to activity as compounds with log D7.4 values ranging from 1–6 showed comparable activity (Fig. 9).
Lipophilicity is one of the most important physicochemical properties to consider when designing drugs as finding the right balance between lipophilicity and hydrophilicity for a compound is key to acquiring good cell penetration either through membrane permeation or via transport proteins as well as influencing drug solubility, activity and toxicity. The trend in this set of data of lipophilicity having an effect on activity is consistent with the trend observed in our previous study identifying active S-MGBs against B16-F10-luc lung cancer cells. Therein, we found the most active compounds tended to have a higher log D7.4 value of 3–5.13
A large lipophilicity is often correlated with greater activity in medicinal chemistry as more hydrophobic compounds tend to bind more efficiently to their desired binding site, which is often hydrophobic as well. Increased lipophilicity can, however, result in compound promiscuity and heightened toxicity through non-specific binding which is a trend also observed in this dataset (Fig. 10). Many of the most active compounds that possess log D7.4 values ranging from 3–6 also do not have a particularly satisfactory SI (SI less than or near to 1). For example, one of the more active compounds, S-MGB-378, has a log D7.4 value of 5.12 but a mean SI of 1.49. This is in contrast to S-MGB-4, which has a log D7.4 value of 2.48 and a mean SI of >9.23. This again asserts that finding the right balance between lipophilicity and hydrophobicity is not only key for imparting compound activity but also compound specificity. With this in mind, the S-MGBs with the best SIs fall in the range of a log D7.4 ∼1–2.5 (Fig. 10 in box).
In summary, an isopentylthiazole heterocycle in conjunction with a dimethylaminopropyl tail group appeared to impart greater anti-cancer activity, as well as enhanced selectivity whereas the specific structure of the head group had no notable effect on activity, as evident in the set of S-MGBs 4, 74 and 317 which contain all these structural components.
2.3. Assessment of DNA damage
In a number of target organisms, including bacteria, parasites and cancer cells, we have demonstrated that fluorescent probe S-MGBs can localise in regions where DNA is found.13,21 Thus it is likely that the structural type of S-MGBs described here do also. However, one limitation of many anti-cancer MGBs reported in the literature is toxicity to normal cells due to DNA damage. The primary reason for this is that the mechanism of action of most reported anti-cancer MGBs is as DNA alkylating agents that covalently modify the DNA; this is a type of DNA damage that can result in DNA strand breaks.11 Importantly, our S-MGBs do not possess any alkylating moieties and we hypothesise that their mechanism of action against cancer cells is similar to that observed in our ongoing studies of anti-bacterial and anti-parasitic S-MGBs which suggest non-covalent interruption of DNA functions.19–21 Moreover, we have previously demonstrated that S-MGBs 4, 74 and 317 do bind to DNA giving ΔTms of 7, 4 and 7 °C, respectively.9 However, it was important to establish whether these lead anti-cancer S-MGBs induce significant DNA damage or not and thus whether these might suffer from similar toxicity issues as alkylating anti-cancer MGBs.
To investigate this, HCT116 cancer cells were exposed to a range of doses of S-MGB-4 for 24 h and assessed for the possible induction of DNA double strand breaks using γH2AX phosphorylation at serine 139 as a sensitive biomarker of double strand breaks.22 As a positive control, the topoisomerase II inhibitor doxorubicin, which is known to induce DSBs, was also tested. To determine whether MGB4 might induce other types of DNA damage to DSBs, levels of the tumour suppressor p53 protein were also assessed. p53 protein levels rapidly increase in response to multiple types of DNA damage through its primary cellular role as ‘guardian of the genome’. p53 is a major cellular stress sensor and coordinates the cellular response to DNA damage and other types of stress. As Fig. 11 shows, doxorubicin treatment caused a substantial increase in levels of both H2AX phosphorylation and of p53. In contrast, S-MGB-4 treatment (24 h 0.25–5 μM) failed to induce either H2AX phosphorylation or induction of p53 protein above levels of the DMSO (0.1%) solvent control. These results suggest that the mechanism of action of S-MGB-4 does not involve the induction of cellular DNA damage consistent with its non-alkylating structure.
Fig. 11. Western blot analysis of the effect of S-MGB-4 (24 hour drug exposure) on γH2AX phosphorylation and p53 induction in HCT116 cancer cells.

3. Conclusion
A suite of 24 S-MGBs was synthesised and these were screened for in vitro anti-cancer activity against a human colon carcinoma cell line and cisplatin-sensitive versus cisplatin-resistant ovarian cancer cell lines. A human non-cancerous retinal epithelial cell line was used to measure selectivity of any response.
Significant single digit micromolar activities were measured for the majority of the compounds tested with S-MGBs 4, 74 and 317 showing promising potency and selectivity; this was particularly the case for S-MGB-4 whereas S-MGB-74 was active against the ovarian cancer cells but inactive towards the colon cancer cells. A clear structural activity was determined from these results and it is evident that a lipophilic heterocycle such as an isopentylthiazole is key to imparting high activity and also that the dimethylaminopropyl tail influences specificity. For these reasons, S-MGBs 4, 74 and 317 will be subject to further structural modifications at the isopentylthiazole heterocycle subunit and modifications to the dimethylaminopropyl tail will also be investigated.
Furthermore, an initial investigation into the mechanism of action of one compound, S-MGB-4, was carried out. This has shown that DNA double strand breaks are not induced and neither is the DNA damage stress sensor protein p53, indicating that the action of these S-MGBs is likely not through an alkylating or DNA damage response mechanism.
4. Experimental
4.1. Chemistry
4.1.1. General experimental methods
1H and 13C NMR spectra were measured on a Bruker DPX-400 MHz spectrometer with chemical shifts given in ppm (d values), relative to proton and carbon traces in solvent. Coupling constants are reported in Hz. IR spectra were recorded on a Perkin Elmer, FT-IR spectrometer. Mass spectra were obtained on a Jeol JMS AX505. Anhydrous solvents were obtained from a Puresolv purification system, from Innovative Technologies, or purchased as such from Aldrich. Melting points were recorded on a Reichert hotstage microscope, and are uncorrected. Chromatography was carried out using 200–400 mesh silica gels, or using reverse-phase HPLC on a waters system using a C18 Luna column (Luna SuC18(2), 100A, AXIA, 50 × 21.20 mm, 5 micron Phenomenex) with the gradient given below and using a detection wavelength of 254 nm.
HPLC method used for the purification of final MGBs:
| Time (min) | Flow rate (mL min–1) | % water (0.1% TFA) | % acetonitrile (0.1% TFA) |
| 0 | 6 | 90 | 10 |
| 25 | 6 | 60 | 40 |
| 35 | 6 | 50 | 50 |
| 40 | 6 | 30 | 70 |
| 44 | 6 | 90 | 10 |
4.1.2. Lipophilicity
The log DpH values at 7.4, 6.5 and 5.0 were estimated using the software MarvinSketch (version 15.6.29.0, ChemAxon, ; http://www.chemaxon.com), using default parameters as previously described.13
4.1.3. Synthesis
4.1.3.1. Synthesis of S-MGBs: 371, 372, 373, 374
5-Isopentyl-2-{[(1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrol-2-yl)carbonyl]amino}-N-[2-(4-morpholinyl)ethyl]-1,3-thiazole-4-carboxamide (170 mg, 0.283 mmol) was suspended in methanol (25 mL). Pd/C-10% (90 mg) was added portionwise at 0 °C with stirring under nitrogen. The reaction mixture was hydrogenated for 4 h. The catalyst was removed over Kieselguhr and the solvent was removed under reduced pressure. The amine so formed was dissolved in DMF (8 mL, dry) and divided into four equal portions.
2-{[(4-{[(4-{[(3-Fluorophenyl)(imino)methyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-5-isopentyl-N-[2-(4-morpholinyl)ethyl]-1,3-thiazole-4-carboxamide bis(trifluoroacetate) (S-MGB-371)
The amine solution (2 mL) was added to methyl 3-fluorobenzenecarbimidothioate hydroiodide (21 mg, 0.071 mmol) at room temperature with stirring. The stirring was continued overnight at room temperature. The solution was subjected to HPLC purification (without work up). Fractions containing the required product were collected and freeze dried. The required product was obtained as white solid (33 mg, 50%), RT = 21.5 min with no distinct melting point.
IR: 718, 797, 831, 891, 1005, 1059, 1107, 1126, 1177, 1196, 1236, 1271, 1368, 1400, 1431, 1464, 1547, 1586, 1647 cm–1.
1H NMR (DMSO-d6): 12.09 (1H, s), 11.26 (1H, s), 10.14 (1H, s), 9.89 (1H, s), 9.68 (1H, br), 8.95 (1H, s), 8.10 (1H, br), 7.80–7.67 (1H, m), 7.48 (1H, d, J = 1.9 Hz), 7.40 (1H, d, J = 1.9 Hz), 7.31 (1H, d, J = 1.9 Hz), 7.10 (1H, d, J = 1.9 Hz), 4.03–4.00 (2H, m), 3.97 (3H, s), 3.92 (3H, s), 3.66–3.55 (8H, m), 4.23–3.14 (4H, m), 1.62–1.51 (3H, m), 0.94 (6H, d, J = 6.4 Hz).
HRESIMS: found: ½ M/Z: 346.6603 calculated for C34H44O4N9FS = 346.6605.
2-{[(4-{[(4-{[(4-Fluorophenyl)(imino)methyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-5-isopentyl-N-[2-(4-morpholinyl)ethyl]-1,3-thiazole-4-carboxamide bis(trifluoroacetate) (S-MGB-372)
The amine solution (2 mL) was added to methyl 4-fluorobenzenecarbimidothioate hydroiodide (21 mg, 0.071 mmol) at room temperature with stirring. The stirring was continued overnight at room temperature. The solution was subjected to HPLC purification (without work up). Fractions containing the required product were collected and freeze dried. The required product was obtained as white solid (31 mg, 47%), RT = 21.2 min with no distinct melting point.
IR: 718, 739, 781, 799, 831, 893, 1007, 1059, 1105, 1126, 1171, 1196, 1242, 1271, 1368, 1400, 1435, 1464, 1508, 1545, 1586, 1609, 1653 cm–1.
1H NMR (DMSO-d6): 12.09 (1H, s), 11.16 (1H, s), 10.13 (1H, s), 9.82 (1H, s), 9.66 (1H, br), 8.84 (1H, s), 8.10 (1H, br), 7.98 (1H, d, J = 5.2 Hz), 7.96 (1H, d, J = 5,2 Hz), 7.59 (1H, d, J = 8.8 Hz), 7.56 (1H, d, J = 8.8 Hz), 7.48 (1H, d, J = 1.8 Hz), 7.39 (1H, d, J = 1.8 Hz), 7.30 (1H, d, J = 1.8 Hz), 7.09 (1H, d, J = 1.8 Hz), 4.40–4.01 (2H, m), 3.97 (3H, s),3.92 (3H, s), 3.69–3.55 (6H, m), 3.23–3.14 (4H, m), 2.53–2.52 (2H, m),1.64–1.52 (3H, m), 0.94 (6H, d, J = 6.4 Hz).
HRESIMS: ½ M/Z found: 346.6603 C34H44O4N9FS = 346.6605.
2-{[(4-{[(4-{[(3-Chlorophenyl)(imino)methyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-5-isopentyl-N-[2-(4-morpholinyl)ethyl]-1,3-thiazole-4-carboxamide bis(trifluoroacetate) (S-MGB-373)
The amine solution (2 mL) was added to methyl 3-chlorobenzenecarbimidothioate hydroiodide (22 mg, 0.071 mmol) at room temperature with stirring. The stirring was continued overnight at room temperature. The solution was subjected to HPLC purification (without work up). Fractions containing the required product were collected and freeze dried. The required product was obtained as white solid (33 mg, 49%), RT = 21.7 min with no distinct melting point.
IR: 716, 797, 829, 893, 1007, 1059, 1128, 1175, 1196, 1275, 1368, 1400, 1429, 1466, 1508, 1547, 1655 cm–1.
1H NMR (DMSO-d6): 12.08 (1H, s), 11.27 (1H, s), 10.14 (1H, s), 9.89 (1H, s), 9.66 (1H, br), 8.94 (1H, s), 8.09 (1H, br), 7.99 (1H, t, J = 1.9 Hz), 7.89–7.82 (2H, m), 7.74 (1H, t, J = 8.0 Hz), 7.49 (1H, d, J = 1.8 Hz), 7.40 (1H, d, J = 1.8 Hz), 7.30 (1H, d, J = 1.8 Hz), 7.09 (1H, d, J = 1.8 Hz), 4.03–4.00 (2H, m), 3.97 (3H, s), 3.92 (3H, s), 3.69–3.55 (8H, m), 3.23–3.15 (4H, m), 1.64–1.51 (3H, m), 0.94 (6H, d, J = 6.4 Hz).
HRESIMS: ½ M/Z: found: 354.6455 calculated for C34H44O4N9ClS 354.6457.
2-{[(4-{[(4-{[(4-Chlorophenyl)(imino)methyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-1-methyl-1H-pyrrol-2-yl)carbonyl]amino}-5-isopentyl-N-[2-(4-morpholinyl)ethyl]-1,3-thiazole-4-carboxamide bis(trifluoroacetate) (S-MGB-374)
The amine solution (2 mL) was added to methyl 4-chlorobenzenecarbimidothioate hydroiodide (22 mg, 0.071 mmol) at room temperature with stirring. The stirring was continued overnight at room temperature. The solution was subjected to HPLC purification (without work up). Fractions containing the required product were collected and freeze dried. The required product was obtained as white solid (33 mg, 50%), RT = 21.9 min with no distinct melting point.
IR: 718, 797, 831, 893, 1013, 1059, 1128, 1177, 1198, 1287, 1368, 1400, 1427, 1466, 1549, 1661 cm–1.
1H NMR (DMSO-d6): 12.08 (1H, s), 11.22 (1H, s), 10.14 (1H, s), 9.86 (1H, s), 9.68 (1H, br), 8.89 (1H, s), 8.09 (1H, br), 7.91 (2H, d, J = 8.7 Hz), 7.80 (2H, d, J = 8.7 Hz), 7.48 (1H, d, J = 1.8 Hz), 7.40 (1H, d, J = 1.8 Hz), 7.30 (1H, d, J = 1.8 Hz), 7.09 (1H, d, J = 1.8 Hz), 4.04–4.00 (2H, m), 3.97 (3H, s), 3.92 (3H, s), 3.69–3.50 (8H, m), 3.23–3.15 (4H, m), 1.64–1.51 (3H, m), 0.94 (6H, d, J = 6.4 Hz).
HRESIMS: ½ M/Z found: 354.6455 calculated for C34H44O4N9ClS 354.6457.
4.1.3.2. Synthesis of S-MGBs: 376, 377, 378 & 379
N-[3-(Dimethylamino)propyl]-5-isopentyl-2-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1,3-thiazole-4-carboxamide (234 mg, 0.519 mmol) was dissolved in methanol (25 mL) to which Pd/C-10% (104 mg) was added at 0 °C under nitrogen with stirring. The reaction mixture was hydrogenated for 4 h at atmospheric pressure and room temperature. The catalyst was removed over Kieselguhr and the solution was divided into four equal portions.
N-[5-({[4-({[3-(Dimethylamino)propyl]amino}carbonyl)-5-isopentyl-1,3-thiazol-2-yl]amino}carbonyl)-1-methyl-1H-pyrrol-3-yl]-6-[(E)-2-(4-fluorophenyl)ethenyl]nicotinamide trifluoroacetate (S-MGB-376)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 6-[(E)-2-(4-fluorophenyl)ethenyl]nicotinic acid (32 mg, 0.13 mmol) and HBTU (99 mg, 0.26 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as orange solid (32 mg, 32%) with no distinct melting point. RT = 26 min.
IR: 720, 797, 826, 893, 968, 1007, 1061, 1125, 1161, 1179, 1196, 1238, 1285, 1400, 1427, 1466, 1508, 1549, 1593, 1655 cm–1.
1H NMR (DMSO-d6): 12.14 (1H, s), 10.60 (1H, s), 9.27 (1H, br), 9.10 (1H, d, J = 2.2 Hz), 8.31 (1H, dd, J = 2.2 Hz, J = 8.2 Hz), 7.98 (1H, t, J = 6.2 Hz), 7.83–7.77 (3H, m), 7.70 (1H, d, J = 8.2 Hz), 7.55 (1H, d, J = 1.7 Hz), 7.46 (1H, d, J = 1.7 Hz), 7.46 (1H, s), 7.40 (1H, s), 7.37 (1H, d, J = 8.8 Hz), 7.28 (1H, d, J = 8.8 Hz), 3.93 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 646.2967 calculated for C34H41O3N7FS 646.2970.
N-[3-(Dimethylamino)propyl]-5-isopentyl-2-[({1-methyl-4-[(4-{(E)-2-[4-(trifluoromethyl)phenyl]ethenyl}benzoyl)amino]-1H-pyrrol-2-yl}carbonyl)amino]-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-377)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 4-{(E)-2-[4-(trifluoromethyl)phenyl]ethenyl}benzoic acid (38 mg, 0.13 mmol) and HBTU (99 mg, 0.26 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as pale yellow solid (40 mg, 38%) with no distinct melting point. RT = 35 min.
IR: 720, 797, 839, 895, 951, 968, 999, 1015, 1065, 1107, 1165, 1198, 1254, 1283, 1323, 1398, 1464, 1505, 1549, 1613, 1653 cm–1.
1H NMR (DMSO-d6): 12.02 (1H, s), 10.45 (1H, s), 9.26 (1H, br), 8.01 (2H, d, J = 8.4 Hz), 7.88–7.87 (1H, m), 7.82–7.77 (2H, m), 7.79–7.77 (2H, m), 7.53–7.52 (3H, m), 7.46 (1H, d, J = 1.7 Hz), 3.92 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 695.2994 calculated for C36H42O3N6F3S 695.2986.
N-[3-(Dimethylamino)propyl]-2-({[4-({4-[(E)-2-(3-fluorophenyl)ethenyl]benzoyl}amino)-1-methyl-1H-pyrrol-2-yl]carbonyl}amino)-5-isopentyl-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-378)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 4-[(E)-2-(3-fluorophenyl)ethenyl]benzoic acid (32 mg, 0.13 mmol) and HBTU (99 mg, 0.26 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as pale yellow solid (25 mg, 25%) with no distinct melting point. RT = 36 min.
IR: 7120, 777, 829, 858, 893, 963, 1055, 1128, 1177, 1198, 1238, 1281, 1391, 1441, 1466, 1506, 1545, 1580, 1609, 1655 cm–1.
1H NMR (DMSO-d6): 12.12 (1H, s), 10.44 (1H, s), 9.32 (1H, br), 8.00–7.95 (3H, m), 7.77–7.72 (3H, m), 7.54–7.43 (6H, m), 7.16–7.13 (1H, m), 3.92 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 645.3028 calculated for C35H42O3N6FS 645.3018.
N-[3-(Dimethylamino)propyl]-2-({[4-({4-[(E)-2-(4-fluorophenyl)ethenyl]benzoyl}amino)-1-methyl-1H-pyrrol-2-yl]carbonyl}amino)-5-isopentyl-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-379)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 4-[(E)-2-(4-fluorophenyl)ethenyl]benzoic acid (32 mg, 0.13 mmol) and HBTU (99 mg, 0.26 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as pale yellow solid (24 mg, 24%) with no distinct melting point. RT = 36 min.
IR: 720, 783, 799, 841, 893, 964, 1007, 1059, 1126, 1159, 1173, 1198, 1233, 1283, 1398, 1429, 1466, 1506, 1547, 1597, 1649 cm–1.
1H NMR (DMSO-d6): 12.12 (1H, s), 10.42 (1H, s), 9.27 (1H, br), 7.99–7.94 (3H, m), 7.76–7.70 (4H, m), 7.54 (1H, d, J = 1.8 Hz), 7.46 (1H, d, J = 1.8 Hz), 7.40 (1H, s), 7.33 (1H, s), 7.28–7.24 (2H, m), 3.93 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 645.3028 calculated for C35H42O3N6FS 645.3018.
4.1.3.3. Synthesis of S-MGBs: 380, 381 & 382
N-[3-(Dimethylamino)propyl]-5-isopentyl-2-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1,3-thiazole-4-carboxamide (150 mg, 0.333 mmol) was dissolved in methanol (25 mL) to which Pd/C-10% (93 mg) was added at 0 °C under nitrogen with stirring. The reaction mixture was hydrogenated for 4 h at atmospheric pressure and room temperature. The catalyst was removed over Kieselguhr and the solution was divided into three equal portions.
N-[5-({[4-({[3-(Dimethylamino)propyl]amino}carbonyl)-5-isopentyl-1,3-thiazol-2-yl]amino}carbonyl)-1-methyl-1H-pyrrol-3-yl]-6-{(E)-2-[3-(trifluoromethyl)phenyl]ethenyl}nicotinamide trifluoroacetate (S-MGB-380)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 6-{(E)-2-[3-(trifluoromethyl)phenyl]ethenyl}nicotinic acid (33 mg, 0.111 mmol) and HBTU (84 mg, 0.222 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as orange solid (38 mg, 42%) with no distinct melting point. RT = 30 min.
IR: 698, 698, 698, 698, 720, 779, 797, 831, 893, 964, 1001, 1072, 1123, 1167, 1198, 1287, 1327, 1402, 1433, 1462, 1508, 1545, 1655 cm–1.
1H NMR (DMSO-d6): 12.14 (1H, s), 10.62 (1H, s), 9.26 (1H, br), 9.13 (1H, d, J = 2.2 Hz), 8.34 (1H, dd, J = 2.2 Hz and J = 8.2 Hz), 8.10 (1H, s), 8.05 (1H, d, J = 7.6 Hz), 7.98 (1H, t, J = 6.3 Hz), 7.92 (1H, d, J = 16.0 Hz), 7.74–7.63 (3H, m), 7.63 (1H, d, J = 16.0 Hz), 7.55 (1H, d, J = 1.6 Hz), 7.47 (1H, d, J = 1.6 Hz), 3.94 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 696.2949 calculated for C35H41O3N7F3S 696.2938.
N-[5-({[4-({[3-(Dimethylamino)propyl]amino}carbonyl)-5-isopentyl-1,3-thiazol-2-yl]amino}carbonyl)-1-methyl-1H-pyrrol-3-yl]-6-[(E)-2-(4-fluoro-3-methoxyphenyl)ethenyl]nicotinamide trifluoroacetate (S-MGB-381)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 6-[(E)-2-(4-fluoro-3-methoxyphenyl)ethenyl]nicotinic acid (30 mg, 0.111 mmol) and HBTU (84 mg, 0.222 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as orange solid (27 mg, 30%) with no distinct melting point. RT = 26.6 min.
IR: 720, 743, 779, 799, 831, 893, 964, 1005, 1028, 1061, 1125, 1196, 1265, 1289, 1370, 1402, 1422, 1466, 1514, 1551, 1595, 1663 cm–1.
1H NMR (DMSO-d6): 12.14 (1H, s), 10.60 (1H, s), 9.27 (1H, br), 9.11 (1H, d, J = 2.0 Hz), 8.32 (1H, dd, J = 2.2 Hz and J = 8.2 Hz), 7.98 (1H, t, J = 6.3 Hz), 7.80 (1H, d, J = 16.0 Hz), 7.70 (1H, d, J = 8.2 Hz), 7.57–7.55 (2H, m), 7.46 (1H, d, J = 1.7 Hz), 7.45 (1H, d, J = 16.0 Hz), 7.28–7.27 (2H, m), 3.94 (3H, s), 3.93 (3H, s), 3.37–3.33 (2H, m), 3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 676.3086 calculated for C35H43O4N7FS 676.3076.
N-[3-(Dimethylamino)propyl]-2-({[4-({4-[(4-fluorobenzoyl)amino]benzoyl}amino)-1-methyl-1H-pyrrol-2-yl]carbonyl}amino)-5-isopentyl-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-382)
The solvent was removed under reduced pressure and then the residue was dissolved in DMF (2 mL, dry) to which 4-[(4-fluorobenzoyl)amino]benzoic acid (29 mg, 0.111 mmol) and HBTU (84 mg, 0.222 mmol) were added at room temperature with stirring. The stirring was continued overnight at room temperature. HPLC purification followed by freeze-drying of the appropriate fractions gave the required product as white solid (28 mg, 32%) with no distinct melting point. RT = 26 min.
IR: 720, 760, 799, 833, 851, 895, 1011, 1059, 1128, 1163, 1196, 1234, 1285, 1323, 1400, 1433, 1466, 1505, 1547, 1599, 1643, 1651 cm–1.
1H NMR (DMSO-d6): 12.11 (1H, s), 10.51 (1H, s), 10.34 (1H, s), 9.26 (1H, br), 8.09 (2H, dd, J = 5.5 Hz and J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz and 1H, m), 7.93 (2H, d, J = 8.8 Hz), 7.52 (1H, d, J = 1.7 Hz), 7.45 (1H, d, J = 1.7 Hz), 7.42 (2H, t, J = 8.8 Hz), 3.92 (3H, s), 3.37–3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 662.2930 calculated for C34H41O4N7FS 662.2919.
4.1.3.4. Synthesis of S-MGBs: 385, 386 & 387
N-[3-(Dimethylamino)propyl]-5-isopentyl-2-{[(1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrol-2-yl)carbonyl]amino}-1,3-thiazole-4-carboxamide (110 mg, 0.192 mmol) was suspended in methanol (25 mL) to which was added Pd/C-10% (65 mg) at 0 °C with stirring. The reaction mixture was hydrogenated for 3 h at room temperature and atmospheric pressure. The catalyst was removed over Kieselguhr and the solvent was removed under reduced pressure. The amine so formed was dissolved in DMF (6 mL, dry) and the solution was divided into three equal portions.
N-[3-(Dimethylamino)propyl]-2-[({4-[({4-[(4-fluorobenzoyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-5-isopentyl-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-385)
4-Fluorobenzoic acid (9 mg, 0.064 mmol), and HBTU (48 mg, 0.128 mmol) were added to the amine (2 mL, 0.064 mmol) at room temperature with stirring. The reaction mixture was left stirring overnight and then purified by HPLC. Fractions containing the required material were collected and freeze-dried to give the desired product as white solid (16 mg, 31%) with no distinct melting point.
IR: 720, 762, 777, 801, 891, 1009, 1057, 1130, 1161, 1179, 1198, 1238, 1271, 1400, 1437, 1464, 1505, 1547, 1643 cm–1.
1H NMR (DMSO-d6): 12.06 (1H, s), 10.37 (1H, s), 10.08 (1H, s), 9.25 (1H, br), 8.05 (2H, dd, J = 5.5 Hz and J = 8.9 Hz), 7.98 (1H, t, J = 6.2 Hz), 7.45 (1H, d, J = 1.8 Hz), 7.42 (1H, d, J = 1.8 Hz), 7.39–7.34 (3H, m), 7.14 (1H, d, J = 1.8 Hz), 3.91 (3H, s), 3.90 (3H, s), 3.37–3.32 (2H, m), 3.22–3.18 (2H, m), 3.12–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 665.3038 calculated for C33H42O4N8FS 665.3028.
N-[3-(Dimethylamino)propyl]-2-[({4-[({4-[(3-fluorobenzoyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-1-methyl-1H-pyrrol-2-yl}carbonyl)amino]-5-isopentyl-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-386)
3-Fluorobenzoic acid (9 mg, 0.064 mmol), and HBTU (48 mg, 0.128 mmol) were added to the amine (2 mL, 0.064 mmol) at room temperature with stirring. The reaction mixture was left stirring overnight and then purified by HPLC. Fractions containing the required material were collected and freeze-dried to give the desired product as white solid (17 mg, 34%) with no distinct melting point.
IR: 720, 777, 799, 831, 889, 943, 1007, 1059, 1128, 1175, 1198, 1269, 1289, 1400, 1435, 1464, 1547, 1582, 1643 cm–1.
1H NMR (DMSO-d6): 12.07 (1H, s), 10.44 (1H, s), 10.10 (1H, s), 9.25 (1H, br), 7.98 (1H, t, J = 6.1 Hz), 7.83 (1H, d, J = 8.0 Hz), 7.78–7.74 (2H, m), 7.46–7.41 (3H, m), 7.36 (1H, d, J = 1.8 Hz), 7.16 (1H, d, J = 1.8 Hz), 3.91 (3H, s), 3.90 (3H, s), 3.37–3.32 (2H, m), 3.22–3.18 (2H, m), 3.12–3.07 (2H, m), 2.80 (6H, d, J = 4.9 Hz), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 665.3038 calculated for C33H42O4N8FS 665.3028.
N-[3-(Dimethylamino)propyl]-5-isopentyl-2-{[(1-methyl-4-{[(1-methyl-4-{[4-(trifluoromethyl)benzoyl]amino}-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrol-2-yl)carbonyl]amino}-1,3-thiazole-4-carboxamide trifluoroacetate (S-MGB-387)
4-(Trifluoromethyl)benzoic acid (12 mg, 0.064 mmol), and HBTU (48 mg, 0.128 mmol) were added to the amine (2 mL, 0.064 mmol) at room temperature with stirring. The reaction mixture was left stirring overnight and then purified by HPLC. Fractions containing the required material were collected and freeze-dried to give the desired product as off-white solid (14 mg, 26%) with no distinct melting point.
IR: 720, 770, 799, 833, 856, 891, 1015, 1065, 1117, 1169, 1200, 1275, 1289, 1325, 1402, 1439, 1464, 1510, 1547, 1574, 1643 cm–1.
1H NMR (DMSO-d6): 12.11 (1H, s), 10.51 (1H, s), 10.34 (1H, s), 9.24 (1H, br), 8.17 (2H, d, J = 8.4 Hz), 7.98 (1H, t, J = 6.0 Hz), 7.93 (2H, d, J = 8.4 Hz), 7.46 (1H, d, J = 2.0 Hz), 7.42 (1H, d, J = 2.0 Hz), 7.38 (1H, d, J = 2.0 Hz), 7.17 (1H, d, J = 2.0 Hz), 3.91 (6H, s), 3.37–3.22 (2H, m), 3.11–3.07 (2H, m), 2.80 (3H, s), 2.79 (3H, s), 1.91–1.85 (2H, m), 1.63–1.51 (3H, m), 0.93 (6H, d, J = 6.4 Hz).
HRESIMS: found: 715.3005 calculated for C34H42O4N8F3S 715.2996.
4.2. Biological evaluation
4.2.1. Cell lines
Cell lines used in this study were all from ATCC or ECACC and of low passage. HCT116 p53+/+ human colorectal cancer cells were cultured in DMEM supplemented with 10% foetal bovine serum and 2 mM l-glutamine. A2780 and A2780cis human ovarian carcinoma cells were cultured in RPMI 1640 media with 10% foetal bovine serum and 2 mM l-glutamine. For maintenance of cisplatin resistance of A2780cis cancer cells, every 2–3 passages 1 μM cisplatin was added to the cell culture media. ARPE19 non-cancer cells were cultured as described.18
4.2.2. Chemosensitivity studies
Cells were seeded in round bottom 96-well plates at 2000 cells per well. After 24 hours incubation at 37 °C, cells were treated with varying doses of compounds or vehicle control for 96 h. The MTT assay (2,5-diphenyltetrazolium bromide) was performed after 96 hours by addition of MTT at a final concentration of 0.5 mg ml–1 and after 4 hours incubation at 37 °C, any formazan crystals formed were dissolved in DMSO. The absorbance of each well was measured at 540 nm and the resulting absorbance values were used to calculate the 50% inhibitory concentrations (IC50) for each of the compounds as is previously described.15,16
4.2.3. Antibodies and Western blot analysis
Primary antibodies used were: phosphorylated S139 γH2AX (1 : 1000 dilution, 9F3 clone, Abcam) p53 (DO-1 clone, 1 : 1000, Santa Cruz) and β-actin (1 : 40 000, C4 clone, Millipore). Cell lysates were prepared by lysing cells in RIPA buffer (Sigma) following 24 h treatment with MGB4 or doxorubicin. 40 μg of cell lysates were resolved on 15% SDS-PAGE gels and transferred to nitrocellulose for immunoblotting as previously described.18 HRP-conjugated secondary antibodies were used with visualisation of bound antibodies carried out by enhanced chemiluminescence (ECL).
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was, in part, funded through a Rosetrees Trust Research Grant (CM528), and an EPSRC Impact Acceleration Account administered by the University of Strathclyde. The authors wish to thank Gavin Bain, Patricia Keating and Craig Irving for their technical assistance during this study.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00268e
References
- WHO Global Cancer report, http://www.who.int/cancer/en/, 2015.
- Kerru N., Singh P., Koorbanally N., Raj R., Kumar V. Eur. J. Med. Chem. 2017;15:179–212. doi: 10.1016/j.ejmech.2017.07.033. [DOI] [PubMed] [Google Scholar]
- Schmittel A., Sebastian M., Fischer von Weikersthal L., Martus P., Gauler T. C., Kaufmann C., Keilholz U. Ann. Oncol. 2011;22(8):1798–1804. doi: 10.1093/annonc/mdq652. [DOI] [PubMed] [Google Scholar]
- Devarakonda S., Govindan R. Clin. Cancer Res. 2018;24:6112–6114. doi: 10.1158/1078-0432.CCR-18-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C., Puschendorf B., Grunicke H., Chandra P., Venner H. Eur. J. Biochem. 1971;21:269–278. doi: 10.1111/j.1432-1033.1971.tb01466.x. [DOI] [PubMed] [Google Scholar]
- Barret M. P., Gemmell C. G., Suckling C. J. Pharmacol. Ther. 2013;139:12–23. doi: 10.1016/j.pharmthera.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Khalaf A. I., Bourdin C., Breen D., Donoghue G., Scott F. J., Suckling C. J., MacMillan D., Clements C., Fox K., Sekibo D. A. T. Eur. J. Med. Chem. 2012;56:39–47. doi: 10.1016/j.ejmech.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Scott F. J., Khalaf A. I., Giordani F., Wong P. E., Duffy S., Barrett M., Avery V. M., Suckling C. J. Eur. J. Med. Chem. 2016;116:116–125. doi: 10.1016/j.ejmech.2016.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott F. J., Nichol R. J. O., Khalaf A. I., Giordani F., Gillingwater K., Ramu S., Elliot A., Zuegg J., Duffy P., Rosslee M. J., Hlaka L., Kumar S., Ozturk M., Brombacher F., Barretr M., Guler R., Suckling C. J. Eur. J. Med. Chem. 2017;136:561–572. doi: 10.1016/j.ejmech.2017.05.039. [DOI] [PubMed] [Google Scholar]
- Dasari S., Tchounwou P. B. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Incalci M., Sessa C. Expert Opin. Invest. Drugs. 1997;6:875–884. doi: 10.1517/13543784.6.7.875. [DOI] [PubMed] [Google Scholar]
- Dervan P. B., Edelson B. S. Curr. Opin. Struct. Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- Scott F. J., Puig-Sellart M., Khalafa A. I., Henderson C. J., Westrop G., Watson D. G., Carter K., Grant M. H., Suckling C. J. Bioorg. Med. Chem. Lett. 2016;15:3478–3486. doi: 10.1016/j.bmcl.2016.06.040. [DOI] [PubMed] [Google Scholar]
- Allison S. J., Cooke D., Davidson F. S., Elliott P. I. P., Faulkner R. A., Griffiths H. B. S. Angew. Chem., Int. Ed. 2018;57:9799–9804. doi: 10.1002/anie.201805510. [DOI] [PubMed] [Google Scholar]
- Basri A. M., Lord R. M., Allison S. J., Rodríguez-Bárzano A., Lucas S. J., Janeway F. D., Shepherd H. J., Pask C. M., Phillips R. M., McGowan P. C. Chem. – Eur. J. 2017;23:6341–6356. doi: 10.1002/chem.201605960. [DOI] [PubMed] [Google Scholar]
- Allison S. J., Sadiq M., Baronou E., Cooper P. A., Dunnill C., Georgopoulos N. T., Latif A., Shepherd S., Shnyder S. D., Stratford I. J., Wheelhousel R. T., Willans C. E., Phillips R. M. Cancer Lett. 2017;403:98–107. doi: 10.1016/j.canlet.2017.04.041. [DOI] [PubMed] [Google Scholar]
- Allison S. J., Knight J. R. P., Granchi C., Rani R., Minutolo F., Milner J., Phillips R. M. Oncogenesis. 2014;3:e201. doi: 10.1038/oncsis.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison S. J., Milner J. Mol. Ther.--Nucleic Acids. 2014;2:e141. doi: 10.1038/mtna.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlaka L., Rosslee M.-J., Ozturk M., Kumar S., Parihar S. P., Brombacher F., Carter K., Scott F. J., Suckling C. J., Guler R. J. Antimicrob. Chemother. 2017;72:3334–3341. doi: 10.1093/jac/dkx326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckling C. J., Khalaf A. I., Scott F., Gillingwater K., Morrison L., De Koning H. P., Giordani F. and Barrett M., 3rd International Electronic Conference on Medicinal Chemistry, 2017.
- Suckling C. J., Hunter I., Khalaf A. I., Scott F., Tucker N., Niemenen L. and Lemonidis K., 3rd International Electronic Conference on Medicinal Chemistry, 2017.
- Kuo L. J., Yang L. X. In Vivo. 2008;22:305–310. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.