

Visual Abstract

Keywords: hepatic cyst, somatostatin analog, polycystic kidney disease, liver disease
Abstract
Background and objectives
We assessed safety and efficacy of another somatostatin receptor analog, pasireotide long-acting release, in severe polycystic liver disease and autosomal dominant polycystic kidney disease. Pasireotide long-acting release, with its broader binding profile and higher affinity to known somatostatin receptors, has potential for greater efficacy.
Design, setting, participants, & measurements
Individuals with severe polycystic liver disease were assigned in a 2:1 ratio in a 1-year, double-blind, randomized trial to receive pasireotide long-acting release or placebo. Primary outcome was change in total liver volume; secondary outcomes were change in total kidney volume, eGFR, and quality of life.
Results
Of 48 subjects randomized, 41 completed total liver volume measurements (n=29 pasireotide long-acting release and n=12 placebo). From baseline, there were −99±189 ml/m absolute and −3%±7% change in annualized change in height-adjusted total liver volume (from 2582±1381 to 2479±1317 ml/m) in the pasireotide long-acting release group compared with 136±117 ml/m absolute and 6%±7% increase (from 2387±759 to 2533±770 ml/m) in placebo (P<0.001 for both). Total kidney volumes decreased by −12±34 ml/m and −1%±4% in pasireotide long-acting release compared with 21±21 ml/m and 4%±5% increase in the placebo group (P=0.05 for both). Changes in eGFR were similar between groups. Among the n=48 randomized, adverse events included hyperglycemia (26 of 33 [79%] in pasireotide long-acting release versus four of 15 [27%] in the placebo group; P<0.001), and among the 47 without diabetes at baseline, 19 of 32 (59%) in the pasireotide long-acting release group versus one of 15 (7%) in the placebo group developed diabetes (P=0.001).
Conclusions
Another somatostatin analog, pasireotide long-acting release, slowed progressive increase in both total liver volume/total kidney volume growth rates without affecting GFR decline. Participants experienced higher frequency of adverse events (hyperglycemia and diabetes).
Clinical Trial registry name and registration number
Pasireotide LAR in Severe Polycystic Liver Disease, NCT01670110
Podcast
This article contains a podcast at https://www.asn-online.org/media/podcast/CJASN/2020_08_28_CJN13661119.mp3
Introduction
Liver cysts are the most common extrarenal manifestation of autosomal dominant polycystic kidney disease (ADPKD) and are also seen in autosomal dominant polycystic liver disease (ADPLD) (1,2). As polycystic liver disease (PLD) progresses, there is expansion of liver cysts and liver parenchyma. The enlarged liver compresses adjacent organs (heart, kidneys, spleen, diaphragm, gastrointestinal tract, and vasculature) and may lead to infectious complications (cholelithiasis, cyst infection, and cholangitis), which is debilitating in a subgroup (especially women) (3–7). Treatment interventions include cyst aspiration; fenestration or sclerotherapy; liver resection; octreotide; and, in severe cases, liver or combined liver-kidney transplantation (8–10).
Multiple mechanisms underlie growth of hepatic and kidney cysts controlled by intracellular cAMP, which is markedly increased in cystic kidney epithelial cells and cholangiocytes (11–14). Activation of any of five known somatostatin receptors (SSTRs) inhibits cAMP production, whereas inhibition of cell proliferation occurs predominantly via SSTR2 and SSTR5 and, in some cases, SSTR1 and SSTR3.
Kidney tubular epithelial cells express all five receptors. SSTR1 and SSTR2 are expressed in the thick ascending limb of Henle, distal tubule, and collecting duct, and SSTR3, SSTR4, and SSTR5 are expressed in proximal tubules (15–17). All five are present in cholangiocytes lining bile duct in healthy individuals and are differentially expressed in cholangiocytes lining liver cysts in patients with PLD (18). SSTR1 and SSTR2 levels were decreased in cystic cholangiocytes, whereas SSTR3 and SSTR5 levels did not change compared with controls. Pasireotide more effectively decreased cAMP levels in cholangiocytes than octreotide and more effectively reduced proliferation of cystic cholangiocytes and kidney epithelial cells and decreased hepatic and kidney cystic and fibrotic scores in rodents with PLD/polycystic kidney disease (PKD) (19).
Preclinical studies and randomized clinical trials have confirmed the role of somatostatin analogs (which reduce cAMP production by binding to multiple SSTRs in arresting the progressive liver enlargement seen in individuals, especially women, with severe PLD) (18,20–25). Two long-term studies confirmed durability of the positive effects of somatostatin analogs (26,27). PLD is currently a listed indication for use of octreotide long-acting release (LAR) treatment in Micromedex (26,27). There is also continued interest in studying all available somatostatin analogs, as they are the first effective medical therapy for PLD (and are a possible treatment in PKD) with their emergence as a treatment option (especially in young individuals with symptomatic PLD). They are currently being used for this indication in the United States, Japan, Italy, France, Belgium, The Netherlands, and the United Kingdom.
Pasireotide is available as an LAR intramuscular formulation. In order to test our hypothesis that pasireotide LAR, a second-generation somatostatin analog with its broader binding profile and higher affinity to known SSTRs, had potential for greater efficacy in symptomatic PLD, we performed a randomized, double-blind, phase 2 study in patients with ADPKD and ADPLD.
Materials and Methods
Study Design
The Mayo Clinic Institutional Review Board approved this study, which was conducted in adherence to the Declaration of Helsinki, Mayo Clinic institutional policies, and regulations for protection of human subjects. Trial rationale, design, eligibility criteria, and implementation (NCT01670110) are described in a prespecified protocol (Supplemental Material).
Study Population
Eligible patients were individuals with severe PLD (TLV>4000 ml or symptomatic disease) associated with ADPKD or isolated ADPLD and not a candidate for, or were, declining surgical intervention. Severe PLD was defined as a liver volume >4000 ml or symptomatic disease due to mass effects from hepatic cysts. Patients had to be willing to travel to the Mayo Clinic. All women of child-bearing age had a pregnancy test at enrollment, and all patients were required to use contraception or were postmenopausal. Recruitment started on November 13, 2012, with the last subject enrolling on August 3, 2015. All patients completed year 1 on July 12, 2016. All authors had access to the study data and reviewed and approved the final manuscript.
Intervention
This trial consisted of a 1-year, randomized, placebo-controlled, double-blinded study of pasireotide LAR intramuscular injection with a 2:1 randomization of all participants versus placebo injection stratified on ADPKD/ADPLD status. All patients were seen at the Mayo Clinic and evaluated by the principal investigator (M.C.H.) or coinvestigator (V.E.T.) every 6 months. Evaluations included a physical examination, vital signs, and clinical laboratory parameters (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, bilirubin, electrolytes, BUN, creatinine, fasting glucose, hemoglobin A1C, complete blood count, activated partial thromboplastin time, prothrombin time, and electrocardiogram). Magnetic resonance imaging (MRI) of liver and kidneys (without gadolinium) was obtained at the beginning of the study and at the end of the first year. Monthly injection visits and follow-up monitoring were performed by a study coordinator. An unblinded nurse mixed the injection and administered therapy, where the identities of the treatments were concealed by the use of study treatments (stored at 4°C in the Mayo Clinic Research Pharmacy) that were all identical in packaging, labeling, schedule of administration, and appearance after reconstitution. Patients were treated with pasireotide LAR 60 mg intramuscularly once every 28 days or placebo. Each cycle was 28±2 days. There was no dose titration above the 60-mg dose, but dose reduction to 20 mg was permitted if patients did not tolerate 60 or 40 mg. The placebo dose was monitored and adjusted monthly in a similar fashion. If drug side effects were identified during a phone or study visit, a decision was made by the study team whether to reduce the next injection dose by 20 mg, hold, or discontinue therapy. Therapy and randomization arm were administered by the Mayo Clinic Research Pharmacy. All injections were administered at the Mayo Clinic. Novartis US supplied pasireotide LAR and funded the study.
Outcomes
Primary outcome was the absolute and percent change in TLV (assessed by MRI at baseline and 12-month follow-up). Patients were classified as having “slow progression” if TLV decreased, did not change, or increased by <2% over the study period. Otherwise, the patient was classified as having “rapid progression.” Secondary outcomes included absolute and percent change in TKV, GFR, and other laboratory parameters, including vital signs, creatinine, liver function tests, APTT, PT, and glucose values, from baseline to 12 months. Acquisition of MRIs was performed using the protocol as described in a previous clinical trial (21,24). Total liver volume and total kidney volume measurements were performed blindly (M.E.) in the Imaging Core of the Mayo PKD Translational Center as previously described (24). Comparability of volumetric measurements from MRI and computed tomography and low interobserver variability of total liver volume and total kidney volume measurements have been previously established (24). Data from patients with ADPLD or patients after transplant were excluded from kidney outcome subanalyses.
Quality of life outcomes as well as safety and toxicity were also investigated. Changes in quality of life at baseline and 12 months were measured by the SF36 QOL questionnaire and a gastrointestinal questionnaire.
Safety was ascertained by reported adverse events, vital signs, and clinical laboratory tests that were all evaluated at the same time points. Side effects were classified according to the Common Terminology Criteria for Adverse Events and Common Toxicity Criteria (version 3). The study coordinator recorded monthly drug and dose administration.
Power Statement
A sample size of 16 patients on placebo and 32 patients on pasireotide LAR was determined in order to achieve >80% power (α=0.05, two sided) to detect a 7% point difference on the basis of an estimated growth rate of 5%±3% in liver volume per year in untreated patients and after allowing for estimated dropouts (one patient on placebo and two patients on pasireotide LAR). Stratified randomization by clinical diagnosis was performed by the study statistician using software to generate random numbers from the uniform distribution using a block size of three. The randomization list was then provided to pharmacy.
Statistical Analyses
Clinical and laboratory characteristics, total liver volume, total kidney volume, adverse events, and survey measures at baseline and 12 months were summarized as mean (SD) for continuous variables and n (%) for categorical variables. P values for comparisons between treatment groups were calculated using the equal variance t test for continuous measures and the chi-squared test or Fisher exact test for categorical measures. P values for comparisons within treatment groups were calculated using the paired t test or the McNemar test. Changes in total liver volume and total kidney volume were assessed using height-adjusted annualized absolute and percent change. Additional analysis of covariance analysis was also carried out on the liver and kidney volume measures, with baseline volumes included as covariates. Change in clinical and laboratory characteristics were assessed using percent change.
This study was carried out using a modified intention to treat analysis with all available data. Secondary analyses included comparing the proportion of patients with slow progressions between treatment groups—with patients without MRIs available at 1 year considered to have rapid progression to account for dropouts in the analysis—as well as evaluating quality of life measures among those with slow progression in the treatment group. All calculated P values were two sided, and P=0.05 was considered statistically significant.
Data Safety and Monitoring Board
Two independent medical experts served on the data safety and monitoring board, and analyses were supplied by the statistician at 6-month intervals. Serious adverse events and adverse events were reviewed at these meetings conducted every 6 months.
Results
Baseline Characteristics
Fifty-four patients were assessed for eligibility, and 48 underwent randomization (Figure 1). Of these, 33 patients were randomized to the masked drug (pasireotide LAR), and 15 were randomized to placebo. Of the 48 randomized, four (2%) had prior intervention for kidney disease (cyst drainage, kidney procedure, or kidney transplant), and 12 (25%) had prior intervention for PLD (liver cyst drainage, liver resection, listed for liver transplant, or told they had inoperable PLD).
Figure 1.
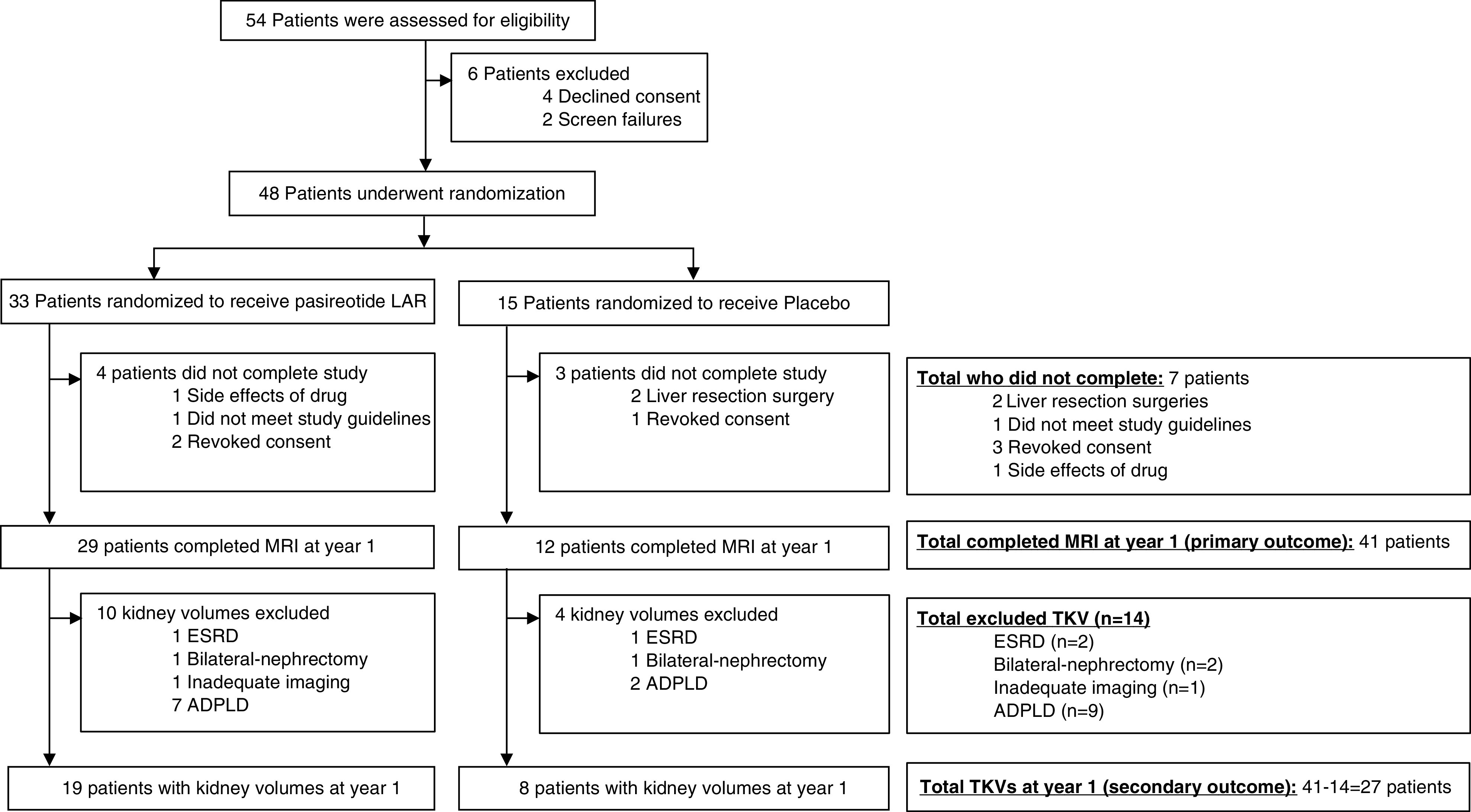
Study flow diagram. ADPLD, autosomal dominant polycystic liver disease; LAR, long-acting release; MRI, magnetic resonance imaging; TKV, total kidney volume.
Among the 33 patients randomized to the pasireotide LAR treatment arm, four patients (12%) did not complete the study (one due to travel time, one due to not meeting study guidelines, and two due to drug side effects and disease progression). Of the 15 participants randomized to the placebo group, three patients (20%) did not complete the study (two patients opted for liver resection, and one did not continue due to the travel burden to the study center). Forty-one individuals completed the 1-year trial and were included in the primary analysis (n=29 on drug and n=12 on placebo). Baseline patient characteristics were similar between the two groups (Table 1). Thirty-two patients had ADPKD, and nine patients had ADPLD. Twenty-two patients had mutations in PKD1, six patients had mutations in PKD2, four patients had a mutation in SEC63, one patient had a PRKCSH mutation, two patients had no mutation detected, and six patients did not have a sample collected. Our goal from randomization was to have patients equally distributed between ADPKD and ADPLD genotypes and phenotypes across pasireotide LAR and placebo groups (Table 1).
Table 1.
Baseline characteristics of participants in a 1-year, randomized, placebo-controlled, double-blinded study of pasireotide long-acting release
| Variable | Placebo, n=15 | Pasireotide Long-Acting Release, n=33 |
|---|---|---|
| Demographics | ||
| Women, n (%) | 12 (80%) | 31 (94%) |
| Age, yr | 51±8 | 50±9 |
| Weight, kg | 76±12 | 72±16 |
| BMI, kg/m2 | 26.1±3.6 | 26.0±5.0 |
| Clinical diagnosis, n (%) | ||
| ADPKD | 13 (87%) | 26 (79%) |
| ADPLD | 2 (13%) | 7 (21%) |
| Genotyping results, n (%) | ||
| PKD1 | 6 (40%) | 19 (58%) |
| PKD2 | 1 (7%) | 5 (15%) |
| SEC63 | 2 (13%) | 2 (6%) |
| PRKCSH | 0 (0%) | 1 (3%) |
| NMD | 1 (7%) | 2 (6%) |
| Unknown | 5 (33%) | 4 (12%) |
| Primary outcome | ||
| Total liver volume, ml/ma | 2387±759 | 2582±1381 |
| Secondary outcomes | ||
| Total kidney volume, ml/ma,b | 397±159 | 534±343 |
| eGFR (CKD-EPI)c | 76±17 | 74±24 |
| Blood glucose, mg/dla | 90±11 | 92±12 |
| Hemoglobin A1C, % | 5.3±0.3 | 5.4±0.5 |
| Urinary albumin-creatinine ratio, mg/g | 4.3±0.2 | 4.4±0.3 |
| Serum creatinine, mg/dl | 1.0±0.3 | 1.0±0.3 |
| Heart rate, beats per minute | 62±10 | 65±12 |
| QT calculated Fridericia, ms | 419±22 | 414±18 |
Unless indicated otherwise, data are mean ± SD or n (%). P values were calculated using the equal variance t test for continuous measures and the chi-squared or Fisher exact test for categorical measures. BMI, body mass index; ADPKD, autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; NMD, no mutation detected; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; QT, interval on EKG.
Height-adjusted volumes were calculated by dividing volume by height (meters) at enrollment.
Total kidney volume: 27 patients with ADPKD completed magnetic resonance imaging and did not have ESKD or kidney transplant by year 1, and so, they had total kidney volume available for analysis.
Fasting glucose obtained at screening visit.
Primary Outcomes: Liver Volumes
Total liver volume in the pasireotide LAR group decreased from 2582±1381 to 2479±1317 ml/m at baseline and 12 months, respectively (annualized absolute change −99±189 ml/m; % change −3%±7%) (Table 2). Total liver volume in the placebo group increased from 2387±759 ml/m at baseline to 2533±770 ml/m at 12 months (annualized absolute change 136±117; % change 6%±7%; P<0.001 between groups for both absolute and percent change); this reduction did not seem to vary by genotype or clinical diagnosis (Figure 2, A and B). Twenty-three of 29 pasireotide LAR–treated patients (79%) were deemed to have slow total liver volume progression during the 12 months of treatment (defined as a reduction, no change, or change of <2% in total liver volume from baseline to 12 months) compared with two of 12 patients with slow progression (17%) in the placebo group (Figure 2C).
Table 2.
Primary and secondary outcomes among 41 participants who completed the study
| Outcome | Pasireotide Long-Acting Release, n=29 | Placebo, n=12 | P Value |
|---|---|---|---|
| Primary outcome | |||
| Total liver volume, ml/ma | — | — | <0.001e |
| Baseline | 2582±1381 | 2387±759 | — |
| 12 mo | 2479±1317 | 2533±770 | — |
| Absolute changeb | −99±189 | 136±117 | <0.001e |
| Percent changec | −3±7 | 6±7 | <0.001e |
| Secondary outcome | |||
| Total kidney volume, ml/ma,d | — | — | 0.02e |
| Baseline | 534±343 | 397±159 | — |
| 12 mo | 523±325 | 417±177 | — |
| Absolute changeb | −11±34 | 21±21 | 0.02e |
| Percent changec | −1±4 | 4±5 | 0.003e |
Additional analysis of covariance (ANCOVA) on the liver volume measure was performed with baseline liver volume included as a covariate and found that the estimated treatment effect was still statistically significant (after adjusting for height-adjusted liver volume at baseline, the estimated change in height-adjusted liver volume at 1 year is estimated to be −239 ml/m in patients on pasireotide long-acting release [LAR] compared with patients on placebo; P<0.001). Results were also similar after performing an ANCOVA on the kidney volume measure (estimated difference: −26 ml/m in patients on pasireotide LAR compared with patients on placebo; P=0.03). Mean ± SD. P values were calculated using the equal variance t test. —, not significant.
Height-adjusted volumes were calculated by dividing volume by height (meters) at enrollment.
Annualized absolute change was calculated for liver and kidney volumes using the equation =(X month value – baseline value) ×12/X month.
Annualized percent change was calculated for liver and kidney volumes using the equation =[(X month value – baseline value)/baseline value] ×100×12/X month.
Twenty-seven patients with autosomal dominant polycystic kidney disease completed magnetic resonance imaging and did not have ESKD or kidney transplant by year 1, and so, they had total kidney volume available for analysis.
Denotes significance at the 0.05 α-level.
Figure 2.

Liver volume changes over time. (A) Height-adjusted liver volumes from 0 to 12 months by study group and gene mutation. (B) Annualized percent change in liver volumes by study group and clinical diagnosis. ADPLD is denoted by circles, and autosomal dominant polycystic kidney disease (ADPKD) is denoted by triangles. Plus signs represent means. P values were derived from the equal variance t test comparing annualized percent change in liver volume in patients on pasireotide LAR versus patients on placebo. (C) Annualized percent change in liver volume by study group.
Secondary Outcomes: Kidney Volumes
Fourteen individuals were excluded from kidney volume analysis (two who previously developed ESKD and underwent kidney transplants, two with bilateral nephrectomies, one with inadequate imaging, and nine with ADPLD) (Figure 1). Total kidney volumes in the pasireotide LAR group were 534±343 and 523±325 ml/m at the baseline and 12-month visits, respectively, whereas total kidney volumes in the placebo group were 397±159 and 417±177 ml/m at baseline and 12-month visits, respectively (Figure 3A, Table 2). Total kidney volume changed by 21±21 ml/m and 4%±5% in the placebo group compared with −11±34 ml/m and −1%±4% in the pasireotide LAR group (P=0.05 for both), and it did not seem to be affected by gene mutation (Figure 3A). Absolute changes in total kidney volume from baseline to 12 months did not seem to be greater in patients with larger kidneys at baseline. Of 19 pasireotide LAR–treated patients, 18 (95%) were deemed as having slow progression, whereas three (38%) of eight individuals in the placebo group were classified as having slow progression during the 12 months of treatment (Figure 3B).
Figure 3.

Kidney volume changes over time. (A) Height-adjusted kidney volumes from 0 to 12 months by study group and mutation. (B) Annualized change in kidney volume by study group.
Secondary Analyses
As a secondary analysis, we evaluated total liver volume and total kidney volume among those with slow progression between treatment groups, considering withdrawals as treatment failures to account for dropouts; results were similar as for the main analysis. For total liver volume, 23 (70%) of 33 pasireotide LAR–treated patients had slow progression, whereas only two (13%) of 15 in the placebo group progressed slowly; for total kidney volume, 18 (72%) of pasireotide LAR–treated patients had slow progression compared with three (23%) patients on placebo (P=0.003 and P=0.004, respectively). We assessed their disease severity using the Mayo ADPKD Imaging Classification only for participants with ADPKD (n=27); pasireotide group (n=19): Class 1A (n=5); Class 1B (n=9); Class 1C (n=4); Class 1E (n=1) and similarly milder kidney disease in the placebo group (n=8): as reflected by few severe cases with Class 1A (n=2); Class 1B (n=5); Class 1C (n=1) using this classification.
Kidney Function.
eGFRs using the Chronic Kidney Disease Epidemiology Collaboration equation were 74±24 and 73±22 ml/min per 1.73 m2 at the baseline and 12-month visits, respectively, in the pasireotide LAR group and 74±18 and 73±22 ml/min per 1.73 m2 at baseline and 12 months, respectively, in the placebo group (Table 3). eGFR changed by −0.3%±14% in the pasireotide LAR group compared with −2%±18% in the placebo group (P=0.79 between groups) (Supplemental Figure 1). Similarly, there was no significant difference observed in the percent change in serum creatinine between the pasireotide LAR (2%±12%) and the placebo (3%±16%) groups (P=0.79) (Table 3).
Table 3.
Changes in clinical and laboratory data among 41 participants who completed the study
| Secondary Outcomes | Pasireotide Long-Acting Release, n=29 | Placebo, n=12 | P Value for Percent Change between Groups | ||||
|---|---|---|---|---|---|---|---|
| Baseline | 12 mo | Percent Changea | Baseline | 12 mo | Percent Changea | ||
| Blood glucose, mg/dlb | 93±12 | 127±27 | 39±30 | 91±13 | 92±11 | 2±15 | <0.001d |
| Hemoglobin A1C, %b | 5.5±0.5 | 6.4±0.6 | 18±11 | 5.29±0.28 | 5.4±0.3 | 2±3 | <0.001d |
| Serum creatinine, mg/dlb | 1.0±0.3 | 1.0±0.3 | 2±12 | 1.00±0.28 | 1.1±0.5 | 3±16 | 0.79 |
| eGFR (CKD-EPI), ml/min per 1.73 m2b | 74±24 | 73±22 | −0.3±14 | 74±18 | 73±22 | −2±18 | 0.79 |
| Albumin-creatinine ratio, mg/gb | 4.4±0.2 | 4.3±0.3 | −1±6 | 4.34±0.21 | 4.4±0.2 | 1±3 | 0.23 |
| Heart rate, BPMc | 65±13 | 61±16 | −0.2±0.2 | 62.8±11.1 | 58±11 | −0.1±0.1 | 0.73 |
| QT calculated, msc | 416±18 | 417±24 | 0.0±0.1 | 416±17 | 418±19 | 0.0±0 | 0.97 |
Unless indicated otherwise, data are mean ± SD or n (%). P values were calculated using the equal variance t test for continuous measures and the chi-squared or Fisher exact test for categorical measures. CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; BPM, beats per minute; QT, time from the beginning of the QRS complex, representing ventricular depolarization, to the end of the T wave.
Percent change was calculated for clinical and laboratory parameters using the equation =[(X month value – baseline value)/baseline value] ×100.
Laboratory measurements: one patient was missing laboratory measurements at 12 mo and so, was not included in the analysis. Fourteen patients had their serum creatinine and eGFR measures excluded due to autosomal dominant polycystic liver disease genotype, ESKD, or kidney transplant.
EKG measurements: QT calculated (Fridericia).
Denotes significance at the 0.05 α-level.
Quality of Life.
No quality of life subdomains changed significantly over the course of the study, either within or between treatment groups (Supplemental Table 1). Results were similar after subsetting to patients with liver disease and patients with kidney disease with slow progression in the treatment group (results not shown). Within the abdominal symptom severity subdomains, shortness of breath was found to be improved among patients receiving pasireotide from baseline to 12 months (P=0.05) (Supplemental Table 2).
Tolerability.
Most adverse events were grades 1 and 2 in severity (88%). The most common adverse events included hyperglycemia: 26 of 33 (79%) in the pasireotide LAR group versus four of 15 (27%) in the placebo group (P<0.001) (Table 4). Plasma glucose levels increased 39% from baseline to 12 months in patients on pasireotide LAR compared with a 2% increase in patients on placebo (P<0.001) (Table 3). One patient had diabetes at baseline. Among the 47 without diabetes at baseline, there were 19 of 32 (59%) in the pasireotide LAR group versus one of 15 (7%) in the placebo group who developed diabetes (P<0.001) (Table 4). The one patient in the pasireotide LAR group with prediabetes at baseline later progressed to diabetes. The one individual in the placebo group with prediabetes at baseline did not develop diabetes at 1 year. Diarrhea occurred in 17 of 33 (52%) patients on pasireotide LAR and eight of 15 (53%) patients on placebo (P=0.91) (Table 4). Dosing was adjusted on the basis of reports of side effects at monthly injection visits (Figure 4).
Table 4.
Adverse events
| Adverse Event Type | Placebo, n=15 | Pasireotide Long-Acting Release, n=33 | P Value |
|---|---|---|---|
| Most frequently occurring adverse events | |||
| Hyperglycemia | 4 (27%) | 26 (79%) | <0.001b |
| Diarrhea | 8 (53%) | 17 (52%) | 0.91 |
| Dizziness | 3 (20%) | 10 (30%) | 0.46 |
| Pain abdomen | 1 (7%) | 11 (33%) | 0.05b |
| Fatigue | 3 (20%) | 13 (39%) | 0.19 |
| Diabetesa | 1/15 (7%) | 19/32 (59%) | 0.001b |
| Nausea | 3 (20%) | 9 (27%) | 0.59 |
| Bradycardia | 1 (7%) | 10 (30%) | 0.07 |
| Alopecia | 0 (0%) | 9 (27%) | 0.03b |
| Headache | 3 (20%) | 5 (15%) | 0.68 |
| Serious adverse event | 2 (13%) | 4 (12%) | 0.91 |
P values were derived using the chi-squared test.
We used standard definitions for diagnosis of diabetes: a fasting plasma glucose (no caloric intake ×8 hours) ≥126 on two occasions; hemoglobin A1C >6.5; or in a patient with classic symptoms of hyperglycemia or hyperglycemic crisis, a random plasma glucose ≥200 mg/dl were equally appropriate for diagnostic testing. The numbers above are on the basis of the 47 patients without diabetes at baseline.
Denotes significance at the 0.05 α-level.
Figure 4.
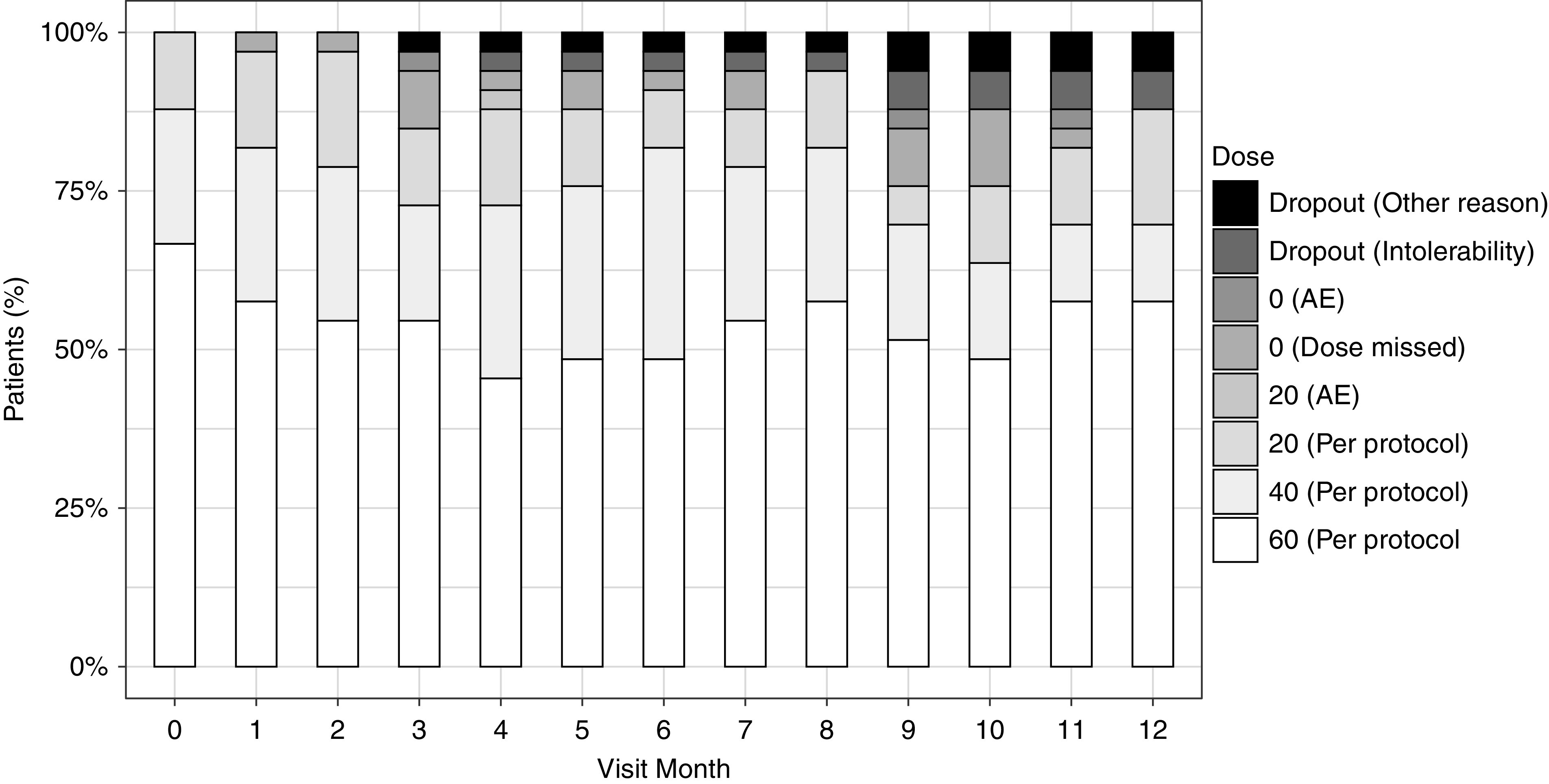
Percentage of patients on pasireotide LAR on each dose level by visit month among 33 randomized patients on pasireotide LAR. AE, adverse event.
Safety
Over the 12-month study period, one patient receiving pasireotide LAR was hospitalized with a ruptured liver cyst and elevated alkaline phosphatase felt not to be related to the study drug. Another individual on pasireotide LAR developed a prolonged QTc >480 ms, and pasireotide LAR was temporarily withheld; she did not require hospitalization. Another individual on pasireotide LAR developed severe right upper quadrant pain felt not to be due to pasireotide LAR but to her severe PLD; the patient later discontinued the study due to inability to travel to the study site. She did not require hospitalization. Another pasireotide LAR recipient developed abdominal pain and ascites and had the treatment temporarily withheld. Two were hospitalized in the placebo group, one for abdominal hernia repair and a second who died following postoperative surgical complications after undergoing liver resection at another medical center. No significant fluctuations in cyclosporin or tacrolimus levels were observed in kidney transplant recipients receiving both medications. QT lengthening was not different between groups.
Discussion
The primary results of our study show that in patients with severe PLD, pasireotide LAR reduced total liver volume and total kidney volume compared with patients who received placebo. We also observed that 25 of 41 (61%) participants (with respect to total liver volume) and 21 of 27 (78%) participants (with respect to total kidney volume) had slow progression. Pasireotide-treated individuals experienced high rates of hyperglycemia. In our previous octreotide LAR study over 2 years, similar reductions in liver volume were associated with improvements of quality of life (physical role, bodily pain, and vitality); however, in this study, there was no reported improvement in quality of life, likely due to the high rates of diabetes and hyperglycemia, despite very similar reductions in liver volume (24).
PLD is characterized by the progressive growth of cholangiocyte-derived fluid-field cysts that gradually replace liver tissue as a result of mutations in at least nine causative genes. The kidney and liver disease is linked to mutations in one of several genes: PKD1, PKD2, GANAB, DNAJB11, and ALG9 (28–36). PKD1 mutations are responsible for approximately 85% of clinically detected ADPKD cases. ADPLD is a genetically distinct disease with few or no kidney cysts associated with PRKCSH, SEC63, LRP5, and GANAB mutations (30,37–40). Mutations in PRKCSH and SEC63 account for 35% of clinically diagnosed cases. In addition, LRP5, ALG8, SEC61B, and PKHD1 are responsible for hepatic cystogenesis in ADPLD (41). Substantial experimental evidence suggests that hepatic cystogenesis is associated with disturbances in multiple cellular mechanisms. Elevated levels of intracellular cAMP in kidney epithelial cells and in cholangiocytes are considered to be one of the major forces that underlie cyst growth in PLD and PKD. Recent advances in the understanding of cAMP signaling pathway led to development of cAMP-based therapies for these disorders (42). Somatostatin and its synthetic analogs (such as octreotide and pasireotide) have been shown to decrease levels of intracellular cAMP in different cell types by activating SSTRs (18). Octreotide binds to three SSTRs (i.e., SSTR2, SSTR3, and SSTR5), whereas pasireotide had a broader spectrum of affinity binding to SSTR1, SSTR2, SSTR3, and SSTR5 (43). The median inhibitory concentration of pasireotide is much lower, and this somatostatin analog is more stable (43).
Our data from this trial using pasireotide LAR and findings from previous octreotide and lanreotide studies suggest that in individuals with ADPKD/ADPLD, somatostatin analogs have a significant and clinically relevant effect of arresting growth of both liver and kidney cysts. Because of their favorable safety and tolerability profile, even long-term somatostatin analog therapy seems to be a viable option for chronic therapy of patients with ADPKD and symptomatic PLD. Their effects on GFR and total kidney volume are less impressive. In the DIPAK study, which enrolled individuals with ADPKD with eGFR of 30–60 ml/min per 1.73 m2, although total kidney volume and total liver volume growth were lower, GFR decline was not altered in the lanreotide-treated versus placebo group (25,44). The 3-year octreotide LAR ALADIN-2 trial (GFR=15–40 ml/min per 1.73 m2 at enrollment) also reported no difference in slope of GFR decline when compared with placebo. In the ALADIN-2 trial, of 63 participants, three on octreotide LAR versus eight on placebo progressed to ESKD (P=0.04) (45). In our study, we also did not detect any significant effect on arresting GFR decline (indeed, most ADPKD enrollees had mild kidney disease on the basis of their Mayo imaging classification); however, because our study was only powered to detect significant differences in the primary outcome of liver volume, it is possible that the size of our remaining cohort after removing those with ADPLD genotype, ESKD, or kidney transplant for assessment of our secondary kidney outcomes was too small to be able to detect statistically significant differences between treatment groups. This could also pose an issue with our assessment of quality of life outcomes. Since the implementation of this clinical trial, a validated PLD-specific quality of life tool has become available (46). Although pasireotide LAR was effective in arresting organ growth in both kidney and liver, treated individuals experienced more hyperglycemia and diabetes compared with patients on placebo. It is a potent inhibitor of insulin secretion due to its broader SSTR binding profile and because SSTRs are also expressed on other tissues, including pancreatic islet cells. Because it has high binding affinity for SSTR1 to -3 and SSTR5, its effects on glucose metabolism are mediated through effects on both insulin-producing β-cells and the glucagon-producing α-cells (47). Despite medical and dietary interventions, these adverse effects were considered by some participants receiving pasireotide LAR as too untoward to continue therapy, leading to a higher withdrawal rate than seen in prior somatostatin trials. We favor continued use of first-generation somatostatin analogs octreotide LAR and lanreotide LAR in selected patients with symptomatic PLD.
Disclosures
M.C. Hogan is an investigator who receives funding from Novartis US for studies of somatostatin analogs in polycystic kidney and liver disease and an investigator of an Otsuka-funded study of tolvaptan in polycystic kidney disease. N.F. LaRusso and T.V. Masyuk have filed a patent for use of somatostatin analogs in polycystic liver disease. P.C. Harris reports receiving grants from Otsuka Pharmaceuticals and other from Amgen, Inc.; Bayer AG; EMD Millipore Corporation (also known as EMD., Merck KGaA); Genzyme Corporation; GlaxoSmithKline LLC (GSK); Mitobridge, Inc.; Otsuka Pharmaceuticals; Regulus; and Vertex Pharmaceuticals, outside the submitted work. M.C. Hogan reports receiving grants from Bristol Myers Squibb, Palladio Biosciences, and Regulus, outside the submitted work. W.K. Kremers reports receiving grants from AstraZeneca, Biogen, and Roche, outside the submitted work. N.F. LaRusso reports receiving National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant DK24031, outside the submitted work. V.E. Torres reports receiving grants from Acceleron Pharma Inc., grants from Blueprint Medicines, grants and other from Mironid, grants and other from Otsuka Pharmaceuticals, grants and other from Palladio Biosciences, other from Reata, and other from Sanofi Genzyme, outside the submitted work. All remaining authors have nothing to disclose.
Funding
This publication was made possible by Mayo Translational Polycystic Kidney Disease Center grant DK090728; the Pirnie Family Foundation; National Center for Research Resources grant UL1 RR024150, a component of the National Institutes of Health; a National Institutes of Health Roadmap for Medical Research award (to M.C. Hogan); the Mayo Foundation for Medical Education and Research; resources in the Mayo Center for Translational Science Activities Center; and Novartis Pharma grant CSOM230XUS30T (to M.C. Hogan). This project was also supported by National Center for Advancing Translational Sciences grant UL1 TR002377.
Supplementary Material
Acknowledgments
We are indebted to Data Safety and Monitoring members (Dr. Gary L. Schwartz and Dr. Patrick S. Kamath), the patients who participated in the study, their families and physicians, and the Mayo Clinic Department of Internal Medicine Clinical Trials Unit that provided assistance with study implementation.
The contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Center for Research Resources or the National Institutes of Health.
Data Sharing Statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymized data may be granted following review. The study protocol is available in Supplemental Material.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.13661119/-/DCSupplemental.
Supplemental Material. Methods and references.
Supplemental Figure 1. Percent change in eGFR by study group.
Supplemental Table 1. SF-36 scores by study group.
Supplemental Table 2. Abdominal symptom severity scores by study group.
References
- 1.Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF Jr., Wetzel LH, Kenney PJ, Brummer ME, Bennett WM, Klahr S, Meyers CM, Zhang X, Thompson PA, Miller JP; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) : Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: The consortium for radiologic imaging studies of polycystic kidney disease cohort. Clin J Am Soc Nephrol 1: 64–69, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Hogan MC, Abebe K, Torres VE, Chapman AB, Bae KT, Tao C, Sun H, Perrone RD, Steinman TI, Braun W, Winklhofer FT, Miskulin DC, Rahbari-Oskoui F, Brosnahan G, Masoumi A, Karpov IO, Spillane S, Flessner M, Moore CG, Schrier RW: Liver involvement in early autosomal-dominant polycystic kidney disease. Clin. Gastroenterol. Hepatol 13[1]: 155–64.e6, 2015. 10.1016/j.cgh.2014.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lantinga MA, de Sévaux RGL, Gevers TJG, Oyen WJG, de Fijter JW, Soonawala D, Zietse R, Salih M, Casteleijn NF, Spithoven EM, Meijer E, Gansevoort RT, Drenth JPH; On Behalf Of The Dipak Consortium : Clinical predictors of escalating care in hepatic and renal cyst infection in autosomal dominant polycystic kidney and liver disease. Neth J Med 76: 226–234, 2018. [PubMed] [Google Scholar]
- 4.Neijenhuis MK, Kievit W, Verheesen SM, D’Agnolo HM, Gevers TJ, Drenth JP: Impact of liver volume on polycystic liver disease-related symptoms and quality of life. United European Gastroenterol J 6: 81–88, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryu H, Kim H, Park HC, Kim H, Cho EJ, Lee KB, Chung W, Oh KH, Cho JY, Hwang YH, Ahn C: Total kidney and liver volume is a major risk factor for malnutrition in ambulatory patients with autosomal dominant polycystic kidney disease. BMC Nephrol 18: 22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Judge PK, Harper CHS, Storey BC, Haynes R, Wilcock MJ, Staplin N, Goldacre R, Baigent C, Collier J, Goldacre M, Landray MJ, Winearls CG, Herrington WG: Biliary tract and liver complications in polycystic kidney disease. J Am Soc Nephrol 28: 2738–2748, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin WP, Vaughan LE, Yoshida K, Takahashi N, Edwards ME, Metzger A, Senum SR, Masyuk TV, LaRusso NF, Griffin MD, El-Zoghby Z, Harris PC, Kremers WK, Nagorney DM, Kamath PS, Torres VE, Hogan MC: Bacterial cholangitis in autosomal dominant polycystic kidney and liver disease. Mayo Clin Proc Innov Qual Outcomes 3: 149–159, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coquillard C, Berger J, Daily M, Shah M, Mei X, Marti F, Gedaly R: Combined liver-kidney transplantation for polycystic liver and kidney disease: Analysis from the united network for organ sharing dataset. Liver Int 36: 1018–1025, 2016. [DOI] [PubMed] [Google Scholar]
- 9.Chandok N, Uhanova J, Marotta P: Clinical outcomes of liver transplantation for polycystic liver disease: A single center experience. Ann Hepatol 9: 278–281, 2010. [PubMed] [Google Scholar]
- 10.Saidi RF, Jabbour N, Shah SA, Li Y, Bozorgzadeh A: Improving outcomes of liver transplantation for polycystic disease in MELD era. Int J Organ Transplant Med 4: 27–29, 2013. [PMC free article] [PubMed] [Google Scholar]
- 11.Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, Banales JM: Polycystic liver diseases: Advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol 11: 750–761, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masyuk TV, Masyuk AI, LaRusso NF: Therapeutic targets in polycystic liver disease. Curr Drug Targets 18: 950–957, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larusso NF, Masyuk TV, Hogan MC: Polycystic liver disease: The benefits of targeting cAMP. Clin Gastroenterol Hepatol 14: 1031–1034, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF: The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology 49: 160–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates CM, Kegg H, Grady S: Expression of somatostatin in the adult and developing mouse kidney. Kidney Int 66: 1785–1793, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Balster DA, O’Dorisio MS, Summers MA, Turman MA: Segmental expression of somatostatin receptor subtypes sst(1) and sst(2) in tubules and glomeruli of human kidney. Am J Physiol Renal Physiol 280: F457–F465, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Reubi JC, Horisberger U, Studer UE, Waser B, Laissue JA: Human kidney as target for somatostatin: High affinity receptors in tubules and vasa recta. J Clin Endocrinol Metab 77: 1323–1328, 1993. [DOI] [PubMed] [Google Scholar]
- 18.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF: Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology 132: 1104–1116, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, Masyuk AI, Hogan MC, Torres VE, Larusso NF: Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology 58: 409–421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MGH, Hoffmann AL, Dekker HM, de Man RA, Drenth JPH: Lanreotide reduces the volume of polycystic liver: A randomized, double-blind, placebo-controlled trial. Gastroenterology 137: 1661-1668.e1-2, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR 3rd, Rossetti S, Harris PC, LaRusso NF, Torres VE: Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 21: 1052–1061, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P: Reducing polycystic liver volume in ADPKD: Effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol 5: 783–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gevers TJ, Drenth JP: Somatostatin analogues for treatment of polycystic liver disease. Curr Opin Gastroenterol 27: 294–300, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Hogan MC, Masyuk TV, Page L, Holmes DR 3rd, Li X, Bergstralh EJ, Irazabal MV, Kim B, King BF, Glockner JF, Larusso NF, Torres VE: Somatostatin analog therapy for severe polycystic liver disease: Results after 2 years. Nephrol Dial Transplant 27: 3532–3539, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Aerts RMM, Kievit W, D’Agnolo HMA, Blijdorp CJ, Casteleijn NF, Dekker SEI, de Fijter JW, van Gastel M, Gevers TJ, van de Laarschot LFM, Lantinga MA, Losekoot M, Meijer E, Messchendorp AL, Neijenhuis MK, Pena MJ, Peters DJM, Salih M, Soonawala D, Spithoven EM, Visser FW, Wetzels JF, Zietse R, Gansevoort RT, Drenth JPH; DIPAK-1 Investigators : Lanreotide reduces liver growth in patients with autosomal dominant polycystic liver and kidney disease. Gastroenterology 157: 481–491.e7, 2019. [DOI] [PubMed] [Google Scholar]
- 26.Hogan MC, Masyuk T, Bergstralh E, Li B, Kremers WK, Vaughan LE, Ihrke A, Severson AL, Irazabal MV, Glockner J, LaRusso NF, Torres VE: Efficacy of 4 years of octreotide long-acting release therapy in patients with severe polycystic liver disease. Mayo Clin Proc 90: 1030–1037, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pisani A, Sabbatini M, Imbriaco M, Riccio E, Rubis N, Prinster A, Perna A, Liuzzi R, Spinelli L, Santangelo M, Remuzzi G, Ruggenenti P; ALADIN Study Group : Long-term effects of octreotide on liver volume in patients with polycystic kidney and liver disease. Clin Gastroenterol Hepatol 14: 1022–1030.e4, 2016 [DOI] [PubMed] [Google Scholar]
- 28.The European Polycystic Kidney Disease Consortium : The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium [published correction appears in Cell 78: 725, 1994]. Cell 77: 881–894, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, Gamble V, Harris PC: The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151–160, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR, Banks CJ, Baheti S, Reddy B, Herrero JI, Bañales JM, Hogan MC, Tasic V, Watnick TJ, Chapman AB, Vigneau C, Lavainne F, Audrézet MP, Ferec C, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet 98: 1193–1207, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S: PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339–1342, 1996. [DOI] [PubMed] [Google Scholar]
- 32.Reynolds DM, Falk CT, Li A, King BF, Kamath PS, Huston J 3rd, Shub C, Iglesias DM, Martin RS, Pirson Y, Torres VE, Somlo S: Identification of a locus for autosomal dominant polycystic liver disease, on chromosome 19p13.2-13.1. Am J Hum Genet 67: 1598–1604, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, Audrézet MP, Hopp K, Porath B, Shi B, Baheti S, Senum SR, Arroyo J, Madsen CD, Férec C, Joly D, Jouret F, Fikri-Benbrahim O, Charasse C, Coulibaly JM, Yu AS, Khalili K, Pei Y, Somlo S, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinstein M, Schollen E, Matthijs G, Neupert C, Hennet T, Grubenmann CE, Frank CG, Aebi M, Clarke JT, Griffiths A, Seargeant L, Poplawski N: CDG-IL: An infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am J Med Genet A 136: 194–197, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Davis K, Webster D, Smith C, Jackson S, Sinasac D, Seargeant L, Wei XC, Ferreira P, Midgley J, Foster Y, Li X, He M, Al-Hertani W: ALG9-CDG: New clinical case and review of the literature. Mol Genet Metab Rep 13: 55–63, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, Hartzel DN, Mane S, Torres VE, Somlo S, Mirshahi T; Regeneron Genetics Center : ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol 30: 2091–2102, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, King BF, Torres VE, Somlo S: Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet 72: 691–703, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB: Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet 33: 345–347, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, Li A, Cai Y, Kamath PS, King BF, Azurmendi PJ, Tahvanainen P, Kääriäinen H, Höckerstedt K, Devuyst O, Pirson Y, Martin RS, Lifton RP, Tahvanainen E, Torres VE, Somlo S: Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet 36: 575–577, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Cnossen WR, te Morsche RH, Hoischen A, Gilissen C, Chrispijn M, Venselaar H, Mehdi S, Bergmann C, Veltman JA, Drenth JP: Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc Natl Acad Sci U S A 111: 5343–5348, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, Gallagher AR, Huang EB, Gulati A, Knight J, Mane S, Tahvanainen E, Tahvanainen P, Sanna-Cherchi S, Lifton RP, Watnick T, Pei YP, Torres VE, Somlo S: Isolated polycystic liver disease genes define effectors of polycystin-1 function. [published correction appears in J Clin Invest 127: 1772–1785, 2017]. J Clin Invest 127: 3558, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torres VE, Harris PC: Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol 25: 18–32, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novartis: SOM230 Investigator Brochure. Edition 9, October 24, 2008
- 44.Meijer E, Visser FW, van Aerts RMM, Blijdorp CJ, Casteleijn NF, D’Agnolo HMA, Dekker SEI, Drenth JPH, de Fijter JW, van Gastel MDA, Gevers TJ, Lantinga MA, Losekoot M, Messchendorp AL, Neijenhuis MK, Pena MJ, Peters DJM, Salih M, Soonawala D, Spithoven EM, Wetzels JF, Zietse R, Gansevoort RT; DIPAK-1 Investigators : Effect of lanreotide on kidney function in patients with autosomal dominant polycystic kidney disease: The DIPAK 1 randomized clinical trial. JAMA 320: 2010–2019, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perico N, Ruggenenti P, Perna A, Caroli A, Trillini M, Sironi S, Pisani A, Riccio E, Imbriaco M, Dugo M, Morana G, Granata A, Figuera M, Gaspari F, Carrara F, Rubis N, Villa A, Gamba S, Prandini S, Cortinovis M, Remuzzi A, Remuzzi G; ALADIN 2 Study Group : Octreotide-LAR in later-stage autosomal dominant polycystic kidney disease (ALADIN 2): A randomized, double-blind, placebo-controlled, multicenter trial. PLoS Med 16: e1002777, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neijenhuis MK, Gevers TJ, Hogan MC, Kamath PS, Wijnands TF, van den Ouweland RC, Edwards ME, Sloan JA, Kievit W, Drenth JP: Development and validation of a disease-specific questionnaire to assess patient-reported symptoms in polycystic liver disease. Hepatology 64: 151–160, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry RR, Ciaraldi TP, Armstrong D, Burke P, Ligueros-Saylan M, Mudaliar S: Hyperglycemia associated with pasireotide: Results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab 98: 3446–3453, 2013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.