

SUMMARY
Most loci identified by GWAS have been found in populations of European ancestry (EUR). In trans-ethnic meta-analyses for 15 hematological traits in 746,667 participants, including 184,535 non-EUR individuals, we identified 5,552 trait-variant associations at P<5×10−9, including 71 novel loci not found in EUR populations. We also identified 28 additional novel variants in ancestry-specific, non-EUR meta-analyses, including an IL7 missense variant in South Asians associated with lymphocyte count in vivo and IL7 secretion levels in vitro. Fine-mapping prioritized variants annotated as functional, and generated 95% credible sets that were 30% smaller when using the trans-ethnic as opposed to the EUR-only results. We explored the clinical significance and predictive value of trans-ethnic variants in multiple populations, and compared genetic architecture and the impact of natural selection on these blood phenotypes between populations. Altogether, our results for hematological traits highlight the value of a more global representation of populations in genetic studies.
In Brief
Delineation of the genetic architecture of hematological traits in a multi-ethnic cohort allows for the identification of rare variants with strong effect specific to non-European populations and improved fine mapping of GWAS variants using the trans-ethnic approach.
Graphical Abstract
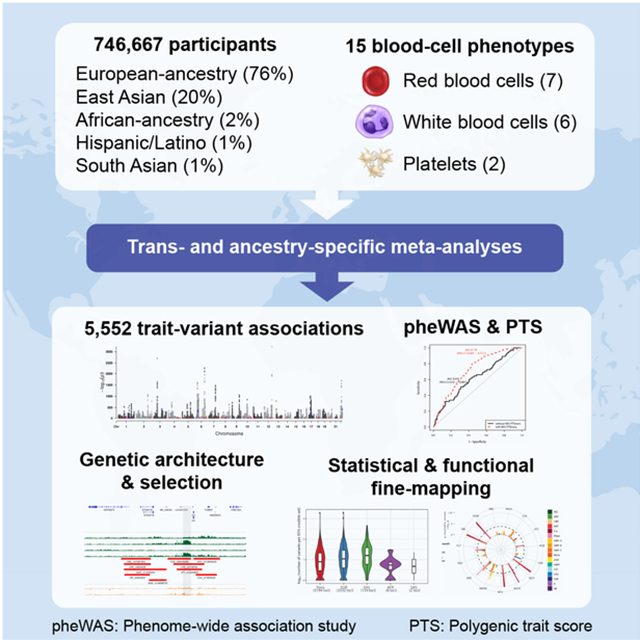
INTRODUCTION
Blood-cell counts and indices are quantitative clinical laboratory measures that reflect hematopoietic progenitor cell production, hemoglobin synthesis, maturation and release from the bone marrow, and clearance of mature or senescent blood cells from the circulation. Quantitative red blood cell (RBC), white blood cell (WBC) and platelet (PLT) traits exhibit strong heritability (h2~30–80%) (Evans et al., 1999; Hinckley et al., 2013) and have been the subject of various genome-wide association studies (GWAS), including a large study that identified >1000 genomic loci in ~150,000 individuals of European-ancestry (EUR) (Astle et al., 2016).
Importantly, the distribution of hematologic traits and prevalence of inherited hematologic conditions differs by ethnicity. For example, the prevalence of anemia and microcytosis is higher among African-ancestry (AFR) individuals compared to EUR individuals in part due to the presence of globin gene mutations (e.g. sickle cell, α/β-thalassemia) more common among African, Mediterranean and Asian populations (Beutler and West, 2005; Raffield et al., 2018; Rana et al., 1993). AFR individuals tend to have lower WBC and neutrophil counts partly because of the Duffy/DARC null variant (Rappoport et al., 2019). Among Hispanics/Latinos (HA), a common Native American functional intronic variant of ACTN1 is associated with lower PLT count (Schick et al., 2016).
Despite these observations, non-EUR populations have been severely under-represented in most blood-cell genetic studies to date (Popejoy and Fullerton, 2016; Popejoy et al., 2018; Wojcik et al., 2019). Multiethnic GWAS have been recognized as more powerful for gene mapping due to ancestry-specific differences in allele frequency, linkage disequilibrium (LD), and effect size of causal variants (Li and Keating, 2014). Since blood cells play a key role in pathogen invasion, defense and inflammatory responses, hematologic-associated genetic loci are particularly predisposed to be differentiated across ancestral populations as a result of population history and local evolutionary selective pressures (Ding et al., 2013; Lo et al., 2011; Raj et al., 2013). Given the essential role of blood cells in tissue oxygen delivery, inflammatory responses, atherosclerosis, and thrombosis (Byrnes and Wolberg, 2017; Chu et al., 2010; Colin et al., 2014; Tajuddin et al., 2016), factors that contribute to such interpopulation differences in blood-cell traits may also play appreciable roles in the pathogenesis of chronic diseases and health disparities between populations.
RESULTS
Trans-ethnic and ancestry-specific blood-cell traits genetic associations
We analyzed genotype-phenotype associations at up to 45 million autosomal variants in 746,667 participants, including 184,424 individuals of non-EUR descent, for 15 traits (Figure 1, Figure S1, Tables S1A–D and S2, and Methods). The association results of the EUR-specific meta-analyses are reported separately in a companion paper (Vuckovic et al., 2020). In the trans-ethnic meta-analyses, we identified 5,552 trait-variant associations at P<5×10−9, including 71 novel associations not reported in the EUR-specific manuscript (Table S3A). Of the 5,552 trans-ethnic loci, 128 showed strong evidence of allelic effect heterogeneity across populations (Pancestry.hetero <5×10−9) (Table S3A). Ancestry-specific meta-analyses revealed 28 additional novel trait-variant associations (Figure 1b and Table S3B–F). However, 21 of these 28 novel loci were identified in AFR-ancestry participants, and 19 of these 21 novel AFR-specific associations map to chromosome 1 and are associated with WBC or neutrophil counts, therefore reflecting long-range associations due to the admixture signal at the Duffy/DARC locus that confers resistance to Plasmodium vivax infections (Reich et al., 2009). We attempted to replicate all novel trans-ethnic or ancestry-specific genetic associations in the Million Veteran Program (MVP) cohort (Gaziano et al., 2016). Of the 88 variant-trait associations that we could test in MVP, 85 had a consistent direction of effect (binomial P=6×10−24), 83 were confirmed with a false discovery rate <5%, and 44 met the Bonferroni-adjusted significance threshold of P<6×10−4 (Table S3G).
Figure 1.

Trans-ethnic and ancestry-specific meta-analyses of blood-cell traits. (a) List of blood-cell phenotypes and analyses that were carried out in this project. Note that RDW and MPV were not available in EAS. (b) Study design of the project. We used a fixed-effect meta-analysis strategy to analyze genetic associations within each of the five populations available, and a mega-regression approach that considers allele frequency heterogeneity for the trans-ethnic association tests. Nmax, maximum sample size in each meta-analysis; Nassoc, number of trait-variant associations. A locus is defined as novel when the 500-kb region surrounding its sentinel variant does not physically overlap with previously identified blood-cell trait-associated variants (for any trait) in the corresponding population. (c) Most blood-cell trait-associated loci physically overlap between populations. For this analysis, a locus associated with several blood-cell traits was counted only once. Despite different sample sizes between populations, we note that few loci are found in a single population, suggesting shared genetic architecture. EUR, European-ancestry; EAS, East Asian; AFR, African-ancestry; HA, Hispanic American; SAS, South Asian. See also Figure S1 and Tables S1A–D, S2 and S3A–F.
For 3,552 loci in which conditional analyses identified a single genome-wide significant variant in EUR, we generated fine-mapping results for each trans-ethnic or ancestry-specific dataset using an approximate Bayesian approach (Methods) (Wellcome Trust Case Control et al., 2012). The 95% credible sets were smaller in the trans-ethnic meta-analyses than in the EUR or EAS meta-analyses (Figure 2a). When comparing loci discovered in both the trans-ethnic and EUR analyses, we found that the 95% credible sets were 30% smaller among the trans-ethnic results (median (interquartile range) number of variants per 95% credible set was 4 (2–13) in trans vs. 5 (2–16) in EUR, Wilcoxon’s P=3×10−4). For instance, a locus on chromosome 9 associated with PLT count included seven variants in the EUR 95% credible set but only one in the trans-ethnic set, an increase in fine-mapping resolution likely driven by limited LD at the locus in EAS (Figure 2b). In the trans-ethnic and EUR results, respectively, we identified 433 and 403 loci with a single variant in the 95% credible sets (Figure 2c), and >300 variants with a posterior inclusion probability (PIP) ≥0.99 (Figure 2d). To determine the reason for the improved resolution in the trans-ethnic results, we sub-sampled the data and re-ran the EUR-only (N=141,636), EAS-only (N=143,085), and trans-ethnic (N=137,702) meta-analyses on similarly sized sample sets for PLT, RBC, WBC and HGB. The resulting 95% credible sets were still smaller in the trans-ethnic meta-analyses, suggesting that the improved resolution was due to LD structure rather than an increase in sample size (Figure S2).
Figure 2.

Fine-mapping of genome-wide significant loci associated with hematological traits. (a) We restricted fine-mapping to loci with evidence for a single association signal in European-ancestry (EUR) populations. There are no such loci in Hispanic Americans. The 95% credible sets in the trans-ethnic meta-analyses are smaller than in the EUR or East-Asian-ancestry (EAS) meta-analyses. (b) Trans-ethnic fine-mapping of a platelet locus. In EUR individuals, the 95% credible set include seven variants with posterior inclusion probability (PIP) >0.04 and strong pairwise linkage disequilibrium (LD) with the sentinel variant rs10758481 (r2>0.93 in British in England and Scotland (GBR) individuals from 1000 Genomes Project, middle panel). LD is similarly strong in African-, Hispanic/South American-, and South-Asian-ancestry populations from the 1000 Genomes Project. However, LD is weaker in East Asians (r2=0.68 in Japanese individuals (JPT) from the 1000 Genomes Project, bottom panel). In the trans-ethnic meta-analysis, rs10758481 has a PIP>0.99 (top panel). In EUR and EAS, LD is color-coded based on pairwise r2 with rs10758481. The dotted line indicates the genome-wide significance threshold (P<5×10−9). ( c) Proportion of 95% credible sets in each population with a defined number of variants. For instance, in the EUR and trans meta-analysis results, we identified 403 and 433 95% credible sets that contain a single variant, respectively. (d) Prioritization of causal variants using fine-mapping PIP. In each population, we provide the proportion of variants with a PIP within a specified range. For instance, in EUR and trans, we found 314 and 327 variants with a PIP ≥99%, respectively. See also Figure S2.
Next, we assessed our fine-mapped 95% credible sets for the presence of functional variants, which we defined as variants with coding consequences or those mapping to accessible chromatin in hematopoietic cells. Genomic annotation of the 95% credible sets of the trans-ethnic, EUR and EAS hematological trait-associated loci revealed that the proportion of likely functional variants was higher among those with high PIP (Figure 3a). The enrichment within high-PIP categories was particularly notable for missense variants, but also observed for intronic and intergenic variants that map to open chromatin regions in progenitor or mature blood cells (Figure 3a)(Corces et al., 2016). We used g-chromVAR to quantify the enrichment of trans, EUR and EAS 95% credible set variants within regions of accessible chromatin identified by the assay for transpose accessible chromatin by sequencing (ATAC-seq) in 18 hematopoietic populations (Ulirsch et al., 2019). We noted 22 significant trait-cell type enrichments using the trans-ethnic credible sets, all of which were lineage specific, including RBC traits in erythroid progenitors, platelet traits in megakaryocytes, and monocyte count in granulocyte-macrophage progenitors (GMP) (Figure 3b and Table S3H). Cell-type enrichments were largely consistent between fine-mapped variants found in the trans, EUR and EAS loci. However, we observed two noteworthy ancestry-specific differences: the EAS results revealed significant enrichments (defined as Bonferroni-corrected threshold P<1.9×10−4) in basophil count for common myeloid progenitors (CMP)(PEAS-BASO-CMP=7.6×10−5) and eosinophil count for GMP (PEAS-EOS-GMP=4.5×10−6), but neither pairing reached significance in the larger EUR meta-analyses (PEUR-BASO-CMP=0.08 and PEUR-EOS-GMP=0.01)(Figure S3). These differences persisted even after controlling for the number of loci tested in each ancestry.
Figure 3.

Functional annotation of possible causal variants associated with blood-cell traits. (a) Annotation of variants in trans, EUR and EAS shows a similar pattern, with a larger proportion of likely functional variants (e.g. missense, intergenic and intronic variants within ATAC-seq peaks) among variants with higher posterior inclusion probability (PIP). (b) g-chromVAR results for trans variants within 95% credible sets for 15 traits. The Bonferroni-adjusted significance level (corrected for 15 traits and 18 cell types) is indicated by the dotted line. Mono, monocyte; HSC, hematopoietic stem cell; Ery, erythroid; Mega, megakaryocyte; CD4, CD4+ T lymphocyte; CD8, CD8+ T lymphocyte; B, B lymphocyte; NK, natural killer cell; mDC, Myeloid dendritic cell; pDC, Plasmacytoid dendritic cell; MPP, multipotent progenitor; LMPP, lymphoid-primed multipotent progenitor; CMP, common myeloid progenitor; CLP, common lymphoid progenitor; GMP, granulocyte–macrophage progenitor; MEP, megakaryocyte–erythroid progenitor. (c) rs115906455 is a novel variant associated with mean corpuscular volume in the trans-ethnic meta-analysis (P=4.2×10−12, PIP=0.57). It maps to an intron of ELL2 and overlaps with ATAC-seq peaks found in CMP, MEP, erythroblasts but not megakaryocytes. (d) rs941616 is a novel variant associated with eosinophil counts in the trans-ethnic meta-analysis (P=2.4×10−9, PIP=0.2). It is a strong eQTL for PTGDR located 112-kb downstream and overlaps with ATAC-seq peaks found in CMP, CD8+ lymphocytes and NK cells. See also Figures S3–4 and Table S3H–I.
Among the novel loci identified in the trans-ethnic meta-analyses, several included excellent candidate causal variants with high fine-mapping PIP and overlap with open chromatin regions found in hematopoietic cells (Figure S4 and Table S3I). For instance, rs115906455, located in an intron of the RNA polymerase II elongation factor ELL2, is strongly associated with MCV (P=4.2×10−12, PIP=0.57) and maps to an accessible chromatin region found in RBC progenitors (CMP, megakaryocyte–erythroid progenitor and erythroblasts) but not megakaryocytes (Figure 3c). This variant is common in AFR populations (minor allele frequency (MAF)=4.7%) but rare or monomorphic in non-AFR populations. A different variant at the ELL2 locus has previously been associated with multiple myeloma and IgG levels (Swaminathan et al., 2015). Another example is rs941616, a common variant in an intergenic region on chromosome 14 that is associated with eosinophil count (P=2.4×10−9, PIP=0.2) and maps to a region of chromatin accessibility in CMP, CD8+ lymphocytes and natural killer cells (Figure 3d). This variant, which is in LD with another eosinophil-associated variant recently identified (Kichaev et al., 2019), is an eQTL for PTGDR (Võsa et al., 2018), which encodes prostaglandin D2 receptor. Prostaglandins can activate eosinophils, which in turn contribute to the etiology of asthma, chronic obstructive pulmonary disease (COPD), and allergies (Brusselle et al., 2016). In the UK Biobank (UKBB), rs941616 is associated with allergic rhinitis (P=5×10−4) but not asthma (P=0.077) (Canela-Xandri et al., 2018).
Phenome-wide association studies (pheWAS)
We queried the 5,552 trans-ethnic genome-wide significant variants associated with blood-cell traits in three ancestrally distinct biobanks including 408,961 EUR individuals from the UKBB with 1,403 disease states, 143,988 individuals of Japanese descent from BioBank Japan (BBJ) with 22 disease states, and 5,275 African Americans from the Vanderbilt University Biobank (BioVU) with 1,403 disease states (Methods). We found 366 variant-disease associations in the UKBB (Table S4A). Of these 366 associations, the BBJ had matching phenotypes for 95, 26 of which were replicated. Only one of these 366 associations was replicated in BioVU. In only three cases did we observe a variant-disease association in UKBB that failed to replicate when BBJ was well-powered (power >80%) and the matching phenotype was available. We found 133 variant-disease associations in BBJ (Table S4A). Of these 133 associations, the UKBB had matching phenotypes for 90, 55 of which were replicated in UKBB and one of which was replicated in BioVU. Almost all of the non-replicated associations were well-powered to replicate in UKBB, suggesting heterogeneity across populations in genetic effects, in clinical definitions of disease states, or in disease prevalence and relevant environmental exposures. Only three of the non-replicated associations were well-powered to replicate in BioVU. Finally, in BioVU we observed 19 variant-disease associations (Table S4A), 18 of which were located at the β-globin locus that reflect the known clinical sequelae of sickle cell disease. Unsurprisingly, these were not replicated in UKBB and BBJ because the variant is monomorphic.
Many of the variant-disease associations we observed were located at well-known highly pleiotropic loci, with signal in multiple biobanks. For instance, rs1260326 in GCKR was associated with diabetes, dyslipidemia, alcohol consumption, gout, and urolithiasis. Multiple variants in TERT were associated with pre-cancerous conditions such as seborrheic keratosis, uterine leiomyoma, and myeloproliferative disease. Unsurprisingly, the MHC region harbored multiple variants associated with a variety of immune-related diseases such as celiac disease, psoriasis, asthma, rheumatoid arthritis, Graves’ disease and type-1 diabetes. Variants in and near ABO were associated with cardiovascular disease phenotypes, as well as gastric cancer, hemorrhoids, and diverticulosis. And variants in and near APOE were associated with cardiovascular diseases and neurological disorders including dementia.
We found two regions with widespread pleiotropy that were specific to a particular ancestry (in addition to the β-globin locus in AFR). Variants in and near SH2B3 were associated with celiac disease, myocardial infarction, hypertension, and hypothyroidism in UKBB. None of these associations were replicated in BBJ, due to these variants having very low MAFs (~0.3%) in EAS. About 2-Mb away from SH2B3, rs11066008 in ACAD10 was associated with angina, myocardial infarction, arrythmia, and colorectal cancer in BBJ. None of these associations were replicated in UKBB, due to very low MAFs in EUR and AFR (0 and 0.08%, respectively). A well-known selective sweep in this region approximately 1200–1700 years ago in European populations may explain why these loci display such large, ancestry-specific effects (Zhernakova et al., 2010).
Trans-ethnic predictions of hematological traits
Polygenic trait scores (PTS) developed in a single ethnically homogeneous population tend to underperform when tested in a different population (Grinde et al., 2019; Marquez-Luna et al., 2017; Martin et al., 2019). We explored whether we could combine the genome-wide significant trans-ethnic variants identified in our analyses into PTS that can predict blood-cell traits in a multi-ethnic setting. First, we used trans-ethnic effect sizes as weights to compute PTStrans for each trait, and tested their performance in independent EUR, AFR and HA participants from the BioMe Biobank (Methods). As expected because our trans-ethnic meta-analyses are dominated by EUR individuals, PTStrans were more predictive in EUR, although their performance in HA was comparable for several traits (lymphocyte and monocyte count, mean PLT volume) (Figure 4a and Table S4B). For neutrophil and WBC counts, the variance explained by the PTStrans was up to three times higher in AFR and HA than in EUR samples due to the inclusion of the strong Duffy/DARC locus (Figure 4a and Table S4B). Because these Duffy/DARC variants would not have been included in PTS derived uniquely from EUR association results, this illustrates an interesting feature of using trans-ethnic variants for building polygenic predictors. Consistent with previous reports for other human diseases, PTStrans improved the precision to predict hematological disorders defined using blood-cell clinical thresholds (Figures 4b–c and Table S4C).
Figure 4.

Phenotypic variance and hematological disease prediction using polygenic trait scores (PTS) in independent participants from the BioMe Biobank. (a) For each blood-cell trait, PTStrans were calculated using genome-wide significant variants identified in the trans-ethnic meta-analyses. Trait-increasing alleles were weighted using effect sizes derived from fixed-effect trans-ethnic meta-analyses. (b) Receiver operating characteristic (ROC) curve and area under the curve (AUC and 95% confidence interval) for neutropenia (defined as <1500 NEU/mL) in BioMe participants of African-ancestry without (black) or with (red) the PTStrans for neutrophil count in the predictive model. Age, sex, and the first 10 principal components were used in the basic prediction model. (c) As for b, but for thrombocytopenia (defined as <150×109 PLT/L) and the PTStrans for platelet count in Hispanic participants from BioMe. See also Figure S5 and Table S4B–C.
Next, we asked if we could increase the variance explained by calculating PTS using the same trans-ethnic variants but weighting these variants using ancestry-specific as opposed to trans-ethnic effect sizes. In contrast to our expectations that a PTS calculated using ancestry-specific weights would be more accurate, we found for most traits that PTStrans outperformed ancestry-specific PTSAFR and PTSHA in BioMe AFR and HA participants, respectively (Figure S5 and Table S4B). This result likely indicates that the discovery sample size for these two populations is still too small to provide robust estimates of the true population-specific effect sizes and that additional ancestry-specific variants have yet to be identified.
Rare coding blood-cell-traits-associated variants
The identification of rare coding variants has successfully pinpointed candidate genes for many complex traits, including blood-cell phenotypes (Auer et al., 2014; Chami et al., 2016; Eicher et al., 2016; Justice et al., 2019; Marouli et al., 2017; Mousas et al., 2017; Tajuddin et al., 2016). Our trans-ethnic and non-EUR ancestry-specific meta-analyses yielded 16 coding variants with MAF <1% (Table S5A–B). This list includes variants of clinical significance (variants in TUBB1, GFI1B, HBB, MPL, and SH2B3) and variants that nominate candidate genes within GWAS loci (ABCA7, GMPR). Our analyses also retrieved a known missense variant in EGLN1 (rs186996510) that is associated with high-altitude adaptation and hemoglobin levels in Tibetans (Lorenzo et al., 2014; Xiang et al., 2013).
We noted a missense variant in IL7 (rs201412253, Val18Ile) associated with increased lymphocyte count in South Asians (SAS)(P=4.4×10−10) (Figure 5a and Table S5C). This variant is low-frequency in SAS (MAF=2.6%) but rare in other populations (MAF <0.4%). This association was replicated in 4,554 British-Pakistani and 10,638 British-Bangladeshi participants from the Genes & Health Study (combined P=5.7×10−5)(Table S5C). IL7 encodes interleukin-7, a cytokine essential for B- and T-cell lymphopoiesis (Lin et al., 2017). In large eQTL datasets such as eQTLgen and GTEx, rs201412253 is monomorphic. However, we found four heterozygote individuals among 75 Gujarati Indians that had genotypes and transcriptomic data from lymphoid cell lines (Stranger et al., 2012): in this limited dataset, rs201412253 was not associated with IL7 expression levels (Figure 5b). IL7 is synthesized as a proprotein that is cleaved prior to secretion, and the IL7-Val18Ile variant localizes to the IL7 signal peptide comprising the first 25 amino acids. To determine if this variant alters IL7 secretion, we engineered HEK293 cells with either IL7 allele (Methods). Although there was no difference in IL7 RNA expression levels (t-test P=0.63), we found that the IL7–18Ile allele, which associates with higher lymphocyte counts in SAS individuals, significantly increased IL7 protein secretion in this heterologous cellular system (+83%, P=2.7×10−5) (Figure 5c).
Figure 5.

A South-Asian-ancestry IL7 missense variant associates with increased lymphocyte count in humans and IL7 secretion in vitro. ( a) Lymphocyte count association results at the IL7 locus in South Asians (SAS), European-ancestry participants (EUR) and East Asians (EAS). In SAS, there are seven genome-wide significant variants near IL7, but only rs201412253 is coding. Linkage disequilibrium (LD) r2 is from 1000 Genomes Project SAS populations. In EUR, the sentinel variant is located downstream of IL7; rs201412253 is rare (minor allele frequency=4×10−4) and not significant (P=0.073). In EAS, the locus is not associated with lymphocyte count. rs201412253 is monomorphic in 1000 Genomes Project EUR and EAS so we could not calculate pairwise LD. (b) Association between genotypes at rs201412253 and normalized IL7 expression levels in lymphoid cell lines from 75 Gujarati Indians from HapMap3. The T-allele frequency is 2.7% and the association is not significant (P=0.62). (c) The 18Ile allele at IL7-rs201412253 increases IL7 secretion in a heterologous cellular system. Our ELISA assay did not detect secreted IL7 in clones generated with an empty vector. We tested eight independent clones for each IL7 alleles. Each experiment was done in duplicate, and we performed the experiments three times. The black dots and vertical lines indicate means and standard deviations. We assess statistical significance by linear regression correcting for experimental batch effects. See also Table S5A–C.
Genetic architecture of blood-cell traits in EUR and EAS populations
The genetic architecture of a trait is defined by the number, the frequency and the effect size of all variants that contribute to phenotypic variation (Hansen, 2006). We used several different approaches to quantify similarities and differences in genetic architecture of hematologic traits across populations. Focusing on the two largest studied populations, EUR and EAS, we calculated heritability for all blood traits and found them to be concordant between ancestries (Pearson’s r=0.75, P=0.0033) (Figure S6 and Table S6A) (Bulik-Sullivan et al., 2015b). Likewise, within-ancestry genetic correlation coefficients (rg) between pairs of hematological traits were highly concordant across ancestries (Pearson’s r=0.97, P<2.2×10−16)(Figure S6)(Bulik-Sullivan et al., 2015a). We then used the Popcorn method to measure genetic correlations for blood-cell traits between EUR and EAS using summary statistics for common variants (Brown et al., 2016). For all 13 traits available in both EUR and EAS, genetic correlations were high (lowest for basophils (rg=0.30) and highest for MCH (rg=0.66)), but significantly different than 1 (P<3×10−6) (Figure S6 and Table S6B). This suggests that although the effect sizes of common variants are correlated between EUR and EAS, there are significant differences between these two populations.
To further contrast the genetic architecture of blood-cell traits between these two populations, we compared effect sizes for 1423 genome-wide significant variants with PIP >0.5 in either EUR or EAS (Figure 6 and Table S6C). Effect sizes were correlated (Pearson’s r=0.46 for variants with PIP >0.5 in EUR and r=0.70 for variants with PIP >0.5 in EAS, P<2.2×10−16 for both) (Figure 6), which indicated largely concordant effect sizes across populations, a result consistent with the Popcorn analyses. But we also noted many interesting differences. We found 70 variants with PIP >0.5 that are common (MAF >5%) and have similar MAF (±5%) in EUR and EAS, but have at least a two-fold difference in effect sizes (Table S6C). For instance, rs34651 is strongly associated with PLT in EUR (P=1.1×10−30, PIP>0.99, effect size=- 0.0428) but the association signal is weaker in EAS (P=2.5×10−7, effect size= −0.0336) despite the fact that the variant is more common in EAS (MAFEUR=8.1%, MAFEAS=12.9%) (Table S23). This variant maps to a region of accessible chromatin in most progenitor and mature hematopoietic cells and is a strong eQTL for FCHO2 (Figure S4)(Võsa et al., 2018). Some variants were also significant in both EUR and EAS even if they had different effect sizes and MAF. This category includes rs77046277, which is strongly associated with LYM in EUR and EAS despite being rare in EUR (MAFEUR=0.1%, MAFEAS=1.2%)(Figure 6). This variant is located near S1PR1 and maps to regions of accessible chromatin found in T lymphocytes (Figure S4). rs78744187 is another example: it is common in EUR and less frequent in EAS (MAFEUR=8.2%, MAFEAS=1.8%), but strongly associated with basophil count in both populations (Figure 6); this variant is an eQTL for CEBPA (Võsa et al., 2018) and is located within an accessible chromatin region in CMPs and prior studies using genome editing of this regulatory element in primary hematopoietic progenitors have validated its role in regulating CEBPA expression to enable basophil production (Figure S4) (Guo et al., 2017). Finally, there were also variants that were ancestry-specific because they were very rare in the other population: this included the known missense variants in SH2B3 (rs78894077) associated with LYM in EAS and in HFE (rs1800562) associated with RBC traits in EUR (Figure 6).
Figure 6.

Comparisons of effect sizes for variants with posterior inclusion probabilities (PIP) >0.5. We retained only variants with an analyzed sample size ≥70,000 in East Asians (EAS) and ≥100,000 in European-ancestry participants (EUR). (a) We retrieved minor allele frequencies (MAF), effect sizes (Beta), P-values (P) and PIP for all variants with PIP >0.5 in EUR. By definition, all these variants are significant in EUR (P<5×10−9). For these variants, we then retrieved the corresponding results in EAS. Effect sizes (standard errors (SE)) in EUR and EAS are plotted on the x- and y-axis, respectively. (b) as in a, but for variants with PIP >0.5 in EAS. In a and b, when we provide detailed information on a specific variant, the first number always corresponds to EUR and the second to EAS (e.g. for rs77046277, BetaEUR=0.712 and BetaEAS=0.348). See also Figure S4 and Table S6C.
Natural selection at blood-cell trait loci
Natural selection can account for differences in association results between populations, as highlighted by our analyses of rare coding variants which includes several loci known to be under selection (CD36, b-globin, EGLN1) ( Table S5A). To further explore this possibility, we assessed whether variants that tag selective sweeps (tagSweeps, variants with the highest integrated haplotype score (iHS)) within continental populations from the 1000 Genomes Project (1000G) are associated with blood-cell phenotypes (Johnson and Voight, 2018). We found a genome-wide enrichment of association results between tagSweeps and hematological traits, particularly within EUR, EAS and AFR populations (Figure S7 and Table S7A). To rule out simple overlaps due to the large number of sweeps and blood-cell trait loci, we compared the number of genome-wide significant tagSweeps in EUR, EAS and AFR with the number of significant variants among 100 sets of matched variants (Methods). We found significant enrichment of selective sweeps for WBC (EUR, EAS, AFR), monocytes (EUR, AFR), eosinophils (EUR), neutrophils (AFR), lymphocytes (EAS), and PLT (EUR, EAS)(Table S7B).
In AFR and HA, the enrichments for WBC, neutrophils and monocytes were entirely driven by selective sweeps on chromosome 1 near Duffy/DARC (Reich et al., 2009). Only three additional loci shared evidence of associations with blood-cell traits and positive selection across populations: HLA, SH2B3 (Zhernakova et al., 2010) and CYP3A5 (Chen et al., 2009). We found eight and 100 non-overlapping selective sweeps with variants associated with hematological traits in EAS and EUR, respectively (Table S7C). Six of the eight EAS-specific tagSweeps are also associated with blood-cell traits in EUR participants, indicating that these regions do not account for population differences in hematological trait regulation (Table S7C). One of the remaining two variants is located at the HBS1L-MYB locus and, although it is not associated with blood-cell traits in EUR, there are many other variants near MYB associated with blood phenotypes in EUR (Table S3B). The remaining selective sweep highlighted by this analysis is located upstream of IL6 (Figure 7). The tagSweep at this locus, rs2188580, is strongly associated with PLT count in EAS (PEAS=2.8×10−9, PEUR=0.0022), is differentiated between EAS and EUR as indicated by the population branch statistic (PBS)(Yi et al., 2010)(C-allele frequency in EAS=44%, 4% in EUR; standardized PBSEAS=7.353), and overlaps selective sweeps identified in several EAS populations from the 1000G(e.g. iHSCHS=3.935) (Figure 7). The IL6 locus has previously been associated with WBC traits in EUR (Astle et al., 2016), but our finding is the first report of its association with PLT. IL6 encodes interleukin-6, a cytokine that is a maturation factor for megakaryocytes (Kimura et al., 1990). Further supporting the role of IL6 signaling in PLT biology, a well-characterized missense variant in the IL6 receptor gene (IL6R-rs2228145) (van Dongen et al., 2014) is also nominally associated with PLT count in EAS(P=4.3×10−6).
Figure 7.

Selective sweep and association with platelet count at the IL6 locus in East Asians. The grey rectangle highlights a genomic region upstream of IL6 that is strongly associated with platelet (PLT) count. This association signal is driven by results from East Asians (EAS), and is absent from other populations, including European- (EUR) and African-ancestry (AFR) individuals (green). The region overlaps several selective sweeps detected in EAS from the 1000 Genomes Project (Chinese Dai in Xishuangbanna (CDX), Southern Han Chinese (CHS), Japanese in Tokyo (JPT)). In orange, we provide standardized population branch site (stdPBS) metrics in EUR and EAS, indicative of allele frequency differentiation at this locus between these two populations. Coordinates are chr7:22–23.5Mb (hg19). See also Figure S7 and Table S7A–C.
DISCUSSION
Our meta-analyses of 15 hematological traits in up to 746,667 individuals represents one of the largest genetic studies of clinically relevant complex human traits across diverse ancestral groups. We have continued to expand the repertoire of loci and genes that contribute to interindividual variation in blood-cell traits, with potential implications for hematological diseases, as well as other conditions such as cancer, immune and cardiovascular diseases.
Differences in clinical definitions, phenotype measurements, gene-gene and gene-environment interactions could account for some of the differences in genetic effects observed between populations. In our analyses of hematological traits in EUR and EAS, we have identified extensive genetic overlaps, but also significant differences in effect sizes between these two populations. Our estimates of trans-ancestry genetic correlations for blood-cell traits are similar to estimates for other complex human phenotypes such as type-2 diabetes, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis (Brown et al., 2016; Liu et al., 2015), although higher genetic correlations have also been reported (Lam et al., 2019; Martin et al., 2019). Despite the shared genetic architecture, we found evidence of heterogeneity at hematological trait-associated variants with high PIP (Figure 6). Similarly, although the genetic correlation for Crohn’s disease between EUR and EAS is high (rg=0.76), heterogeneity was noted at causal variants in NOD2, IL23R, and TNFSF15 due to differences in allele frequency, effect size, or both (Liu et al., 2015). This is in sharp contrast with the recent report that the genetic correlation between EUR and EAS for schizophrenia is near unity (rg=0.98) and that there is no evidence of locus-level heterogeneity (Lam et al., 2019). These observations, largely limited to EUR-EAS comparisons for a handful of phenotypes, already suggest that different complex human diseases and traits have different genetic architecture. These results also highlight a need for large genetic analyses in other populations, and for the development of methodologies amenable to admixture for genetic correlation analyses.
Our results have implications for future human genetic studies. First, we showed that adding even a “modest” number of non-EUR participants to GWAS can yield important biology, such as the identification of a LYM count-associated IL7 missense variants in 8,189 South Asians (Figure 5). Second, loci that underlie variation in blood-cell traits represent a broad mixture of shared associations (i.e. similar allele frequencies and effect sizes across populations) and heterogeneous associations (i.e. dissimilar allele frequencies and effect sizes across populations). This result contributes to mounting evidence that a full accounting of the genetic basis of complex human traits will require a thorough catalog of global genetic and phenotypic variation. Third, because of heterogeneity across populations in both allele frequencies and patterns of LD, fine-mapping of association signals can be substantially aided by including multiple ancestries. This will have a dramatic impact on the success of large-scale efforts aimed at functionally characterizing GWAS findings, but also to develop polygenic predictors that transfer to multiple ancestries. As more studies seek to unravel the causal variants that underlie complex traits associations, we anticipate that genetic evidence from diverse ancestries will play an important role.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Guillaume Lettre (guillaume.lettre@umontreal.ca).
Material Availability
The reagents generated in this study are available without restriction.
Data and Code Availability
The genetic association results (summary statistics), functional annotations, and fine-mapping results are available at: http://www.mhi-humangenetics.org/en/resources.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study design and participants
Cell lines
All participants provided written informed consent and the project was approved by each institution’s ethical committee. Table S1B lists all participating cohorts. The SNPs we identified are available from the NCBI dbSNP database of short genetic variations (https://www.ncbi.nlm.nih.gov/projects/SNP/). No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Flip-In™-293 cells (ThermoFisher Scientific) were grown at 80% confluency in DMEM medium supplemented with 10% Foetal Bovine Serum, 4 mM L-glutamine, 100 IU penicillin, 100 μg/ml streptomycin and 100 μg/ml hygromycin. 293 cells were likely originally derived from a female donor.
METHODS DETAILS
Phenotypes
Complete blood count (CBC) and related blood indices were analyzed as quantitative traits. The descriptive statistics for each phenotype in each cohort analyzed are in Table S1C. Exclusion criteria and phenotype modeling in UKBB (European-ancestry individuals), INTERVAL, and BBJ have been described previously (Astle et al., 2016; Kanai et al., 2018). For all other studies, we followed the protocol developed by the Blood-Cell Consortium (Chami et al., 2016; Eicher et al., 2016; Tajuddin et al., 2016). Briefly, we excluded when possible participants with blood cancer, acute medical/surgical illness, myelodysplastic syndrome, bone marrow transplant, congenital/hereditary anemia, HIV, end-stage kidney disease, splenectomy, and cirrhosis, as well as pregnant women and those undergoing chemotherapy or erythropoietin treatment. We also excluded extreme blood-cell measures: WBC>200×109 cells/L, HGB>20 g/dL, HCT>60%, and PLT>1000×109 cells/L. For WBC subtypes, we analyzed log10-transformed absolute counts obtained by multiplying relative counts with total WBC count. For all phenotypes in all studies, we corrected the blood-cell phenotypes for sex, age, age-squared, the 10 first genetic principal components, and other cohort-specific covariates (e.g. recruitment center) using linear regression analysis. We applied rank-based inverse normal transformation to the residuals form the regression analysis and used the normalized residuals to test for association with genetic variants.
Genotype quality-control and imputation
The genotyping array and quality-control steps used by each cohort as well as their quality-control steps are listed in Table S1D. Unless otherwise specified, all studies applied the following criteria: samples were removed if the genotyping call rate was <95%, if they showed excess heterozygosity, if we identified gender mismatches or sample duplicates, or if they appeared as population outliers in principal component analyses nested with continental populations from the 1000 Genomes Project (Genomes Project et al., 2012). We removed monomorphic variants, as well as variants with Hardy-Weinberg P<1×10−6 and call rate <98%.
Genotype imputation for the UKBB, INTERVAL, and BBJ have been described in details elsewhere (Astle et al., 2016; Bycroft et al., 2018; Kanai et al., 2018). For all other studies, unless specified in Table S1D, we applied the following steps for genotype imputation of autosomal variants. We aligned all alleles on the forward strand of build 37/hg19 of the human reference genome (http://www.well.ox.ac.uk/~wrayner/strand) and converted files into the VCF format. We then applied check VCF (http://genome.sph.umich.edu/wiki/CheckVCF.py) to confirm strand and allele orientation. We carried out genotype imputation using the University of Michigan (https://imputationserver.sph.umich.edu) orthe Sanger Institute (https://imputation.sanger.ac.uk/) imputation servers. We phased genotype data using SHAPEIT (Delaneau et al., 2013), EAGLE (Loh et al., 2016), or HAPI-UR (Williams et al., 2012). For populations of European ancestry, we used reference haplotypes from the Haplotype Reference Consortium (HRC r1.1 2016) for imputation (McCarthy et al., 2016) unless otherwise noted, whereas reference haplotypes from the 1000 Genomes Project (Phase 3, Version 5) (Genomes Project et al., 2012) were used for non-European ancestry participants.
Study-level statistical analyses
We tested an additive genetic model of association between genotype imputation doses and inverse normal transformed blood-cell phenotypes. We analyzed the major ancestry groups (European (EUR), East Asian (EAS), African (AFR), Hispanic-Latino (HA), South Asian (SAS)) separately and used linear mixed-effect models implemented in BOLT-LMM (Loh et al., 2018), EPACTS (https://genome.sph.umich.edu/wiki/EPACTS), or EMMAX (Kang et al., 2010) to account for cryptic and known relatedness. Autosomal single nucleotide variants were analyzed in all contributing studies. For simplicity, we only analyzed insertion-deletion (indel) variants from UKBB and INTERVAL, since a similar reference panel was used for genotype imputation.
Centralized quality-control and meta-analyses
We performed a centralized quality-control check on the association results of each single study using EasyQC (v9.0)(Winkler et al., 2014). By mapping variants of each study to the appropriate ethnicity reference panel (HRC for EUR and 1000 Genomes Project Phase3 for non-EUR participants), we were able to harmonize alleles and markers across all studies. We were also able to assess the presence of flipped alleles per study and check for excessive allele frequency discrepancies using allele frequency reference data. We also inspected quantile-quantile (QQ) plots generated by EasyQC and the corresponding genomic inflation factors as well as SE-N plots (inverse of the median standard error vs. the square root of the sample size) to evaluate potential issues with, for example, trait transformation or unaccounted relatedness. We removed variants with imputation quality metric (INFO score) ≤0.4. Except for three studies, we also removed variants with minor allele count (MAC) ≤5. For UKBB EUR, Women Health Initiative (WHI), and GERA (EUR), we instead applied a MAC ≤20 filter because empirical observations suggested that unusual inflation of the test statistics (i.e. extreme effect sizes and standard errors) was due to rarer variants. To simplify handling of tri-allelic and indel variants, which have the same genomic coordinates but different alleles, we created a unique variant ID for each tested variant. Specifically, we assigned a chromosome:position(hg19)_allele1_allele2 unique ID to each variant, in which the order of the allele in the ID was based on the lexicographical order or the indel length. We performed inverse variance-weighted fixed-effect meta-analyses with GWAMA (v2.2.2)(Magi and Morris, 2010) and trans-ethnic meta-analyses with MR-MEGA (v0.1.5)(Magi et al., 2017). For MR-MEGA, we calculated four axes of genetic variation, the default recommendation, to separate global population groups.
Million Veteran Program (MVP) blood-cell trait analyses for replication
Phenotyping. Phenotyping methods published by the EMERGE Consortium and available on PheKB (https://phekb.org/) were used for retrieving lab data and exclusion criteria for all blood cell indices. This information was pulled from the VA electronic medical records for all MVP participants. Lab data was subject to the Boston Lab Adjudication Protocol. This entails five steps: (i) compile an initial spreadsheet of possible relevant lab tests, (ii) Subject Matter Expert (SME) does an initial review of possible tests, (iii) analyst adds relevant LOINC codes for SME review, (iv) second Subject Matter Expert (SME) review, (v) creation of a Lab Phenotype Table/Data Set. After restricting to only outpatient labs and applying the EMERGE exclusion criteria, for each trait and each person, the minimum, maximum, mean, median, SD, and number of labs was recorded. Values were compared to those from UKBB (Astle et al., 2016).
Genotyping
DNA extracted from whole blood was genotyped using a customized Affymetrix Axiom biobank array, the MVP 1.0 Genotyping Array. With 723,305 total DNA sequence variants, the array is enriched for both common and rare variants of clinical importance in different ethnic backgrounds (Klarin et al., 2018).
Analysis
The median lab value was the trait used for analysis. Linear regression models were run under an additive model in plink2 on 1000G (v3p5) imputed dosages. Analyses were run using models described above within each race/ethnicity stratum (AFR, ASN, EUR, HA) classified based on their genotype data using HARE (Fang et al., 2019). Meta-analyses for the trans-ethnic analyses were completed in METAL (Willer et al., 2010).
Heritabilities and genetic correlations
We calculated heritabilities and genetic correlations between blood-cell traits within the EUR and EAS populations using default parameters implemented in the LD score regression method (Figure S6 and Table S6A) (Bulik-Sullivan et al., 2015a; Bulik-Sullivan et al., 2015b). For genetic correlation of the same phenotype between ancestral populations, we used Popcorn (Brown et al., 2016). Briefly, Popcorn uses a Bayesian framework to estimate, using genome-wide summary statistics, the genetic correlation of the same phenotype but in two different populations (in our case, between EUR and EAS). It reports the trans-ethnic genetic-effect correlation (ρge), i.e. the correlation coefficient of per-allele SNP effect sizes, but also the trans-ethnic genetic impact correlation (ρgi), which includes a normalization of the effect based on allele frequency (Table S6B). To address whether a difference in the sample size for the EUR and EAS meta-analyses could impact the Popcorn results, we repeated our analyses using the current EAS results (Nmax=151,807) and EUR results from preliminary analyses of the UKBB dataset (Nmax=87,265) (Astle et al., 2016). These analyses confirmed that for common variants, cross-ancestry EUR-EAS genetic correlations are significantly different (but non-null). Both LD score regression and Popcorn are not amenable to admixed populations, and cannot handle rare variants. For these reasons, we limited these analyses to the large EUR and EAS populations and focused on common variants (MAF ≥5%) from the 1000 Genomes Project.
Functional annotation
To derive basic functional annotation information, we annotated all variants included in 95% credible sets from ancestry-specific and trans-ethnic meta-analyses with the Variant Effect Predictor (VEP)(https://useast.ensembl.org/info/docs/tools/vep/index.html), compiling both all consequences and the most severe consequence for Ensembl/GENCODE transcripts. We also specifically annotated rare coding variants using VEP (defined as any variant with MAF <1% in a given analysis, with a GC-corrected P-value <5×10−9, and annotated as a missense_variant, stop_gained, stop_lost, splice_donor, or a splice_acceptor, regardless of fine-mapping results). We removed all variants with a GC-corrected P-value <5×10−9 in EUR, in the MHC region, and, in analyses including individuals with at least some African ancestry, on chromosome 1 for neutrophils and total WBC count and for RBC traits near the chromosome 11 β-globin and the chromosome 16 α-globin loci.
Bias-corrected enrichment of blood trait variants for chromatin accessibility of 18 hematopoietic populations was performed using g-chromVAR, which has been previously described in detail (Ulirsch et al., 2019). In brief, this method weights chromatin features by fine-mapped variant posterior probabilities and computes the enrichment for each cell type versus an empirical background matched for GC content and feature intensity. For chromatin feature input, we used a consensus peak set for all hematopoietic cell types with a uniform width of 500 bp centered at the summit. For variant input, we included all fine-mapped variants within 95% credible sets of the trans-ethnic GWAS. We also ran g-chromVAR for each ancestry-specific meta-analysis, keeping all other parameters the same, but using fine-mapped variants with the 95% credible sets of each ancestry-specific study. Finally, to control for the number of loci tested within each ancestry-specific study, we first ranked the loci of the largest cohort (i.e. EUR) by sentinel variant p-value, and then subset only the top n loci, where n equals the number of loci in the smaller cohort (e.g. EAS) for the same trait. We then ran g-chromVAR on the subset of variants falling within these top n loci.
Phenome-wide association study (pheWAS) analysis
UK Biobank (UKBB)
We extracted pheWAS results for a list of 5552 variants in UKBB ICD PheWeb hosted at the University of Michigan (Accessed 21 August 2019). To account for severe imbalance in case-control ratios, we selected the output from the SAIGE analyses (http://pheweb.sph.umich.edu/SAIGE-UKB/) based on 408,961 samples from White British participants (Zhou et al., 2018). In total, 1403 phecodes were tested for association. All results were downloaded using R, and were parsed and organized into data table format using the data.table, rvest, stringr, dplyr and tidyr packages.
BioBank Japan (BBJ)
We performed a pheWAS for the lead variants identified by the trans-ethnic meta-analyses. From the list of all the significantly associated variants with blood cell-related traits, we extracted those genotyped or imputed in the BBJ project (nSNP = 4,255). Next, we curated the phenotype record of the disease status and clinical values for the same individuals analyzed in the discovery phase (nindiv = 143,988). Then, we performed the logistic regression analyses for 22 binary traits (20 diseases and 2 behavioral habits) which had a sufficient number of case samples (ncase = 2,500). Regression models were adjusted for age, sex and 20 principal components as covariates. Trait-specific covariates are described elsewhere (Kanai et al., 2018).
BioVU
BioVU is the biobank of Vanderbilt University Medical Center (VUMC) that houses de-identified DNA samples linked to phenotypic data derived from electronic health records (EHRs) system of VUMC. The clinical information is updated every 1–3 months for the de-identified EHRs. Detailed description of program operations, ethical considerations, and continuing oversight and patient engagement have been published (Roden et al., 2008). DNA samples were genotyped with genome-wide arrays including the Multi-Ethnic Global (MEGA) array, and the genotype data were imputed into the HRC reference panel (McCarthy et al., 2016) using the Michigan imputation server (Das et al., 2016). Imputed data and the 1000 Genome Project data were combined to carry out principal component analysis (PCA) and African-American samples were extracted for analysis based on the PCA plot. PheWAS were carried out for each SNP with the specified allele (Denny et al., 2010). Phenotypes were derived from billing codes of EHRs as described previously (Carroll et al., 2014). Each phenotype (‘phecode’) has defined case, control and exclusion criteria. We required two codes on different visit days to instantiate a case for each phecode. In total, 1815 phecodes were tested for association. Association between each binary phecode and a SNP was assessed using logistic regression, while adjusting for covariates of age, sex, genotyping array type/batch and 10 principal components of ancestry.
Merging across biobanks
We defined statistical significance within each biobank to be a Bonferroni corrected level of 0.05/pq, where p is the number of phecodes tested and q is the number of variants tested. We considered an association to be replicated if the p-value for the association was < 0.05/s with a consistent direction of effect, where s represents the number of associations being replicated. To match phenotypes across biobanks, we merged the UKBB and BioVU by phecode, as these two biobanks used the same phecode system for classifying outcomes. To match with BBJ, we cross-referenced the 22 outcomes in BBJ with the phecode library used by BioVU/UKBB. Matches were determined based on phenotype similarity between the BioVU/UKBB phenotype description and the outcomes described in Nagai et al. (Nagai et al., 2017).
Power analysis
For all variant-disease associations that failed to replicate, we performed power analyses in the replication biobank to determine if the lack of replication was likely due to lack of signal or lack of statistical power. We assumed that the replication biobank would have the same prevalence and odds-ratio as the biobank in which the association was discovered, and we used allele frequencies from the matching population in the 1000 Genomes project. To guard against winner’s curse in our power analyses, we assumed a maximum odds-ratio of 3. Power was assessed at a P-value threshold of 0.05/s, where s represents the number of associations being tested for replication.
Polygenic trait score (PTS) analyses
We restricted these analyses to variant-trait associations that reached genome-wide significance (P<5×10−9) in the trans-ethnic MR-MEGA meta-analyses (Table S3A). For each of these variant-trait pairs, we calculated an effect size – hereafter referred to as trans weights – using the fixed-effect meta-analysis method implemented in GWAMA and all cohorts available (Magi and Morris, 2010). For the same variants, we also retrieved the ancestry-specific effect sizes (or weights). We calculated the PTS using plink2 by summing up the number of trait-increasing alleles (or imputation doses) that were weighted by their corresponding trans (PTStrans) or ancestry-specific (PTSEUR, PTSAFR, PTSHA) weights. The variance explained by the PTS on corrected and normalized blood-cell traits was calculated in R using linear regression. For these analyses, we had access to 2,651 AFR, 5,048 EUR and 4,281 HA BioMe participants that were not used in the discovery effort. For the analyses of hematological diseases, we used the same independent BioMe participants and implemented logistic regression models in R. We used age, sex, and the first four principal components as covariates in all models. We used the PredictABEL package in R to calculate precision metrics. We used the following thresholds to define disease state: anemia (women <12 g/dL, men <13 g/dL), neutropenia (<1500 NEU/μL), thrombocytosis (>450×109 PLT/L), and thrombocytopenia (<150×109 PLT/L).
Analysis of natural selection
To quantify the contribution of positive selection on blood-cell trait variation, we used the recent map of selective sweeps identified in the different populations of the 1000 Genomes Project (Johnson and Voight, 2018). We grouped the sweeps identified in the 26 1000 Genomes Project populations into five larger populations that correspond to our ancestry-specific meta-analyses: Europe-ancestry (CEU, TSI, GBR, FIN, IBS); East-Asian-ancestry (CHB, JPT, CHS, CDX, KHV); African-ancestry (YRI, LWK, GWD, MSL, ESN, ASW, ACB); South-Asian-ancestry (GIH, PJL, BEB, STU, ITU); and Hispanic/Latino-ancestry (MXL, PUR, CLM, PEL). Following the nomenclature by Johnson and Voight(Johnson and Voight, 2018), each selective sweep is summarized by the variant located within the sweep that has the highest iHS value. iHS (Integrated Haplotype Score) is a statistic to quantify evidence of recent positive selection. A high positive iHS score (iHS > 2) means that haplotypes on the ancestral allele background are longer compared to derived allele background. A high negative iHS score (iHS < −2) means that the haplotypes on the derived allele background are longer compared to the haplotypes associated with the ancestral allele.
We retrieved the blood-cell trait association results for these sweep-tagging SNPs from the ancestry-specific meta-analyses (Table S7A). To determine if the inflation observed in the QQ plots was significant, we generated 100 sets of SNPs that match the selective sweep-tagging SNPs based on allele frequency, gene proximity, and the number of LD proxies in European-ancestry, East-Asian-ancestry and African-ancestry individuals using SNPsnap (Pers et al., 2015). For these analyses, we excluded the HLA region and variants in LD (r2>0.5). We computed empirical significance by tallying the number of sets with the same or more genome-wide significant variants than the canonical sets of selective sweep-tagging SNPs (Table S7B).
We also computed the population branch statistic (PBS) using whole-genome sequencing information from the 1000 Genomes Project (Yi et al., 2010). PBS measures the amount of allele frequency change in the population since its divergence from the other two populations. For a target population, PBS is calculated as:
where T = −log=1 - FST) is an estimate of the divergence time between two populations. Here, FST between each pair of populations was estimated using Weir and Cockerham’s estimate (Weir and Cockerham, 1984). We then divided all variants with calculated PBS into 50 bins of equal size by derived allele count in the target population, and then standardized the raw PBS values within each bin. To calculate PBS for Europe-ancestry (CEU, TSI, GBR, and IBS, without FIN), we used YRI as an outgroup and East-Asian-ancestry (CHB, JPT, CHS, CDX, KHV) as a sister population; for East-Asian-ancestry, we used YRI as an outgroup and Europe-ancestry as a sister population; for YRI, we used East-Asian-ancestry as an outgroup and Europe-ancestry as a sister population.
Replication of the association between IL7-rs201412253 and lymphocyte count in Genes & Health
Genes & Health is a population cohort study of British-Bangladeshi and British-Pakistani adult volunteers recruited from London and Bradford UK (www.genesandhealth.org)(Finer et al., 2020). Participant saliva DNA samples (Oragene, DNA Genotek) were genotyped on the Illumina GSAMD-24v3–0-EA genotyping chip. Several rounds of data filtering and quality control were undertaken in Genome Studio using cluster separation scores (<0.57), Gentrain score (≤0.7) and with increasingly stringent per-variant call rate threshold across remaining samples, and per-sample call rate threshold across remaining variants. Final dataset had call rate of >0.992 per female-, and >0.995 per male-sample across all 637,829 variants (which included Y chromosome). PLINK gender calls were compared to self-stated questionnaire gender information and where discordant, samples were removed from analyses. For individuals that had taken part on multiple occasions the sample with highest call rate was retained, whilst all samples were removed for an individual if duplicate samples were not concordant. Where exome data was available, sample genotypes were compared across platforms and highly discordant samples removed for further work.
Genome-wide imputation using the genotype chip data was carried out on the Michigan Imputation Server using reference panel Genome Asia Pilot (GAsP). This panel performed better than other available reference panels in the south Asian samples. Variants with minimac4 imputation Rsq <0.3 were removed, as were variants with MAF <0.1%.
Genotyped volunteer samples with Barts Health NHS Trust hospital clinical pathology laboratory full blood count data - to obtain lymphocyte count data – were selected. This included tests ordered on hospital patients, and also from primary care GP surgeries using the hospital laboratory. We split the data into Pakistani and Bangladeshi populations based on those samples with complete DNA genotype principal component and questionnaire ethnicity agreement (N=5,912 Pakistani Individuals, N=13,611 Bangladeshi Individuals). Intersex individuals, and related individuals (one from each pair of samples with piHat>0.1875) were removed to leave 4,554 Pakistani and 10,638 Bangladeshi samples.
Absolute lymphocyte counts (×109 cells/L) were extracted from Barts Health NHS Trust pathology data warehouse. The median count, and age at test for that measurement, were taken when multiple measurements were available on an individual. Log10 transformation of cell counts was undertaken in RStudio(v1.1.453), before correcting for median age at test, median age at test squared and gender using linear regression analysis on each population separately. Residuals from the regression analyses were extracted and rank-based inverse normalisation was performed. These normalised residuals were used as the phenotype in association analysis which was undertaken in PLINK2.0 (--glm) using bgen files from Imputation and only default settings. Pakistani and Bangladeshi populations were analysed separately.
IL7 functional analyses
We PCR amplified and cloned the IL7 wildtype (rs201412253-Val18) and mutant (rs201412253–18Ile) open reading frame (ORF) in the pcDNA5/FRT vector (ThermoFisher Scientific) using HindIII and BamHI restriction sites (see Table S5D for ORF and primer sequences). We validated the sequences of the two plasmids by Sanger Sequencing. FlipIn™-293 cells (ThermoFisher Scientific) at 80% confluency were transfected with 1:10 mixes of empty pcDNA5 or pcDNA5 derivatives coding for IL7-Val8 or IL7–18Ile and pOG44 FLP recombinase coding vector (ThermoFisher Scientific) using polyethylenimine. Transfectant clones were expanded and selected in DMEM medium supplemented with 10% Foetal Bovine Serum, 4 mM L-glutamine, 100 IU penicillin, 100 μg/ml streptomycin and 100 μg/ml hygromycin. We measured the secretion of IL7 in eight independent clones for each IL7 allele (rs201412253-Val18 and rs201412253–18Ile) as well as in four clones generated with the empty vector by ELISA assay. We used the High Sensitivity Quantikine HS ELISA kit from R & D Systems (Cat # HS750). We seeded 100,000 cells per 12-wells plates and grew them for 6 days in DMEM glutamax plus 10% FBS before doing the ELISA. We measured each supernatant in duplicate and seeded each of the clones in triplicate. The whole experiment was done on three different weeks (three complete biological replicates). We extracted total proteins from cells with RIPA buffer and we quantified the lysates by BCA. We used this quantification to normalize the ELISA assays. We extracted total RNA from ~500,000 cells using the Qiagen RNEasy kit (cat # 74136). We checked the quality of the RNA by Bioanalyzer and quantified its concentration by Nanodrop. We reverse transcribed 1 ug of total RNA into cDNA using the ABI kit (Life Technologies Cat # 4368814). We used two pairs of primers for IL7 and assays for three normalizing genes (HPRT, GAPDH,TBP, Table S5D). We followed the MIQE recommendations and performed the qPCR reactions with the Sybergreen Platinum (Life Technologies Cat # 11733–046) on a Biorad CFX384 thermocycler.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical significance, genomic inflation and locus definition
For each meta-analysis, we calculated the genomic inflation factor (λGC) for all variants, which were modest when considering the large sample sizes (λGC range: 0.9–1.2) (Table S2). We used α ≤5×10−9 after GC-correction to declare statistical significance, accounting for the inflation of the test statistics and the number of blood-cell traits analyzed. To count the number of loci that we discovered, we first identified the most significant variants (with P≤5×10−9) and extended the physical region around that variant 250-kb on each side. Overlapping loci were merged, and we used the most significant variant within the interval as the sentinel variant. In this manuscript, we defined as novel a locus if no variants were previously reported in the literature to be associated with the specific blood-cell trait and if the locus is not reported in the companion manuscript that focuses on EUR-specific genetic discoveries.
Conditional analyses in the UK Biobank European-ancestry population to identify independent variants associated with blood-cell traits
This method is described in details in the companion manuscript (Vuckovic et al., 2020). Briefly, we applied the following four steps: (1) Initialisation step: From the list of all variants in the block, add the variant with the lowest P-value that is also below the significance threshold (8.31×10−9). (2) Dropping: Study the P-values for all variants in the model, if any of these are above the significance threshold we iteratively prune and rebuild model starting with the variant with the highest P-value. Once a variant is pruned it is returned to the list of variants not currently in the parsimonious model and may rejoin at a later iteration. (3) Addition: Test each variant not currently in the block sequentially in the model, add the variant with the lowest P-value which is below the threshold. Any tested variants which have a P-value of higher than 0.01 are not tested again in future iterations. Variants are not permitted to be tested in the model if they have a LD r2 >0.9 with any variant currently in the model. (4) Completion: If the algorithm could neither add a variant into the model nor remove a variant from the model then we abort the iteration with the model at this stage representing the parsimonious model for this block. Following identification of conditionally significant variants in each block, all conditionally significant variants within each chromosome are put into a single linear model and tested with the same multiple stepwise linear regression algorithm as that defined above. The resultant set is the ‘conditionally significant’ list of variants for the blood cell index. Full results from these conditional analyses are described in the companion European focused manuscript. We will note that this conditional analysis model for selecting loci for fine-mapping would not allow for the detection of non-European ancestry specific secondary signals, with these direct conditional analyses only feasible at most loci in a very large single cohort like the UK Biobank.
Statistical fine-mapping
No fine-mapping methods currently exist to handle admixed populations. Furthermore, for some of the ethnic groups analyzed here, we did not have access to a sufficiently large reference panel to properly account for LD, complicating conditional analyses and fine-mapping efforts. For these reasons, we fine-mapped the ancestry-specific fixed-effect meta-analyses by adapting the method proposed by Maller et al. (Wellcome Trust Case Control et al., 2012) in order to assign posterior probability of inclusion (PIP) to each variant and construct 95% credible sets.
This method makes the strong assumption that there is a single independent causal variant at the tested locus. For this reason, we limited our Bayesian fine-mapping to loci where we identified a single independent association signal by conditional analysis in EUR individuals from the UKBB (Vuckovic et al., 2020). Because EUR represented the largest group, we then inferred that there was also a single association signal in the other populations at these loci, an inference that may not always be right. Briefly, we added 250-kb on either side of genome-wide significant variants (P<5×10−9) and merged loci when they overlapped. For the loci identified in the ancestry-specific meta-analyses, we converted P-values into approximate Bayes factors (aBF) using (Wakefield, 2009; Wellcome Trust Case Control et al., 2012):
where β and SE are the variant’s effect size and standard error, respectively, and ω denotes the prior variance in allelic effects, taken here to be 0.04 (Wakefield, 2007). For the trans-ethnic results, we directly used Bayes factors calculated by MR-MEGA (Magi et al., 2017). We calculated PIP of each variant by dividing the variant’s aBF by the sum of the aBF for all the variants within the locus. We generated the 95% credible sets by ordering all variants in a given locus from the largest to the smallest PIP and by including variants until the cumulative sum of the PIP ≥95% (Mahajan et al., 2018). All variants that map to 95% credible sets are available online (http://www.mhi-humangenetics.org/en/resources).
Supplementary Material
Highlights.
Blood-cell traits differ by ancestry and are subject to selective pressures.
We assessed 15 blood-cell traits in 746,667 participants from 5 global populations.
We identified >5,500 associations, including ~100 variants not found in Europeans.
These analyses improved risk prediction and identified potential causal variants.
ACKNOWLEDGMENTS
We thank all participants, as well as Dr. John D. Rioux for providing the IL7 ORF. A full list of acknowledgments appears in the Table S1F. Part of this work was conducted using the UK Biobank resource (Projects number 11707 and 13745).
Footnotes
DECLARATION OF INTERESTS
Competing financial interests are declared in the Table S1F.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. (2016). The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167, 1415–1429 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer PL, Teumer A, Schick U, O'Shaughnessy A, Lo KS, Chami N, Carlson C, de Denus S, Dube MP, Haessler J, et al. (2014). Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler E, and West C (2005). Hematologic differences between African-Americans and whites: the roles of iron deficiency and alpha-thalassemia on hemoglobin levels and mean corpuscular volume. Blood 106, 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BC, Asian Genetic Epidemiology Network Type 2 Diabetes, C, Ye CJ, Price AL, and Zaitlen N (2016). Transethnic Genetic-Correlation Estimates from Summary Statistics. Am J Hum Genet 99, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusselle GG, Provoost S, and Maes T (2016). Prostaglandin D2 receptor antagonism: a novel therapeutic option for eosinophilic asthma? Lancet Respir Med 4, 676–677. [DOI] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control, C, Duncan L., et al. (2015a). An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics, C., Patterson N, Daly MJ, Price AL, and Neale BM (2015b). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes JR, and Wolberg AS (2017). Red blood cells in thrombosis. Blood 130, 1795–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela-Xandri O, Rawlik K, and Tenesa A (2018). An atlas of genetic associations in UK Biobank. Nat Genet 50, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RJ, Bastarache L, and Denny JC (2014). R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chami N, Chen MH, Slater AJ, Eicher JD, Evangelou E, Tajuddin SM, Love-Gregory L, Kacprowski T, Schick UM, Nomura A, et al. (2016). Exome Genotyping Identifies Pleiotropic Variants Associated with Red Blood Cell Traits. Am J Hum Genet 99, 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang H, Zhou G, Zhang X, Dong X, Zhi L, Jin L, and He F (2009). Molecular population genetics of human CYP3A locus: signatures of positive selection and implications for evolutionary environmental medicine. Environ Health Perspect 117, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SG, Becker RC, Berger PB, Bhatt DL, Eikelboom JW, Konkle B, Mohler ER, Reilly MP, and Berger JS (2010). Mean platelet volume as a predictor of cardiovascular risk: a systematic review and meta-analysis. J Thromb Haemost 8, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin Y, Le Van Kim C, and El Nemer W (2014). Red cell adhesion in human diseases. Current opinion in hematology 21, 186–192. [DOI] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, and Marchini J (2013). Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 10, 5–6. [DOI] [PubMed] [Google Scholar]
- Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, and Crawford DC (2010). PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K, de Andrade M, Manolio TA, Crawford DC, Rasmussen-Torvik LJ, Ritchie MD, Denny JC, Masys DR, Jouni H, Pachecho JA, et al. (2013). Genetic variants that confer resistance to malaria are associated with red blood cell traits in African-Americans: an electronic medical record-based genome-wide association study. G3 (Bethesda) 3, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher JD, Chami N, Kacprowski T, Nomura A, Chen MH, Yanek LR, Tajuddin SM, Schick UM, Slater AJ, Pankratz N, et al. (2016). Platelet-Related Variants Identified by Exomechip Meta-analysis in 157,293 Individuals. Am J Hum Genet 99, 40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DM, Frazer IH, and Martin NG (1999). Genetic and environmental causes of variation in basal levels of blood cells. Twin Res 2, 250–257. [DOI] [PubMed] [Google Scholar]
- Fang H, Hui Q, Lynch J, Honerlaw J, Assimes TL, Huang J, Vujkovic M, Damrauer SM, Pyarajan S, Gaziano JM, et al. (2019). Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity in Genome-wide Association Studies. Am J Hum Genet 105, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finer S, Martin HC, Khan A, Hunt KA, MacLaughlin B, Ahmed Z, Ashcroft R, Durham C, MacArthur DG, McCarthy MI, et al. (2020). Cohort Profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int J Epidemiol 49, 20–21i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, et al. (2016). Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–223. [DOI] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, and McVean GA (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinde KE, Qi Q, Thornton TA, Liu S, Shadyab AH, Chan KHK, Reiner AP, and Sofer T (2019). Generalizing polygenic risk scores from Europeans to Hispanics/Latinos. Genetic epidemiology 43, 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo MH, Nandakumar SK, Ulirsch JC, Zekavat SM, Buenrostro JD, Natarajan P, Salem RM, Chiarle R, Mitt M, Kals M, et al. (2017). Comprehensive population-based genome sequencing provides insight into hematopoietic regulatory mechanisms. Proc Natl Acad Sci U S A 114, E327–E336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TF (2006). The Evolution of Genetic Architecture. Annu Rev Ecol Evol Syst 37, 123–157. [Google Scholar]
- Hinckley JD, Abbott D, Burns TL, Heiman M, Shapiro AD, Wang K, and Di Paola J (2013). Quantitative trait locus linkage analysis in a large Amish pedigree identifies novel candidate loci for erythrocyte traits. Mol Genet Genomic Med 1, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, and Voight BF (2018). Patterns of shared signatures of recent positive selection across human populations. Nat Ecol Evol 2, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice AE, Karaderi T, Highland HM, Young KL, Graff M, Lu Y, Turcot V, Auer PL, Fine RS, Guo X, et al. (2019). Protein-coding variants implicate novel genes related to lipid homeostasis contributing to body-fat distribution. Nat Genet 51, 452–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, et al. (2018). Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet 50, 390–400. [DOI] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, and Eskin E (2010). Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, and Price AL (2019). Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Hum Genet 104, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Ishibashi T, Uchida T, Maruyama Y, Friese P, and Burstein SA (1990). Interleukin 6 is a differentiation factor for human megakaryocytes in vitro. European journal of immunology 20, 1927–1931. [DOI] [PubMed] [Google Scholar]
- Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, et al. (2018). Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 50, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M, Chen CY, Li Z, Martin AR, Bryois J, Ma X, Gaspar H, Ikeda M, Benyamin B, Brown BC, et al. (2019). Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet 51, 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, and Keating BJ (2014). Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med 6, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zhu Z, Xiao H, Wakefield MR, Ding VA, Bai Q, and Fang Y (2017). The role of IL-7 in Immunity and Cancer. Anticancer Res 37, 963–967. [DOI] [PubMed] [Google Scholar]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KS, Wilson JG, Lange LA, Folsom AR, Galarneau G, Ganesh SK, Grant SF, Keating BJ, McCarroll SA, Mohler ER 3rd, et al. (2011). Genetic association analysis highlights new loci that modulate hematological trait variation in Caucasians and African Americans. Hum Genet 129, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Kichaev G, Gazal S, Schoech AP, and Price AL (2018). Mixed-model association for biobank-scale datasets. Nat Genet 50, 906–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Palamara PF, and Price AL (2016). Fast and accurate long-range phasing in a UK Biobank cohort. Nat Genet 48, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo FR, Huff C, Myllymaki M, Olenchock B, Swierczek S, Tashi T, Gordeuk V, Wuren T, Ri-Li G, McClain DA, et al. (2014). A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet 46, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magi R, Horikoshi M, Sofer T, Mahajan A, Kitajima H, Franceschini N, McCarthy MI, Cogent-Kidney Consortium TDGC, and Morris AP (2017). Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet 26, 3639–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magi R, and Morris AP (2010). GWAMA: software for genome-wide association meta-analysis. BMC bioinformatics 11, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, Fine RS, Lu Y, Schurmann C, Highland HM, et al. (2017). Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez-Luna C, Loh PR, South Asian Type 2 Diabetes, C., Consortium STD, and Price AL (2017). Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol 41, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, and Daly MJ (2019). Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousas A, Ntritsos G, Chen MH, Song C, Huffman JE, Tzoulaki I, Elliott P, Psaty BM, Blood-Cell C, Auer PL, et al. (2017). Rare coding variants pinpoint genes that control human hematological traits. PLoS Genet 13, e1006925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, Kiyohara Y, Ninomiya T, Tamakoshi A, Yamagata Z, Mushiroda T, et al. (2017). Overview of the BioBank Japan Project: Study design and profile. J Epidemiol 27, S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers TH, Timshel P, and Hirschhorn JN (2015). SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popejoy AB, and Fullerton SM (2016). Genomics is failing on diversity. Nature 538, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popejoy AB, Ritter DI, Crooks K, Currey E, Fullerton SM, Hindorff LA, Koenig B, Ramos EM, Sorokin EP, Wand H, et al. (2018). The clinical imperative for inclusivity: Race, ethnicity, and ancestry (REA) in genomics. Hum Mutat 39, 1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffield LM, Ulirsch JC, Naik RP, Lessard S, Handsaker RE, Jain D, Kang HM, Pankratz N, Auer PL, Bao EL, et al. (2018). Common alpha-globin variants modify hematologic and other clinical phenotypes in sickle cell trait and disease. PLoS Genet 14, e1007293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj T, Kuchroo M, Replogle JM, Raychaudhuri S, Stranger BE, and De Jager PL (2013). Common risk alleles for inflammatory diseases are targets of recent positive selection. Am J Hum Genet 92, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana SR, Sekhsaria S, and Castro OL (1993). Hemoglobin S and C traits: contributing causes for decreased mean hematocrit in African-American children. Pediatrics 91, 800–802. [PubMed] [Google Scholar]
- Rappoport N, Simon AJ, Amariglio N, and Rechavi G (2019). The Duffy antigen receptor for chemokines, ACKR1,- 'Jeanne DARC' of benign neutropenia. Br J Haematol 184, 497–507. [DOI] [PubMed] [Google Scholar]
- Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, Patterson N, Mullikin J, Hsueh WC, Cheng CY, Coresh J, et al. (2009). Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS genetics 5, e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, and Masys DR (2008). Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick UM, Jain D, Hodonsky CJ, Morrison JV, Davis JP, Brown L, Sofer T, Conomos MP, Schurmann C, McHugh CP, et al. (2016). Genome-wide Association Study of Platelet Count Identifies Ancestry-Specific Loci in Hispanic/Latino Americans. Am J Hum Genet 98, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. (2012). Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8, e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan B, Thorleifsson G, Joud M, Ali M, Johnsson E, Ajore R, Sulem P, Halvarsson BM, Eyjolfsson G, Haraldsdottir V, et al. (2015). Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat Commun 6, 7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajuddin SM, Schick UM, Eicher JD, Chami N, Giri A, Brody JA, Hill WD, Kacprowski T, Li J, Lyytikainen LP, et al. (2016). Large-Scale Exome-wide Association Analysis Identifies Loci for White Blood Cell Traits and Pleiotropy with Immune-Mediated Diseases. Am J Hum Genet 99, 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, et al. (2019). Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet 51, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen J, Jansen R, Smit D, Hottenga JJ, Mbarek H, Willemsen G, Kluft C, Collaborators A, Penninx BW, Ferreira MA, et al. (2014). The contribution of the functional IL6R polymorphism rs2228145, eQTLs and other genome-wide SNPs to the heritability of plasma sIL-6R levels. Behav Genet 44, 368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S, et al. (2018). Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv, 447367. [Google Scholar]
- Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, Chen M-H, Raffield LM, Tardaguila M, Huffman JE, et al. (2020). The Polygenic and Monogenic Basis of Blood Traits and Diseases. medRxiv, 2020.2002.2002.20020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J (2007). A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet 81, 208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J (2009). Bayes factors for genome-wide association studies: comparison with P-values. Genet Epidemiol 33, 79–86. [DOI] [PubMed] [Google Scholar]
- Weir BS, and Cockerham CC (1984). Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control, C., Maller JB, McVean G, Byrnes J, Vukcevic D, Palin K, Su Z, Howson JM, Auton A, Myers S et al. (2012). Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet 44, 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, and Abecasis GR (2010). METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AL, Patterson N, Glessner J, Hakonarson H, and Reich D (2012). Phasing of many thousands of genotyped samples. Am J Hum Genet 91, 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Magi R, Ferreira T, Fall T, Graff M, Justice AE, et al. (2014). Quality control and conduct of genome-wide association meta-analyses. Nature protocols 9, 1192–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, Sorokin EP, Avery CL, et al. (2019). Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Ouzhuluobu Peng Y, Yang Z, Zhang X, Cui C, Zhang H, Li M, Zhang Y, Bianba et al. (2013). Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation. Mol Biol Evol 30, 1889–1898. [DOI] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, et al. (2010). Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhernakova A, Elbers CC, Ferwerda B, Romanos J, Trynka G, Dubois PC, de Kovel CG, Franke L, Oosting M, Barisani D, et al. (2010). Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am J Hum Genet 86, 970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, LeFaive J, VandeHaar P, Gagliano SA, Gifford A, et al. (2018). Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genetic association results (summary statistics), functional annotations, and fine-mapping results are available at: http://www.mhi-humangenetics.org/en/resources.