

Key Points
PI3Kδ inhibition reduces FL proliferation,Treg and TFH recruitment, whereas angiogenesis and dissemination are hampered in selected cases.
Microenvironment renders FL dependent on BCL-XL, MCL-1, and BFL-1; idelalisib restores FL dependence on BCL-2 and venetoclax cytotoxicity.
Abstract
Despite idelalisib approval in relapsed follicular lymphoma (FL), a complete characterization of the immunomodulatory consequences of phosphatidylinositol 3-kinase δ (PI3Kδ) inhibition, biomarkers of response, and potential combinatorial therapies in FL remain to be established. Using ex vivo cocultures of FL patient biopsies and follicular dendritic cells (FDCs) to mimic the germinal center (n = 42), we uncovered that PI3Kδ inhibition interferes with FDC-induced genes related to angiogenesis, extracellular matrix formation, and transendothelial migration in a subset of FL samples, defining an 18-gene signature fingerprint of idelalisib sensitivity. A common hallmark of idelalisib found in all FL cases was its interference with the CD40/CD40L pathway and induced proliferation, together with the downregulation of proteins crucial for B–T-cell synapses, leading to an inefficient cross talk between FL cells and the supportive T-follicular helper cells (TFH). Moreover, idelalisib downmodulates the chemokine CCL22, hampering the recruitment of TFH and immunosupressive T-regulatory cells to the FL niche, leading to a less supportive and tolerogenic immune microenvironment. Finally, using BH3 profiling, we uncovered that FL–FDC and FL–macrophage cocultures augment tumor addiction to BCL-XL and MCL-1 or BFL-1, respectively, limiting the cytotoxic activity of the BCL-2 inhibitor venetoclax. Idelalisib restored FL dependence on BCL-2 and venetoclax activity. In summary, idelalisib exhibits a patient-dependent activity toward angiogenesis and lymphoma dissemination. In all FL cases, idelalisib exerts a general reshaping of the FL immune microenvironment and restores dependence on BCL-2, predisposing FL to cell death, providing a mechanistic rationale for investigating the combination of PI3Kδ inhibitors and venetoclax in clinical trials.
Visual Abstract

Introduction
The pathogenesis of follicular lymphoma (FL), the most common indolent B-cell non-Hodgkin lymphoma characterized by the presence of t(14;18) and BCL-2 overexpression, is thought to be the result of a close collaboration between recurrent somatic mutations and a permissive microenvironment.1,2 FL tumor cells are dependent on their microenvironment for proliferation and survival, remaining dependent on signals through the B-cell receptor (BCR).3 In this context, 2 populations present in the tumor niche are fundamental: follicular dendritic cells (FDCs) and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin–expressing macrophages (Mϕ). Both cell types interact with stereotypic mannosylated residues of the BCR, in a T cell–independent manner, activating pathways such as nuclear factor-κB or phosphatidylinositol 3-kinase (PI3K)-Akt, which support tumor growth.4-6 Moreover, FL cells are intermixed in tight contact with supportive T-follicular helper cells (TFH) that contribute to FL lymphomagenesis through tumor necrosis factor-α (TNF-α), lymphotoxin α, interleukin-4 (IL-4), and CD40L7,8 and regulatory T cells (Treg) facilitating immune evasion.9 In this cross talk, PI3K behaves as a hub that integrates different signals coming from the microenvironmental milieu. Among the existing PI3K isoforms, PI3Kδ is restricted to leukocytes, and early studies showed an important role for in nontransformed B cells, with an almost exclusive dependence of the BCR signaling on PI3Kδ over other PI3K isoforms.10 In the last years, specific inhibitors of PI3Kδ have been developed, and idelalisib has been the first to enter the clinic, reaching US Food and Drug Administration approval in 2014 for relapsed small lymphocytic lymphoma, chronic lymphocyctic leukemia (CLL), and FL.11,12
The mechanism of action of idelalisib has been studied mainly in CLL, where this PI3Kδ inhibitor acts by targeting stroma–cancer cell interactions, abrogating BCR-derived survival signals, and inducing a redistribution of CLL cells from the tissue compartments into the blood. In addition, idelalisib inhibits CLL cell signaling pathways in response to CD40L, B-cell activating factor (BAFF), or TNF-α and interfered with integrin-mediated CLL cell adhesion to endothelial and bone marrow stroma cells.13-15 Moreover, recent findings indicate that PI3Kδ inhibitors could also be used to target the immune system by dampening Treg activity and facilitating host–antitumor responses.16,17
Surprisingly, despite the approval of idelalisib for relapsed FL, the current knowledge about its specific mechanism of action is mostly restricted to CLL. Our purpose in the current study is to characterize the immune microenvironment remodeling exerted by idelalisib together with its mechanisms of sensitivity and resistance in ex vivo cocultures of FL-FDC ad FL-Mϕ established with FL patient samples.
We also addressed how FL microenvironment modulates the FL dependence on antiapoptotic members of the BCL-2 family different from BCL-2, potentially explaining the limited success of ABT-199/venetoclax in the clinical setting.18 Moreover, we demonstrated how idelalisib restores BCL-2 dependence and venetoclax activity, setting up the mechanistical basis for the initiation of clinical trials of PI3Kδ inhibitiors and venetoclax.
Methods
Patient samples
Primary FL cells were isolated from tumoral lymph nodes from a cohort of 42 patients, diagnosed according to the World Health Organization classification criteria in the Hospital Clinic of Barcelona. Patient clinical features are summarized in supplemental Table 1. The study was approved by the Ethics Committee of Hospital Clinic (IRB# HCB/2018/0397), and patients signed written informed consent.
FL microenvironment models
To prepare FL cocultures, we used HK cells, a nonimmortalized FDC cell line generated from normal tonsils, which was kindly provided by Yong Sung Choi and has been extensively used to mimic the germinal center (GC) microenvironment.19-21 Mϕ were derived from peripheral blood mononuclear cells (PBMCs) of healthy donors (Banc de Sang i Teixits Barcelona). Blood samples were enriched in monocyte population using Rosette Sep (human monocyte enrichment cocktail; STEMCELL Technologies, Grenoble, France) and then cultured for 7 days with 100 ng/mL macrophage colony-stimulating factor (Thermo Fisher Scientific, Waltham, MA). FL-FDC and FL-Mϕ cocultures were performed in RPMI 10% fetal bovine serum (FBS) at ratios of 1:20 (FDC:FL) and 1:4 (Mϕ:FL), respectively, for the times indicated in the presence or absence of idelalisib provided by Gilead Sciences (Seattle, WA) and/or venetoclax (Selleck Chemicals, Houston TX).
Intracellular BH3 (iBH3) profiling
FL cells were cocultured with FDC or Mϕ with or without idelalisib for 24 hours at a cell density of 2 × 106 cells/mL. First, isolated FL cells were stained using Live/Dead Fixable Aqua Dead Cell Stain kit and labeled with CD19-PE (BD Biosciences, San Jose, CA). After washing, cells were placed in 96-well plates permeabilized and exposed to peptides (Gene Script, Piscataway, NJ), which mimic the BH3 (BCL-2 Homology 3) domain of several proapoptotic members of the BCL-2 family and proceed according to the standardized protocol from the Anthony Letai laboratory (http://letailab.dana-farber.org/bh3-profiling.html). Cytochrome C release in viable and CD19+ cells was used as a read-out.
Detailed description of additional methods is included in supplemental Methods.
Results
Idelalisib modulates FDC-induced gene sets in selected FL patients
To examine the molecular effect of idelalisib in a relevant ex vivo FL model, we established FL cocultures (n = 5) with cells isolated from lymph node biopsies from FL patients and FDC generated from normal tonsils to mimic the GC microenvironment and maintain FL cell viability.22 FL patient characteristics are summarized in supplemental Table 1. FL-FDC cocultures significantly (P = .0002) increased the viability of FL cells, and idelalisib induced moderate cytotoxicity at 72 hours on tumor cells that was maintained in the coculture (supplemental Figure 1). Gene expression profiling (GEP) of magnetically purified CD20+ B cells from FL-FDC cocultures treated with or without idelalisib indicated that FDC significantly changed the FL transcriptome. A differential gene expression analysis identified 306 genes significantly upregulated (P < .05 and fold change [fc] > 2) in patients FL1-FL4 (mean fc = 6.1), whereas FL5 was not responsive to FDC coculture (mean fc = 1.6; Figure 1A). We uncovered a differential gene regulation by idelalisib of these FDC upregulated genes, leading to 2 different patterns of response: FL1 and FL4 sensitive to idelalisib with 133 FDC-induced genes downmodulated by idelalisib (average fc < 0.5), whereas FL2 and FL3 appeared resistant to the inhibitor, with no downmodulated genes and some of them even upregulated (FL2; Figure 1A). Of note, the PI3K/AKT pathway was significantly blocked by idelalisib in the presence or absence of FDC, even in the resistant case FL35 (supplemental Figure 2). No significant changes in FL cells viability were observed at 48 hours in idelalisib-treated samples (supplemental Figure 3).
Figure 1.
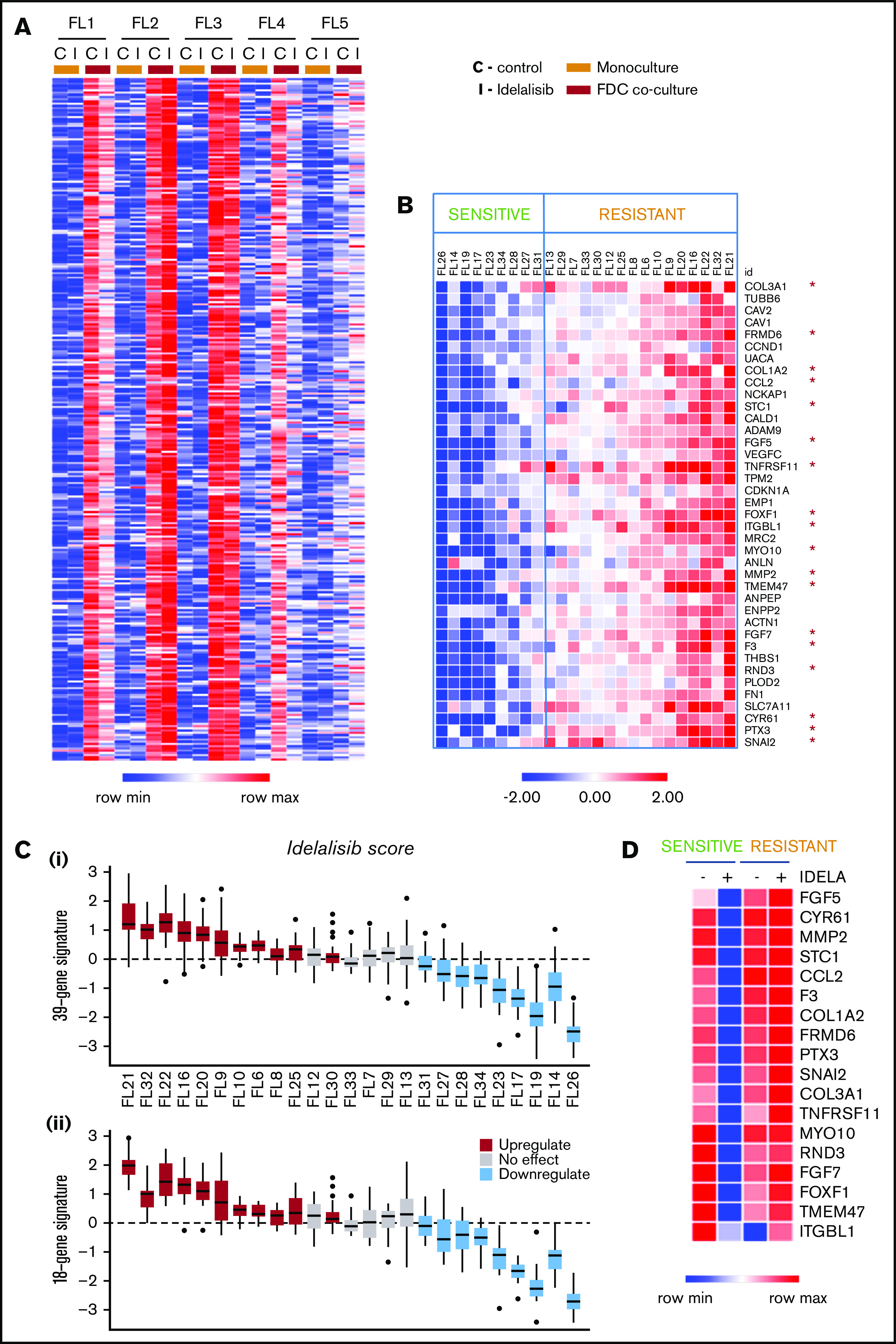
Idelalisib modulates FDC-induced gene sets in selected FL patients. (A) FL primary cells (n = 5) were isolated from monocultures or FL-FDC cocultures, treated with and without idelalisib (500 nM, 48 hours), RNA extracted, and subjected to GEP. Robust multiarray average data were analyzed with a paired (monoculture vs coculture) Limma algorithm (false discovery rate < 0.05, fc > 2). Heatmap represents the genes significantly upregulated in FL cells by FDCs in 4 of 5 patients. A differential regulation by idelalisib was acknowledged. (B) FL primary cells were isolated from FL-FDC (n = 25) cocultures treated with and without idelalisib (500 nM, 48 hours), and RNA was extracted and subjected to a multiplex reverse transcription-polymerase chain reaction by Fluidigm to characterize 39 selected genes upregulated in the FL-FDC coculture (fc > 2) and differently modulated by idelalisib (fc < 0.5 in sensitive patients). Heatmap displays fc in response to idelalisib referred to the untreated control. The cutoff to consider idelalisib sensitive was established using a paired t test comparing the expression −/+ idelalisib adjusted with the Benjamini-Hochberg method (details in supplemental Methods). (C) Boxplots for each patient of the log2 fc expression in response to idealisib. (i) Full 39-gene signature. (ii) Reduced 18-gene signature (details in supplemental Methods). The median represented in each box plot corresponds to the idelalisib score. (D) Heatmap displaying the mean expression of the selected 18 genes in either sensitive (n = 9) and resistant (n = 16) patients classified as in panel B, referred to the baseline control without coculture.
To validate these differential idelalisib responses in a larger patient cohort, 39 genes were selected with the following criteria: upregulation by FDC coculture (fc > 2, P < .05) and differential regulation by idelalisib (fc < 0.5 just in sensitive patients). The effect of idelalisib in this custom gene signature was analyzed in 25 FL-FDC cocultures. Figure 1B and supplemental Figure 4 illustrate the power of this gene signature to identify idelalisib-sensitive and -resistant FL-FDC cultures. To generate a more manageable predictor, we reduced this signature to 18 genes (supplemental Methods), virtually maintaining the same predictive power (Figure 1C-D). This gene signature provides an easy and manageable fingerprint to identify new sensitive FL patients showing idelalisib score ≤ −0.4 (maximum idealisib score for the 18-gene signature in the sensitive patient cohort):
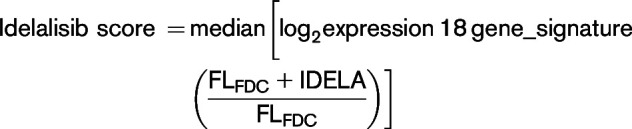 |
To further characterize possible patterns of clinical responses to idelalisib, we analyzed the mutational status of 10 commonly mutated genes in the series of FL with distinctive molecular responses to idelalisib according to the signature established in Figure 1D. These genes included CREBBP, KMT2D, TNFRFS14, EP300, EZH2, MEF2B, TNFAIP3, TP53, RRAGC, and EPHA7 (supplemental Figure 5; supplemental Table 2). The frequency of these mutations in our patient series was in accordance with published results in larger patient cohorts.2
We did not observe any correlation between molecular responses to idelalisib and mutational load, similar to previous reports.23 Of note, RRAGC24 (FL9 and FL16) and P53 (FL10) mutations were just present in resistant FL cases, whereas MEF2B mutations were restricted to sensitive cases (supplemental Figure 5). However, the low number of cases precluded extracting definitive conclusions in this regard.
Idelalisib reduces FDC-induced angiogenesis and transendothelial migration in sensitive patients
We then sought to determine the functional consequences of this differential gene regulation by idelalisib between sensitive and resistant patients. Gene set enrichment analysis (GSEA) of the whole expression data set of FL-FDC cocultures vs FL monocultures uncovered a striking enrichment of genes related to angiogenesis, transendothelial migration (TEM), cell-cell/cell-matrix adhesion, extracellular matrix formation, and cell migration among others, in accordance with previous results.22 Table 1 summarizes GSEA results of the total enriched gene sets associated with a specific biologic process. Enrichment plots of representative upregulated pathways in FL-FDC cocultures are provided in Figure 2A. Likewise, these gene sets were downregulated by idelalisib in FL1- and FL4-sensitive cases, whereas FL2 and FL3 showed no modulation (Figure 2B). Next, we proceed to validate these results at functional level. We focused on 2 processes of paramount importance in lymphoma: angiogenesis and cell dissemination.
Table 1.
Gene sets regulated by idelalisib treatment in FL-FDC cocultures in sensitive patients
| Gene sets | No. of enriched gene sets | NES | FDR, q |
|---|---|---|---|
| Custom gene sets | |||
| Human angiogenesis | 1 | 2.63 | <.0001 |
| IRF4 pathway | 1 | 1.98 | .0045 |
| Cell cycle regulation | 2 | 1.98 | .0039 |
| Integrin pathway | 1 | 1.92 | .0067 |
| Serum response | 1 | 1.85 | .0131 |
| Canonical pathways (C2CP) | |||
| Focal adhesion-integrins | 12 | 2.80 | <.0001 |
| Extracellular matrix formation | 7 | 2.76 | <.0001 |
| Angiogenesis (VEGF/PDGF pathways) | 5 | 2.29 | <.0001 |
| Cell adherent junctions–ECadherin | 5 | 2.08 | .0009 |
| Cell cycle G1-M | 8 | 2.01 | .0027 |
| Transendothelial cell migration | 2 | 1.90 | .0115 |
| Motif gene sets (C3 TFT) | |||
| SRF | 6 | 2.31 | <.0001 |
| IRF | 2 | 1.85 | .0080 |
| NFAT | 1 | 1.73 | .021 |
| NF-κB | 1 | 1.64 | .041 |
| Hallmark gene sets (H) | |||
| Epithelial mesenchymal transition | 1 | 3.25 | <.0001 |
| Angiogenesis | 1 | 2.26 | <.0001 |
| mTOR | 1 | 1.95 | .0002 |
| Interferon α and γ responses | 2 | 1.6 | .015 |
| GO gene sets (C5) | |||
| Extracellullar matrix organization | 8 | 2.9 | <.0001 |
| Adhesion-integrins | 9 | 2.56 | <.0001 |
| Vasculature-angiogenesis-EC growth | 15 | 2.49 | <.0001 |
| Cell cycle G1-S and G2-M | 6 | 2.09 | .0007 |
Gene sets regulated by idelalisib were identified by GSEA using custom gene sets experimentally derived (http://lymphochip.nih.gov/signaturedb/index.html), C2 canonical pathways, C3 motif gene sets, Hallmark, and C5-GO signatures obtained from the Molecular Signature Database (v2.5). Threshold: FDR < 0.05 and NES > 1.5. The number of enriched gene sets and the best FDR and NES scores are indicated for each biological process. A complete list of the enriched gene set is detailed in supplemental Table 3.
EC, endothelial cell; IRF, interferon response factor; IRF4, interferon regulatory factor 4; FDR, false discovery rate; mTOR, mammalian target of rapamycin; NES, normalized enriched score; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor κB; SRF, serum response factor.
Figure 2.
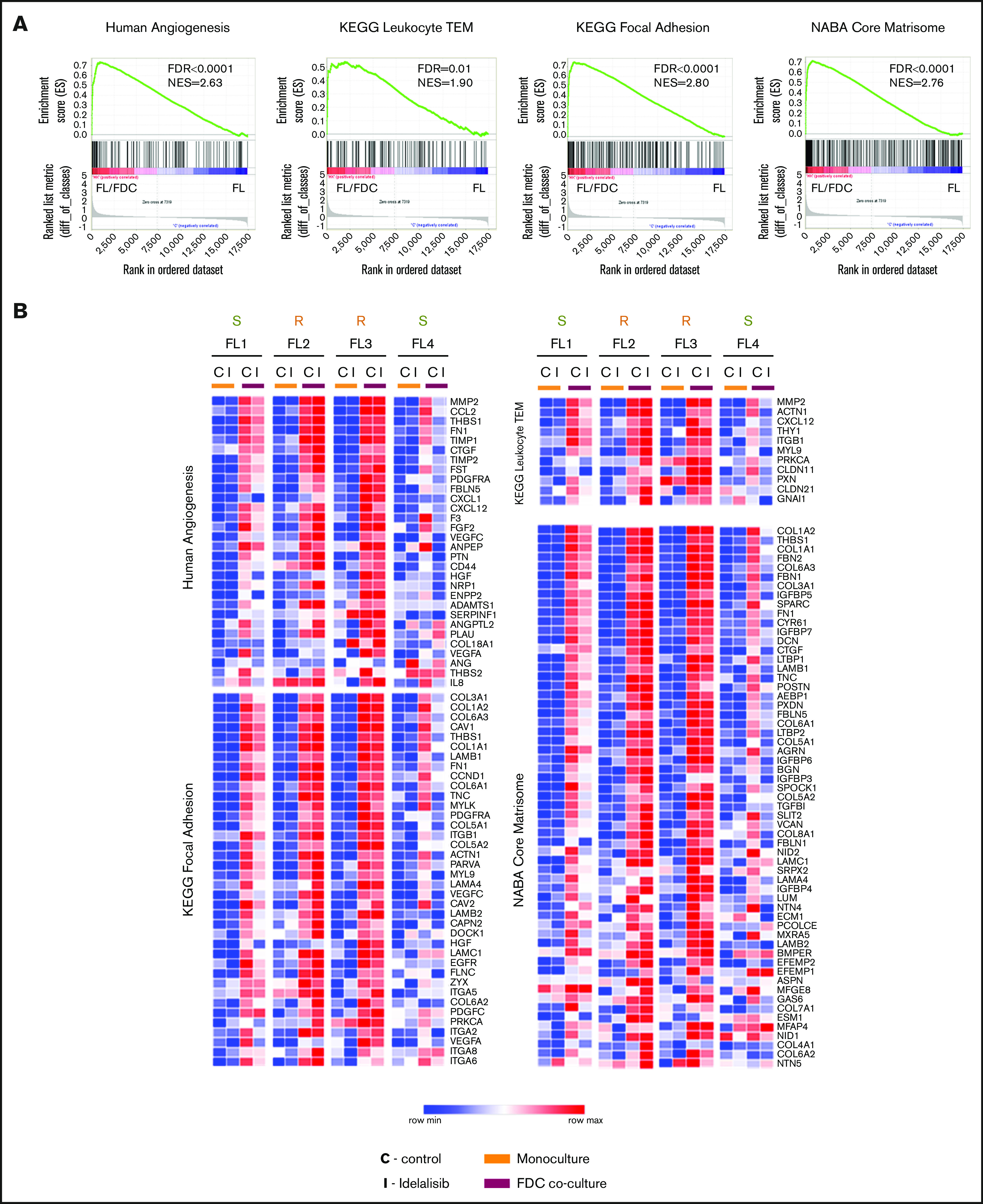
Idelalisib modulates biological pathways in FL-FDC cocultures. FL primary cells were isolated from monocultures or FL-FDC cocultures with/without idelalisib (500 nM, 48 hours) and subjected to GEP. Gene sets regulated by idelalisib were identified by GSEA using custom genes sets, C2 canonical pathways, C3 motifs, Hallmark, and C5-GO signatures. Enrichment plots (A) and heatmaps (B) of the corresponding leading edges of selected gene sets are shown.
It is well documented that PI3K/AKT plays a key role in angiogenesis, both through regulation of vascular endothelial growth factor-A (VEGF-A) expression and as a transducer of VEGF-A–VEGFR1 and VEGF-C–VEGFR2 downstream signaling.25 Analysis of VEGF-A and VEGF-C secretion by enzyme-linked immunosorbent assay (ELISA) on supernatants from FL-FDC cocultures with or without idelalisib uncovered a significant (P = .0033 and P = .0222, respectively) downregulation of both proangiogenic factors mostly in idelalisib-sensitive patients (Figure 3A) in accordance with GEP results (Figure 2B). We then used these supernatants in a tube formation assay with human umbilical vein endothelial cells (HUVECs; Figure 3B). Supernatants from FL-FDC cocultures significantly increased the number of nodes and junctions compared with those from FL monocultures (nodes: P = .0056 in sensitive patients, P = .0076 in resistant patients; junctions: P = .0067 in sensitive patients, P = .0058 in resistant patients). Importantly, the presence of idelalisib diminished the proangiogenic potential of those supernatants exclusively in idelalisib-sensitive patients (nodes: P = .0138; junctions: P = .0091), in accordance with the results obtained for VEGF-A and VEGF-C.
Figure 3.
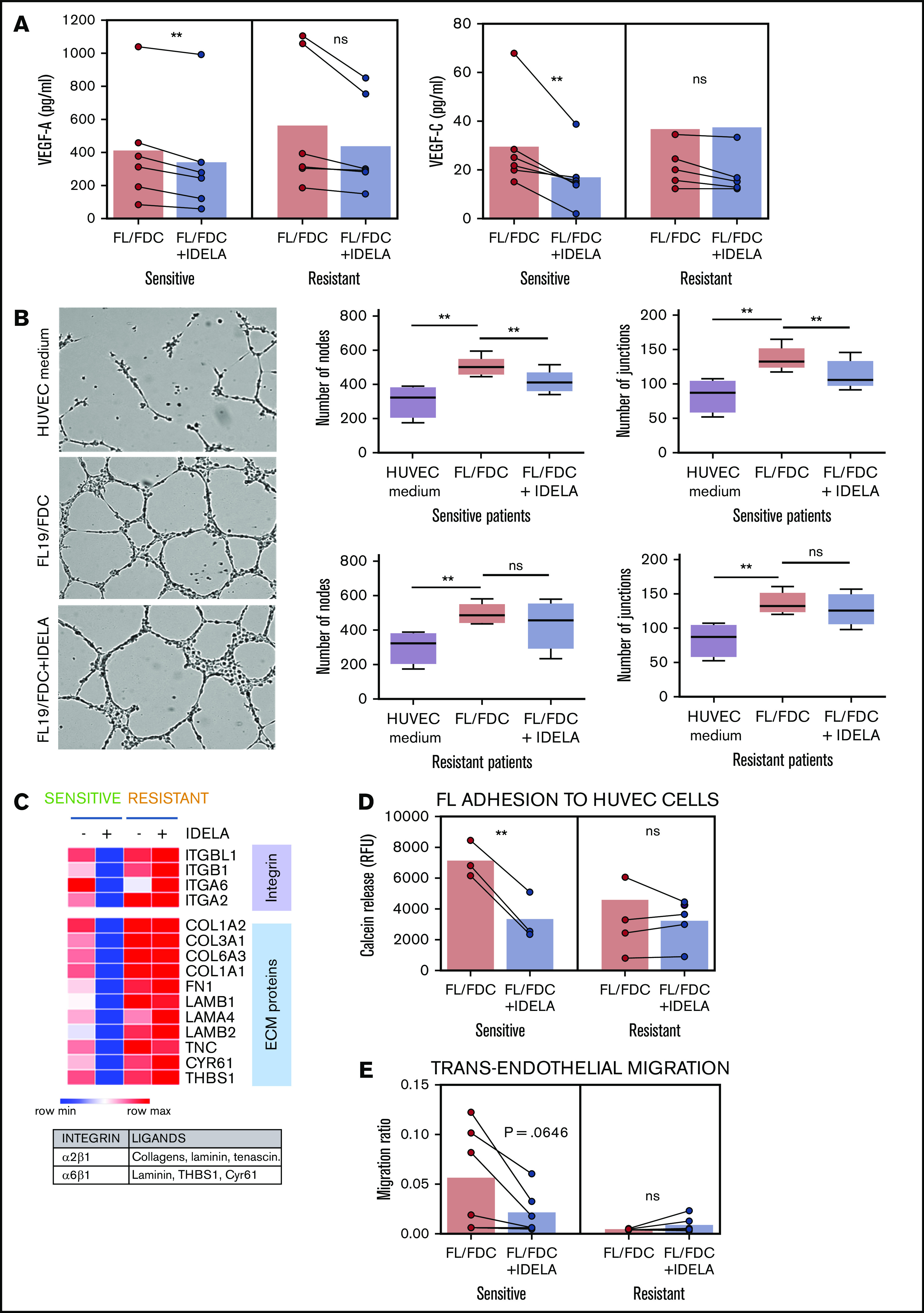
Idelalisib reduces FDC-induced angiogenesis and TEM in sensitive patients. FL-FDC coculture supernatants with or without idelalisib (IDELA, 500 nM, 48 hours) were used to determine VEGF-A and VEGF-C protein secretion by ELISA in sensitive (n = 6) and resistant (n = 6) patients (A) and tube formation assay of endothelial HUVEC cells cultured for 24 hours with their own media alone or mixed with the corresponding supernatants (ratio 1:1) (B) (magnification ×40). Five representative images of each condition were captured using a phase-contrast microscope and analyzed by FIJI-ImageJ (angiogenesis analyzer plug-in). Representative images from a sensitive patient are shown. Node and junction numbers from sensitive (n = 5) and resistant (n = 5) patients are displayed. (C) Heatmap displaying the regulation induced by idelalisib (IDELA) in the expression of integrins and their ligands in FL cells from FL-FDC cocultures of sensitive (FL1 and FL4) and resistant patient samples (FL2 and FL3). (D) After IDELA treatment (500 nM, 48 hours) FL cells from FL-FDC cocultures with or without idelalisib (500 nM, 48 hours) of sensitive and resistant patients (n = 8) were stained with calcein and allowed to adhere for 3 hours to HUVECs. After extensive washing, the cells that remained attached were lysed, and fluorescence was measured in a Synergy HT microplate reader. (E) FL cells (n = 12) from FL-FDC cocultures with or without idelalisib (500 nM, 48 hours) were challenged to migrate for 6 hours in a gradient of FBS through trans-wells coated with HUVECs seeded on gelatin 0.1% coated + TNF-α (10 ng/mL). CD20+ cells crossing the HUVEC barrier were counted by flow cytometry. *P < .05, **P < .01. ns, not significant.
As described in Figure 2, FL-FDC coculture significantly modulated the expression of some adhesion-related molecules and extracellular matrix components, with a differential regulation by idelalisib between sensitive and resistant patients as displayed in Figure 3C. The main integrins upregulated were ITGA2, ITGA6, ITGB1, and ITGBL1, whereas the main corresponding ligands were the extracellular matrix component collagens (COL1A2, COL3A1, COL6A3, and COL1A1), fibronectin (FN1), laminin (LAMB1, LAMA4, LAMB2), tenascin (TNC), cysteine-rich angiogenic inducer 61 (CYR61), and the glycoprotein thrombospondin 1 (THBS1), which are involved in angiogenesis, cell-to-cell interaction, and cell-to-matrix interaction. We then validated the functional consequences of this gene regulation. As adhesion represents a precedent step for cell migration onset, FL cells from FL-FDC cocultures with or without idelalisib were challenged to cell adhesion experiments to HUVECs. Idelalisib significantly (P = .0046) reduced this event only in sensitive patients (Figure 3D). Likewise, the simultaneous reduction observed in both integrins and their ligands in sensitive patients may indicate a decrease in the migratory capacity of these cells inside the lymph node and through the blood vessel wall. To examine that hypothesis, we carried out a TEM assay. FL cells from FL-FDC cocultures with and without idelalisib were challenged to migrate through transwells coated with a HUVEC monolayer toward a gradient of FBS. In line with the adhesion assay results, idelalisib exhibited a trend to reduced TEM in sensitive patients (P = .0646), whereas it did not affect this phenomenon in resistant patients (Figure 3E).
These results taken together uncovered antiangiogenic and antidissemination properties of idelalisib in sensitive FL patients.
Idelalisib interferes with FL–T cells cross talk through CD40/CD40L and affects Treg and TFH recruitment through CCL22 downregulation
In addition to the modulatory effects of idelalisib on FDC-induced genes, idelalisib displays a more general downregulatory action over a group of genes that were mostly not induced by FDC coculture and similarly downmodulated by idelalisib independently of the presence of FDCs in all patients tested. GSEA revealed that these genes were related to CD40L signaling (Figure 4A) and to the GC program including B-lymphocyte–induced maturation protein 1 (BLIMP)–regulated genes (supplemental Table 4; supplemental Figure 6). The CD40-CD40L pathway lies at the cross talk between B and T cells in the GC and is fundamental for B-cell survival, proliferation, and differentiation into plasma cells.26 Likewise, the mTORC1 pathway was also downregulated by idelalisib, validating the inhibition of PI3K/AKT/mTOR pathway by idelalisib both in monoculture and in FL-FDC coculture (supplemental Table 4; supplemental Figure 6), and idelalisib decreased ribosomal protein S6 phosphorylation (supplemental Figure 7).
Figure 4.
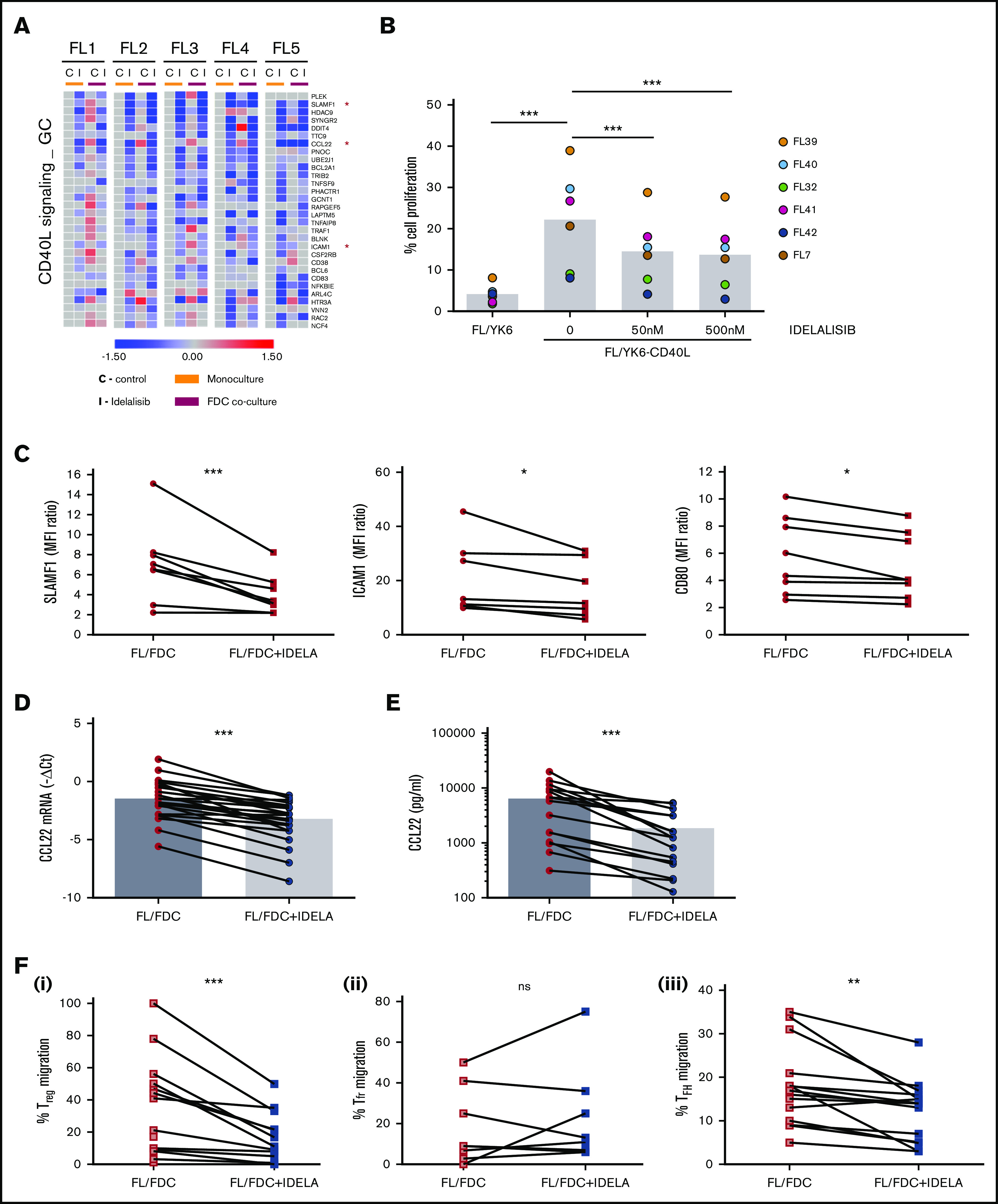
Idelalisib interferes with FL–T cells cross talk through CD40/CD40L and affects Treg and TFH recruitment through CCL22 downregulation. (A) FL cells (n = 5) were cultured for 48 hours with idelalisib (500 nM), and B cells were purified and subjected to GEP. Gene sets regulated by idelalisib (IDELA) in the presence or absence of FDC coculture were identified by GSEA using custom genes set (http://lymphochip.nih.gov/signaturedb/index.html). A heatmap of the leading edge corresponding to the CD40L_signaling_GC gene set is shown and represents the relative gene expression of FL cells cultured with and without IDELA compared with the untreated control. (B) FL cells (n = 7) were labeled with carboxyfluorescein succinimidyl ester (CFSE; 0.5μM) and cocultured for 5 days with and without 500 nM idelalisib on pre-established layers of FDCs engineered or not for CD40L expression (YK6 and YK6-CD40L). The percentage of viable CD19+ cells with low CFSE fluorescence was used as a read-out of proliferation. (C) SLAMF1, ICAM1, and CD80 membrane expression was evaluated by flow cytometry in FL-FDC cocultures (n = 8) with and without IDELA (500 nM, 48 hours). FL-FDC coculture supernatants with and without IDELA were used to assess CCL22 gene expression by real-time polymerase chain reaction (n = 26) using GUSB, ACTB, and B2M as houskeeping genes (D), CCL22 protein expression by ELISA (n = 16) (E), and migration of Treg cells (CD4+CD25+FoxP3+; n = 14) obtained from PBMCs of healthy donors (Fi) or migration of Tfr (CD4+CXCR5+FoxP3+; n = 9) (Fii) and TFH cells (CD4+CXCR5+CD25−; n = 14) (Fiii) obtained from normal tonsils.
The FL patient samples used in this study were obtained from patient biopsies of lymph nodes and contained a significant percentage of T cells (supplemental Table 1), although their full functionality ex vivo, in the absence of an adequate microenvironment, may be compromised.27 For this reason, we chose to validate the interference of idelalisib with the CD40L pathway using an engineered FDC cell line (YK6-CD40L) that overexpresses CD40L on the cell membrane. FL cells labeled with Carboxy Fluorescein Succinimidyl Ester (CFSE) proliferated in the presence of YK6-CD40L (FL/YK6 vs FL/YK6-CD40L: P = .0084; Figure 4B), as attested by the loss of their fluorescent signal, and idelalisib inhibited their proliferation at concentrations of just 50 nM (untreated vs 50 nM: P = .003398; untreated vs 500 nM: P = .001338; supplemental Figure 8; Figure 4B), thus validating GEP results. We also validated at a protein level the downregulation by idelalisib of several CD40L target genes directly involved in this B–T immunological synapse, such as the activation molecule SLAMF1, the intercellular adhesion molecule ICAM1, or the costimulatory protein CD80 from the GC gene set. In all FL cases analyzed, idelalisib induced a moderate but constant decrease in their expression (Figure 4C).
Among the genes regulated by idelalisib in the CD40L pathway, the chemokine CCL22 stood out. This chemokine is secreted by many tumor types including FL.9,28,29 CCL22 and CCL17 are the ligands for CCR4 receptor used by circulating effector/memory lymphocytes, especially Treg and T helper cells, for their recruitment.30 To gain insights into the consequences of CCL22 downregulation by idelalisib, we first validated the GEP results, analyzing its expression (P < .0001) in an expanded series of FL-FDC cocultures (n = 25; Figure 4D) followed by its quantification at a protein level. Analysis of CCL22 by ELISA in supernatants from FL-FDC cocultures treated with or without idelalisib (500 nM, 48 hours) demonstrated that CCL22 is secreted in the FL-FDC niche, and idelalisib induced a significant reduction of this chemokine (Figure 4E, P = .003). Then, we checked whether FL-FDC coculture supernatants were able to effectively recruit Treg cells from blood. To this aim, PBMCs from healthy donors, enriched in the T-cell fraction, were allowed to migrate toward those FL-FDC supernatants (with or without idelalisib), where CCL22 was determined previously, and counted by flow cytometry (CD4+/CD25+/FoxP3+). FL-FDC supernatant favored Treg recruitment, and idelalisib reduced this event (Figure 4Fi; P = .0009) in agreement with a recent study.31 In contrast, using PBMCs from fresh tonsils to mimic the follicular origin, we demonstrated that the recruitment of T-follicular regulatory cells (Tfr) (CD4+/CXCR5+/FoxP3+) related to good prognosis32 and was not affected by idelalisib (Figure 4Fii). We finally analyzed the ability of those coculture supernatants to recruit T-follicular helper cells (TFH), a T-cell subpopulation fundamental for FL survival.7,33 Likewise, using PBMCs from fresh tonsils enriched in the T-cell fraction, we quantified the effect of idelalisib on TFH cells (CD4+CXCR5+CD25−) migration and demonstrated that FL-FDC supernatants recruited TFH and idelalisib (P = .002) diminished this migration (Figure 4Fiii).
In summary, idelalisib reshapes the immune FL microenvironment by decreasing the levels of the CCL22 that limits the recruitment of immunosuppressive Treg and the supportive TFH to the FL niche, together with the disruption of FL–TFH cross talk cells via CD40L.
Immune microenvironment modulates BCL-2 dependence of FL cells
BCL-2 inhibition by venetoclax has proven to be a good partner for BCR kinase inhibitors in several lymphoid malignancies different from FL.34-36 Surprisingly, despite the fact that BCL-2 overexpression is a hallmark of FL, the BCL-2 inhibitor venetoclax has shown limited activity in this lymphoma.18 To better understand this apparent contradiction, we characterized the BCL-2 dependency in a set of FL patient samples by BH3 profiling.37 This technique is a functional assay designed to interrogate mitochondrial apoptotic machinery as a whole. Using a BH3 peptide derived from the proapoptotic protein BIM, which interacts with all major antiapoptotic proteins, we measured the proximity of FL cells to the threshold of apoptosis, a property called mitochondrial priming,38 and found that FL cells showed a different degree of mitochondrial priming (Figure 5A). BH3 profiling was also able to determine precisely the dependence of FL cells on specific antiapoptotic BCL-2 family proteins, such as BCL-XL, BCL-2, or BFL-1 (Figure 5B). In the absence of coculture, we found that FL cells primarily showed patterns of either BCL-2 (n = 6) or BCL-XL dependence (n = 6), whereas only 1 case manifested dependence on MCL-1/BFL-1 (Figure 5C shows a representative example of each pattern). BCL2 pattern was characterized by high sensitivity to both venetoclax (used as a specific peptide against BCL-2) and BCL-2–associated agonist of cell death (BAD), whereas BCL-XL pattern was associated to higher sensitivity to Harakiri (HRK) peptide than for BCL-2 (venetoclax). Finally, BFL-1 dependence, just found in 1 case, was linked to high sensitivity for phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1/NOXA) and FS2 peptides. In the FL samples tested, we did not identify a case with MCL-1 pattern (high sensitivity for NOXA and low for FS2).
Figure 5.

Immune microenvironment modulates FL dependence on BCL-2. (A) Mitochondrial priming with BIM peptide in FL samples. (B) Binding preferences of BH3-only proteins/ peptides with antiapoptoctic BCL-2 proteins. (C) Examples of BH3 profiles from 3 individual FL patients showing patterns of relative dependence on BCL-2 (FL13), BCL-XL (FL12), and MCL1/BFL-1 (FL14). (D) BCL-2 family protein dependence was assessed by BH3 profiling using venetoclax (VEN, n = 9), BAD (n = 13), HRK (n = 13), NOXA (n = 13), and FS2 (n = 6) in FL-FDC and FL-Mϕ cocultures. *P < .05, **P < .01.
Next, we sought to determine how microenvironment modulates this BCL-2 dependency. For this purpose, we set FL cocultures with FDC or human Mϕ, as they are also fundamental in FL pathogenesis.4,39 Interestingly, we identified that FL-FDC coculture significantly protected from venetoclax and BAD-induced apoptosis, whereas it increased the sensitivity to HRK (P = .031) and NOXA (P = .042) peptides, uncovering that BCL-XL and MCL-1 dependence raises at the expense of BCL-2. Furthermore, Mϕ also protected FL cells from venetoclax and BAD peptide apoptosis, whereas it sensitized them to a synthetic peptide specific for BFL-1 (FS2; P = .047),40 indicating a higher dependence on this antiapoptotic protein (Figure 5D). In summary, immune microenvironment renders FL more dependent on apoptotic proteins different from BCL-2, reducing their priming for apoptosis.37 This fact may lie at the basis of the reduced clinical benefit observed with venetoclax in FL patients.18
Idelalisib bypasses microenvironment derived resistance of FL cells to venetoclax
We next wondered if idelalisib, despite inducing limited cell death, may be able to increase FL overall mitochondrial priming or modify their specific BH3 profile. We uncovered that idelalisib did not significantly increase FL priming either in monoculture or in FL-FDC and FL-Mϕ cocultures (supplemental Figure 9) but restored BCL-2 dependence over BCL-XL (Figure 6A). Venetoclax, used as a specific peptide for BCL-2, exhibited minimal response in FL-FDC and FL-Mϕ cocultures, whereas a recovery of venetoclax activity was observed in the presence of idelalisib both in FL-FDC (P = .0393) and FL-Mϕ (P = .0001) cocultures (Figure 6A). Likewise, FL cells displayed lower response to BAD peptide (which would simulate combined BCL-2/BCL-XL inhibition) in FL-FDC (P = .0085) and FL-Mϕ (P = .0455) cocultures, and idelalisib treatment allowed the recovery of BAD activity in both FL-FDC (P = .0014) and FL-Mϕ (P = .0279) cocultures (supplemental Figure 10). On the contrary, idelalisib did not modify the overall priming of HRK peptide activity in these cocultures (supplemental Figure 10), implying changes in BCL-XL dependence, reinforcing the notion that idelalisib activity relies on restoring FL dependence on BCL-2 (Figure 6A).
Figure 6.
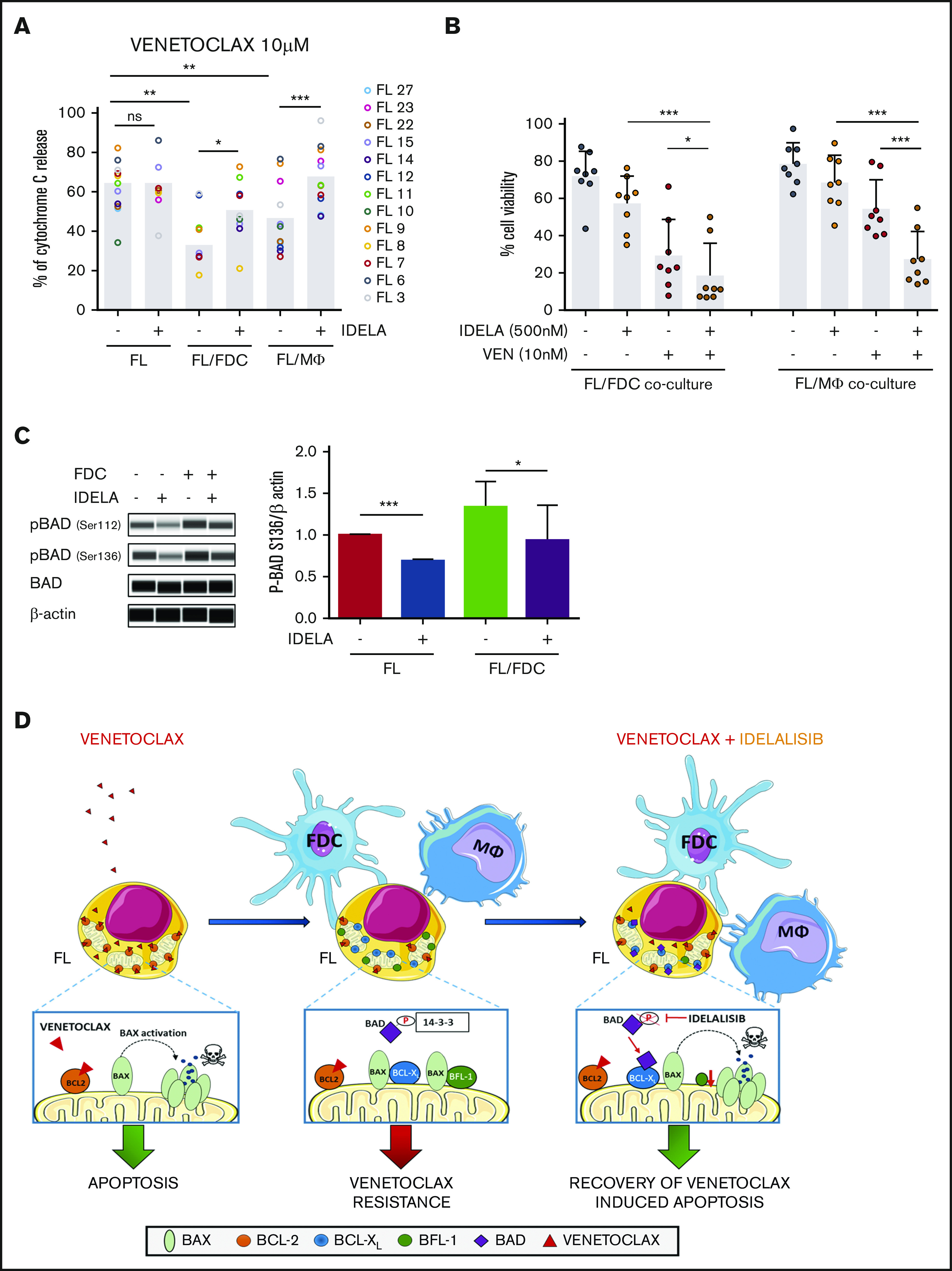
Idelalisib bypasses microenvironment-derived resistance to venetoclax. (A) FL cells from monocultures (FL) or FL-FDC and FL-Mϕ cocultures treated with or without idelalisib (500 nM, 24 hours) were permeabilized and incubated for 1 hour with 10 μM venetoclax fixed/stained for intracellular cytochrome C and evaluated by flow cytometry as a read-out of apoptosis priming (n = 13). (B) Cell viability (AnnexinV−/7AAD−) was assessed in FL cells from FL-FDC and FL-Mϕ with and without idelalisib (500 nM) and with and without venetoclax (10 nM) after 72 hours of treatment (n = 8). (C) FL cells from monocultures or FL-FDC cocultures (n = 3) with or without idelalisib (IDELA, 500 nM, 3 hours) were lysed, and the phosphorylation of BAD at Ser112 and Ser136 was analyzed by Peggy Sue simple Western blotting and quantified by densitometry. Images from a representative case (FL3) are shown. (D) Rational of venetoclax PI3Kδ combined therapy in FL. *P < .05, **P < .01, ***P < .001.
One possible contributory mechanism of this idelalisib activity in FL-FDC cocultures may be explained by the reduction of BAD phosphorylation on the direct PI3K target Ser13641 that may allow proapoptotic BAD relocation to the mitochondrial membrane and apoptosis induction42 (Figure 6C). Another contributory mechanism could be the increase in the proapoptic HRK expression in response to idelalisib in FL-FDC cocultures (supplemental Figure 11). In contrast, in FL-Mϕ cocultures, the downregulation of BFL-1 expression by idelalisib may have a role (supplemental Figure 12).
The therapeutic cooperation of idelalisib with venetoclax on apoptosis induction was further assessed by flow cytometry measuring the percentage of viable cells after 3-day coculture with each agent alone or in combination at therapeutic doses (ie, venetoclax in the nanomolar range). We concluded that the treatment of FL cells with the combination resulted in significant reduction of cell viability compared with the single agents both in the FL-FDC cocultures (P = .0135) and in FL-Mϕ cocultures (P = .0006; Figure 6B). Additionally, this positive combinatorial effect was observed in both idelalisib-sensitive and -resistant FL samples (supplemental Figure 13), classified according to the 18-gene signature defined in Figure 1. Neither venetoclax nor idelalisib displayed significant cytotoxic activity on FDC or Mϕ (supplemental Figure 14).
Thus, these results provide the molecular basis for a bench-to-bedside translation of this new therapeutic combination. A schematic rationale is provided in Figure 6D.
Discussion
The microenvironment of FL, an incurable B-cell non-Hodgkin lymphoma, is thought to play a major role in its pathogenesis and clinical outcome, and a number of therapies targeting FL–microenvironment cross talk have reached the clinic. Idelalisib is a first-in-class PI3Kδ inhibitor approved for the treatment of relapsed/refractory FL.11,43 Despite its introduction into the clinic, a precise characterization of the interference of idelalisib with the FL–microenvironment cross talk remains poorly defined. In the present study, using a meaningful ex vivo coculture system composed of FL patient cells and supportive FDCs from normal tonsils, we discovered that idelalisib interferes with specific biologic processes including angiogenesis and TEM (Table 1; supplemental Table 3), exclusively in a selected group of patients. This discovery allows us to define a gene signature to discriminate between idelalisib-sensitive and -resistant FL primary cultures validated in an expanded cohort of patients. Ideally, the predictive value of this “idelalisib score” should be further validated in pretreatment samples from FL patients enrolled in idelalisib clinical trials to correlate in vivo responses with this in vitro predictor.
In those patients defined as sensitive based on the idelalisib score, idelalisib reduced the secretion of the proangiogenic factors VEGF-A and VEGF-C. These supernatants were significantly less efficient in the generation of endothelial HUVEC microtubules, used as a read-out of their proangiogenic potential. This is of key importance in FL, because vascularization predicts overall survival and risk of transformation.44
FL patients usually present disseminated disease at diagnosis, indicating the high mobility properties of these tumor cells. To enter lymphoid organs, B cells must adhere to the endothelium and transmigrate across the endothelial barrier. Both processes are mediated through selectin ligands, integrins, or CD44.45 Importantly, in several models of lymphoma, including FL, the expression of several β-integrins has been associated with disease dissemination and patient prognosis.46 Thus, the regulation of this process is of paramount importance to control the disease, and idelalisib has shown significant activity in sensitive patients. In this regard, studies of the interference of idelalisib and others BCR inhibitors (ie, ibrutinib) with adhesion and migration have been used as a read-out of antitumor activity.47 FL is characterized by a strong infiltration of diverse T-cell subpopulations. Our results uncovered the modulation of the CD40/CD40L pathway at the B–T interface by idelalisib as a general phenomenon, decreasing CD40L-induced proliferation. Idelalisib also downregulates the expression of several membrane proteins critical for B–T cell synapses (CD80, SLAMF1, and ICAM1). The net balance of these effects might result in an inefficient cross talk between FL cells and the supportive TFH cells. Of the specific genes regulated by the CD40L-CD40 system, CCL22 stood out as a chemokine fundamental for the migration of diverse T-cell subpopulations.30 The decrease in CCL22 secretion by idelalisib may contribute to changes in the composition of FL microenvironment. By means of in vitro migration assays, we observed a significant decrease in the recruitment of Treg and TFH when these cells were challenged to migrate toward supernatants from FL-FDC cocultures treated with idelalisib. PI3Kδ is fundamental for the generation of TFH,48 and the presence of these supportive TFH has been associated with poor prognosis in a number of hematologic malignancies.49 Intratumoral TFH cells induce production of CCL22 by FL tumor cells and facilitate active recruitment of Tregs and IL-4–producing T cells, which, in turn, may stimulate more chemokine production in a feedforward cycle. Likewise, based on previous studies,50 the decrease in the activation receptor SLAMF-1 observed in our system may reduce IL-4 production by TFH. Moreover, the cross talk between FL and TFH contributes to FL pathogenesis and promotes immune evasion in FL microenvironment.9 Thus, the coordinated decrease in TFH and Treg recruitment may allow the host to mount superior immune responses against the tumor.
BCL-2 overexpression is the genetic hallmark of FL. However, BCL-2 antiapoptotic proteins extend well beyond BCL-2 and are known to be regulated by the microenvironment.51 We uncovered that FL-FDC cocultures augmented tumor addiction to BCL-XL and MCL-1, whereas BFL-1 was relevant in FL-Mϕ cocultures. The consequence of these changes was a decrease in the activity of the BCL-2 inhibitor venetoclax. These results are in agreement with those reported by several groups in CLL and MCL34,52-55 and may well be the basis of the reduced clinical benefit observed in FL patients treated with venetoclax.18 Idelalisib restored FL dependence on BCL-2 and venetoclax activity by several mechanisms including BAD serine dephosphorylation, an increase in HRK, and a decrease in BFL-1 expression, thus providing a mechanistic rationale for investigating the combination of PI3Kδ inhibitors and venetoclax in clinical trials, following the success in CLL.56
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ariadna Giró and Fabian Arenas for technical assistance and the IDIBAPS genomics facility for gene expression data and NGS. This work was carried out at the Esther Koplowitz Center, Barcelona, Spain.
Gilead Science Pharmaceuticals funded part of this study. Additional grants that contributed to this work include the following: Spanish Ministry of Economy and Competitiveness & European Regional Development Fund (ERDF) “Una manera de hacer Europa” for SAF2014/57708R and SAF2017/88275R (P.P.-G.), SAF2015/31242R (D.C.), CIBERONC (CB16/12/00334 and CB16/12/00225), and Generalitat de Catalunya support for AGAUR 2017SGR1009 (D.C.).
Portions of this manuscript were from the thesis “Targeting tumor microenvironment cross talk through GPCR receptors and PI3K,” by M.G.-H., Faculty of Medicine, University of Barcelona, 12 February 2019; supervisors: P.P.-G. and Jordi Camps.
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE130918).
Authorship
Contribution: N.S. and M.G.-H. conducted molecular and cellular assays, performed data analysis, and contributed to study design and manuscript writing; A.M.-C. and A.Y. conducted molecular and cellular assays and performed data analysis; J.G.V. contributed to data analysis and discussion; F.N. and A.E. performed NGS analysis; G.C. and S.B. provided statistics support; M.D.R. and D.J.H. provided reagents; M.C.-B. and M.C.C. provided reagents and protocols; L.M., A.R.-D., and A.L.-G. provided patient clinical data; J.M. supervised BH3 profiling studies; E.C. and D.C. provided economic support and study guidance; S.T. supervised the study; P.P.-G. designed the study, analyzed data, provided economic support, and wrote the paper; and all authors revised and approved the manuscript.
Conflict-of-interest disclosure: A.Y. and S.T. were Gilead Sciences employees and P.P.-G. received research funding from Gilead Sciences. The remaining authors declare no competing financial interests.
Correspondence: Patricia Pérez-Galán, Department of Hemato-Oncology, IDIBAPS, Rosselló 149-153, 08036 Barcelona, Spain; e-mail: pperez@clinic.cat.
References
- 1.Huet S, Sujobert P, Salles G. From genetics to the clinic: a translational perspective on follicular lymphoma. Nat Rev Cancer. 2018;18(4):224-239. [DOI] [PubMed] [Google Scholar]
- 2.Lackraj T, Goswami R, Kridel R. Pathogenesis of follicular lymphoma. Best Pract Res Clin Haematol. 2018;31(1):2-14. [DOI] [PubMed] [Google Scholar]
- 3.Amé-Thomas P, Tarte K. The yin and the yang of follicular lymphoma cell niches: role of microenvironment heterogeneity and plasticity. Semin Cancer Biol. 2014;24:23-32. [DOI] [PubMed] [Google Scholar]
- 4.Amin R, Mourcin F, Uhel F, et al. DC-SIGN-expressing macrophages trigger activation of mannosylated IgM B-cell receptor in follicular lymphoma. Blood. 2015;126(16):1911-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linley A, Krysov S, Ponzoni M, Johnson PW, Packham G, Stevenson FK. Lectin binding to surface Ig variable regions provides a universal persistent activating signal for follicular lymphoma cells. Blood. 2015;126(16):1902-1910. [DOI] [PubMed] [Google Scholar]
- 6.Coelho V, Krysov S, Ghaemmaghami AM, et al. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci USA. 2010;107(43):18587-18592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pangault C, Amé-Thomas P, Ruminy P, et al. Follicular lymphoma cell niche: identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia. 2010;24(12):2080-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amé-Thomas P, Le Priol J, Yssel H, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia. 2012;26(5):1053-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawal S, Chu F, Zhang M, et al. Cross talk between follicular Th cells and tumor cells in human follicular lymphoma promotes immune evasion in the tumor microenvironment. J Immunol. 2013;190(12):6681-6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okkenhaug K, Bilancio A, Farjot G, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031-1034. [DOI] [PubMed] [Google Scholar]
- 11.Gopal AK, Kahl BS, de Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller BW, Przepiorka D, de Claro RA, et al. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015;21(7):1525-1529. [DOI] [PubMed] [Google Scholar]
- 13.Herman SEM, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lannutti BJ, Meadows SA, Herman SEM, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong S, Harrington BK, Hu EY, et al. PI3K p110δ inactivation antagonizes chronic lymphocytic leukemia and reverses T cell immune suppression. J Clin Invest. 2019;129(1):122-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abu-Eid R, Samara RN, Ozbun L, et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2(11):1080-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davids MS, Roberts AW, Seymour JF, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J Clin Oncol. 2017;35(8):826-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park C-S, Yoon S-O, Armitage RJ, Choi YS. Follicular dendritic cells produce IL-15 that enhances germinal center B cell proliferation in membrane-bound form. J Immunol. 2004;173(11):6676-6683. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Yoon S-O, Fu D-D, Zhang X, Choi YS. Novel follicular dendritic cell molecule, 8D6, collaborates with CD44 in supporting lymphomagenesis by a Burkitt lymphoma cell line, L3055. Blood. 2004;104(3):815-821. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Zhang X, Kovacic S, et al. Identification of a human follicular dendritic cell molecule that stimulates germinal center B cell growth. J Exp Med. 2000;191(6):1077-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matas-Céspedes A, Rodriguez V, Kalko SG, et al. Disruption of follicular dendritic cells-follicular lymphoma cross-talk by the pan-PI3K inhibitor BKM120 (Buparlisib). Clin Cancer Res. 2014;20(13):3458-3471. [DOI] [PubMed] [Google Scholar]
- 23.Scheffold A, Jebaraj BMC, Tausch E, et al. IGF1R as druggable target mediating PI3K-δ inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood. 2019;134(6):534-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okosun J, Wolfson RL, Wang J, et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma [published correction appears in Nat Genet. 2016;48:700]. Nat Genet. 2016;48(2):183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soler A, Angulo-Urarte A, Graupera M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol Cell Oncol. 2015;2(2):e975624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawabe T, Naka T, Yoshida K, et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1(3):167-178. [DOI] [PubMed] [Google Scholar]
- 27.Brady MT, Hilchey SP, Hyrien O, Spence SA, Bernstein SH. Mesenchymal stromal cells support the viability and differentiation of follicular lymphoma-infiltrating follicular helper T-cells. PLoS One. 2014;9(5):e97597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gobert M, Treilleux I, Bendriss-Vermare N, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000-2009. [DOI] [PubMed] [Google Scholar]
- 29.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942-949. [DOI] [PubMed] [Google Scholar]
- 30.Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol. 2015;27(1):11-20. [DOI] [PubMed] [Google Scholar]
- 31.Chellappa S, Kushekhar K, Munthe LA, et al. The PI3K p110δ isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J Immunol. 2019;202(5):1397-1405. [DOI] [PubMed] [Google Scholar]
- 32.Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108(9):2957-2964. [DOI] [PubMed] [Google Scholar]
- 33.Ochando J, Braza MS. T follicular helper cells: a potential therapeutic target in follicular lymphoma. Oncotarget. 2017;8(67):112116-112131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel VM, Balakrishnan K, Douglas M, et al. Duvelisib treatment is associated with altered expression of apoptotic regulators that helps in sensitization of chronic lymphocytic leukemia cells to venetoclax (ABT-199). Leukemia. 2017;31(9):1872-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia. 2017;31(10):2075-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bojarczuk K, Wienand K, Ryan JA, et al. Targeted inhibition of PI3Kα/δ is synergistic with BCL-2 blockade in genetically defined subtypes of DLBCL. Blood. 2019;133(1):70-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter DS, Letai A. To prime, or not to prime: that is the question. Cold Spring Harb Symp Quant Biol. 2016;81:131-140. [DOI] [PubMed] [Google Scholar]
- 38.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351-365. [DOI] [PubMed] [Google Scholar]
- 39.Küppers R, Stevenson FK. Critical influences on the pathogenesis of follicular lymphoma. Blood. 2018;131(21):2297-2306. [DOI] [PubMed] [Google Scholar]
- 40.Jenson JM, Ryan JA, Grant RA, Letai A, Keating AE. Epistatic mutations in PUMA BH3 drive an alternate binding mode to potently and selectively inhibit anti-apoptotic Bfl-1. eLife. 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231-241. [DOI] [PubMed] [Google Scholar]
- 42.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278(5338):687-689. [DOI] [PubMed] [Google Scholar]
- 43.Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res. 2015;21(7):1537-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farinha P, Kyle AH, Minchinton AI, Connors JM, Karsan A, Gascoyne RD. Vascularization predicts overall survival and risk of transformation in follicular lymphoma. Haematologica. 2010;95(12):2157-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drillenburg P, Pals ST. Cell adhesion receptors in lymphoma dissemination. Blood. 2000;95(6):1900-1910. [PubMed] [Google Scholar]
- 46.Terol MJ, López-Guillermo A, Bosch F, et al. Expression of beta-integrin adhesion molecules in non-Hodgkin’s lymphoma: correlation with clinical and evolutive features. J Clin Oncol. 1999;17(6):1869-1875. [DOI] [PubMed] [Google Scholar]
- 47.de Rooij MFM, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood. 2015;125(14):2306-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rolf J, Bell SE, Kovesdi D, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol. 2010;185(7):4042-4052. [DOI] [PubMed] [Google Scholar]
- 49.Zhou DM, Xu YX, Zhang LY, et al. The role of follicular T helper cells in patients with malignant lymphoid disease. Hematology. 2017;22(7):412-418. [DOI] [PubMed] [Google Scholar]
- 50.Yusuf I, Kageyama R, Monticelli L, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol. 2010;185(1):190-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leverson JD, Cojocari D. Hematologic tumor cell resistance to the BCL-2 inhibitor venetoclax: a product of its microenvironment? Front Oncol. 2018;8:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jayappa KD, Portell CA, Gordon VL, et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL [published correction appears in Blood Adv. 2017;1(19):1537]. Blood Adv. 2017;1(14):933-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oppermann S, Ylanko J, Shi Y, et al. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood. 2016;128(7):934-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thijssen R, Slinger E, Weller K, et al. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica. 2015;100(8):e302-e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiron D, Dousset C, Brosseau C, et al. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015;6(11):8750-8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095-2103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.