

Abstract
Type 2 diabetes (T2D) is one of the most escalating global metabolic diseases, which is highly associated with insulin resistance (IR) and risk of combination with nonalcoholic fatty liver disease (NAFLD). Previous studies suggest that soluble klotho (sKL) could serve as a circulating hormone to mediate energy metabolism, but the detailed mechanism is poorly understood. In this study, we generated T2D models of wild-type (WT), sKL heterozygous (KL+/−), and sKL transgenic (TgKL) mice continuously fed a high-fat diet (HFD) and constructed L02 cell lines that stably overexpress sKL to investigate the effect of sKL on hepatic glucose and lipid metabolism. Surprisingly, we discovered that sKL deficiency resulted in exacerbated diabetic phenotypes and hepatic glucolipid metabolism disorders in HFD-fed KL+/− diabetic mice (KL+/− DM), whereas TgKL diabetic mice (TgKL DM) exhibited ameliorated diabetic phenotypes and decreased IR. Mechanistic studies in vitro and in vivo demonstrated that sKL could inhibit the PI3K/AKT/mTORC1 signaling to upregulate peroxisome proliferator-activated receptor α (PPARα) expression by directly interacting with type 1 insulin-like growth factor receptor (IGF1R) in HFD-fed T2D mice. Thus, sKL could improve hepatic glucolipid homeostasis to ameliorate diabetic phenotypes and lipid accumulation and may function as a potential therapeutic target for the treatment of T2D and reduce the risk of NAFLD.
Keywords: sKL, T2D, IR, NAFLD, glucose and lipid homeostasis, PPARα
Graphical Abstract
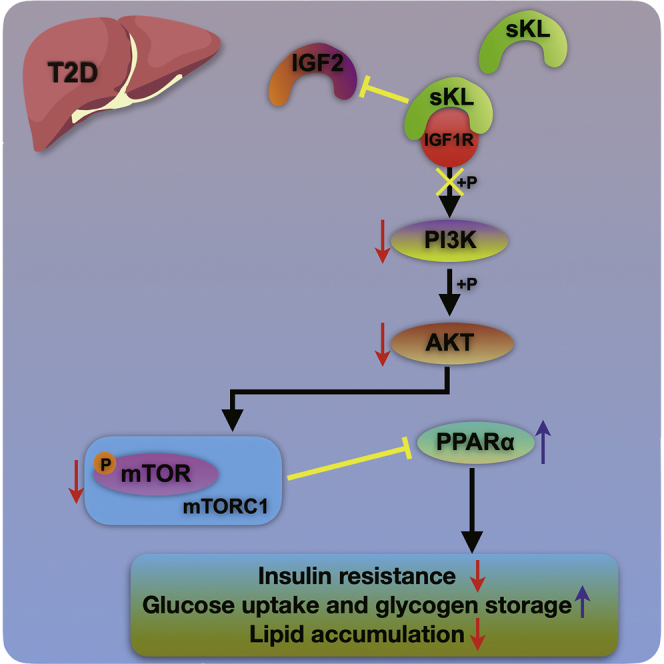
Zheng and colleagues elucidated that soluble klotho (sKL) could improve hepatic glucose and lipid homeostasis by regulating the IGF1R/PI3K/AKT/mTORC1/PPARα signal axis in T2D, which indicated that sKL may function as a potential therapeutic target for the progression of T2D and NAFLD.
Introduction
Type 2 diabetes (T2D), the most common metabolic disease characterized by persistent hyperglycemia, affects approximately one-quarter of the general population worldwide.1,2 A strong association between T2D and nonalcoholic fatty liver disease (NAFLD) has been shown, as T2D is commonly accompanied by NAFLD in patients.3, 4, 5 With the development of T2D, the risk of NAFLD progression from simple steatosis to nonalcoholic steatohepatitis (NASH) could increase.4,5 However, the exact mechanism by which T2D induces NAFLD is poorly understood, and efforts to ameliorate T2D or NAFLD with effective pharmacotherapies have been limited.
In mice, the loss of function of klotho, also called a-klotho, results in a syndrome that resembles premature human aging and the generation of lean mice with decreased white adipose tissue accumulation.6,7 Klotho exists in several forms, including a single-pass transmembrane protein with an extracellular domain composed of two tandem domains (KL1 and KL2) that are present primarily in the kidneys and a soluble form (KL1), named soluble klotho (sKL), that is present in the blood circulation.6,7 The overexpression of sKL inhibits the aging process through the induction of insulin resistance (IR), as indicated by the disrupted insulin and type 1 insulin-like growth factor (IGF1) signaling in sKL-deficient mice.7, 8, 9 sKL plays an important role in kidney diseases that involve hyperphosphatemia, vascular calcification, inflammation, and oxidative stress.10, 11, 12 Previous studies suggest that sKL is also linked to metabolic diseases, e.g., NASH and T2D, and the regulation of adipogenesis.13,14 In light of some studies, sKL may function as a circulating hormone that represses intracellular signals to mediate energy metabolism.13,15 But the detailed molecular mechanisms require further investigation, especially its effect on glycolipid metabolism in hepatocytes in the context of T2D.
In this study, we aimed to explore the role of sKL on hepatic glucose and lipid metabolism in T2D. Our results may highlight a potential therapeutic target for the progression of T2D and NAFLD.
Results
sKL Could Induce IR and Be Associated with Glucose and Lipid Metabolism
Overexpression of sKL induces IR to slow the aging process, as previously shown.7,9 Inspired by the previous studies, we generated sKL heterozygous (KL+/−)mice and sKL transgenic (TgKL) mice, and the level of sKL in serum of each group was confirmed by the enzyme-linked immunosorbent (ELISA) assay (Figure 1A). We further tested the effect of sKL on glucose and lipid metabolism. In particular, our results demonstrated that compared to WT and KL+/− mice, the TgKL mice exhibited an increase in body weight and fasting insulin levels (Figures 1B and 1C), no difference in serum glucose levels (Figure 1D), a decrease in insulin sensitivity measured by the quantitative insulin sensitivity check index (QUICKI), and an impairment of glucose intolerance measured by a glucose tolerance test (GTT) (Figures 1E and 1F). In addition, we also found that sKL induced increases in the serum lipid profiles (total cholesterol [TC], high-density lipoprotein cholesterol [HDL], low-density lipoprotein cholesterol [LDL], and triglyceride [TG]) of the TgKL mice compared to those of the wild-type (WT) and KL+/− mice (Figure 1G). Overexpression of sKL also induced an increase in hepatic TG as compared to KL+/− mice (Figure 1H). However, there were no obvious changes in the pathology or glycogen contents among the three groups, as confirmed by hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining (Figure 1I). In addition, immunohistochemistry (IHC) staining was performed and indicated that sKL could be observed in the hepatocytes of the TgKL mice (Figure 1I). Taken together, the results suggested that the overexpression of sKL could induce IR and seemed to be associated with glucose and lipid metabolism.
Figure 1.

Comparison of the Characteristics of WT, KL+/−, and TgKL Mice
WT, KL+/−, and TgKL mice were fed regular chow for 14 weeks (n = 8/group). (A–F) Serum sKL levels (A), BW (B), fasting blood insulin levels (C), fasting blood glucose levels (D), QUICKI (E), and GTT (F) were determined. (G and H) The serum lipid profiles (TC, HDL, LDL, and TG) (G) and hepatic lipid contents (TG and TC) (H) were tested enzymatically. (I) H&E, PAS staining, and IHC staining of sKL of liver sections from three groups. Scale bars, 50 μm. For all statistical plots, the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (F) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; †p < 0.05, ††p < 0.01, †††p < 0.001; KL+/− mice, TgKL mice compared with WT mice, respectively.
sKL Could Improve Insulin Sensitivity and Hepatic Glucose Homeostasis in T2D
Therefore, we wondered whether sKL could also cause IR and aggravate diabetic phenotypes in T2D mice. To this end, we further generated T2D models of WT, KL+/−, and TgKL mice continuously fed a high-fat diet (HFD). The concentration of serum sKL of three groups was tested by ELISA assays, and the results indicated that the serum sKL concentration was significantly higher in TgKL diabetic mice (DM) and was significantly decreased in KL+/− DM as compared to that in WT DM (Figure 2A). Interestingly, compared to WT DM, KL+/− DM exhibited an aggravation of diabetic phenotypes, as indicated by an increase in body weight (Figure 2B), significant elevation in fasting blood glucose levels (Figure 2C), impairment of fasting insulin levels (Figure 2D), insulin sensitivity, and glucose intolerance (Figures 2E and 2F). In contrast, TgKL DM exhibited opposite results on these indexes (Figures 2B–2F). Additionally, overexpression of sKL could partly inhibit a diabetes-induced high level of inflammatory factors (tumor necrosis factor α [TNF-α] and interleukin [IL]-1β) (Figure 2G).
Figure 2.

Comparison of the Characteristics of T2D Models of WT, KL+/−, and TgKL Mice
T2D models of WT, KL+/−, and TgKL male mice were given a continuous HFD (n = 8/group). (A–I) SerumsKL levels (A), BW change (B), fasting blood glucose levels (C), fasting blood insulin levels (D), QUICKI (E), GTT (F), serum levels of inflammatory factors (TNF-α and IL1-β) (G), hepatic glycogen content (H), and hepatic 2-deoxyglucose (2-DG) uptake levels (I) were determined. (J) H&E staining, PAS staining, Masson staining, and IHC staining of sKL of liver sections from the three groups. Scale bars, 50 μm. (K) The protein levels of sKL, GCK, and PEPCK in livers of the three groups. (L–N) Protein levels of sKL (L), hepatic 2-DG uptake levels (M), and protein levels of GCK and PEPCK (N) were determined in HG-FFA-treated L02 cells with overexpression of sKL or not. For all statistical plots, the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (F) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; †p < 0.05, ††p < 0.01, †††p < 0.001; KL+/− DM, TgKL DM compared with WT DM, respectively.
As the liver is the key organ for maintaining glucose homeostasis, we wondered whether sKL regulates hepatic glucose metabolism. Of note, compared with WT DM, KL+/− DM displayed decreased glycogen contents (Figure 2H) and reduced hepatic glucose uptake (HGU) (Figure 2I). Histological staining showed much larger and more numerous hepatocyte vacuoles, moderate steatosis, and mild inflammation but no obvious liver fibrosis with reduced sKL expression (Figure 2J). However, accompanied by increased sKL expression, these indicators exhibited opposite trends, except no obvious liver fibrosis either in TgKL DM (Figures 2H–2J). In addition, the expression levels of glucokinase (GCK) and phosphoenolpyruvate carboxykinase (PEPCK), the key enzymes related to glucose metabolism, were also measured in the livers. And the results showed that sKL expression was positively associated with GCK expression but significantly negatively correlated with PEPCK expression (Figure 2K). Furthermore, for the in vitro experiments, we constructed a L02 cell line that stably overexpress sKL, called the pLV-sKL L02 cell line, and the level of sKL was confirmed by western blot analysis (Figure 2L). When the cells were treated with high glucose and free fatty acid (HG-FFA), we found that compared to HG-FFA-treated L02 cells, sKL could facilitate HGU (Figure 2M), and the expression level of GCK was upregulated, whereas the level of PEPCK was downregulated (Figure 2N) in HG-FFA-treated pLV-sKL L02 cells, which suggests that sKL could promote glycogen storage but suppress gluconeogenesis. Taken together, these data suggested that sKL could improve insulin sensitivity and hepatic glucose homeostasis in T2D.
sKL Could Inhibit Hepatic Lipid Accumulation in T2D
As we showed, sKL could favor HGU and glycogen storage in T2D. Therefore, we sought to determine if sKL in hepatocytes was critical for T2D-driven lipid accumulation. WT DM, KL+/− DM, and TgKL DM were further employed and identified. Importantly, compared with WT DM, KL+/− DM exhibited an increase in the serum lipid profiles (TC, HDL, LDL, and TG) (Figure 3A), ratio of liver weight to body weight (LW/BW) (Figure 3B), activities of the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure 3C), and hepatic lipid content (TG, TC, and nonesterified FAs [NEFAs]) (Figures 3D and 3E). Nevertheless, the overexpression of sKL reversed these phenomena (Figures 3A–3E). Oil red O staining was performed in hepatic tissues and HG-FFA-treated pLV-sKL L02 cells, and the results showed that sKL could significantly ameliorate lipid accumulation in the hepatocytes (Figure 3F). Furthermore, the transcription levels of representative genes that regulate lipid metabolism were also measured by quantitative real-time PCR in the three groups and HG-FFA-treated pLV-sKL L02 cells. The in vivo and in vitro experiments indicated that the overexpression of sKL could downregulate the levels of the genes related to lipogenesis and fatty acid uptake but could partially upregulate the levels of the genes related to fatty acid oxidation (Figures 3G and 3H). Therefore, these data suggested that sKL may improve liver function and inhibit hepatic lipid accumulation in T2D.
Figure 3.

Comparison of the Lipid Metabolism-Related Indicators of T2D Models of WT, KL+/−, and TgKL Mice
WT DM, KL+/− DM, and TgKL DM were fed with a HFD for 8 weeks (n = 8/group). (A–E) Serum lipid profiles (TC, HDL, LDL, and TG) (A), LW/BW (B), serum ALT and AST levels (C), hepatic lipid contents (TG, TC) (D), and NEFA (E) of the three groups were determined. (F) Oil red O staining of liver sections from the three groups and HG-FFA-treated L02 cells with overexpression of sKL or not. Scale bar, 50 μm. (G and H) mRNA levels of genes related to lipogenesis, fatty acid uptake, and fatty acid oxidation in livers of the three groups (G) and HG-FFA-treated L02 cells (H). For all statistical plots, the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Peroxisome Proliferator-Activated Receptor α (PPARα) Mediates the Regulation of sKL on Hepatic Glucose and Lipid Metabolism in T2D
We noticed significant changes in the transcriptional levels of PPARα in vitro and in vivo due to sKL expression (Figures 3G and 3H). Previous studies have shown that hepatocyte-restricted PPARα deletion impairs liver function and whole-body fatty acid homeostasis seriously and is sufficient to cause fatty liver.16, 17, 18 In addition, PPARα plays a dual role in the crosstalk between hepatic glucose and lipid metabolism related to T2D and NAFLD, is mainly highly expressed, and improves insulin sensitivity in the liver.19
To investigate the association between sKL and PPARα in depth, western blot analysis of PPARα in the livers of the three groups was performed, and the representative results showed that PPARα expression was substantially inhibited in the liver tissues of KL+/− DM mice and partially restored in TgKL DM compared to its expression in WT DM (Figure 4A), and immunofluorescence (IF) staining was further performed to confirm the results (Figure 4B). In addition, to determining whether sKL targeted PPARα to regulate hepatic glucose and lipid metabolism, in vitro experiments were performed. We observed that the inhibition of gluconeogenesis by sKL was effectively eliminated by GW6471 (an inhibitor of PPARα) in TgKL DM primary hepatocytes (Figure 4C). We also found that the contribution of sKL to HGU was effectively inhibited by GW6471 in HG-FFA-treated pLV-sKL L02 cells (Figure 4D). Oil red O staining demonstrated that GW6471 significantly abolished the sKL-mediated inhibition of lipid accumulation in HG-FFA-treated pLV-sKL L02 cells (Figure 4E). Similarly, overexpression of sKL could effectively inhibit the inflammatory factors (TNF-α and IL-1β), and the inhibition could partially eliminate by GW6471 in HG-FFA-treated pLV-sKL L02 cells (Figure 4F). In addition, consistent with the results obtained by western blot analysis and quantitative real-time assays in vitro, the regulation of sKL on the expression of GCK and PEPCK was inhibited by GW6471 (Figures 4G–4I). Collectively, sKL could improve hepatic glucose and lipid metabolism in T2D by inhibiting PPARα.
Figure 4.

PPARα Mediates the Regulation of sKL on Hepatic Glucose and Lipid Metabolism in T2D
WT DM, KL+/− DM, and TgKL DM were fed with a HFD for 8 weeks (n = 8/group). (A) The protein levels of PPARα in the livers of the three groups were subjected to western blot analysis. (B) IF staining of PPARα (green) in liver sections from the three groups. Nuclei were labeled in blue using 4′,6-diamidino-2-phenylindole (DAPI). (C) Hepatic glucose production (HGP) in mouse primary hepatocytes derived from TgKL DM treated with glucagon applied with GW6471 or not. (D) 2-DG uptake levels of cells with GW6471 or not were tested. (E) Oil red O staining of cells with GW6471 or not were performed. (F) Cellular supernatant levels of inflammatory factors (TNF-α and IL-1β) in cells were determined. (G–I) Protein levels of GCK and PEPCK (G) and mRNA levels of GCK (H) and PEPCK (I) in cells with GW6471 or not. Scale bars, 50 μm. For all statistical plots, the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
sKL Inhibits the PI3K/AKT/ mTORC1 Signaling Pathway, Thereby Regulating PPARα Expression in T2D
PI3K and its downstream targets, including Akt and mTOR, which are known to be substrates of insulin or IGF, play a vital role in the regulation of glucose and lipid metabolism.20 Therefore, to examine whether PI3K/AKT/mTOR was involved in the signaling pathway by which sKL modulated the PPARα function to regulate glucose and lipid metabolism in T2D, we determined the relationship of the PI3K/AKT/mTOR axis and PPARα with sKL. We first measured the activation of the PI3K/AKT/mTOR pathway by quantifying the expression of PI3K and the phosphorylation ratios of AKT and mTOR in the hepatic tissues of each group by western blot analysis. Interestingly, the results showed a negative correlation with the expression of PPARα. The levels of PI3KP110α and PI3KP85α, the levels of AKT phosphorylated at serine 473, and the levels of mTOR phosphorylated at serine 2448 actually increased due to the absence of sKL in KL+/− DM but were reduced in TgKL DM compared to the levels in WT DM (Figure 5A). In addition, the downstream targets of mTORC1, P70S6K, and 4EBP1 were also analyzed by western blot analysis. Consistent results were observed, and the results indicated that sKL may inhibit the phosphorylation of P70S6K and 4EBP1 (Figure 5B).
Figure 5.

The PI3K/AKT/mTORC1 Signaling Pathway Mediated the Regulation of sKL on PPARα Expression
WT DM, KL+/− DM, and TgKL DM were fed with a HFD for 8 weeks (n = 8/group). (A and B) The levels of PI3K (PI3KP85α and PI3KP110α), p-AKT, AKT, p-mTOR, and mTOR (A) and phosphorylation levels of P70S6K and 4EBP1 (B) in livers of the three groups were subjected to western blot analysis. (C) Levels of AKT, mTOR, P70S6K, 4EBP1, and PPARα in HG-FFA-treated pLV-sKL L02 cells were tested. (D) Levels of p-AKT, p-mTOR, p-P70S6K, p-4EBP1, and PPARα in HG-FFA-treated pLV-sKL L02 cells treated with 740 Y-P or NV-5138 or not were determined. (E) Oil red O staining of HG-FFA-treated pLV-sKL L02 cells treated with 740 Y-P or NV-5138 or not were performed. Scale bar, 50 μm. For all statistical plots, the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Based on the results of the above in vivo experiments, in vitro experiments were conducted to further test the hypothesis. The results showed that the levels of phosphorylated (p)-AKT, mTOR, and its targets (p70S6K and 4EBP1) were upregulated in HG-FFA-treated L02 cells (Figure 5C). However, HG-FFA-activated PI3K/AKT/mTORC1 signaling could partially be inhibited, and the expression of PPARα was partially rescued in HG-FFA-treated pLV-sKL L02 cells (Figure 5C). Furthermore, both 740 Y-P (an agonist of PI3K) and NV-5138 (an activator of mTORC1) were employed. When 740 Y-P or NV-5138 was applied, the increased PPARα expression in HG-FFA-treated pLV-sKL L02 cells was almost offset, as the levels of p-AKT, mTOR, P70S6K, and 4EBP1 increased (Figure 5D). In addition, oil red O staining also demonstrated that 740 Y-P and NV-5138 significantly abolished the sKL-mediated inhibition of HG-FFA-induced lipid accumulation in pLV-sKL L02 cells (Figure 5E). Therefore, we believe that sKL could promote PPARα expression by modulating the PI3K/AKT/mTORC1 signaling pathway in T2D.
sKL Could Interact with the IGF1 Receptor (IGF1R) to Target the PI3K/AKT/mTOR Signaling Pathway
IR contributes to the pathogenesis of T2D and is exacerbated by crosstalk with the homologous IGF1R.21,22 Intracellular lipid accumulation is associated with increased IGF2 levels and decreased IGF1 levels in the serum, and both of these molecules are ligands of IGF1R.22,23 Our experiments suggested that sKL could improve insulin sensitivity in HFD-fed T2D mice, prompting us to determine whether IGF1R acts as a regulator linking sKL to the PI3K/AKT/mTOR/PPARα signaling pathway. The IGF1R protein levels were first analyzed by western blot analysis. The results showed that IGF1R expression in the hepatic tissues was increased from KL+/− DM and decreased from TgKL DM when compared to that from WT DM (Figure 6A). Representative IF staining further confirmed that IGF1R was observed with higher intensity in the hepatic tissues of KL+/− DM and lower intensity in TgKL DM (Figure 6B). Additionally, to further investigate the effect of sKL on hepatic IGF1 and IGF2 expression, the protein levels of IGF1 and IGF2 were measured using western blot analysis. Notably, results showed that sKL deficiency promoted the expression of IGF2 but inhibited the expression of IGF1 in livers; these results were in contrast to those observed from TgKL DM (Figure 6C). Thus, we further explored the effect of IGF2/IGF1R on sKL-mediated hepatic glucolipid metabolism in vivo. We treated KL+/− DM with picropodophyllin (PPP), an inhibitor of IGF1R, and TgKL DM with recombinant mouse IGF2 (rmIGF2) by intraperitoneal injection. The representative H&E staining of liver sections showed that when KL+/− DM were treated with PPP, the hepatocyte vacuoles were much smaller and less numerous with no presence of inflammation, and the hepatocyte vacuoles, observed in rmIGF2-treated TgKL DM, were larger and more numerous and showed mild inflammation and moderate steatosis compared to those in TgKL DM (Figure 6D). Importantly, the stimulatory effect of PPP increased hepatic glycogen in KL+/− DM, whereas rmIGF2-treated TgKL DM exhibited decreased hepatic glycogen, as determined by PAS staining (Figure 6D). However, we did not observe significant fibrosis in each group (Figure 6D). Oil red O staining demonstrated that PPP could improve the sKL deficiency-mediated increase in hepatic lipid accumulation and that IGF2 could significantly abolish the sKL overexpression-mediated inhibition of lipid accumulation in T2D (Figure 6D). Consistent results of representative western blots were observed in vitro. Treatment with PPP or overexpression of sKL partially alleviated HG-FFA-induced activation of the PI3K/AKT/mTOR signaling in L02 cells, as indicated by the reduced levels of p-AKT, mTOR, P70S6K, and 4EBP1 following the increased expression of PPARα (Figure 6E). However, the effects of sKL on the PI3K/AKT/mTOR pathway were substantially blocked by the administration of recombinant human IGF2 (rhIGF2) (Figure 6E). Besides, the results of oil red O staining on L02 cells were consistent with that observed in mice (Figure 6F).
Figure 6.

sKL Could Interact with IGF1R to Target the PI3K/AKT/mTOR Signaling Pathway
(A) The expression of IGF1R in the livers of three groups was tested. (B) IF assays of IGF1R (green) and sKL (red) in liver sections from the three groups were performed. Nuclei were labeled in blue using DAPI. (C) The expression of hepatic IGF1 and IGF2 was measured. (D) Representative images of H&E-stained (top row), PAS-stained (second row), oil red O-stained (third row), and Masson-stained (bottom row) liver sections from KL+/− DM, PPP-treated KL+/− DM, TgKL DM, and rmIGF2-treated TgKL DM (n = 8/group). (E) The protein levels of PI3K, p-AKT, p-mTOR, p-P70S6K, p-4EBP1, and PPARα of cells were determined. (F) Oil red O staining of treated cells was performed. (G and H) coIP assay (G) and analysis (H) of yeast two-hybrid system for protein interaction between sKL and IGF1R were determined. Scale bars, 50 μm. For the statistical plot of (G), the data are presented as the mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Having demonstrated the role of sKL on IGF2-mediated IGF1R/AKT/mTORC1 signaling, we wondered whether sKL is directly involved in its regulation. As Figure 5B suggests that sKL could colocalize with IGF1R on the hepatic membrane, the relationship between sKL and IGF1R was further studied. Coimmunoprecipitation (coIP) experiments were performed, and the results confirmed that sKL could bind to IGF1R in mouse hepatocytes (Figure 6G). To further strengthen this result, a yeast two-hybrid experiment was also performed. To eliminate false positives, a series of controlled trials were designed, and the results showed that sKL could interact with IGF1R (Figure 6H; Tables S2 and S3), which was consistent with the results of the coIP.
Taken together, these findings suggested that sKL could mediate the activity of IGF1R by interacting with this receptor to inhibit the PI3K/AKT/mTORC1 signaling pathway, which eventually improved insulin sensitivity and hepatic glucose and lipid metabolism in T2D, indicating that sKL may function as a potential target for the progression of T2D and NAFLD.
Discussion
NAFLD is emerging as the most common liver disease and represents a hepatic metabolic syndrome that is highly associated with the development of T2D.4,5 IR appears to induce lipid accumulation in hepatocytes and renders the liver more susceptible to diseases with the development of T2D.1 The molecular mechanisms underlying the induced formation of fatty liver from T2D and the later deterioration to NASH are not well understood. Accumulating evidence indicates that deregulation of the PI3K/AKT pathway in hepatocytes is a common molecular event associated with metabolic dysfunctions.24, 25, 26 Therefore, changes in the PI3K/AKT signaling pathway may contribute to specific therapeutic strategies for the development of T2D.
Klotho is mainly expressed in mammalian kidneys and is the major source of sKL, which is released through proteolytic cleavage of the transmembrane form and through alternative gene transcription.8,27 Genetic studies have demonstrated that klotho deficiency results in phenotypes prone to the development of kidney diseases, and renal dysfunction has been demonstrated to reduce klotho levels in kidney tissue and reduce circulating sKL.10,27,28 sKL is reported to have antiaging properties that mediate multiple systemic effects, including regulation of insulin signaling and prevention of vascular calcium deposits, oxidative stress, and fibrosis.11,27,28 However, research on the functions of sKL in liver diseases is limited. Current research mainly focuses on the function of β-klotho (KLB). KLB, which is mainly expressed in the liver, has been identified by homology with α-klotho and acts as a coreceptor of fibroblast growth factor (FGF)21 to improve hepatic insulin action and maintain glucose homeostasis in T2D.29,30 Recently, Dongiovanni et al.31 observed a significant reduction of KLB plasma levels in children with NAFLD, and the KLB plasma levels were associated with the lobular inflammation, ballooning, and fibrosis. In vitro data confirmed that lipid accumulation, after FFA treatment, causes a reduction of cellular and soluble expression of the KLB protein in human hepatoma cell lines. Moreover, the reduction of KLB in the same cells increases lipid accumulation and induces upregulation of lipotoxic and proinflammatory genes. So they speculate that KLB protein may exert a protective role against lipotoxicity and inflammation in hepatocytes. Combined with our findings, it seems to indicate that KLB and sKL may have synergistic or redundant functions on the hepatic lipid metabolism.
In this study, we found that sKL deficiency resulted in aggravated IR, exacerbated diabetic phenotypes, and increased hepatic lipid accumulation in T2D, whereas the overexpression of sKL in TgKL DM enhances glycogen synthesis and storage but suppresses hepatic gluconeogenesis and lipogenesis by elevating hepatic insulin sensitivity. Further mechanism research suggested that sKL could localize in the liver and interacts with IGF1R targeting the IGF1R/PI3K/AKT/mTORC1 signaling pathway to upregulate PPARα in T2D, resulting in improved insulin sensitivity and hepatic glucose and lipid homeostasis. However, the novel function of sKL in regulating insulin sensitivity is different from the other molecular mechanisms described in current reports and prompts us that sKL functions as a soluble hormone and may have a wide spectrum of direct targets depending on the disease states.
Previous research revealed nonconservative mutations in the Wnt coreceptor LRP6, which led to the activation of IGF1, AKT, and both mTORC1 and mTORC2 and also led to elevated plasma LDL and TG levels and fatty liver accompanied by hepatic de novo lipogenesis and lipid and cholesterol biosynthesis.32 The overexpression of IGF1R might be the molecular mechanism underlying the development of liver cirrhosis.33,34 As one ligand of IGF1R, serum IGF1 was reduced in both diabetes and NAFLD, and the overexpression of IGF1 could alleviate the progression of the two diseases.23,35 It is worth noting that IGF2, another ligand of IGF1R, was increased in both diabetes and NAFLD and could contribute to the development of T2D and NAFLD.36 In our study, we showed that HFD-fed KL+/− DM exhibited lower levels of IGF1 but higher levels of IGF1R and IGF2. However, we found that the overexpression of sKL could upregulate IGF1 but downregulate both IGF2 and IGF1R in HFD-fed DM. We also demonstrated that sKL negatively regulates IGF2/IGF1R to inhibit gluconeogenesis and lipid accumulation in the livers of TgKL DM. This finding seemed to indicate that IGF2 substitution for IGF1 acts as a negative regulatory factor of insulin sensitivity and that the effect could be inhibited by sKL. The combination of the findings of previous studies with our findings suggested a more detailed molecular function of IGF1/IGF1R in regulating insulin sensitivity in diabetes or fatty liver.
In conclusion, the principal findings of this study revealed an important role of sKL in the regulation of hepatic glucose and lipid metabolism in T2D. The findings highlight the possibility of developing novel therapeutic strategies to attenuate T2D and reduce the risk of NAFLD.
Materials and Methods
Animals and Treatment
Male C57BL/6 mice were purchased from Beijing HFK Biologic Technology (Beijing, China), and the transgenic mice with overexpression of sKL, named TgKL mice, were purchased from Cyagen Biosciences (Suzhou, China). The TgKL mice were generated by microinjection of mouse sKL cDNA fused with an elongation factor 1α (EF1α) promoter (TgKL, pRP[Exp]-EF1A > sKL [AB010088.1]) into fertilized mouse eggs that were obtained from C57BL/6 females mated with C57BL/6 males. The genotype-specific primers for identification were listed below: TF1: 5′-TTTGCCCTTTTTGAGTTTGGATCTT-3′, TR1: 5′-GTGATGGGTGAAAGTGTCCCAGAT-3′; TF2: 5′-ACCAAAAGCTGATAGAGGACAATG-3′, TR2: 5′-GTGATGGGTGAAAGTGTCCCAGAT-3′; ICF: 5′-GCAGAAGAGGACAGATACATTCAT-3′, ICR: 5′-CCTACTGAAGAATCTATCCCACAG-3′. The expected PCR product size of TgKL was 411 bp (use primers TF1 and TR1) and 383 bp (use primers TF2 and TR2), respectively, and internal control was 689 bp (use primers ICF and ICR). The sKL deficiency (KL+/−, C57BL/6 background) mice6 were provided by our lab by disturbing the transcription enhancer element of sKL and generated by mating pairs of KL+/−. The genotype-specific primers for identification were listed below: WF: 5′-TTGTGGAGATTGGAAGTGGACGAAAGAG-3′, WR: 5′-CTGGACCCCCTGAAGCTGGAGTTAC-3′; KF: 5′-TTGTGGAGATTGGAAGTGGACGAAAGAG-3′, and KR: 5′-CGCCCCGACCGGAGCTGAGAGTA-3′. The expected PCR product sizes of the sKL mutant were 815 bp and 419 bp when using the four primers. All of the genotypes were verified by the Mouse Direct PCR Kit (B40015; Bimake, Selleck), and the conditions of the PCR reaction were as follows: denaturation at 94°C for 5 min, 30 cycles of 94°C for 30 s, annealing at 60°C for 1 min, extension at 72°C for 45 s, and a final extension at 72°C for 10 min.
All mice were housed with a standard 12-h dark/light cycle with food and water ad libitum. All mice experiments were performed in accordance with protocols approved by the Laboratory Animal Welfare and Ethics Committee of the Third Military Medical University.
Establishment of T2D models: WT (C57BL/6), KL+/−, and TgKL male mice were fed with regular chow for 6 weeks and mice were switched to a HFD and sustained for 8 weeks starting at 7 weeks of life. The three groups were intraperitoneal injected with a low dose of streptozocin (STZ) (30 mg/kg at weekly intervals for 2 weeks) at 10 weeks of life. The regular chow consisting of 5% fat, 53% carbohydrate, and 23% protein, with total calorific value 25 kJ/kg, and HFD consisting of 22% fat, 48% carbohydrate, and 20% protein, with total calorific value 44.3 kJ/kg, were ordered from the Animal Experimental Center of Third Military Medical University, and STZ was purchased from Sigma (Chongqing, China). Sixteen KL+/− DM were selected and randomly divided into two groups (i.e., PPP group and control group). Each mouse received an intraperitoneal injection of either PPP (20 mg/kg; PPP, MedChemExpress [MCE]; HY1594) or solvent mixture (10% DMSO plus 90% corn oil), twice a day for a continuous 2 weeks at the last 2 weeks. Sixteen TgKL mice were also selected and randomly divided into two groups (i.e., rmIGF2 group and PBS control group), and each mouse received an intraperitoneal injection of either rmIGF2 protein (200 mg/kg; IGF2, R&D; 792-MG) or diluted PBS (same volume as that of rmIGF2), once weekly for 2 weeks at the last 2 weeks.
Isolation of Primary Hepatocyte and Cell Culture
Primary hepatocytes were isolated from the diabetic group mice using a two-step collagenase perfusion method.37 Livers were perfused with 40 mL of wash buffer, followed with 40 mL of collagenase buffer. Briefly, livers were disrupted and washed twice with serum-free Dulbecco’s modified Eagle’s medium (DMEM; Gibco; 21885108). Viable hepatocytes were then isolated by spinning through a 70-μm cell strainer (Falcon; 352350) and centrifugation at 50 × g for 2 min to obtain primary hepatocytes, which were further purified with a 50% Percoll solution (Sigma; P4937). Cells were plated onto collagen-coated plates (Sigma; A1142802) in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco; 10099141), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco; 15140122).
The L02 cell line was purchased from ATCC and cultured in DMEM (Gibco; 21885108), supplemented with 10% FBS in a 5% CO2 incubator at 37°C. Cells were treated with HG (30 mM) and FFA (0.5 mM palmitate acid [PA]; Sigma; P0500), along with 1.0 mM oleic acid (OA) (Sigma; O1008) for 24 h before harvested.
Generation of the pLV-sKL L02 Cell Line
The L02 cell line that stably overexpresses human sKL (1–1,647 nt in coding DNA sequence [CDS]; GenBank: AB009667.2) in this study was generated by a lentiviral expression system. In brief, full-length human sKL cDNA was made by PCR cloning using designed primers and then subcloned into the pLVX-AcGFP-N1-Fluc vector, and the cDNA was confirmed by sequencing. The lentiviral vector and packaging vectors, pMD2.G and psPAX2, were cotransfected into 293T cells using Lipofectamine 2000 (Invitrogen; 11668) in Opti-MEM reduced serum medium (Thermo Fisher Scientific; 31985062), and supernatant was collected after 48 h of culture and used to infect L02 cells along with polybrene (EMD Millipore; TR-1003-G); then, the cells were selected with puromycin (Life Technologies; A1113803).
Western Blot Analysis
Liver tissue or cell samples were extracted and quantified. Samples were then separated by 8%–15% Bis-Tris PAGE electrophoresis and transferred to the polyvinylidene fluoride (PVDF) membrane for detection. Western blots were probed overnight at 4°C, with specific primary antibodies in Tris-buffered saline Tween 20 (TBST) containing 5% skim milk. After washing 3 times with TBST, the membranes were incubated for 1 h at room temperature with a respective immunoglobin G (IgG)-horseradish peroxidase (HRP)-labeled second antibody (1:5,000) in TBST containing 5% skim milk. Antigens were revealed using a chemiluminescence assay (Pierce, Rockford, MD, USA). Quantification of bands was achieved by densitometry using the ImageJ software. The antibodies used were list below: klotho (Abcam, ab181373; Invitrogen, PA5-88303), GCK (Abcam; ab88056), PEPCK (Bioss; bs-5002R), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam; ab181602), β-actin (Abcam; ab8226), PPARα (Bioss; bs-23398R), p-mTOR (phospho S2448) (Abcam; ab109268), mTOR (Abcam; ab2732), p-p70S6K (phospho T389) (Cell Signaling Technology [CST]; 9234), p70S6K (CST; 2708), 4EBP1 (CST; 9644), p-4EBP1 (phospho T37/46) (CST; 2855), mTOR (CST; 2972), p-mTOR (phospho S2448) (CST; 2971), AKT (CST; 9272), p-AKT (phospho S473) (CST; 4060), PI3KP85α (CST; 4292), PI3KP110α (CST; 4255), IGF1R (Abcam; ab182408), IGF1 (Abcam; ab182408), and IGF2 (Abcam; ab9574).
Quantitative Real-Time PCR
Briefly, total mRNA was isolated from cultured cells or tissue samples using TRIzol (Takara) and reverse transcribed into cDNA with RT-MLV reverse transcriptase (Promega), according to the manufacturer’s instructions. Fast SYBR Green Master Mix (Thermo Scientific; 4385610) was applied to quantify PCR amplification. The mRNA expression levels of the target genes were normalized to the expression of β-actin. Expression levels were calculated using the 2−ΔΔCT method. The primer pairs used for quantitative real-time in this study are described in Table S1.
ELISA Assay
The blood samples from mice and cellular supernatants were collected and centrifuged for 10 min at 3,000 rpm (4°C), and the sera were stored at −80°C until additional analysis. Serum sKL was measured in duplicate using a mouse klotho ELISA kit, according to the manufacturer’s protocol (Cusabio, Cologne, Germany).
Histological Analysis and Staining
Tissue or L02 cells fixed in neutral-buffered formalin was embedded in paraffin or optimal cutting temperature (OCT; 4583, Sakura, USA) compound by using standard procedures. Frozen sections were stained with oil red O and IF. Paraffin sections were stained with H&E, PAS, Masson, IHC, and IF, according to the manufacturer’s instructions, respectively. Digital images were obtained with a light microscope (Olympus). Immunofluorescent staining and images were obtained by a LSM780 laser-scanning confocal microscope (Zeiss, Germany) system.
Yeast Two-Hybrid System Experiment
The inserts containing DNA sequences encoding human sKL (1–1,647 nt in CDS; GenBank: AB009667.2) and IGF1R (4,110 nt; GenBank: NM_010513.2) were cloned and then cloned into the vectors pGADT7 (Clontech; 630442) and pGBKT7 (Clontech; 631604) using homologous recombination, respectively (ClonExpress II One Step Cloning Kit; Vazyme, Nanjing, China) The yeast strain Y187 (Clontech; 630457) was transformed with pGADT7, and Y2HGold (Clontech; 630498) was transformed with pGBKT7; the fusion plasmids were transformed into yeast strains Y187 and Y2HGold, respectively, following a lithium acetate protocol.38 Yeast cotransformed with pGBKT7-Lam and pGADT7-T was used as a negative control, and yeast cotransformed with pGBKT7-53 and pGADT7-T was used as a positive control. The transformants were grown on synthetically defined double dropout (DDO; SD/-Leu/-Trp) medium plates for 2–3 days. The yeast cells with cotransformation of the fusion plasmids were further dropped on QDO/X/A (SD/-Ade/-His/-Trp/-Leu/-Trp/X-α-gal/AbA) dropout medium plates with a series of 10-fold dilutions (from 106 to 104) for checking the interactions. Exclusion of recombinant plasmids was nontoxic to yeast strains and had no self-transcriptional activation activity. The primer pairs used for yeast two-hybrid experiments in this study are described in Table S2.
Statistical Analyses
All data were analyzed using GraphPad Prism 7 (Macintosh). Quantitative values are presented as the mean ± SD, with p <0.05 being considered statistically significant. Statistical differences between two experimental groups were analyzed by 2-tailed Student’s t test. For multiple comparison analysis, one-way ANOVA, followed by Tukey’s multiple comparison tests, was performed.
Author Contributions
H.G., W.J., N.Y., X.H., Y.L., Z.W., and Y.Z. conducted the experiments and analyzed the data. H.G., W.J., and N.Y. wrote the manuscript and interpreted the data. R.D., X.P., and L.Z. analyzed the data and revised the manuscript. J.L. and L.Z. designed the experiments and supervised the research.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (no. 81672902) and the Natural Science Foundation Project of Chongqing (nos. cstc2017jcyj-yszxX0002 and cstc2017jcyjAX0348).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.08.002.
Contributor Information
Jing Li, Email: xqyylj@163.com.
Lu Zheng, Email: xqyyzl1@163.com.
Supplemental Information
References
- 1.Czech M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017;23:804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ling C., Rönn T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019;29:1028–1044. doi: 10.1016/j.cmet.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams L.A., Waters O.R., Knuiman M.W., Elliott R.R., Olynyk J.K. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am. J. Gastroenterol. 2009;104:861–867. doi: 10.1038/ajg.2009.67. [DOI] [PubMed] [Google Scholar]
- 4.Younossi Z., Tacke F., Arrese M., Chander Sharma B., Mostafa I., Bugianesi E., Wai-Sun Wong V., Yilmaz Y., George J., Fan J., Vos M.B. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology. 2019;69:2672–2682. doi: 10.1002/hep.30251. [DOI] [PubMed] [Google Scholar]
- 5.Loomba R., Abraham M., Unalp A., Wilson L., Lavine J., Doo E., Bass N.M., Nonalcoholic Steatohepatitis Clinical Research Network Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56:943–951. doi: 10.1002/hep.25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T., Ohyama Y., Kurabayashi M., Kaname T., Kume E. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 7.Kurosu H., Yamamoto M., Clark J.D., Pastor J.V., Nandi A., Gurnani P., McGuinness O.P., Chikuda H., Yamaguchi M., Kawaguchi H. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y., Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr. Rev. 2015;36:174–193. doi: 10.1210/er.2013-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unger R.H. Klotho-induced insulin resistance: a blessing in disguise? Nat. Med. 2006;12:56–57. doi: 10.1038/nm0106-56. [DOI] [PubMed] [Google Scholar]
- 10.Kuro-O M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019;15:27–44. doi: 10.1038/s41581-018-0078-3. [DOI] [PubMed] [Google Scholar]
- 11.Hum J.M., O’Bryan L.M., Tatiparthi A.K., Cass T.A., Clinkenbeard E.L., Cramer M.S., Bhaskaran M., Johnson R.L., Wilson J.M., Smith R.C., White K.E. Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho. J. Am. Soc. Nephrol. 2017;28:1162–1174. doi: 10.1681/ASN.2015111266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang W., Xiao T., Han W., Xiong J., He T., Liu Y., Huang Y., Yang K., Bi X., Xu X. Klotho inhibits PKCα/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol. Cell. Endocrinol. 2019;494:110490. doi: 10.1016/j.mce.2019.110490. [DOI] [PubMed] [Google Scholar]
- 13.Razzaque M.S. The role of Klotho in energy metabolism. Nat. Rev. Endocrinol. 2012;8:579–587. doi: 10.1038/nrendo.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori K., Yahata K., Mukoyama M., Suganami T., Makino H., Nagae T., Masuzaki H., Ogawa Y., Sugawara A., Nabeshima Y., Nakao K. Disruption of klotho gene causes an abnormal energy homeostasis in mice. Biochem. Biophys. Res. Commun. 2000;278:665–670. doi: 10.1006/bbrc.2000.3864. [DOI] [PubMed] [Google Scholar]
- 15.Chen G., Liu Y., Goetz R., Fu L., Jayaraman S., Hu M.C., Moe O.W., Liang G., Li X., Mohammadi M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553:461–466. doi: 10.1038/nature25451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montagner A., Polizzi A., Fouché E., Ducheix S., Lippi Y., Lasserre F., Barquissau V., Régnier M., Lukowicz C., Benhamed F. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65:1202–1214. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bougarne N., Weyers B., Desmet S.J., Deckers J., Ray D.W., Staels B., De Bosscher K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018;39:760–802. doi: 10.1210/er.2018-00064. [DOI] [PubMed] [Google Scholar]
- 18.Kim H., Haluzik M., Asghar Z., Yau D., Joseph J.W., Fernandez A.M., Reitman M.L., Yakar S., Stannard B., Heron-Milhavet L. Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes. 2003;52:1770–1778. doi: 10.2337/diabetes.52.7.1770. [DOI] [PubMed] [Google Scholar]
- 19.Larter C.Z., Yeh M.M., Van Rooyen D.M., Brooling J., Ghatora K., Farrell G.C. Peroxisome proliferator-activated receptor-α agonist, Wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2012;27:341–350. doi: 10.1111/j.1440-1746.2011.06939.x. [DOI] [PubMed] [Google Scholar]
- 20.Taniguchi C.M., Kondo T., Sajan M., Luo J., Bronson R., Asano T., Farese R., Cantley L.C., Kahn C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Petersen M.C., Shulman G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018;98:2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menting J.G., Whittaker J., Margetts M.B., Whittaker L.J., Kong G.K., Smith B.J., Watson C.J., Záková L., Kletvíková E., Jiráček J. How insulin engages its primary binding site on the insulin receptor. Nature. 2013;493:241–245. doi: 10.1038/nature11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamek A., Kasprzak A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018;19:1308. doi: 10.3390/ijms19051308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C., Chi Y., Li J., Miao Y., Li S., Su W., Jia S., Chen Z., Du S., Zhang X. FAM3A activates PI3K p110α/Akt signaling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology. 2014;59:1779–1790. doi: 10.1002/hep.26945. [DOI] [PubMed] [Google Scholar]
- 25.Liu T.Y., Shi C.X., Gao R., Sun H.J., Xiong X.Q., Ding L., Chen Q., Li Y.H., Wang J.J., Kang Y.M., Zhu G.Q. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. (Lond.) 2015;129:839–850. doi: 10.1042/CS20150009. [DOI] [PubMed] [Google Scholar]
- 26.Rodríguez A., Catalán V., Gómez-Ambrosi J., García-Navarro S., Rotellar F., Valentí V., Silva C., Gil M.J., Salvador J., Burrell M.A. Insulin- and leptin-mediated control of aquaglyceroporins in human adipocytes and hepatocytes is mediated via the PI3K/Akt/mTOR signaling cascade. J. Clin. Endocrinol. Metab. 2011;96:E586–E597. doi: 10.1210/jc.2010-1408. [DOI] [PubMed] [Google Scholar]
- 27.Drew D.A., Katz R., Kritchevsky S., Ix J., Shlipak M., Gutiérrez O.M., Newman A., Hoofnagle A., Fried L., Semba R.D., Sarnak M. Association between Soluble Klotho and Change in Kidney Function: The Health Aging and Body Composition Study. J. Am. Soc. Nephrol. 2017;28:1859–1866. doi: 10.1681/ASN.2016080828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu M.C., Shi M., Zhang J., Addo T., Cho H.J., Barker S.L., Ravikumar P., Gillings N., Bian A., Sidhu S.S. Renal Production, Uptake, and Handling of Circulating αKlotho. J. Am. Soc. Nephrol. 2016;27:79–90. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.So W.Y., Cheng Q., Chen L., Evans-Molina C., Xu A., Lam K.S., Leung P.S. High glucose represses β-klotho expression and impairs fibroblast growth factor 21 action in mouse pancreatic islets: involvement of peroxisome proliferator-activated receptor γ signaling. Diabetes. 2013;62:3751–3759. doi: 10.2337/db13-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Q., Hu Z., Zhang F., Cui A., Chen X., Jiang H., Gao J., Chen X., Han Y., Liang Q. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology. 2016;64:425–438. doi: 10.1002/hep.28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dongiovanni P., Crudele A., Panera N., Romito I., Meroni M., De Stefanis C., Palma A., Comparcola D., Fracanzani A.L., Miele L. β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 2020;72:411–419. doi: 10.1016/j.jhep.2019.10.011. [DOI] [PubMed] [Google Scholar]
- 32.Go G.W., Srivastava R., Hernandez-Ono A., Gang G., Smith S.B., Booth C.J., Ginsberg H.N., Mani A. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab. 2014;19:209–220. doi: 10.1016/j.cmet.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu W., Li J., Cai Y., Wu Q., Pan Y., Chen Y., Chen Y., Zheng X., Li W., Zhang X., e C. Hepatic IGF-1R overexpression combined with the activation of GSK-3β and FOXO3a in the development of liver cirrhosis. Life Sci. 2016;147:97–102. doi: 10.1016/j.lfs.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 34.Rufinatscha K., Ress C., Folie S., Haas S., Salzmann K., Moser P., Dobner J., Weiss G., Iruzubieta P., Arias-Loste M.T. Metabolic effects of reduced growth hormone action in fatty liver disease. Hepatol. Int. 2018;12:474–481. doi: 10.1007/s12072-018-9893-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kajstura J., Fiordaliso F., Andreoli A.M., Li B., Chimenti S., Medow M.S., Limana F., Nadal-Ginard B., Leri A., Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 36.Devedjian J.C., George M., Casellas A., Pujol A., Visa J., Pelegrín M., Gros L., Bosch F. Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J. Clin. Invest. 2000;105:731–740. doi: 10.1172/JCI5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong J., Han C.J., Zhang J.Z., He W.Z., Zhao G.J., Cheng X., Zhang L., Deng K.Q., Liu Y., Fan H.F. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology. 2019;69:2471–2488. doi: 10.1002/hep.30559. [DOI] [PubMed] [Google Scholar]
- 38.Gietz R.D., Schiestl R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.